
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe interface microenvironment mediates the emission of a semiconductor nanocluster via surface-dopant-involving direct charge transfer†
Zhiqiang
Wang‡
ab,
Hao
Ma‡
a,
Jiaxu
Zhang‡
ab,
Yingjia
Lan‡
b,
Jia-Xing
Liu
a,
Shang-Fu
Yuan
 a,
Xiao-Ping
Zhou
a,
Xiaohong
Li
b,
Chaochao
Qin
c,
Dong-Sheng
Li
d and
Tao
Wu
*ab
a,
Xiao-Ping
Zhou
a,
Xiaohong
Li
b,
Chaochao
Qin
c,
Dong-Sheng
Li
d and
Tao
Wu
*ab
aCollege of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, China. E-mail: wutao@jnu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, China
cHenan Key Laboratory of Infrared Materials & Spectrum Measures and Applications, School of Physics, Henan Normal University, Xinxiang 453007, China
dCollege of Materials and Chemical Engineering, Hubei Provincial Collaborative Innovation Center for New Energy Microgrid, Key Laboratory of Inorganic Nonmetallic Crystalline and Energy Conversion Materials, China Three Gorges University, Yichang, Hubei 443002, China
First published on 5th September 2023
Abstract
The interface microenvironment of doped quantum dots (QDs) is crucial in optimizing the properties associated with the photogenerated excitons. However, the imprecision of QDs' surface structures and compositions impedes a thorough understanding of the modulation mechanism caused by the complex interface microenvironment, particularly distinguishing the contribution of surface dopants from inner ones. Herein, we investigated interface-mediated emission using a unique model of an atomically precise chalcogenide semiconductor nanocluster containing uniform near-surface Mn2+ dopants. Significantly, we discovered that Mn2+ ions can directly transfer charges with hydrogen-bonding-bound electron-rich alkylamines with matched molecular configurations and electronic structures at the interface. This work provides a new pathway, the use of atomically precise nanoclusters, for analyzing and enhancing the interface-dependent properties of various doped QDs, including chalcogenides and perovskites.
Introduction
Doped quantum dots (QDs) are widely used in solar cells,1 photoelectric sensors,2 and photocatalytic systems.3 This is because of the intriguing optical, electronic, and catalytic properties of these nanostructures.4–6 It has been demonstrated that the intentional doping of QDs creates new midgap/intermediate energy states, which can serve as energy/charge accepting centers and improve the intrinsic performance of the QDs and even result in new functionalities.7–9 However, effectively manipulating the coupling interactions (i.e., external and internal energy/charge transfer among the excitons in the photoexcited hosts, the dopants in the hosts, and the interfacial molecules attached to the hosts) while identifying their respective roles (i.e., donor or acceptor) during these processes remains a challenge.10–12 Early studies mainly focused on internal energy/charge transfer (IET/ICT) from the host to the dopants (or defects).13,14 In the last decade, however, external energy/charge transfer (EET/ECT) between the host and interfacial molecules (IMs) has also been investigated because the interface microenvironment of QDs is essential for tuning the properties associated with the photoexcited excitons.15 Nevertheless, there has been little research on the direct coupling interactions between the dopants and IMs, especially for the dopant-dominated direct ECT. This may be attributed to the following factors: (1) the dopants in QDs are usually randomly distributed within the QDs and/or on their surfaces (Scheme 1a), and a lack of precise information about the structure and location distribution of the dopants prevents an analysis of the above-described processes;16 (2) ECT initiated by a small amount of surface dopants may be masked to a significant degree by EET arising from a large amount of inner dopants; (3) it is difficult to completely prevent surface vacant defects in QDs through surface passivation, making it challenging to study the excited-state dynamics of surface dopants (Scheme 1b); and (4) there exists an ambiguous interface microenvironment for QDs with an uneven surface, which usually obscures the bonding interactions with IMs.17 Therefore, it is necessary to adopt atomically precise homogeneous doping models in order to overcome this research bottleneck. | ||
| Scheme 1 (a) Schematic of a classical QD with randomly distributed dopant/defect sites. (b) Transection of a doped QD showing IET/ICT from the host to the dopant (or defect) and EET/ECT between the host (or dopant/defects at different sites) and IMs. (c) Structure of the atomically precise semiconductor model, namely, supertetrahedral T4-Mn clusters with Mn2+ ions uniformly distributed in the near-surface region (IET, internal energy transfer; ICT, internal charge transfer; EET, external energy transfer; ECT, external charge transfer; IM, interfacial molecule). | ||
In addition, organic amines play a significant role in the synthesis of QDs as well as in determining their applicability.18,19 Neutral organic amines and their derived cations are frequently employed for passivating (or modifying) the crystal surface of metal-chalcogenide- and halide-perovskite-based QDs.20,21 In addition, organic amines are also used in photocatalytic reactions that occur at the surfaces/interfaces of QDs. For example, triethanolamine is used as a hole-sacrificing reagent to ensure high photocatalytic performance during the hydrogen evolution reaction as well as to improve the stability of the catalyst.22 Furthermore, interfacial organic amine cations of halide-perovskite-based QDs can modulate the excited-state dynamics of the host. For example, Yang et al. reconfigured the near-edge state of perovskite QDs using organic amine cations with different degrees of steric hindrance and could effectively enhance the mobility of holes in the host material and improve the stability of the resulting devices.23 Adachi et al. claimed that organic amine cations can quench the triplet energy of perovskites and hence reduce the emission efficiency of the triplet excitons.24 Given these facts, we believe that an in-depth understanding of direct EET/ECT between dopants and interfacial amine molecules (or cations) would be beneficial for further understanding the modulation mechanism caused by the interface microenvironment and optimizing the performance of doped QDs for practical applications.
In this work, atomically precise T4-MnInS molecular nanoclusters ([Mn4In16S35]14−, denoted as T4-Mn in Scheme 1c), consisting of four uniformly distributed near-surface Mn2+ ions and thirty-four surface S2− ions in a supertetrahedron, were employed as a model to study the direct coupling interactions between Mn2+ ions and IMs and specifically the direct ECT process with amine molecules.25,26 To begin with, the photoluminescence (PL) properties of the surfactant-assisted T4-Mn dispersions were explored. This was followed by a study of the direct coupling interactions. The results of PL and 1H nuclear magnetic resonance (NMR) spectroscopies, electrochemical characterizations, and femtosecond transient absorption measurements confirmed the occurrence of direct ECT between the Mn2+ ions and the interfacial neutral organic amines attached to the surfaces of the clusters via S⋯H–N hydrogen bonding. Furthermore, considering that the geometric configuration of amine molecules and their intrinsic electronic structure have a determining effect on their coupling strength, we also modulated the intensity of the Mn2+ emission by changing the interface microenvironment, i.e., the coupling interactions of the clusters with different types of interfacial amines and other small molecules (e.g., formamide and water).
Results and discussion
PL characteristics of T4-Mn crystals and dispersions
T4-Mn cluster-based microcrystals used here were synthesized and subsequently dispersed well in chloroform with the aid of the surfactant of dimethyldioctadecylammonium bromide (DODA·Br) (Fig. S1–S4†). T4-Mn dispersions (denoted as dn, n = 0–6) with different surfactant/cluster molar ratios were prepared to optimize the dispersity of the clusters for the following investigation (see more details in the Experimental section and ESI†). Analyzing the PL characteristics of the clusters attached by the IMs would be an effective approach for probing the coupling interactions between the dopant and IMs.27 To this end, we first determined the PL excitation (PLE) and PL spectra of the T4-Mn crystals and dn dispersions. It can be seen from Fig. 1a that the Mn2+ emission from the T4-Mn crystals is attributable to a dominant indirect excitation pathway (i.e., host-to-Mn2+ IET) and a secondary direct excitation one (i.e., excitation of the Mn2+ ions).28 Moreover, the Mn2+ emission from the T4-Mn crystals was centered at 604 nm for all the excitation wavelengths. The Mn2+ emission of dispersion d0 exhibited a peak at 604 nm, which is the same as that of the T4-Mn crystals. However, the Mn2+ emissions of dispersions d1–d6 were slightly red-shifted compared with that of the T4-Mn crystals (the data on dispersion d4 is shown in Fig. 1b and those of the others are in Fig. S5†); this may be ascribed to the pressure-induced strain experienced by the clusters because of the surrounding DODA+ molecules (Fig. S6†). The PLE spectra of the dispersions were distinct from that of the T4 crystals (Fig. 1b and S5†), where the Mn2+ emission caused by the direct excitation of the Mn2+ ions was to some extent restricted. However, importantly, the direct-excitation properties of the Mn2+ ions in the dispersed clusters remained, similar to those of the clusters in the crystals. This may be caused by the aggregation of Mn2+ ions in the form of [Mn4S] units within the clusters, wherein the spin-coupling Mn⋯Mn interactions make the spin-prohibited d–d transition of the Mn2+ ions possible.29 It is important to mention that this structure allows the Mn2+ ions in the clusters in the dispersion to be directly excited by visible light, thus facilitating the analysis of the direct coupling interactions between the IMs and Mn2+ ions. | ||
| Fig. 1 (a) Steady-state PLE and PL spectra of T4-Mn crystals at various excitation wavelengths. (b) Steady-state PLE and PL spectra (λex = 356 nm) of dispersion d4. (c) PL decay curves of dn dispersions. All decay curves correspond to maximum emission under excitation at 356 nm. (d) Histograms of average decay lifetimes of Mn2+ emission from dn dispersions. Insets in (a) and (b) show optical photographs of T4-Mn crystals and dispersion d4 under ambient and UV light, respectively. A magnified partial PLE spectrum is also shown in inset (b). Inset in (c) shows the PL decay curve of T4-Mn crystals. | ||
To further elucidate the changes in the PL properties of the excited-state Mn2+ ions, the decay curves of the T4-Mn crystals and dispersions were measured at 365 nm (i.e., the host clusters were excited) (Fig. 1c, d, and Table S1†). The T4-Mn crystals and dispersion d0 exhibited single-exponential decay with emission lifetimes of ∼128.0 (inset in Fig. 1c) and ∼121.7 μs (Fig. S7†), respectively. In contrast, the PL decay modes for dispersions d1–d6 were different from those of each other, and their corresponding decay lifetimes were much shorter than those of the large-sized T4-Mn crystals and cluster-based NPs in dispersion d0. The average lifetime of the Mn2+ emission from the surfactant-assisted dispersions initially decreased and then increased slightly with an increase in the amount of DODA·Br added (Fig. 1d). For dispersions d1 and d2, the decay curves of the Mn2+ emission could be fitted using a double-exponential function with a long component (similar to that of the T4-Mn crystals) and a short component (Table S1†). However, for dispersions d3–d6, the decay curves could be fitted using a single-exponential function, and their lifetimes increased slightly with an increase in the amount of DODA·Br used. Note that DODA·Br showed weak luminescence intensity and did not affect the Mn2+ emission measurements (Fig. S8†). Given the TEM results and the fact that the lifetimes of the Mn2+ emission of dispersions d3–d6 gradually increased with an increase in the amount of DODA·Br used, we concluded that the solvent molecules surrounding the clusters in the dispersions were responsible for the reduction in the emission lifetime, as collisions between these clusters and the chloroform molecules would consume the excited-state energy of the clusters and hence lower the PL lifetimes.30 The long lifetime component of dispersion d2 probably originates from the inner parts of the surfactant-encapsulated cluster-based nanoparticles (SENPs) (Fig. S2c†), which undergo negligible interactions with the chloroform molecules. Correspondingly, the outer parts of the SENPs exhibit strong interactions with the interfacial solvent molecules, resulting in the short lifetime component.31 Owing to the single-exponential decay mode of dispersions d3–d6, we conjectured that each T4-Mn cluster in the 2–3 nm-sized SENPs interacts with the interfacial solvent molecules (Fig. S2d and e†). The presence of higher amounts of DODA·Br on the cluster surfaces partially prevents collisions between the clusters and solvent molecules, resulting in a slight increase in the Mn2+ emission decay lifetime. In addition, under the direct excitation of the Mn2+ ions, the Mn2+ emission intensity of dispersion d4 increased with the addition of DODA·Br (Fig. S9†). This further confirmed that chloroform can directly quench the excited-state energy of the Mn2+ ions in the clusters to a certain extent.
Effects of interfacial neutral amine molecules on Mn2+ emission
Considering that the interfacial triplet oxygen (3O2) molecules close to QDs can quench the emission of the QDs,32–34 control experiments were performed to investigate the possible coupling interactions of 3O2 with the clusters (see more details in Fig. S10–S13† and the related description). Based on the observation of near-infrared PL of 1O2, direct energy transfer from excited-state Mn2+ to 3O2 proved to occur in this case. To further probe the coupling interactions between the clusters with organic IMs, PL measurements were performed on dispersion d4 while adding other amine molecules, such as ethylenediamine (EDA) and triethylamine (TEA), to it (Fig. 2). The amine molecules were added in increasing quantities to a certain amount (3 mL) of the dispersion for qualitatively monitoring the variation in the Mn2+ emission with addition of the amines. The Mn2+ emission intensity decreased with the addition of the amines, with the trends for the two amines being different. The intensity of the Mn2+ emission decreased sharply with an increase in the amount of EDA, which has a linear structure, and was completely quenched when 50 μL of EDA was added (Fig. 2a). However, the decrease in the Mn2+ emission intensity was less rapid when the nonlinear-structured tertiary amine TEA was added (Fig. 2b). Fig. S14† shows the Mn2+ emission intensity as a function of the amounts of EDA and TEA added. Finally, Fig. 2c shows the decay dynamics of the Mn2+ emission of d4 with EDA and TEA added. The addition of EDA and TEA decreased the Mn2+ emission lifetime from 24.5 μs to 7.1 μs and 17.9 μs, respectively. Optical photographs of d4 before and after the addition of amines are shown in the inset. These changes in the lifetimes are in line with changes in the Mn2+ emission intensity. Moreover, with the addition of the amines, dispersion d4 exhibited a broad electron paramagnetic resonance signal (originating from the Mn–Mn coupling interactions within the clusters),35 which was similar to that seen in the case of the pristine dispersion d4 (Fig. S15†). This suggested that the amine molecules did not cause the collapse of the clusters.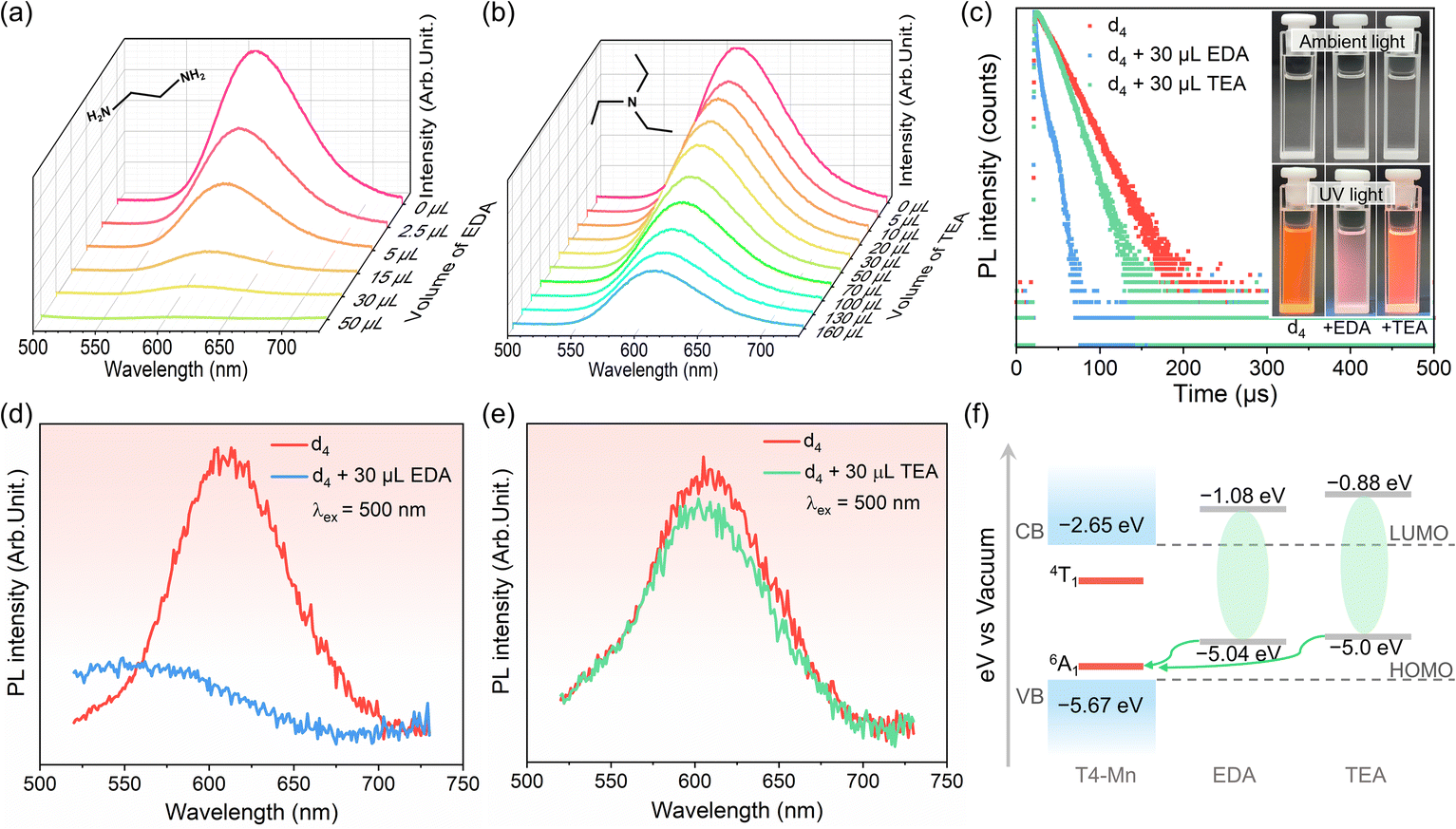 | ||
| Fig. 2 (a and b) PL spectra of dispersion d4 samples (3 mL samples were used) containing EDA and TEA in different amounts, respectively. (c) Decay curves of Mn2+ emission from dispersion d4 before and after addition of 30 μL EDA and TEA (λex = 356 nm and λem = 612 nm); the inset shows the optical images of dispersion d4 samples with and without an amine added (30 μL of EDA and TEA) under ambient light and 365 nm excitation. (d and e) PL spectra of dispersion d4 (λex = 500 nm) with and without addition of 30 μL EDA and TEA, respectively. (f) Qualitative alignment of energy levels of clusters, EDA, and TEA, showing favored electron transfer from the HOMO level of amines to the ground state (6A1) of the Mn2+ ion. | ||
The results described above confirmed the existence of coupling interactions between the T4-Mn clusters and the amine molecules. Mn2+ emission intensity is usually dependent on the host-to-Mn2+ IET efficiency or the newly formed relaxation pathways involving the excited Mn2+ ions.36,37 To further elucidate the interface modulation mechanism of the Mn2+ emission owing to the addition of the amines, PL measurements were performed by directly exciting the Mn2+ ions. It is assumed that, in this situation, Mn2+ emission intensity would be directly related to the population of the excited-state Mn2+ ions and not the host clusters. Notably, under 500 nm excitation, the Mn2+ emission intensity decreased dramatically with the addition of EDA (Fig. 2d) but only slightly with the addition of TEA (Fig. 2e). The changes in the PL spectra were similar to those for the case where the Mn2+ ions were indirectly excited through host-to-Mn2+ IET. Based on this result, it was confirmed that direct coupling interactions had occurred between the Mn2+ ions and organic amines. Furthermore, according to UPS and electrochemical tests, the energy levels (Fig. 2f) of the cluster and amine molecules were determined to match each other for allowing direct electron transfer from EDA/TEA to excited Mn2+ ions (see more details in Fig. S16–S18† and the related description).
Hydrogen bonding between T4-Mn clusters and amine molecules
1H-NMR measurements were performed to elucidate the interface microenvironment, i.e., the interactions between the clusters and interfacial amine molecules. Given the abundant S2− sites on the surfaces of the clusters, it was reasonable to assume that the hydrogen atoms on the N atom of the amines would form links with the surfaces of the clusters via hydrogen bonding.38 This would cause the 1H-NMR characteristics (chemical shift, integration intensity, or coupling splitting) of the H atoms on a given amine to change because of the formation of S⋯H–N hydrogen bonds.39 A series of 1H-NMR control experiments were performed on the amines, dispersion d4, and their mixtures to confirm this (Fig. 3a and b). The 1H-NMR spectrum of dispersion d4 is shown in Fig. S2.† When EDA was added to the dispersion, the peak related to the –NH2 group (2) at 1.17 ppm disappeared, and that related to the –CH2 group (1) was downshifted from 2.75 to 2.73 ppm, with the shape of the peak also changing (Fig. 3a).40 This indicated the formation of hydrogen bonds (N–H⋯S) between the dispersed clusters and EDA molecules. However, no obvious changes were detected in the 1H-NMR signal of the TEA molecules in dispersion d4 (Fig. 3b), suggesting that hydrogen bonds were not formed.41 These results as well as the PL spectra and energy levels indicated that the effect of the amines on the Mn2+ emission intensity may be related to their ability to form hydrogen bonds. The unbranched structure of the EDA molecule allows it to diffuse readily and pass through the surfactant molecules attached to the clusters. Thus, the EDA molecules can come close to the cluster surfaces (Scheme 2a); this is beneficial for forming hydrogen bonds and aids highly efficient ECT between the Mn2+ ions and EDA, thus resulting in the efficient quenching of the Mn2+ emission. In the case of the TEA molecules, the 1H-NMR results indicated that they cannot form tight hydrogen bonds with the clusters (Scheme 2b), which is in keeping with the observation that the Mn2+ emission intensity was not reduced efficiently after the addition of TEA. Thus, it can be concluded that the coupling interactions between the Mn2+ ions and amines are dependent on the structural characteristics of the amine used. To ensure charge balance between the negatively charged clusters and positively charged DODA+ ions, the DODA+ ions should always be close to the clusters, which contained 35 surface S sites, owing to electrostatic interactions. However, the encapsulation phenomenon allows some of the clusters to form hydrogen bonds with the organic amines. It was expected that the coupling interactions between the Mn2+ ions and organic amines can be tuned by selecting an amine with the appropriate molecular structure. | ||
| Fig. 3 (a) 1H-NMR spectra of dispersion d4, EDA, and their mixture. (b) 1H-NMR spectra of d4, TEA, and their mixture. CDCl3 and tetramethylsilane were used as solvent and internal standard, respectively. Numbers in figures represent the H atom of a specific group. Inset in (b) shows magnified chemical peaks of the –CH2 group (1). | ||
 | ||
| Scheme 2 Suggested hydrogen bonds between the cluster and amines. (a) Schematic of a single SENP in dispersion, showing the relative locations of EDA and TEA molecules close to a cluster surface. (b) Strong hydrogen bonding aids ECT between Mn2+ ions and EDA. (c) ECT between Mn2+ ions and TEA is not effective owing to the relatively long distance (no N–H⋯S hydrogen bonds formed). | ||
Transient absorption (TA) spectra
Based on the results discussed above, we assumed that there must exist direct ECT between the near-surface Mn2+ ions in the clusters and the interfacial organic amine molecules. To further elucidate the underlying photophysical processes, ultrafast transient absorption spectroscopy was performed. A pump laser with a wavelength of 365 nm was used to excite the host clusters in d4 samples with and without EDA, and the visible range of 400–650 nm was used for the probe beam. The time window was set to 7.39 ns. While the TA spectra of the surfactant-containing chloroform solution did not contain any positive excited-state-related absorption signals (Fig. S19†), those of dispersion d4 did (Fig. 4a); these were attributable to the clusters in the dispersion. The relaxation of the excited-state absorption can be divided into two phases: a rapid phase and a slow one. The excited-state absorption spectrum decays rapidly initially (within 10 ps). However, in the subsequent stage, the decay rate was much slower, especially for the absorption band around 430 nm (Fig. 4a). Given the spin-forbidden nature of the 4T1 → 6A1 transition of Mn2+,42 it was assumed that the slower relaxation in the second stage was attributable to the excited-state energy level of the Mn2+ ions. Hence, the excited-state absorption relaxation with a longer delay time can be ascribed to the decay of the excited-state Mn2+ ions. A positive excited-state absorption signal was also observed after the addition of EDA (Fig. 4b). However, the rate of decrease of the fast-decay component was smaller, and the relaxation process during the longer time window (of the order of nanoseconds) was a little faster compared with that of the pristine dispersion d4 (i.e., without EDA). This should be ascribed to the ECT between the excited-state Mn2+ ions and EDA, namely, electron transfer from the HOMO of EDA to the 6A1 state of the Mn2+ ions (or hole transfer from the 6A1 state of Mn2+ ions to the HOMO of EDA). Electron transfer from the HOMO level of EDA to the ground-state (6A1) of the Mn2+ ions is likely followed by synchronous electron transfer from the excited-state (4T1) of the Mn2+ ions back to the HOMO level of EDA for restoring the ground state of the amine molecules.43 | ||
| Fig. 4 fs-TA spectra (λex = 365 nm) of dispersion d4 (a) with and (b) without 30 μL EDA for several representative time delays. Decay-associated spectra (DAS) of dispersion d4 (c) with and (d) without 30 μL EDA. (e and f) Schematic illustration of the photophysical mechanism with (e) and without (f) involving ECT between Mn2+ ions and EDA. | ||
Considering the decay kinetics corresponding to the different wavelengths with respect to the excited-state absorption spectrum, a global analysis was performed using Glotaran, a graphical user interface for the R-package TIMP (Vrije Universiteit Amsterdam, Amsterdam, Netherlands). The obtained decay-associated spectra (DAS) are shown in Fig. 4c and d. For dispersion d4, three kinetics components were observed: 0.69 ps (τ1), 15.4 ps (τ2), and >7 ns (τ3). According to the literature, the 0.69 ps component can be ascribed to internal conversion within the host from the high-lying excited state to the low-lying excited state, while the 15.4 ps component is related to a host-to-Mn2+ IET.44–47 The >7 ns component is associated with the 4T1 → 6A1 radiative transition and nonradiative pathways, in keeping with the results described above.48 For the d4 dispersion sample with EDA, four kinetics components were observed: 0.27 ps (τ1), 11.8 ps (τ2), 998 ps (τ3), and >7 ns (τ4). The 0.27 ps and 11.8 ps components were associated with the internal conversion process and the host-to-Mn2+ IET, respectively, as was also the case for the pristine d4 dispersion. On the other hand, the third kinetics component of the pristine d4 dispersion was divided into two components with time constants of 998 ps and >7 ns in the case of the d4 dispersion sample with EDA. The >7 ns component was related to exciton recombination, including radiative and nonradiative Mn2+ ion decay. Thus, based on the results of this study and those reported previously, the 998 ps component can be ascribed to ECT between the Mn2+ ions and EDA.49 The photophysical process is shown schematically in Fig. 4e and f. Essentially, electron transfer from the HOMO of EDA to the 6A1 state of the Mn2+ ions is identical to hole transfer from the 6A1 state of the Mn2+ ions to the HOMO of EDA. For the d4 dispersion sample with TEA, there were no differences in the relaxation stages of the TA spectrum. In addition, a global analysis showed minor differences in the kinetics of the three components, with a weak ECT occurring between the Mn2+ ions and TEA in the case of the third component, which was difficult to dissociate (Fig. S20†).
Control of coupling interactions between Mn2+ ions and IMs
Based on the results described above, it can be concluded that the strength of the coupling interaction between the Mn2+ ions and amine molecules depends on the structure of the amines, as also shown in Scheme 2. Thus, proof-of-concept experiments were performed using several types of amines, including linear ones, such as ethanolamine (EA) and diethylenetriamine (DETA), and branched secondary amines, such as diethylamine (DEA). As expected, when the EA (or DETA) molecules were added into dispersion d4, the Mn2+ emission (including its intensity and decay lifetime) decreased sharply, similar to the case for EDA (Fig. S21a, b, and d†). In the case of DEA, the decrease in the Mn2+ emission was lower and resembled that seen in the case of TEA (Fig. S21c and d†). Furthermore, on directly exciting the Mn2+ ions at 500 nm, a similar decrease in the Mn2+ emission intensity was detected (Fig. S22†). Finally, using optical and electrochemical measurements, direct ECT between the Mn2+ and the above amines was confirmed (Fig. S23–S27†). 1H-NMR analysis also confirmed the formation of hydrogen bonds between the clusters and EA (or DETA) molecules but not with the DEA molecules (Fig. S28–S30†). The organic amines mentioned above are good electron donors. We wished to determine how Mn2+ emission would be affected if the electron-donating ability of the organic amine used were to be reduced. Thus, formamide (FA), which contains an electron-withdrawing carbonyl group, was used to investigate the coupling interactions with the clusters. With the addition of FA, both the Mn2+ emission intensity and decay lifetime increased (Fig. 5a and b). The results of electron paramagnetic resonance measurements confirmed the stability of the T4-Mn clusters after the addition of FA (Fig. S31†). This is the first report to suggest that the Mn2+ emission can be modulated using organic amines to realize PL turn-on behavior without having to modify the host clusters. Moreover, when the Mn2+ ions were excited directly, the intensity of the Mn2+ emission from the clusters increased after the addition of FA (Fig. 5c). To further elucidate the PL brightening mechanism, 1H-NMR measurements were performed. Fig. 5d shows that the two groups of signals observed in the case of d4 shifted with the addition of FA, and the doublet peaks (group 2, 5.8 ppm and 6.24 ppm) combined into a single one (6.36 ppm). The peak at 8.19 ppm shifted downfield to 8.14 ppm.50 Hence, FA also formed hydrogen bonds with the clusters, resulting in an increase in the Mn2+ emission intensity. CV and relevant optical tests were performed to qualitatively analyze the enhancement in the Mn2+ emission intensity (Fig. 5e and S32†). The Förster resonance EET process was excluded, given the lack of spectral overlap. In addition, the oxidation potential of FA as determined from the CV curves is remarkably different from those of the other amines, owing to the poor electron-donating ability of FA. The energy levels of the clusters and FA are shown in Fig. 5f. Apparently, ECT did not occur between the clusters and FA as per the analysis above. Moreover, based on the fact that the excited Mn2+ ions may be de-excited because of the collisions of the clusters with the chloroform and 3O2 molecules, we speculated that the increase in the PL intensity was caused by hydrogen bonding between the clusters and FA molecules, which prevented collisions between the clusters and the solvent molecules to a certain degree. Furthermore, it was found that the molecule of dimethylformamide with a structure similar to that of FA, that is, one in which the two H atoms on the N atom in FA were replaced by two –CH3 groups, did not increase the Mn2+ emission intensity (Fig. S33†). This further confirmed the significance of interfacial hydrogen bonding in controlling the Mn2+ emission. | ||
| Fig. 5 (a) PL spectra of dispersion d4 samples containing different amounts of FA. (b) PL decay lifetimes of d4 before and after addition of 30 μL FA (λex = 356 nm and λem = 612 nm). Inset shows optical images of d4 samples with and without FA under ambient light and 365 nm excitation. (c) PL spectra of d4 samples with and without FA (λex = 500 nm). (d) 1H-NMR spectra of d4, FA, and their mixture formed using CDCl3 as solvent. (e) CV curve of FA. (f) Qualitative energy level alignment of clusters and FA. | ||
Based on the results described above, it can be concluded that the ability to form hydrogen bonds without aiding the ECT process with the clusters is responsible for the “turn-on” behavior of the PL from the Mn2+ ions. From a structural point of view, one of the most stable molecules, namely, H2O, which consists of two H atoms attached to an O atom and is similar to the –NH2 group, should readily form hydrogen bonds with the surface S sites of the clusters and thus enhance the Mn2+ emission intensity. To confirm this, we performed proof-of-concept experiments. Indeed, the Mn2+ emission properties of dispersion d4, including the intensity and decay lifetime, increased with the addition of H2O (Fig. S34†). In addition, the addition of H2O can also recover the quenched Mn2+ emission of d4 by EDA (Fig. S35†), which suggests H2O molecules can strongly repel EDA molecules attached to the clusters.
Conclusions
In this work, the direct coupling interactions (energy and charge transfer processes) between Mn2+ ions and interfacial molecules were investigated systematically using well-defined clusters of a chalcogenide semiconductor, T4-MnInS. The T4-MnInS clusters dispersed well in chloroform using the surfactant DODA·Br exhibited excited-state energy consumption, owing to the collisions between them (and the excited-state Mn2+ ions) and the chloroform molecules. Direct external energy transfer from the excited-state Mn2+ ions to the 3O2 molecules, leading to the formation of 1O2, was also confirmed. More importantly, using steady/transient-state optical measurements for the excited-state Mn2+ ions as well as electrochemical and 1H-NMR analyses, direct external charge transfer between the Mn2+ ions and the interfacial organic amines was also demonstrated. It was found that the coupling strength between the Mn2+ ions and amines is dependent on the structure and electronic configuration of the amine in question. Based on this fact, the coupling interactions between the clusters and organic amines could be tuned. By rationally selecting an organic amine with poor electron-donating ability, the Mn2+ emission intensity could be increased by reducing the amount of energy consumed by the solvent molecules. This, in turn, allowed the PL “turn-on” effect to be realized in the case of other small molecules, such as H2O. It is worth to be mentioned that current work is not easy to explore in the system of traditional Mn2+-doped QDs. This work provides new insights into the interface-microenvironment-mediated coupling interactions (specifically, the direct external charge transfer process) that occur between well-defined, near-surface metal-doped clusters and interfacial molecules, which should help increase the applicability of metal-doped QDs in photophysical and optoelectronic devices.Experimental section
Chemicals
Manganese acetate tetrahydrate (Mn(Ac)2·4H2O, AR, 99%) was purchased from Aladdin Industrial Corporation. Indium powder (In, 99.9%), dimethyldioctadecylammonium bromide (DODA·Br, 98%), and sublimed sulfur (S, 99.9%, powder) were supplied by Meryer Chemical Technology Co. Ltd. 2-(2-Aminoethylamino)ethanol (AEAE, ≥99%, liquid), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 99.0%, liquid), and hexamethyleneimine (98%, liquid) were purchased from Alfa Aesar company. Ethylenediamine (EDA, 99%, liquid), diethylenetriamine (DETA, 99%, liquid), ethanolamine (EA, 99%, liquid), triethylamine (TEA, >99.5%, liquid), and diethylamine (DEA, >99%, liquid) were commercially supplied by Macklin Biochemical Co. Ltd. Formamide (FA, ≥99.5%, liquid), N,N-dimethylformamide (DMF, 99%, liquid), chloroform (AR, 99%), acetonitrile (AR, 99%), and deionized water were supplied by National Pharmaceutical Group Corporation. All reagents and solvents were used as supplied without further purification.Synthesis of T4-Mn crystals
The synthetic method of T4-Mn crystals was based on previous report with a slight modification.51,52 Typically, Mn(Ac)2·4H2O (61 mg, 0.25 mmol), indium powder (115.0 mg, 1.0 mmol), and sublimed sulfur (96.0 mg, 3.0 mmol) were mixed with AEAE (2.0 mL), DBU (1.0 mL), and hexamethyleneimine (1.5 mL) in a 23 mL Teflon-lining stainless steel autoclave, and stirred intensely for 0.5 h. The vessel was then sealed and heated up to 150 °C for 7 days, and then taken out from the oven and cooled naturally to room temperature. 50 mg of irregular micrometer-sized orange crystals were obtained after washing with ethanol several times and drying in air.Synthesis of T4-Cd crystals
The synthesis method of T4-Cd crystals was similar to that of T4-Mn crystals with Mn(Ac)2·4H2O (61.0 mg, 0.25 mmol) replaced by Cd(Ac)2·2H2O (66.0 mg, 0.25 mmol). 60 mg of irregular micrometer-sized yellow crystals were finally obtained.Preparation of T4-Mn dispersions (dn, n = 0–6)
2.0 mg of T4-Mn crystals were mixed with different amounts (0 mg, 63 mg, 126 mg, 252 mg, 378 mg, 504 mg, and 882 mg) of surfactant DODA·Br, and then they were separately added into a glass vial with 8 mL of chloroform solvent, followed by magnetic stirring for 4 h, which accordingly resulted in the formation of T4-Mn dispersions with different concentrations of cluster and different concentration of DODA·Br (0 mmoL L−1, 12.5 mmoL L−1, 25 mmoL L−1, 50 mmoL L−1, 75 mmoL L−1, 100 mmoL L−1, and 175 mmoL L−1) (denoted as d0, d1, d2, d3, d4, d5, and d6, respectively). It was clearly observed that a larger amount of surfactant can lead to better dispersity of clusters. The crystals were found to be fully dispersed in chloroform when the amount of DODA·Br reached 378 mg, and a limpid dispersion was observed by the naked eye. However, the dispersions with a small amount of DODA·Br showed a turbid suspension. After centrifugation with a rotating rate of 8000 rpm for 4 minutes, the supernatants of dispersions were collected and used for subsequent study. Note: For all spectroscopic tests on dispersions throughout this work, 3 mL of dispersion was utilized.X-ray diffraction (XRD)
Powder XRD measurements were carried out on medium-sized diffraction equipment (D2 PHASER, Bruker, Germany) with Cu Kα (λ = 1.54056 Å) radiation with 30 kV acceleration voltage and 10 mA working electric current.Ultraviolet-visible spectroscopy (UV-Vis)
UV-Vis spectra were measured on a UV3600 UV-Vis-NIR spectrophotometer.Transmission electron microscopy (TEM)
TEM measurements were carried out on an FEI TECNAI G2 F20 transmission electron microscope operated at 200 kV.Ultraviolet photoelectron spectroscopy (UPS)
UPS measurements were performed on an Escalab 250Xi (Thermo Fisher Scientific) apparatus.Cyclic voltammetry (CV)
CV measurements were carried out on a CHI 760e electrochemical analyzer with a three-electrode configuration at room temperature. An acetonitrile solution containing tetra-n-butylammonium perchlorate (0.1 M) was used as electrolyte. A glassy carbon electrode, Pt wire, and saturated calomel electrode (SCE) were employed as the working electrode, counter electrode, and reference electrode, respectively. The glassy carbon electrode was polished using 0.5 μm alumina powder, and rinsed with deionized water before CV tests. A scan speed of 100 mV s−1 was used during the working process. The CV curves of organic amines were calibrated with ferrocene/ferrocenium (Fc/Fc+) as a standard tested under the same conditions. The energy level of Fc/Fc+ was set to be −4.8 eV with respect to the vacuum level.1H-NMR
1H-NMR spectra of dispersions were obtained from an Agilent DD2-600 instrument, and tetramethylsilane (TMS) was used as an internal standard.Femtosecond transient absorption (fs-TA)
The fs-TA spectra were measured using a Helios pump–probe system. A 365 nm pulse laser was used as the pump beam, and visible light in the range of 400–650 nm was used as the probe beam. The time window was set to 7.39 ns.Photoluminescence (PL)
Room-temperature steady-state PL and photoluminescence excitation (PLE) spectra were obtained on a HORIBA Scientific Fluorolog-3 fluorimeter equipped with a 450 W xenon lamp. PL decay data were recorded on a Fluorolog-3 spectrophotometer using a time-correlated single-photon counting (TCSPC) method.Electron paramagnetic resonance (EPR)
Room-temperature X-band EPR spectra were obtained on a JES-X320 spectrometer with a 9.15 GHz magnetic field.Data availability
All the data supporting this article have been included in the ESI.†Author contributions
Z. Wang and T. Wu conceived and designed the project. Z. Wang, J. Zhang, Y. Lan, and J.-X. Liu synthesized the materials and carried out the PL, UV-Vis, UPS, CV, TEM, PXRD and EPR characterizations. Z. Wang, Y. Lan and X. Li performed 1H-NMR characterization and analyzed the corresponding data. Z. Wang, J. Zhang and C. Qin performed TA measurements, and Z. Wang, H. Ma, J. Zhang, C. Qin, S.-F. Yuan, X.-P. Zhou, and D.-S. Li analyzed TA data. The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 22071165 and 92261205) and the 111 Project (D20015).References
- P. K. Santra and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 2508–2511 CrossRef CAS PubMed.
- Q. Kong, B. Yang, J. Chen, R. Zhang, S. Liu, D. Zheng, H. Zhang, Q. Liu, Y. Wang and K. Han, Angew. Chem., Int. Ed., 2021, 60, 19653–19659 CrossRef CAS PubMed.
- W. Baek, D. R. Gamelin, M. S. Bootharaju, K. M. Walsh and T. Hyeon, Nat. Mater., 2021, 20, 650–657 CrossRef CAS PubMed.
- B. Su, G. Zhou, J. Huang, E. Song, A. Nag and Z. Xia, Laser Photonics Rev., 2020, 15, 2000334 CrossRef.
- F. Meinardi, Q. A. Akkerman, F. Bruni, S. Park, M. Mauri, Z. Dang, L. Manna and S. Brovelli, ACS Energy Lett., 2017, 2, 2368–2377 CrossRef CAS PubMed.
- H. Zhang, J. Yu, C. Sun, W. Xu, J. Chen, H. Sun, C. Zong, Z. Liu, Y. Tang and D. Zhao, J. Am. Chem. Soc., 2020, 142, 16177–16181 CrossRef CAS PubMed.
- J. Jang, J.-K. Kim, J. Shin, J. Kim, K.-Y. Baek, J. Park, S. Park, Y. D. Kim, S. S. P. Parkin, K. Kang, K. Cho and T. Lee, Sci. Adv., 2022, 8, eabn3181 CrossRef CAS PubMed.
- M. Makkar, L. Dheer, A. Singh, L. Moretti, M. Maiuri, S. Ghosh, G. Cerullo, U. V. Waghmare and R. Viswanatha, Nano Lett., 2021, 21, 3798–3804 CrossRef CAS PubMed.
- K. R. Pradeep, A. Debdipto, J. Priyanka, G. Kushagra, Y. Anur, C. Andrea, Z. Margherita, C. Giulio, N. Chandrabhas and N. Shobhana, ACS Energy Lett., 2019, 5, 353–359 Search PubMed.
- E. Hofman, R. J. Robinson, Z. Li, B. Dzikovski and W. Zheng, J. Am. Chem. Soc., 2017, 139, 8878–8885 CrossRef CAS PubMed.
- R. D. Harris, S. B. Homan, M. Kodaimati, C. He, A. B. Nepomnyashchii, N. K. Swenson, S. Lian, R. Calzada and E. A. Weiss, Chem. Rev., 2016, 116, 12865–12919 CrossRef CAS PubMed.
- W. Lee, J. Oh, W. Kwon, S. H. Lee, D. Kim and S. Kim, Nano Lett., 2019, 19, 308–317 CrossRef CAS PubMed.
- B. Su, M. S. Molokeev and Z. J. Xia, J. Phys. Chem. Lett., 2020, 11, 2510–2517 CrossRef CAS PubMed.
- F. Muckel, S. Delikanli, P. L. Hernández-Martínez, T. Priesner, S. Lorenz, J. Ackermann, M. Sharma, H. V. Demir and G. Bacher, Nano Lett., 2018, 18, 2047–2053 CrossRef CAS PubMed.
- J. Wang, T. Ding, C. Nie, M. Wang, P. Zhou and K. Wu, J. Am. Chem. Soc., 2020, 142, 4723–4731 CrossRef CAS PubMed.
- S. Feldmann, M. K. Gangishetty, I. Bravić, T. Neumann, B. Peng, T. Winkler, R. H. Friend, B. Monserrat, D. N. Congreve and F. Deschler, J. Am. Chem. Soc., 2021, 143, 8647–8653 CrossRef CAS PubMed.
- Q. Wei, M. Li, Z. Zhang, J. Guo, G. Xing, T. C. Sum and W. Huang, Nano Energy, 2018, 51, 704–710 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulovic, ACS Nano, 2014, 8, 5863–5872 CrossRef CAS PubMed.
- B. Dhanabalan, G. Biffi, A. Moliterni, V. Olieric, C. Giannini, G. Saleh, L. Ponet, M. Prato, M. Imran and L. Manna, Adv. Mater., 2021, 33, 2008004 CrossRef CAS PubMed.
- D. P. Morgan and D. F. Kelley, J. Phys. Chem. C, 2018, 122, 25661–25667 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- J. Xue, R. Wang, X. Chen, C. Yao, X. Jin, K.-L. Wang, W. Huang, T. Huang, Y. Zhao, Y. Zhai, D. Meng, S. Tan, R. Liu, Z.-K. Wang, C. Zhu, K. Zhu, M. C. Beard, Y. Yan and Y. Yang, Science, 2021, 371, 636–640 CrossRef CAS PubMed.
- C. Qin, T. Matsushima, W. J. Potscavage Jr, A. S. D. Sandanayaka, M. R. Leyden, F. Bencheikh, K. Goushi, F. Mathevet, B. Heinrich, G. Yumoto, Y. Kanemitsu and C Adachi, Nat. Photonics, 2020, 14, 70–75 CrossRef CAS.
- J. Zhang, P. Feng, X. Bu and T. Wu, Natl. Sci. Rev., 2022, 9, nwab076 CrossRef CAS PubMed.
- J. Zhang, X. Bu, P. Feng and T. Wu, Acc. Chem. Res., 2020, 53, 2261–2272 CrossRef CAS PubMed.
- D. Li, C. Xu, Y. Liao, W. Cai, Y. Zhu and Z. L. Wang, Sci. Adv., 2021, 7, eabj0349 CrossRef CAS PubMed.
- Z. Wang, Y. Liu, J. Zhang, X. Wang, N. Chen, D. Li and T. Wu, J. Mater. Chem. C, 2021, 9, 13680–13686 RSC.
- X. Yang, C. Pu, H. Qin, S. Liu, Z. Xu and X. Peng, J. Am. Chem. Soc., 2019, 141, 2288–2298 CrossRef CAS PubMed.
- M. Zhu, Ji. Zhou, Z. Hu, H. Qin and X. Peng, ACS Photonics, 2018, 5, 4139–4146 CrossRef CAS.
- C. Pu, J. Ma, H. Qin, M. Yan, T. Fu, Y. Niu, X. Yang, Y. Huang, F. Zhao and X. Peng, ACS Cent. Sci., 2016, 2, 32–39 CrossRef CAS PubMed.
- X. Dong, Y. Si, J. Yang, C. Zhang, Z. Han, P. Luo, Z. Wang, S. Zang and T. C. W. Mak, Nat. Commun., 2020, 11, 3678 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- R. Xu, Y. Wang, X. Duan, K. Lu, D. Micheroni, A. Hu and W. Lin, J. Am. Chem. Soc., 2016, 138, 2158–2161 CrossRef CAS PubMed.
- C. Xue, X. Fan, J. Zhang, D. Hu, Xi.-L. Wang, Xi. Wang, R. Zhou, H. Lin, Y. Li, D.-S. Li, X. Wei, D. Zheng, Y. Yang, K. Han and T. Wu, Chem. Sci., 2020, 11, 4085–4096 RSC.
- K. Zhang, T. Yu, F. Liu, M. Sun, H. Yu, B. Liu, Z. Zhang, H. Jiang and S. Wang, Anal. Chem., 2014, 86, 11727–11733 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, B. Han, X. Wang, Z. Wang, C. Xue, G. Bian, D. Hu, R. Zhou, D. Li, Z. Wang, Z. Ouyang, M. Li and T. Wu, J. Am. Chem. Soc., 2020, 142, 6649–6660 CrossRef CAS PubMed.
- J. Wu, B. Jin, X. Wang, Y. Ding, X.-L. Wang, D. Tang, X. Li, J. Shu, D.-S. Li, Q. Lin, Y.-B. Wu and T. Wu, CCS Chem., 2021, 3, 2584–2590 CrossRef CAS.
- Q. Zhang, R. Wang, B. Feng, X. Zhong and K. Ostrikov, Nat. Commun., 2021, 12, 6856 CrossRef CAS PubMed.
- N. S. Nudelman and C. E. S. Alvaro, J. Phys. Org. Chem., 2011, 24, 1067–1071 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 2574–2577 CrossRef CAS PubMed.
- W. R. Algar, A. Khachatrian, J. S. Melinger, A. L. Huston, M. H. Stewart, K. Susumu, J. B. Blanco-Canosa, E. Oh, P. E. Dawson and I. L. Medintz, J. Am. Chem. Soc., 2017, 139, 363–372 CrossRef CAS PubMed.
- D. Luo, S. Yang, Q. Zhang, L. Cha, L. Dang and M. Li, J. Phys. Chem. Lett., 2021, 12, 1838–1846 CrossRef CAS PubMed.
- S. Wang, J. Leng, Y. Yin, J. Liu, K. Wu and S. Jin, ACS Energy Lett., 2020, 5, 328–334 CrossRef CAS.
- J. Yao, J. Ge, B. Han, K. Wang, H. Yao, H. Yu, J. Li, B. Zhu, J. Song and C. Chen, J. Am. Chem. Soc., 2018, 140, 3626–3634 CrossRef CAS PubMed.
- K. J. Babu, G. Kaur, A. Shukla, A. Kaur, T. Goswami, N. Ghorai and H. N. Ghosh, J. Phys. Chem. Lett., 2020, 12, 302–309 CrossRef PubMed.
- R.-T. Liu, X.-P. Zhai, Z.-Y. Zhu, B. Sun, D.-W. Liu, B. Ma, Z.-Q. Zhang, C.-L. Sun, B.-L. Zhu, X.-D. Zhang, Q. Wang and H. L. Zhang, J. Phys. Chem. Lett., 2019, 10, 6572–6577 CrossRef CAS PubMed.
- D. Rossi, D. Parobek, Y. Dong and D. H. Son, J. Phys. Chem. C, 2017, 121, 17143–17149 CrossRef CAS.
- R. J. Abraham, L. Griffiths and M. Perez, Magn. Reson. Chem., 2013, 51, 143–155 CrossRef CAS PubMed.
- Z. Wu, X. Wang, X. Wang, X. Xu, D. Li and T. Wu, Chem. Eng. J., 2021, 426, 131216 CrossRef CAS.
- X.-L. Wang, Z. Wu, X. Wang, C. Xue, C. Liu, J. Zhang, R. Zhou, D.-S. Li and T. Wu, Electrochim. Acta, 2021, 376, 138048 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03091a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |