
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical charge accumulation in a heteroleptic copper(I)-anthraquinone molecular dyad via proton-coupled electron transfer†
Zhu-Lin
Xie
 a,
Nikita
Gupta
ab,
Jens
Niklas
a,
Oleg G.
Poluektov
a,
Vincent M.
Lynch
c,
Ksenija D.
Glusac
ab and
Karen L.
Mulfort
*a
a,
Nikita
Gupta
ab,
Jens
Niklas
a,
Oleg G.
Poluektov
a,
Vincent M.
Lynch
c,
Ksenija D.
Glusac
ab and
Karen L.
Mulfort
*a
aDivision of Chemical Sciences and Engineering, Argonne National Laboratory, USA. E-mail: mulfort@anl.gov
bDepartment of Chemistry, University of Illinois at Chicago, USA
cDepartment of Chemistry, University of Texas at Austin, USA
First published on 1st September 2023
Abstract
Developing efficient photocatalysts that perform multi electron redox reactions is critical to achieving solar energy conversion. One can reach this goal by developing systems which mimic natural photosynthesis and exploit strategies such as proton-coupled electron transfer (PCET) to achieve photochemical charge accumulation. We report herein a heteroleptic Cu(I)bis(phenanthroline) complex, Cu-AnQ, featuring a fused phenazine-anthraquinone moiety that photochemically accumulates two electrons in the anthraquinone unit via PCET. Full spectroscopic and electrochemical analyses allowed us to identify the reduced species and revealed that up to three electrons can be accumulated in the phenazine-anthraquinone ring system under electrochemical conditions. Continuous photolysis of Cu-AnQ in the presence of sacrificial electron donor produced doubly reduced monoprotonated photoproduct confirmed unambiguously by X-ray crystallography. Formation of this photoproduct indicates that a PCET process occurred during illumination and two electrons were accumulated in the system. The role of the heteroleptic Cu(I)bis(phenanthroline) moiety participating in the photochemical charge accumulation as a light absorber was evidenced by comparing the photolysis of Cu-AnQ and the free AnQ ligand with less reductive triethylamine as a sacrificial electron donor, in which photogenerated doubly reduced species was observed with Cu-AnQ, but not with the free ligand. The thermodynamic properties of Cu-AnQ were examined by DFT which mapped the probable reaction pathway for photochemical charge accumulation and the capacity for solar energy stored in the process. This study presents a unique system built on earth-abundant transition metal complex to store electrons, and tune the storage of solar energy by the degree of protonation of the electron acceptor.
Introduction
Photocatalysis targeted at CO2 reduction, hydrogen evolution and water oxidation for solar energy conversion has drawn considerable attention due to the finite amount of fossil fuels and the aggravated outcome of their combustion.1–5 Since these reactions involve transferring multiple electrons/holes as energetic products are generated, efficient photocatalysis can be difficult to achieve if traditional catalysts that operate in single-electron transfer mechanism are utilized.6–8 This issue can be addressed by developing catalytic systems that operate via photochemically accumulated charge transfer steps coupled with proton transfers, the strategy employed by natural photosynthesis. In nature, the oxygen evolving center (OEC) in photosystem II (PS II) undergoes four proton-coupled electron transfer (PCET) processes induced by photoexcitation, which accumulates oxidative equivalents for molecular oxygen generation and avoids the formation of high energy intermediates.9–12 The electrons are transferred through a cascade of cofactors, which include quinones, the important electron carriers that accept two pairs of electrons and protons after electron transfer processes.13–15 At the end of the electron transfer chain, NADP+ is reduced with two electrons and one proton, and the resulting NAPH molecules are eventually used in the Calvin cycle for CO2 assimilation.16,17 The combination of charge accumulation and PCET renders the natural photosynthetic system efficient and robust, which converts solar energy into chemical fuels.Biomimetic donor–acceptor assemblies have provided a fundamental understanding of how to integrate and control photochemical charge accumulation and PCET in photocatalytic systems.6,7 The first molecular system that implements photochemical charge accumulation and PCET was reported by Konduri et al.18,19 In these studies, two dinuclear Ru(II) complexes with extended π-conjugated bridging ligands based on phenazine and/or quinone demonstrated reversible charge accumulation of two and four electrons, respectively, upon photoexcitation in the presence of a sacrificial electron donor. Protonation of the nitrogen or oxygen atoms at the central ligand was shown to couple with the formation of the reduced product by the mechanism of two sequential one-electron processes under basic conditions, or a bielectronic process under neutral or slightly acidic conditions.20 Later, the Wenger group developed a covalently-linked unconjugated pentad complex (TAA-Ru-AQ-Ru-TAA) composed of triarylamine (TAA), Ru tris-bipyridine and the anthraquinone (AQ) and demonstrated that the charge-separated state with two electrons stored at the anthraquinone moiety was accessed upon photo-illumination.21,22 Importantly, a considerably longer lived charge-separated state was observed under acidic conditions21 than in neat solvent.22 These studies underscore a key benefit of PCET, namely, stabilization of a highly reduced species which leads to improvement in the lifetime of the charge accumulated state. However, this increased stability of the protonated, charge-separated state is often achieved with a coincident decrease in energy, essentially reducing the possible amount of solar energy that could be used for fuels catalysis. A potential solution to this issue is to store photogenerated electrons at more reducing potentials by judicial design of the structure of the donor–acceptor assembly. This has been manifested, for example, in the [(phen)2Ru(tatpp)Ru(phen)2][PF6]4 system23 by changing the bridging linear tetraazapentacene ligand into one with a 120° bend which resulted in a ∼500 mV negative shift of the reduction potential of the ligand, or in Ru-dipyridonphenazine-pyridoquinolinone system24,25 by modifying the pyridoquinolinone moiety to a pyridoquinolinone-oxime derivative, inducing a negative shift of the first and second reduction potential by −0.24 V and −0.14 V, respectively. These reports exemplify that effective structural modification of the acceptor can alter the molecular electrochemical response so that optimal photochemical charge accumulation systems with high energy conversion efficiency can be achieved.
The examples described above have provided important insight into the molecular design components needed to support PCET-enabled charge accumulation, although these and several other examples are all built upon the Ru(II)tris(bipyridine) photosensitizer module.26–29 The photoactive Ru(II) center has shown outstanding photophysical and photochemical properties, but the use of precious metals prevents the application of such molecular systems to sustainable large-scale catalysis,30 yet there are few reports describing photochemical charge accumulation achieved without precious metals.31,32 Among the first-row photoactive transition metal complexes, Cu(I)diimine complexes are of special interest and have been regarded as a potential substitute for the quintessential Ru(II)poly(pyridyl) photosensitizers.33–35 The d10 Cu(I) center allows the Cu(I)diimine complexes to access the metal-to-ligand charge transfer (MLCT) state by absorbing visible light ranging from 400 to 550 nm.35 The excited state lifetimes of Cu(I)diimine complexes were historically shorter than Ru(II)poly(pyridyl) complexes,33,36 but a large and growing literature has explored ligand designs that can extend their excited state lifetimes well into the microsecond regime.37–41 Furthermore, the ease of oxidation of the Cu(I) center results in these complexes acting as strong electron donors during photochemical processes.42–44 These exceptional electro- and photochemical features of the Cu(I)diimine complexes have gained interest for their applications in dye-sensitized solar cells,30 photocatalysis,45 and light emitting diodes,46 for example.
In this report, we describe the first molecular donor–acceptor assembly, Cu+-AnQ, built upon a Cu(I)diimine chromophore fused with phenazine-anthraquinone (AnQ) moiety for photochemical charge accumulation (Chart 1). We demonstrate that, upon visible light illumination, the extended π-conjugation of the AnQ ligand is capable of storing two electrons at the anthraquinone moiety in the presence of a sacrificial electron donor via a PCET process, which generates the doubly reduced, monoprotonated product Cu+-AnQH− that is stable for at least 24 hours. DFT calculations show that the basicity of Cu+-AnQH− is significantly lower than the basicity of the doubly reduced, unprotonated species Cu+-AnQ2− because of the unique curvature designed into the ligand. As a result, monoprotonation of Cu-AnQ not only stabilizes the photogenerated two-electron reduced state, but also avoids substantial energy loss associated with protonation. This study demonstrates a distinct strategy for improving the yield of solar energy conversion in molecular systems by controlling the protonation state of photoproducts and provides important foundational knowledge for integrating photochemical charge accumulation in photocatalytic systems.
 | ||
| Chart 1 Photoactive heteroleptic Cu(I) complexes studied in this paper, Cu+-AnQ and Cu+-DPPZ. Superscripts indicate charges carried by Cu. The notation of the mesPhen ligand is omitted from the naming system as it is not the focus of this study. | ||
Results
Molecule design, syntheses and structural characterization
To engineer a charge accumulation site with Cu complexes, we designed the dyad Cu+-AnQ by utilizing the Cu HETPHEN strategy, developed by Schmittel et al.47–49 This method is effective for the synthesis of heteroleptic Cu(I)bis(phenanthroline) complexes by using 2,9-dimesityl-1,10-phenanthroline (mesPhen) as a blocking ligand that prevents bis(mesPhen) coordination and allows the ligation of a second, different ligand to the Cu(I) center. Here the second phenanthroline ligand was derivatized to fuse an anthraquinone moiety, forming a π-conjugated dipyridophenazine-anthraquinone ligand (AnQ). The decreasing energy level follows the order of Cu(I) center, phenazine, anthraquinone, and is designed to realize a directional energy cascade from Cu(I) to anthraquinone. The ability of anthraquinone to accept multiple electrons and protons makes Cu+-AnQ a good candidate for studying charge accumulation and PCET, as demonstrated in similar molecular assemblies based on Ru(II) chromophores and anthraquinone, developed by Wenger and coworkers,21,22,50,51 but importantly here we investigating a model system using only earth-abundant elements. We have also synthesized Cu+-DPPZ and 2,17-dimethylnaphtho[2,3-h]dipyrido[3,2-a:2′,3′-c]phenazine-8,13-dione (MeAnQ) as control molecules to compare with the spectroscopic investigation and photochemical inquiry of Cu+-AnQ. MeAnQ is analogous to the free ligand, AnQ, but the methyl groups increase the solubility in acetonitrile from the uncoordinated ligand AnQ, whose strong π–π stacking interaction results in very low solubility and prevented us from obtaining the solution phase properties of the ligand.The synthesis of the Cu+-AnQ dyad is summarized in Scheme 1 and full details are presented in the ESI.† Briefly, a stoichiometric amount of [Cu(MeCN)4](PF6) and mesPhen ligand were mixed in deaerated dichloromethane, producing a yellow solution of [Cu(mesPhen)(MeCN)2](PF6). The intermediate was then treated with one equivalent of 1,10-phenanthroline-5,6-dione (Phendione) to afford the building block [Cu(mesPhen)(Phendione)](PF6) in 96% yield. Complex Cu+-AnQ was formed through a condensation between [Cu(mesPhen)(Phendione)](PF6) and 1,2-diaminoanthraquinone under refluxing conditions in ethanol and a catalytic amount of acetic acid for seven days. Complex Cu+-DPPZ was synthesized according to the Cu HETPHEN method and following literature precedent52 for the DPPZ ligand. The ligand MeAnQ was synthesized according to the reported procedure.53
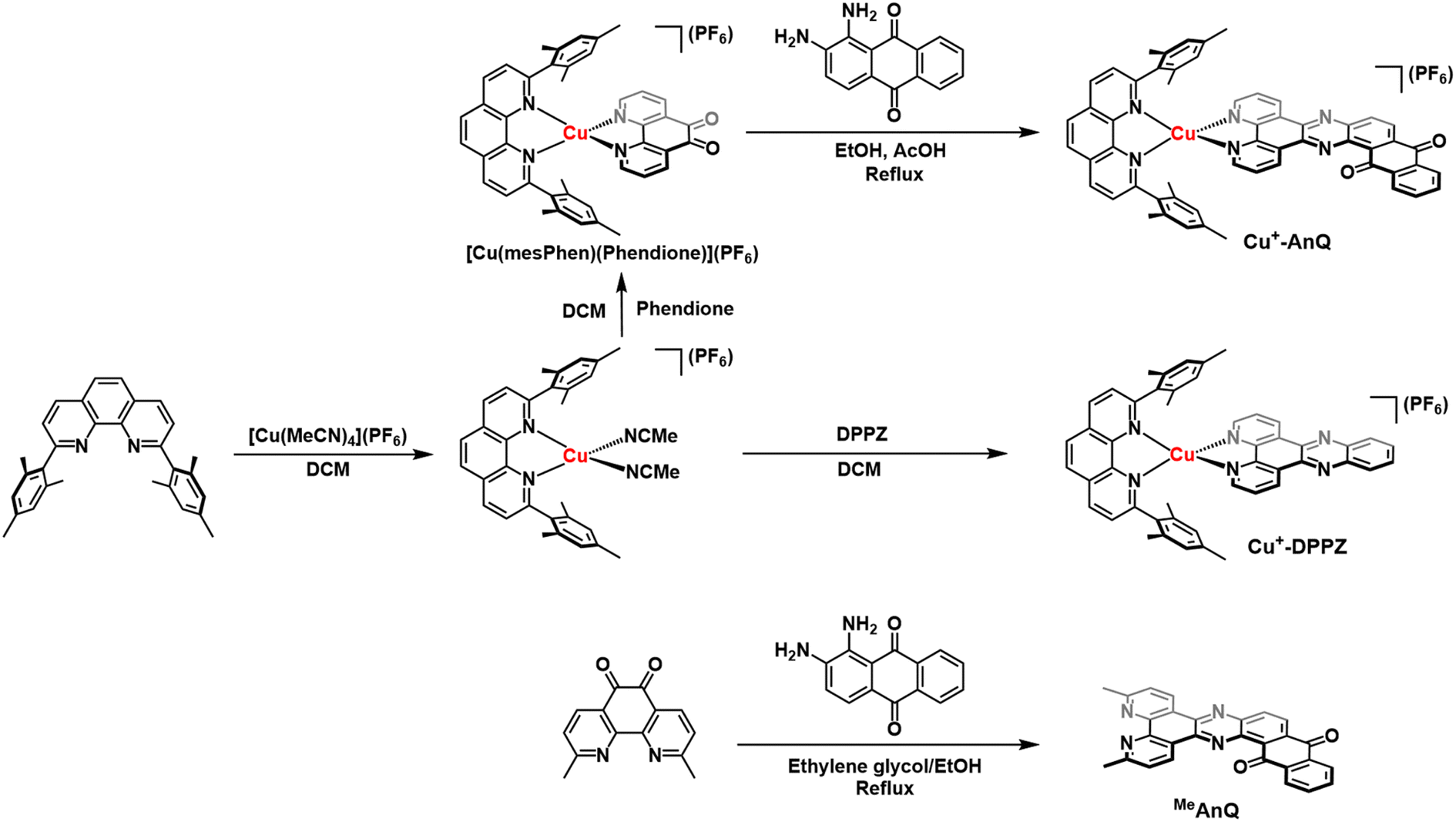 | ||
| Scheme 1 Synthesis of Cu+-AnQ, Cu+-DPPZ and MeAnQ. | ||
Proton NMR spectroscopy was employed to identify the target complexes. The NMR shows that the extension of π-conjugation with an anthraquinone moiety does not impact the local electronic environment of the mesPhen protons, as shown by the fact that the proton signals derived from the mesPhen of Cu+-DPPZ exhibit comparable values with those of Cu+-AnQ (Fig. S5 and S7†). However, the coordination of AnQ to [Cu(mesPhen)]1+ breaks the C2 symmetry of the complex, resulting in a splitting of the proton signals from mesPhen. Also, we observe significant downfield shifts for the protons in the immediate phenanthroline moiety of the AnQ ligand as compared to that for DPPZ (protons H3, H3′, H4, H5 in Fig. S5 and S7†). The change of chemical shift is related to the anthraquinone moiety which acts as an electron withdrawing group that pulls electron density away from the DPPZ moiety.
The crystal structure of Cu+-AnQ was obtained via vapor diffusion of diethyl ether to an acetonitrile solution of Cu+-AnQ. Fig. 1A shows that the Cu(I) center is coordinated with four N atoms from two ligands in a pseudo-tetrahedral geometry with an s4 value of 0.67 (1 for perfect tetrahedral and 0 for perfect square planar54). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distances on the AnQ ligand are 1.223(2) and 1.217(2) Å, confirming the double bond nature of the carbonyl group and are comparable to the literature values for quinone molecules.21 The bond distances of Cu–N range from 1.99 to 2.09 Å, with the two Cu–N bonds to mesPhen being both the shortest and the longest in the structure (1.9967(15) Å, 2.0910(15) Å). This derives from the significant structural distortion of mesPhen coordination, where the mesPhen ligand binds with the Cu(I) center in the “pacman” orientation.55 This preferential orientation in the solid state results in one mesityl aryl ring that stacks with the AnQ ligand plane through π–π interactions and the other aryl ring dangles away, avoiding being parallel with the AnQ ligand. This distortion paves the way for Cu+-AnQ to form dimer structures in crystal lattice, shown in Fig. 1B. The dimers in the solid phase arise through the π–π interactions between the two AnQ ligand planes with a distance of 3.398 Å. Together with the interactions between the mesityl groups and the AnQ ligands, individual complexes are piled forming 1D columns in the crystal lattice (Fig. S14 and S15†).
O bond distances on the AnQ ligand are 1.223(2) and 1.217(2) Å, confirming the double bond nature of the carbonyl group and are comparable to the literature values for quinone molecules.21 The bond distances of Cu–N range from 1.99 to 2.09 Å, with the two Cu–N bonds to mesPhen being both the shortest and the longest in the structure (1.9967(15) Å, 2.0910(15) Å). This derives from the significant structural distortion of mesPhen coordination, where the mesPhen ligand binds with the Cu(I) center in the “pacman” orientation.55 This preferential orientation in the solid state results in one mesityl aryl ring that stacks with the AnQ ligand plane through π–π interactions and the other aryl ring dangles away, avoiding being parallel with the AnQ ligand. This distortion paves the way for Cu+-AnQ to form dimer structures in crystal lattice, shown in Fig. 1B. The dimers in the solid phase arise through the π–π interactions between the two AnQ ligand planes with a distance of 3.398 Å. Together with the interactions between the mesityl groups and the AnQ ligands, individual complexes are piled forming 1D columns in the crystal lattice (Fig. S14 and S15†).
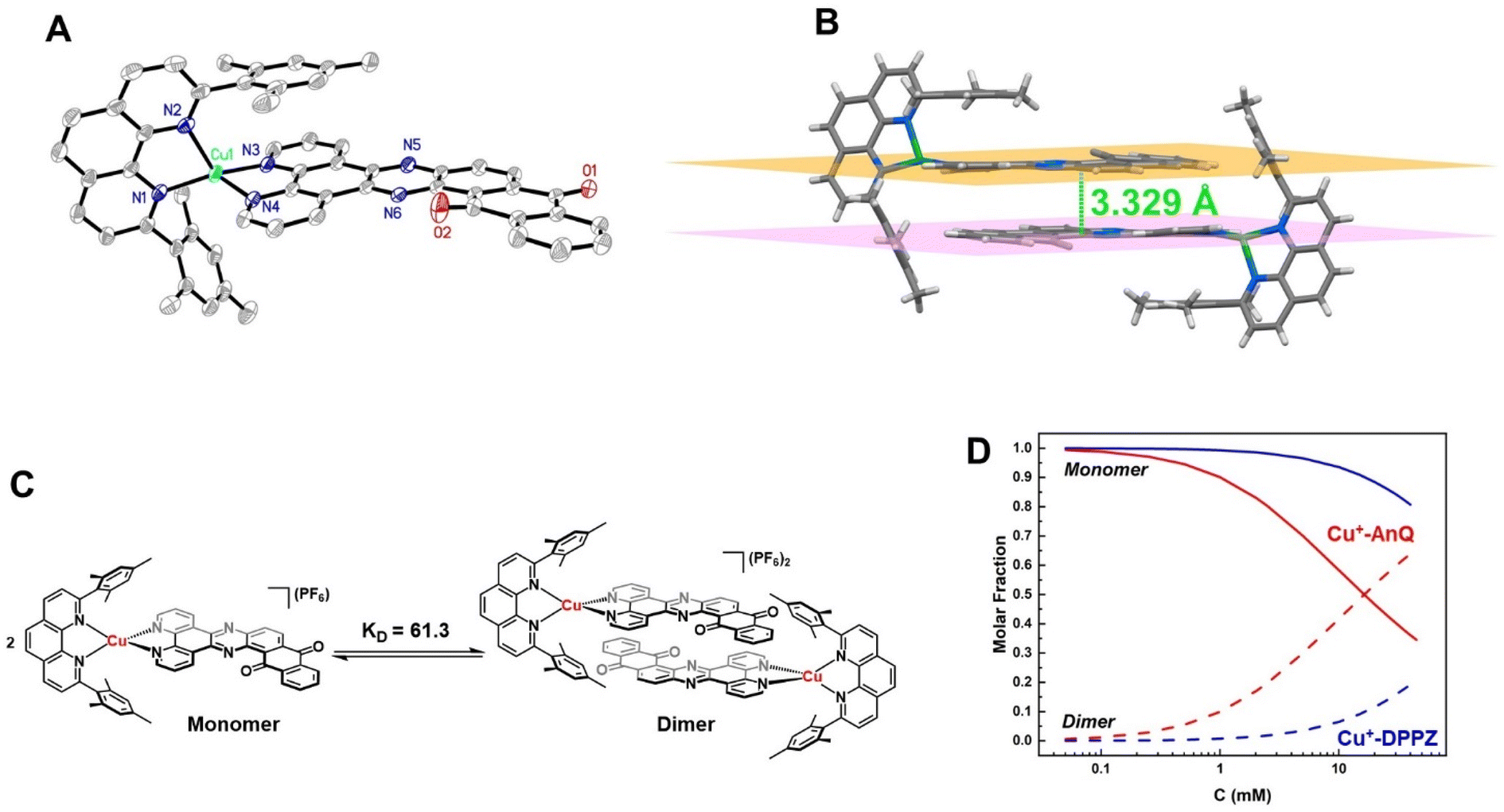 | ||
| Fig. 1 (A) ORTEP diagram of Cu+-AnQ (50% thermal ellipsoids; all hydrogen atoms and the counterion PF6− are omitted for clarity). (B) Dimer structure of Cu+-AnQ in crystal lattice. (C) Schematic presentation of dimerization of Cu+-AnQ in acetonitrile. (D) Calculated molar fractions of dimer and monomer Cu+-AnQ and Cu+-DPPZ in acetonitrile affected by the total concentration of the complex. The calculation was based on the association constant KD. | ||
The dimer formation observed in the solid state structure persists to some extent in the solution phase, as confirmed by a concentration dependence of the 1H NMR peak shifts for both Cu+-AnQ and Cu+-DPPZ. The protons from AnQ and DPPZ show an upfield shift as their concentration in CD3CN increases (Fig. S11 and S12†), which suggests dimerization of individual molecules in the solution state as depicted in Fig. 1C. Such dimerization in the solution state has been observed in Ru(II) and Os(II) complexes coordinated to planar extended π-conjugated ligands, such as TPPHZ56,57 and eilatin.58,59 These complexes tend to stack in head-to-tail fashion via intermolecular π–π interactions by the π-conjugated ligands. The dimer model60,61 was used to fit the shift of the proton signals yielding an association constant KD = 61.3 for Cu+-AnQ and 3.7 for Cu+-DPPZ (for details see ESI†). An important implication of the different ability of the two complexes to form dimers is that for a given concentration, a larger fraction of Cu+-AnQ occurs as a dimer than does Cu+-DPPZ. As shown in Fig. 1D, it is evident that at a concentration of 0.2 mM there is 99% of Cu+-DPPZ exists as monomer and 97% of Cu+-AnQ is in the monomer form. Therefore, we kept both complexes at 0.2 mM for subsequent electro/photochemical studies to avoid the impact of dimerization on these properties.
Steady state optical and electrochemical characterization
The steady state UV-vis spectra of Cu+-AnQ and Cu+-DPPZ in acetonitrile solution exhibit metal-to-ligand charge transfer (MLCT) bands at around 460 nm (Fig. 2A and Table 1). However, Cu+-AnQ displays stronger MLCT absorption, which is about 45% more intense than that of Cu+-DPPZ. In the UV region, we observe moderately structured bands at 385 and 401 nm for Cu+-AnQ and 356 and 374 nm for Cu+-DPPZ, which are assigned to ligand centered (LC) transitions, with Cu+-AnQ presenting higher absorbance in these transitions. The red-shift of the LC transition from Cu+-AnQ clearly indicates that the AnQ ligand has lower energy level than DPPZ due to the extended π-conjugation that includes the anthraquinone moiety. Further, the LC band of Cu+-AnQ aligns with the absorption of the uncoordinated ligand MeAnQ, suggesting that the Cu ligation does not impact the local electronic property of the ligand, the evidence of electronic decoupling between the Cu center and ligand.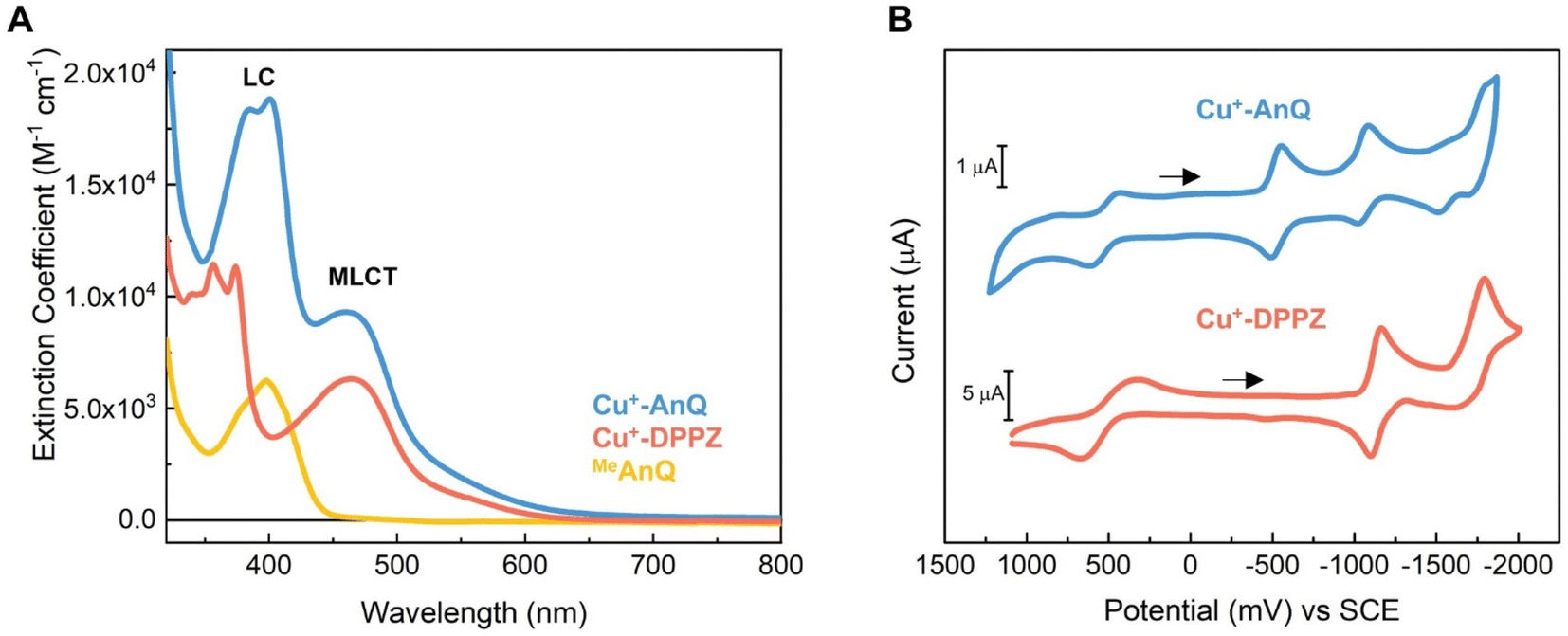 | ||
| Fig. 2 (A) Experimental UV-vis spectra of Cu+-AnQ, Cu+-DPPZ and MeAnQ in acetonitrile (concentrations of the measurements were kept below 0.2 mM); (B) cyclic voltammogram of Cu+-AnQ and Cu+-DPPZ in acetonitrile (0.2 mM for Cu+-AnQ, 1 mM for Cu+-DPPZ, 0.1 M TBAPF6; working: Pt disc, counter: Pt wire, reference: Ag/Ag+ non-aqueous with ferrocene as internal standard; scan rate = 100 mV s−1; arrows indicate the direction of the scan). | ||
| Complexes | λ max/nm (ε/M−1 cm−1) | E (Cu2+/1+) (V vs. SCE) | E (Q0/1−)a (V vs. SCE) | E (Q1−/2−)a (V vs. SCE) | E (Phz0/1−)a (V vs. SCE) | E (BL0/1−)a (V vs. SCE) |
|---|---|---|---|---|---|---|
| Cu+-AnQ | 385 (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 361), 401 (18817), 461 (9304) 361), 401 (18817), 461 (9304) |
+0.52 | −0.53 | −1.05 | −1.55 | −1.77 |
| Cu+-DPPZ | 356 (11435), 374 (11330), 464 (6319) |
+0.50 | −1.13 | −1.70 |
The electrochemical properties of Cu+-AnQ and Cu+-DPPZ were characterized by cyclic voltammetry (CV) in acetonitrile containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the electrolyte (Fig. 2B and Table 1). Both complexes exhibit a quasi-reversible Cu(II/I) redox couple at +0.52 V vs. SCE for Cu+-AnQ and +0.50 V vs. SCE for Cu+-DPPZ. The quasi-reversibility of the Cu oxidation is a result of the pseudo Jahn–Teller distortion of Cu(II) center, which flattens the coordination geometry of Cu from tetrahedral towards square planar.62–64 In addition, the almost identical oxidation potential found in both complexes indicates that the Cu centers of Cu+-AnQ and Cu+-DPPZ have similar electronic structure, which is attributed to the negligible electronic influence from the anthraquinone moiety due to the long distance between the Cu center and anthraquinone moiety. Such weak electronic coupling has been observed in other Ru/Os/Cu bimetallic complexes with two metals bridged by TPPHZ-like ligands;62,65 this further exemplifies that although the π-electrons in the ring system might act as a “wire” for excited state electron transfer, the ground state electronic environment of the metal center remains relatively localized at the first and second coordination sphere.
Turning to the reduction side, Cu+-AnQ exhibits four ligand-based reductions. The first two reductions with E1/2 = −0.53 V and −1.05 V vs. SCE are assigned to the first and second reductions of the anthraquinone moiety, generating the singly reduced Cu+-AnQ− and doubly reduced Cu+-AnQ2− species. These anthraquinone reduction potentials are positively shifted by around 400 mV compared to the free anthraquinone compound in acetonitrile (Fig. S17†) and around 300 mV with respect to the complex featuring an anthraquinone unit linked with Ru(bpy)3 chromophore(s) reported by Orazietti et al.22 The slight positive shift of the first two reduction potentials for Cu+-AnQ is ascribed to the fact that the anthraquinone moiety is fused with the phenazine unit generating a conjugated π-system with a LUMO that occupies lower energy than free anthraquinone or the DPPZ ligand. This is in line with the UV-vis results where the LC band of Cu+-AnQ occurs at a more red, lower energy wavelength than that of Cu+-DPPZ.
The reduction of the phenazine unit in Cu+-AnQ is shown at E1/2 = −1.55 V vs. SCE, which exhibits 400 mV negative shift as compared to the phenazine reduction of Cu+-DPPZ (−1.13 V vs. SCE). The difference in the phenazine reduction potentials between the two complexes is attributed to the negative charges residing on the anthraquinone moiety of Cu+-AnQ2− which makes it more difficult to further reduce the AnQ ligand. Interestingly, both complexes show similar reduction potentials for the blocking ligand mesPhen (−1.77 V vs. SCE for Cu+-AnQ and −1.70 V vs. SCE for Cu+-DPPZ), indicating that in such Cu(I)diimine complexes the reduction of one ligand has minimal impact on the electrochemical behavior of the other ligand. It is important to note that the first three reductions of Cu+-AnQ all occur on the AnQ ligand, demonstrating the capacity of the AnQ ligand to store up to three electrons upon reduction. The moderate reduction potentials for the anthraquinone moiety in AnQ ligand opens up a pathway for (electro)chemical and photochemical charge accumulation on the AnQ ligand.
Spectroelectrochemical study of the reduced Cu anthraquinone complex
To further study the charge accumulation of Cu+-AnQ and obtain the optical signatures of the reduced species, we performed spectroelectrochemical (SEC) studies with Cu+-AnQ and Cu+-DPPZ. The singly reduced species Cu+-AnQ− was generated by poising the potential at −0.80 V vs. SCE, about 270 mV beyond the reduction potential of the anthraquinone moiety (see Fig. 2B). Subtracting the absorption spectrum of Cu+-AnQ from that of Cu+-AnQ−, a difference absorption spectrum (Fig. 3A, orange curve) corresponding to the singly reduced species Cu+-AnQ− is obtained. The spectrum reveals a depletion of the ligand-based band at 380 nm, indicating that the reduction takes place at the AnQ ligand. Surprisingly, a very wide ‘V-shaped’ absorption feature across a broad spectral range from visible to near IR region is observed with the lowest absorption at around 630 nm and a steady growth of absorption above 630 nm. Some distinctive features are captured, which include peaks at 430 nm, 486 nm, 1008 nm and a shoulder at 888 nm. These absorptions are derived from the low energy ligand-based transitions of the reduced AnQ ligand. In addition, the increase of the 400 to 550 region indicates that the copper-based MLCT transition is preserved in the singly reduced species and can be used for pumping a second electron to the anthraquinone center, enabling photochemical charge accumulation.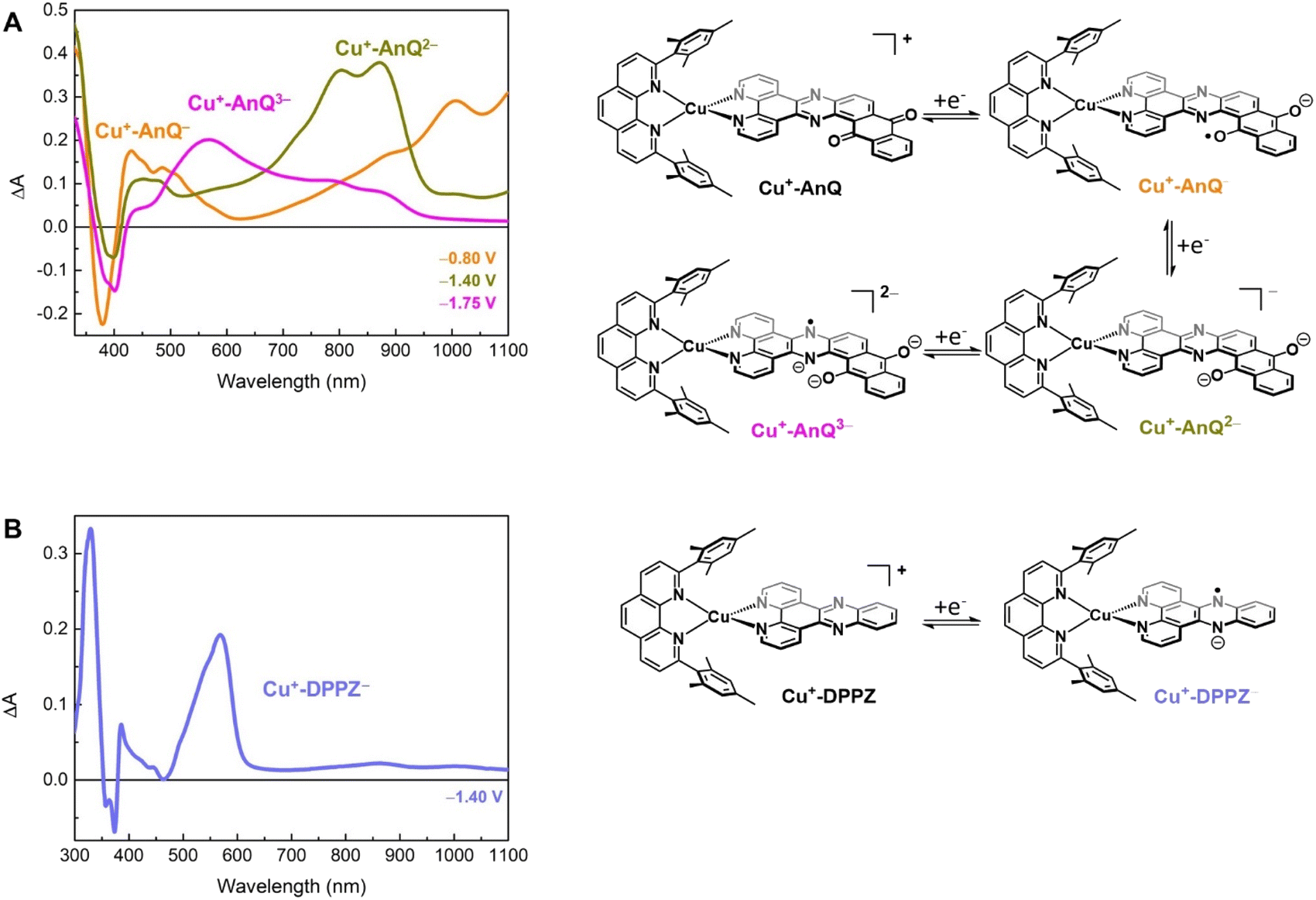 | ||
| Fig. 3 SEC study of (A) Cu+-AnQ and (B) Cu+-DPPZ in 0.1 M TBAPF6/anhydrous acetonitrile (working: Pt mesh; counter: Pt wire; reference: Ag+/Ag pseudoreference electrode). The potential steps shown in the figure are referenced to SCE. | ||
The doubly reduced species Cu+-AnQ2− is afforded after stepping the potential to −1.40 V. As shown by the green trace in Fig. 3A, Cu+-AnQ2− exhibits two equally-intense, relatively narrow peaks centered at 804 nm and 871 nm, a small shoulder at 727 nm, and a broad peak with weak intensity at 452 nm. These features are attributed to the electronic transition taking place on the doubly reduced AnQ ligand of Cu+-AnQ2−. Interestingly, a slightly different bleach feature with respect to that of Cu+-AnQ− is observed. The dip of the depletion of Cu+-AnQ2− appears at 399 nm, which is red-shifted by about 20 nm from that of Cu+-AnQ−. The shift of the bleach band is a response to the difference of the electronic structure between the singly reduced and doubly reduced species and this feature can be used as an additional marker to distinguish the state of reduction of the complex.
Upon further reduction, the difference spectrum at −1.75 V displays a new peak at 571 nm while other absorption features assigned to the doubly reduced species diminish (Fig. 3A magenta curve). This newly detected absorption band arises from the triply reduced complex Cu+-AnQ3−, where the phenazine moiety is the main position that accepts the third electron, forming an AnQ3− ligand. We can make this assignment and confirm the reduction of the phenazine unit by comparison to the SEC response of Cu+-DPPZ held at −1.40 V. As shown in Fig. 3B, the singly reduced Cu+-DPPZ1− shows an intense absorption at 568 nm with a spectral shape similar to that of Cu+-AnQ3−. Comparing the results of Cu+-DPPZ1− with the third reduction of Cu+-AnQ confirms that the first two reduction events are localized on the anthraquinone and the third occurs at the phenazine unit.
Chemical reduction of Cu+-AnQ
The reduced species Cu+-AnQ− and Cu+-AnQ2− can also be afforded by chemical reduction. We observed that an acetonitrile solution of Cu+-AnQ changed color from orange to dark orange and, finally green on titration with cobaltocene (CoCp2) at room temperature under the protection of N2 (Fig. 4). The UV-vis spectra of the titration show that complete conversion of Cu+-AnQ to Cu+-AnQ− was achieved by about two equivalents of CoCp2 and conversion to Cu+-AnQ2− required six equivalents of CoCp2 (Fig. S22†). While reduction with CoCp2 is ideally stoichiometric, excess CoCp2 was required in this case to complete the reaction because of residual oxygen present in our experimental set up. The UV-vis spectra of the chemically generated Cu+-AnQ− and Cu+-AnQ2− are in good agreement with the electrochemically generated species, which are characterized by the increasingly intense absorption from 650 to 1100 nm for Cu+-AnQ− and the two-peak feature from 700 to 1000 nm for Cu+-AnQ2−. We observed that the doubly reduced species is quite stable under inert atmosphere at room temperature, as demonstrated by nearly identical UV-vis spectra between the freshly generated solution and the solution stored under N2 for two days (Fig. S23†).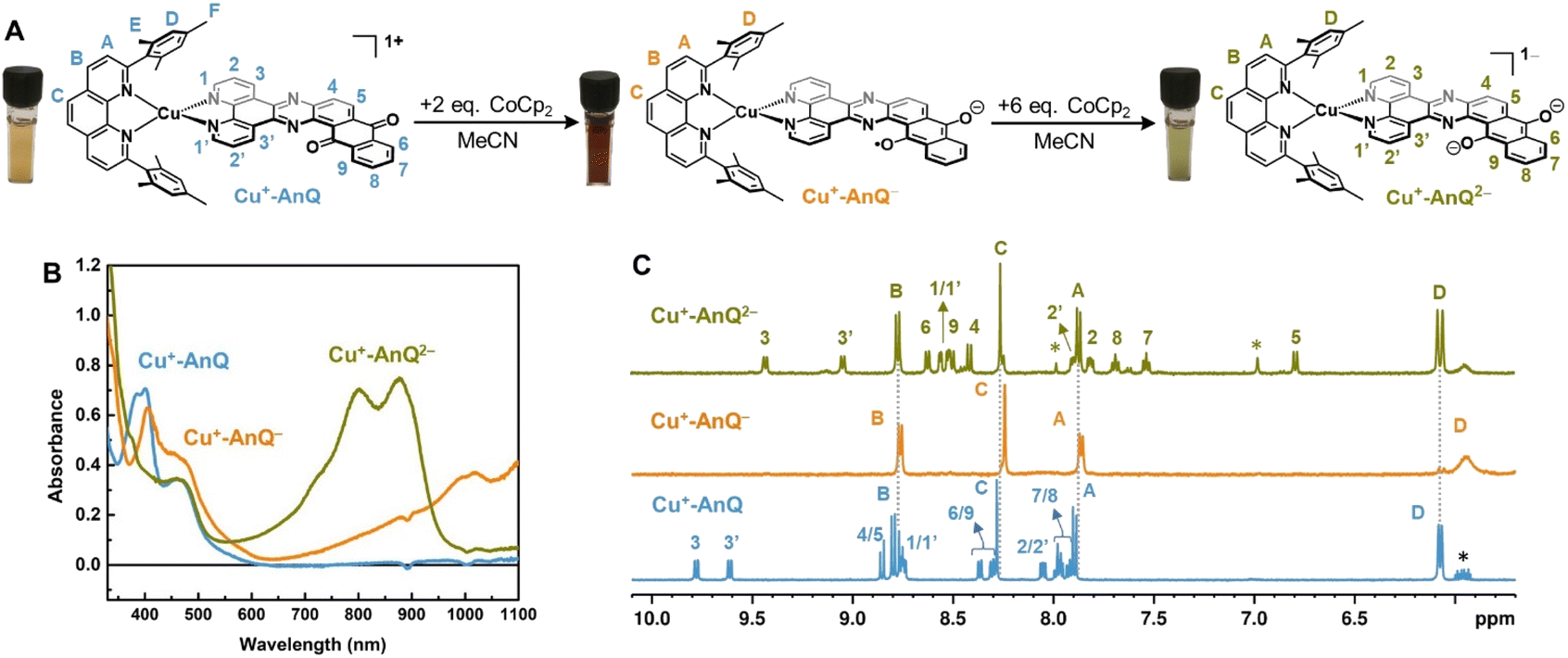 | ||
| Fig. 4 (A) Reduction of Cu+-AnQ. (B) UV-vis spectra of Cu+-AnQ treated with CoCp2 in CH3CN. (C) 1H NMR of the reduced species generated by CoCp2. (Solvent: CD3CN; impurity is denoted by asterisk). | ||
To further characterize the structure of the reduced species, 1H NMR spectroscopy was performed with the reduced species dissolved in CD3CN. In Fig. 4C, the spectra of Cu+-AnQ− and Cu+-AnQ2− are compared with the as-synthesized complex Cu+-AnQ. The singly reduced species Cu+-AnQ− displays only the signals of the mesPhen protons in the aromatic region of the spectrum, whereas the resonances of the protons of AnQ are absent since the unpaired electron is localized on the anthraquinone moiety of Cu+-AnQ−.
The peaks of the mesPhen protons (HA, HB, HC) remain almost unchanged in terms of peak width and position-compared with those from Cu+-AnQ due to the lack of electronic coupling between the mesPhen and AnQ ligand. However, the signals corresponding to the aromatic protons (HD) on the mesityl groups are broadened and shifted upfield to 5.93 ppm with respect to 6.07 ppm in Cu+-AnQ, as a result of the mesityl groups stacking with the AnQ ligand through π–π interaction (see Fig. 1 above). Reduction of the AnQ ligand will accordingly implement a stronger influence on the NMR shifts of the mesityl protons (HD, HE and HF) than those of phenanthroline protons of mesPhen. Indeed, the chemical shift of protons HE′ and HF′ of Cu+-AnQ− are also broadened and shifted upfield (HE′ 1.59 ppm and HF′ 1.27 ppm) compared with the corresponding protons of Cu+-AnQ (HE 1.77 ppm and HF 1.29 ppm) (Fig. S29†).
The 1H NMR spectrum of the doubly reduced Cu+-AnQ2− reveals well-resolved peaks assignable to the protons from both ligands, indicating the two unpaired spins are antiferromagnetically coupled (singlet state) resulting in a diamagnetic complex (green trace, Fig. 4B). Due to the anionic character of the AnQ ligand, all the protons on AnQ are shifted upfield in the NMR spectra compared with Cu+-AnQ, while the chemical shifts of mesPhen protons are relatively unaffected. Looking at the aliphatic region (Fig. S29†), the chemical shift of HF′′ is moved to 1.45 ppm with respect to HF of Cu+-AnQ (1.29 ppm) and HE′′ is observed at 1.74 ppm, a 0.03 shift from 1.77 ppm of HE. The UV-vis and NMR spectroscopy following chemical reduction clearly establish the capacity of Cu+-AnQ to store multiple charges on the ligand while maintaining the structural integrity of the complexes, paving our way into photochemical charge accumulation.
EPR spectroscopy of the reduced species
Additional direct evidence showing that the extra electrons of the singly and doubly reduced species are localized on the anthraquinone unit of the AnQ ligand was obtained by CW X-band (9.7 GHz) EPR spectroscopy. The chemically generated Cu+-AnQ− and Cu+-AnQ2− were prepared in 1:1 MeCN:dichloromethane (DCM) mixture to facilitate glass formation at 100 K. The presence of DCM in the solution does not change the speciation of the complex and the UV-vis spectra of the reduced complexes in this solvent mixture is consistent with that obtained in neat MeCN (Fig. S30†).
A comparison of the EPR spectra of parent complex Cu+-AnQ, and reduced complexes Cu+-AnQ− and Cu+-AnQ2− is presented in Fig. 5A. We detected no signs of paramagnetic Cu2+, another evidence of the excellent stability of the reduced species. The parent compound Cu+-AnQ shows no EPR signal, consistent with the diamagnetic character of the Cu(I) complex.
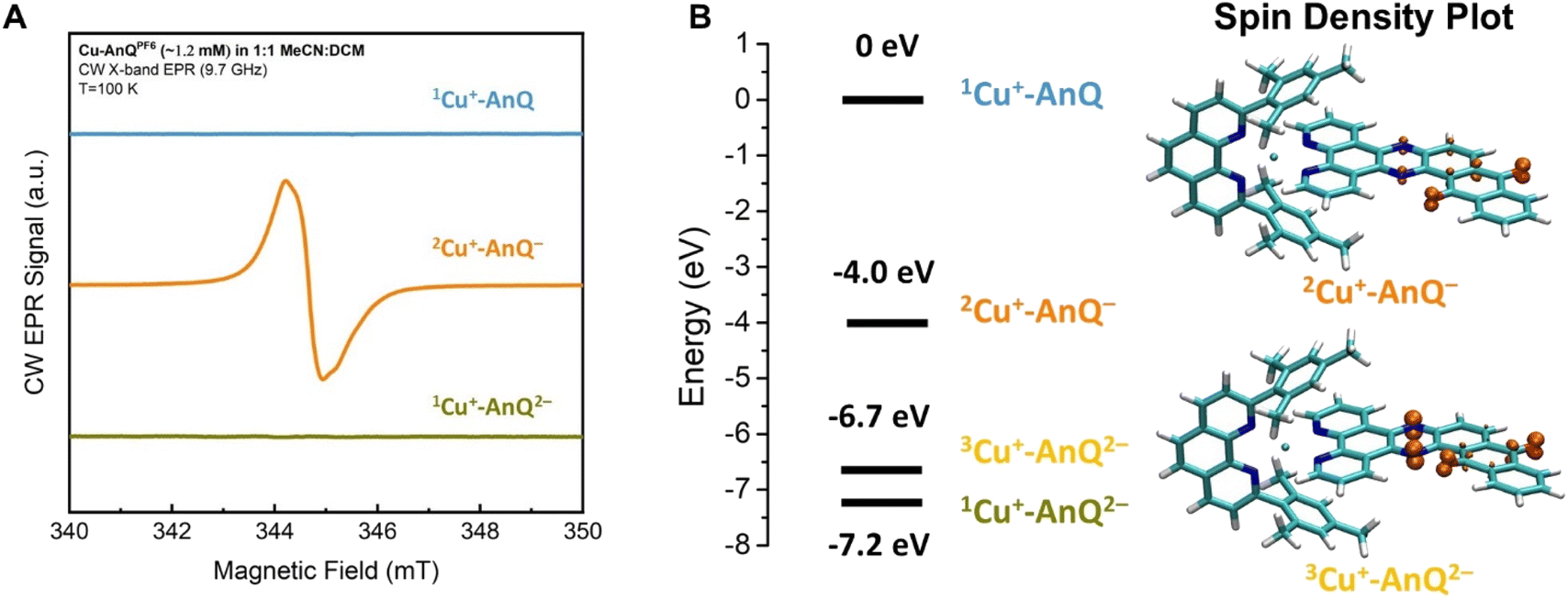 | ||
| Fig. 5 (A) CW EPR spectra of Cu+-AnQ, Cu+-AnQ− and Cu+-AnQ2−. (B) Energy level of Cu+-AnQ, 2Cu+-AnQ−, 3Cu+-AnQ2− and 1Cu+-AnQ2− and the spin density plot of 2Cu+-AnQ− and 3Cu+-AnQ2−. | ||
The singly reduced Cu+-AnQ− shows a strong EPR signal centered at g ≈ 2.004–2.005 (orange spectrum in Fig. 5A), which is in the expected range for an organic anthraquinone radical anion without significant spin density on heavier atoms (like Cu) and is comparable to values reported for similar quinone radical anions (Table S4†).66–68 The slight deviation from the free electron value of ge = 2.002319 is mostly due to spin density on the oxygen atoms.69 Indeed, the DFT calculations revealed that the unpaired electron spin in Cu+-AnQ− is delocalized over the anthraquinone moiety and the nitrogen containing bridging phenazine ring (Fig. 5B). This g-tensor anisotropy is too small to be resolved at X-band frequency. Moreover, the DFT calculation of hyperfine interactions showed three strongly coupled magnetic nuclei, the two nitrogen atoms of the bridging ring and one of the hydrogen atoms in the aryl ring between phenazine and quinone (see ESI Table S4†). This is consistent with the observed partially resolved hyperfine structure in Fig. 5A since the most abundant isotopes of carbon (12C) and oxygen (16O) are not magnetic and can be ignored. Based on the DFT calculation, all other nuclei show weaker hyperfine couplings. In particular, the Cu nucleus shows hyperfine couplings of less than 1 MHz, which is expected since the unpaired spin density is largely localized on the anthraquinone moiety of the molecule.
The doubly reduced Cu+-AnQ2− is EPR silent (green spectrum, Fig. 5A). The DFT calculation (Fig. 5B) reveals that the triplet spin state 3Cu+-AnQ2− is about 0.5 eV higher in energy than the singlet 1Cu+-AnQ2−, corroborating that the two spins on anthraquinone moiety are antiferromagnetically coupled, resulting in a diamagnetic molecule. Interestingly, the spin density plot of higher energy triplet state 3Cu+-AnQ2− presented in Fig. 5B also preserves a major contribution from the oxygen atoms, but greater spin density is located at the phenazine nitrogen atoms than Cu+-AnQ−.
The experimental EPR data and the DFT calculations nicely align with the other results and discussion above, allowing us to confirm that the first and second reduction of Cu+-AnQ occur at the anthraquinone unit. As shown in the spin density plot (Fig. 5B), both phenazine and anthraquinone units contribute to the overall spin and a substantial fraction of the spin resides on the oxygen-containing ring of the anthraquinone.
Photochemical charge accumulation with sacrificial electron donor
The capacity of Cu+-AnQ to accumulate multiple charges via electrochemical and chemical methods motivated us to investigate photochemical charge accumulation with this complex using 1,3-dimethyl-2-phenylbenzimidazoline (BIH) as a sacrificial electron donor (SED). BIH, with the first oxidation potential at +0.32 V vs. SCE,70–72 is a two electron one proton donor (Scheme 2) and has been commonly used as a SED in the photoreduction of CO2 and reduction of organic species.73 Upon losing one electron, the radical cation BIH˙+ is acidic and donates one proton to an acceptor, resulting in the deprotonated BI˙. This radical BI˙ is a potent reductant (E (BI+/BI˙) = −1.67 V vs. SCE) and will easily give away a second electron forming BI+.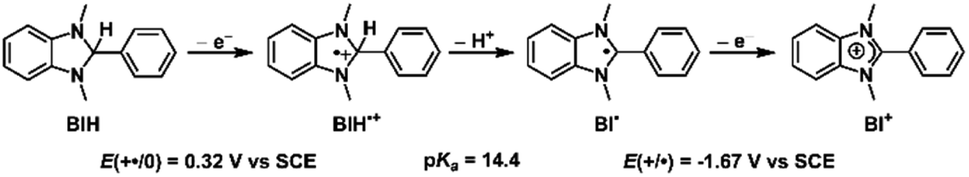 | ||
| Scheme 2 Oxidation and deprotonation process of BIH. | ||
In a continuous photolysis experiment, Cu+-AnQ (0.2 mM) and BIH (0.1 M) were mixed in the dark. Surprisingly, the UV-vis spectrum of the mixture, compared with that of Cu+-AnQ, shows a depletion at 384 nm and increase of absorption at λ > 650 nm, indicating the formation of singly reduced Cu+-AnQ− upon addition of BIH in the dark (Fig. 6). In view of thermodynamics, BIH is not a strong enough reductant to donate one electron and generate the singly reduced Cu+-AnQ− in the dark (Fig. S34†). However, the counterintuitive reduction of Cu+-AnQ by BIH can be rationalized by the extreme excess of BIH compared to the complex (500:1) and possibly the formation of a pre-assembly between BIH and Cu+-AnQ through π–π stacking interactions. Similar phenomena has been observed in previous examples using Ru(II) based photosensitizers.18,50
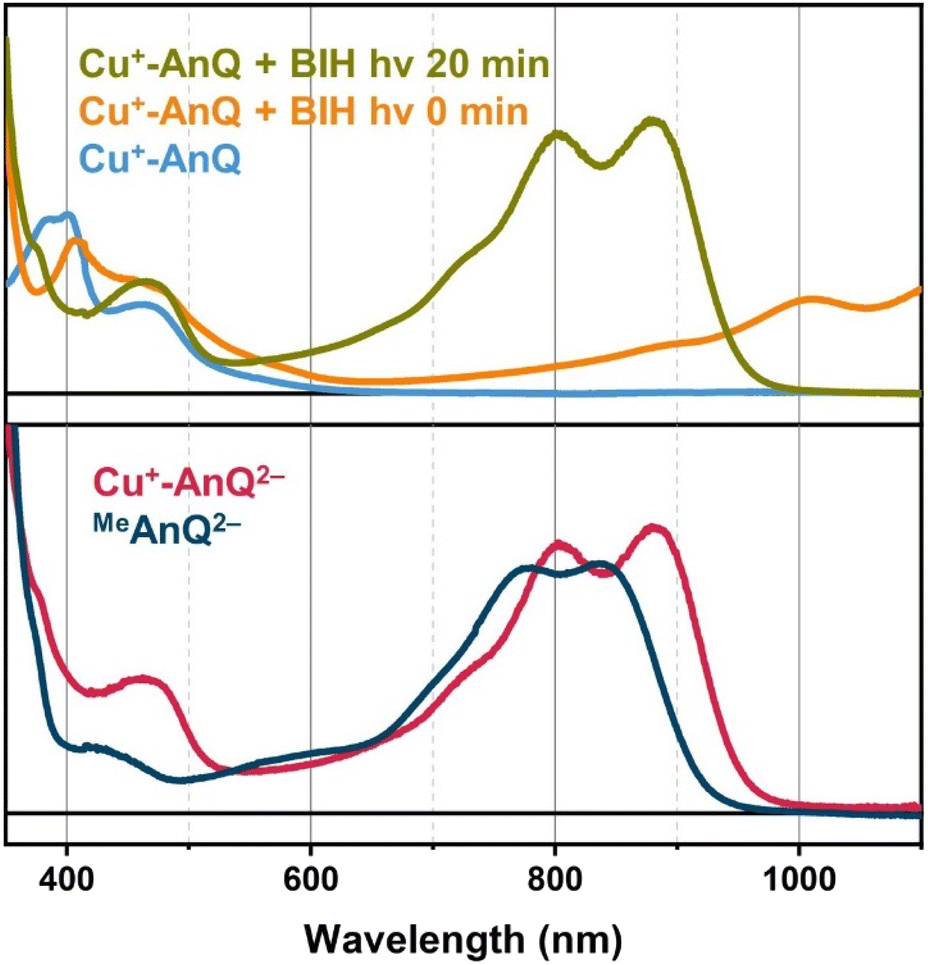 | ||
| Fig. 6 Top: UV-vis spectra of Cu+-AnQ (blue line), Cu+-AnQ + BIH with 0 min illumination (orange line), Cu+-AnQ + BIH with 20 min illumination at 455 nm (green line). Bottom: UV-vis spectra of Cu+-AnQ2− generated from chemical reduction (red line), and MeAnQ2− generated from photoreduction by BIH with 455 nm illumination (dark blue line). For all measurements, the concentrations of the complex, MeAnQ and BIH in acetonitrile were 0.2 mM, ∼0.1 mM and 0.1 M, respectively. | ||
Upon illuminating Cu+-AnQ and BIH with 455 nm light for 20 min, a double-peaked feature at 804 nm and 880 nm, corresponding to Cu+-AnQ2−, was observed in the spectrum (Fig. 6). The conversion of Cu+-AnQ to Cu+-AnQ2− is estimated to be quantitative by comparison with the chemical reduction presented in Fig. 4A. The AnQ ligand stays bound with the Cu center after being doubly reduced, which is supported by the persistent Cu(I) MLCT band in this experiment. The intensity of the absorption band of Cu+-AnQ2− in the photolysis experiment showed only small decrease after being stored under room light for 24 h (Fig. S31A†), demonstrating the stability of the doubly reduced complex in the presence of excessive SED. In contrast, the illumination of a Cu+-AnQ solution without BIH revealed no change in the UV-vis spectrum (Fig. S32†), demonstrating the photostability of Cu+-AnQ and the role of BIH as an electron source.
The control experiment in the dark was performed with the same solution of Cu+-AnQ with excess BIH and after 40 minutes a slight increase in the intensity of features at 804 nm, 880 nm and 1008 nm were observed (Fig. S31A†), which shows that a small amount of Cu+-AnQ− and Cu+-AnQ2− is generated even in the dark. By comparing these spectra to the chemically reduced spectra shown in Fig. 4, the estimated conversion of Cu+-AnQ to Cu+-AnQ2− in the dark, however, is only about 10% and the concentration of the reduced species are largely unchanged after 24 hours in the dark under N2 (Fig. S31A†). We propose that the formation of Cu+-AnQ2− without light derives from either the disproportionation of the singly reduced species into doubly reduced species (see the DFT calculation and discussion below), or hydrogen atom transfer from BIH˙+ species. Therefore, the dark process is also present in the reaction scheme, although it is evident that under illumination Cu+-AnQ can be fully converted into the doubly reduced complex in a short time, confirming photodriven intramolecular charge accumulation. Additionally, this photochemical charge accumulation is fully reversible, as demonstrated by exposure of light- and BIH-generated Cu+-AnQ2− to air for 30 minutes (Fig. S31A†). In this experiment, we observed that the spectral features belonging to Cu+-AnQ2− disappeared and only the features assigned to Cu+-AnQ− remain. The complex was not fully converted back to the Cu+-AnQ ground state, likely because of the large amount of BIH present in solution, but there is no noticeable bleaching in the MLCT band (455 nm), indicating the complex stays intact during oxidation by O2 and the process of photochemical charge accumulation is reversible.
Next, we examined the possibility of achieving photochemical charge accumulation of Cu+-AnQ with decreased amount of BIH. In terms of the thermal reduction process of Cu+-AnQ to Cu+-AnQ− by BIH, we noticed that such process can be depressed by lowering the concentration of BIH. As shown in Fig. S33,† the amount of Cu+-AnQ− generated in the dark process when BIH is at 10 mM is estimated to be about 10% of the total quantity of the complex based on the intensity of the absorption bands. In contrast, there was more than 90% of conversion of Cu+-AnQ into Cu+-AnQ− in the dark when the concentration of BIH was raised to 0.1 M. Looking at the photolysis step, the rate of charge accumulation declined as the concentration of BIH decreases. The time for the photogenerated doubly reduced species to reach maximum is 18 min, 21 min and 40 min with [BIH] = 50 mM, 10 mM and 5 mM, respectively. Notably, when the concentration of BIH is lowered to 1 mM, only the singly reduced species Cu+-AnQ− was generated. This can be accounted for by the decreased BIH concentration leading to lower driving force for photoreduction so that other reaction pathways, such as photodegradation and comproportionation, outperform the charge accumulation. Overall, we can confirm the photodriven nature of the charge accumulation of Cu+-AnQ by BIH although we acknowledge that it is complicated by a variety of thermal processes due to the strong reducing power of BIH, coupled proton transfer, and possible pre-assembly between BIH and Cu+-AnQ.
The important task that remains is to understand the interactions between the Cu(I) center and the ligand of Cu+-AnQ in the observed photochemical charge accumulation to confirm that the Cu(I) center is indeed functioning as a light absorber in this process. To this end, we subjected the methylated ligand MeAnQ to similar photolysis experiments as for the Cu+-AnQ complex. The methyl groups do not significantly change the electronic properties of MeAnQ compared with those of Cu+-AnQ, as is evidenced by the comparable electrochemical and optical behaviors to those derived from the ligand in Cu+-AnQ (Fig. 2A, S18, S20 and S31†). As in the case of Cu+-AnQ, adding BIH to an acetonitrile solution of MeAnQ resulted in the formation of singly reduced MeAnQ−, and illumination of the mixture with 455 nm light led to the doubly reduced species MeAnQ2− (Fig. S31C†). This indicates that the ligand on its own is also photoactive and functions as a chromophore to absorb light which enables photochemical charge accumulation. As both neutral and singly reduced species of MeAnQ exhibit some absorption at around 450 nm in the UV-vis spectra (Fig. 2A, S20†), the ligand can be excited with the 455 nm LED via the red edge of its π → π* electronic transition. The excited state of the ligand presumably has lower energy than BIH and can be reductively quenched by BIH, generating the doubly reduced anthraquinone moiety (Fig. S34†).
To isolate the contribution of the Cu center in the process of the photochemical charge accumulation, we changed the sacrificial electron donor from BIH to triethylamine (TEA), 0.1 M in solution. TEA has a much higher oxidation potential in acetonitrile than BIH (E (TEA+/TEA) ≈ +0.8 V vs. SCE vs. E (BIH+/BIH) = +0.32 V vs. SCE, see Fig. S19 and S34†). As is shown in Fig. S31D,† no thermal reduction of MeAnQ to MeAnQ− was observed upon mixing the ligand with TEA and the features of MeAnQ2− were not present during the photolysis of MeAnQ with TEA, but instead a new band centered at 525 nm and a depletion at 400 nm were noted. Considering the fact that this new feature is slightly blue-shifted with respect to the feature of the phenazine radical (λmax = 591 nm, Fig. S20†) and an almost complete recovery of the spectrum of the starting material was observed after introducing air into the reaction mixture (Fig. S31D†), the absorption at 525 nm is reminiscent of the protonated phenazine anion where the protons are derived from degradation of TEA74,75 or trace water after photoinduced electron transfer. Similar species have been observed by the MacDonnell group with a series of phenazine-containing Ru complexes or ligands, and the protonated doubly reduced species exhibit absorption in the range of 500–600 nm, bluer than the singly reduced phenazine radicals.76,77 Although the thermodynamics of phenazine and anthraquinone in MeAnQ dictates that phenazine anion should not be generated, certain photochemical pathway via a ligand-centered transition relevant to the phenazine moiety could presumably be accessed when coupled with protonation. Nonetheless, a detailed investigation of this process is out of the scope of this study. Switching to the photolysis of Cu+-AnQ with TEA (Fig. S31D†), generation of Cu+-AnQ2− was detected, based on the increase of absorption at 800 nm and 880 nm, but complete formation of the doubly reduced complex required longer illumination times with TEA than for BIH (240 vs. 20 minutes). Other features at 550 nm and 730 nm were also captured in the spectrum of the photolysis, suggesting multiple products of this reaction. The absorption at 730 nm is derived from the doubly reduced species interacting with residual water in the reaction mixture through hydrogen-bonding (the triethylamine was not dried prior to use). The same absorption can be reproduced in the spectroelectrochemical experiment of Cu+-AnQ when a small amount of water was added into the solution (Fig. S21†), which exhibited a broad featureless band at 730 nm and a bleaching at 400 nm. The formation of the doubly reduced species using TEA confirms the role of the Cu center as a chromophore that absorbs visible light and transfers its excited electron to the anthraquinone acceptor toward photochemical charge accumulation.
Protonation state of the doubly reduced species generated in the photochemical charge accumulation with BIH
Since BIH is a proton donor in addition to an electron donor (Scheme 3), and quinones are well-known to be good proton acceptors,13,14 we wanted to specifically investigate the ability of the reduced AnQ ligand to accept the proton from BIH˙+. A mixture of Cu+-AnQ and BIH was dissolved in CD3CN and the resultant mixture was illuminated with 455 nm light for 30 minutes, during which time the reaction was monitored by UV-vis to ensure the complete transformation of the starting material to the doubly reduced species, and NMR spectroscopy was used to identify the final product. Fig. 7 shows that the photoreduced product has comparable NMR signals with that of Cu+-AnQ2− afforded by the chemical reduction of CoCp2, with the addition of a well-resolved singlet peak at 14.88 ppm that integrates to exactly one proton. The same NMR features can be reproduced by treating the chemically generated Cu+-AnQ2− with one equivalent of trifluoroacetic acid (TFA), and the “D2O shake” experiment demonstrates that the proton is exchangeable with the deuterium of D2O as it completely disappears after the addition of D2O. We interpret these results as the newly observed peak arising from a monoprotonated, doubly reduced photoproduct Cu+-AnQH−. Notably, the 14.88 ppm signal of Cu+-AnQH− is similar to the δ(OH) (14.9 ppm in CDCl3) of the structurally-analogous 10-hydroxybenzo[h]quinoline.78,79 The drastic down field shift of the proton signal indicates that the proton of Cu+-AnQH− should bond with the O or N atom inside the bay area of the AnQ ligand forming an intramolecular H bonding of –OH⋯N. Interestingly, the protonation does not shift the other proton resonances from the parent complex, as no apparent change in the peak position of Cu+-AnQH− is observed with respect to the signals from Cu+-AnQ2− which suggests a weak electrostatic interaction of the proton with the reduced ligand leading to little distortion of the electron density of the complex.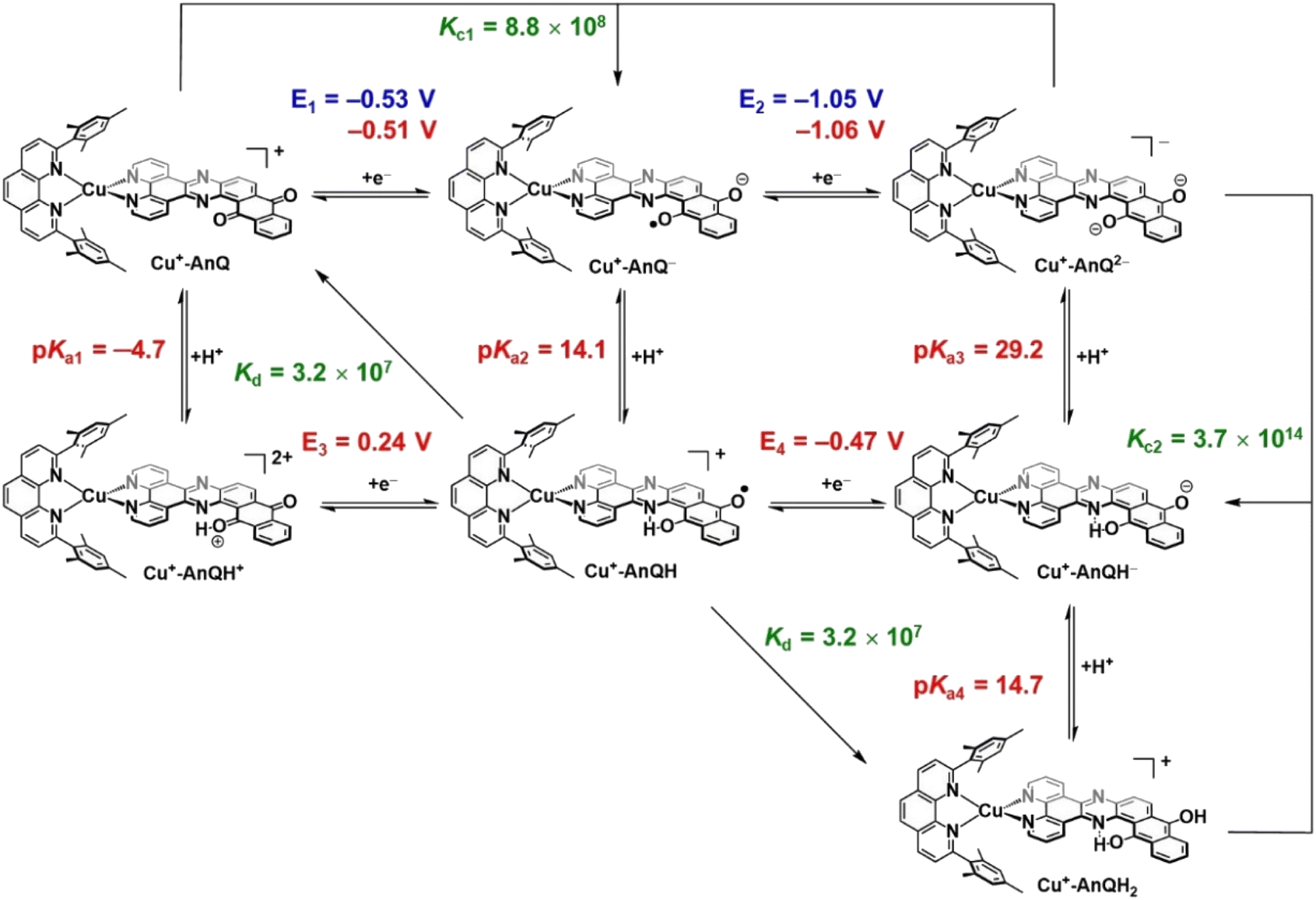 | ||
| Scheme 3 Square diagram of possible protonation and reduction events starting from Cu+-AnQ. Experimental E values in blue; calculated E and pKa in red; calculated comproportionation or disproportionation values in green. | ||
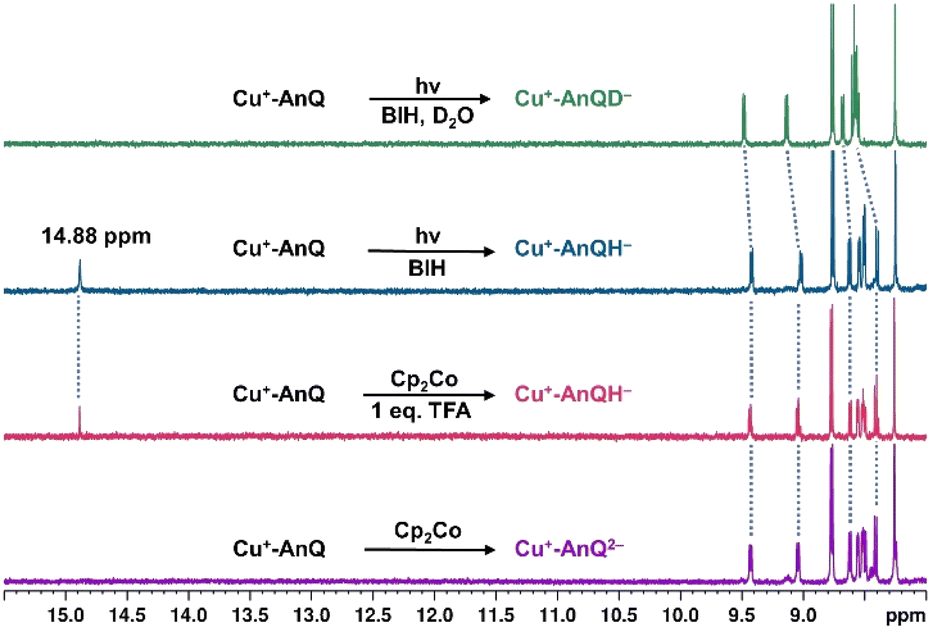 | ||
| Fig. 7 Comparison of the 1H NMR spectra of Cu+-AnQ2− (purple line), Cu+-AnQH− generated by Cp2Co and 1 eq. trifluoroacetic acid (red line), Cu+-AnQH− generated by 30 minutes illumination with BIH (teal line), Cu+-AnQD− generated by BIH with 30 min illumination and then added with one drop of D2O (green line). Illumination with 455 nm light; concentrations of the complex and BIH were kept at 0.2 mM and 0.1 M, respectively, in CD3CN. | ||
The structure of Cu+-AnQH− is further revealed by rotating frame Overhauser effect spectroscopy (ROESY). In our first attempt at this measurement, the acetonitrile-d3 solution of Cu+-AnQ was illuminated using the same conditions for the above experiments, however we observed the crystallization of the photoproduct in the reaction mixture within several hours. The low solubility of the product in acetonitrile prevented us from performing a prolonged NMR measurement. Therefore, we changed the solvent to DMSO-d6 which solubilized the photoreduced product throughout the measurement. The UV-vis and NMR spectra (Fig. S39–S42†) unambiguously demonstrated that the complex undergoes the same photoreduction in DMSO as in acetonitrile, which generates the doubly reduced monoprotonated Cu+-AnQH−. The 2D and 1D ROESY spectra of the resultant mixture in DMSO-d6 reveals that the proton that appears at 14.9 ppm is correlated with the H3 proton, the one close to the bay of the ligand (Fig. 8A and C). Apart from that, no additional coupling of the 14.9 ppm proton with other nuclei was observed. This confirms that the O or N atom in the bay of the ligand accepts the proton during photoreduction and, importantly, intramolecular hydrogen bonding exists between the proton and an acceptor atom leading to a stabilized six-membered ring within the bay.
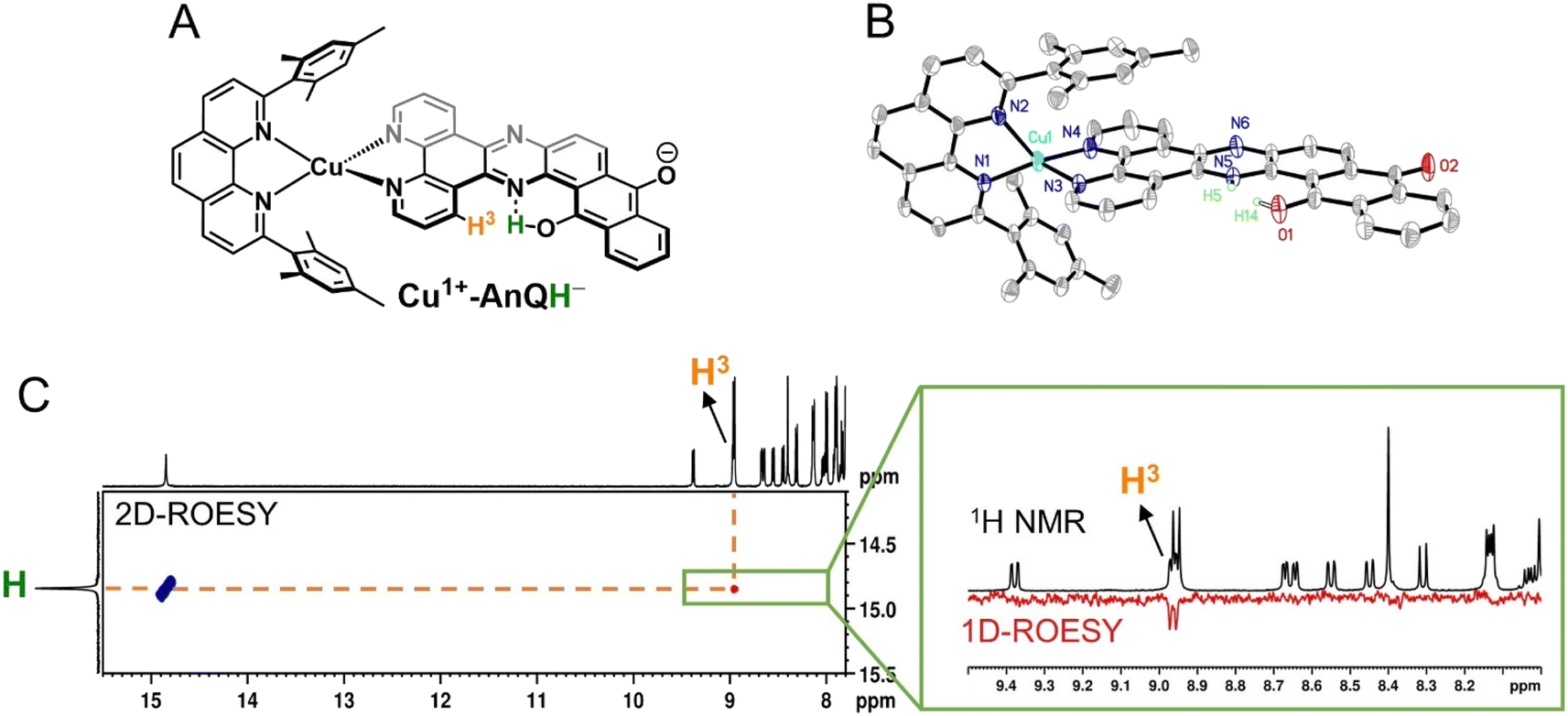 | ||
| Fig. 8 (A) Molecular structure of the enol form of the monoprotonated Cu+-AnQH−. (B) ORTEP diagram of Cu+-AnQH− (50% thermal ellipsoids; all the hydrogen atoms except for H5 and H14 are omitted for clarity). Tautomerization of the proton, affording the keto and enol forms, is exhibited as substitution disorder in the crystal structure where the proton of the keto form is labeled as H5 and the proton of the enol form is labeled as H14. The relative ratio of the keto and enol tautomer is 3:2. (C) ROESY spectrum of photogenerated Cu+-AnQH− in DMSO-d6 showing the correlation of the exchangeable proton (14.9 ppm) with other protons in the parent complex. Inset: comparison of the 1H NMR and 1D ROESY spectra from 8.0 ppm to 9.5 ppm. Complex Cu+-AnQH− was produced by illuminating a DMSO-d6 solution of 0.5 mM Cu+-AnQ and 0.1 M BIH with 455 nm light for 4 hours. | ||
Single crystal X-ray diffraction was used to determine the solid-state structure of the dark crystals obtained from the acetonitrile-d3 solution of the photoreduced species. As is shown in Fig. 8B, the crystal structure clearly shows a tautomerization between the enol form (H14) and keto form (H5) of Cu+-AnQH−, with an approximate 2:3 ratio of the enol and similar energy levels in the energy surface and both the protonation states are produced in the ground state structure. Indeed, DFT calculations (Fig. S44†) were used to investigate the energy of the four possible monoprotonated species (H on N5, N6, O1 or O2), and reveals that Cu+-AnQH−O1 shows the lowest energy although the ΔG between Cu+-AnQH−O1 and Cu+-AnQH−N5 is only 1.5 kcal mol−1. A previous study shows that the installation of electron withdrawing groups on specific positions of HBQ can modulate the relative energy level of the enol and keto isomers.80 By the same token, the similar energetics of the two isomers of Cu+-AnQH− can be attributed to the extended π-conjugation of AnQ which reduces the electron density of the reduced ligand by distributing the extra charge over the π system (see EPR section). Another key observation from the crystal structure is the difference of C–O bond distances in the AnQ ligand. Since only one proton is associated with the anthraquinone unit of the AnQ ligand, the two C–O bonds show very different lengths in the crystal structure. The distance of C–O1 is 1.300(3) Å which is slightly shorter than the normal range of C–O bond lengths for hydroquinones (∼1.35 Å)81,82 but much longer than the CO groups in quinones (∼1.23 Å).21 However, the C–O2 bond is much shorter (1.255(3) Å) than C–O1 due to its deprotonation and falls in the range of carbonyl groups found in quinones.21 Based on the bond distances, the nature of bonding of C–O1 should be single bond and C–O2 be double bond.
DFT calculated pKa and E of the Cu-AnQ intermediates
Photoinitiated charge accumulation by Cu+-AnQ presumably occurs via several proton and electron transfer steps, and we therefore employed DFT calculations to quantify the pKa and reduction potential E values of the protonated intermediates. Full calculation details are presented in the ESI,† and Scheme 3 summarizes the experimental and calculated reduction and protonation processes following photoexcitation of Cu+-AnQ. The calculated reduction potentials of the unprotonated redox couples, Cu+-AnQ0/1− and Cu-AnQ−/2−, are in good agreement with the experimental values, differing by only 10–20 mV. The large difference in the reduction potentials of the two redox couples results in comproportionation of Cu+-AnQ and Cu+-AnQ2−, generating Cu+-AnQ−; the equilibrium constant Kc1 for the comproportionation is calculated as 8.8 × 108.83The vertical direction of Scheme 3 shows protonation of the Cu complexes. In general, as reduction occurs from Cu+-AnQ, we observe an increase in the pKa values, demonstrating an increase of basicity as the complex is reduced. Specifically, the pKa values of the as-synthesized complex Cu+-AnQ, the singly reduced Cu+-AnQ− and the doubly reduced Cu+-AnQ2− are −4.7, 14.1 and 29.2, respectively. Comparing with the pKa of radical BIH˙+ (14.4 (ref. 70–72)), Cu+-AnQ is highly unlikely to be protonated under the conditions in which the photoreduction experiment was conducted. The proton transfer from BIH˙+ to Cu+-AnQ− should have lower thermodynamic barrier, but due to the similar pKa values, this process is still uphill which results in slow proton transfer and generating only small amount of Cu+-AnQH. The doubly reduced species should be easily protonated by BIH˙+, producing Cu+-AnQH−.
The DFT calculations show that the reduction potentials of the protonated complexes are shifted positively compared to their unprotonated analogs. Specifically, the E (Cu+-AnQH+/Cu+-AnQH) and E (Cu+-AnQH/Cu+-AnQH−) are calculated as 0.24 V and −0.47 V vs. SCE, respectively, which are positively shifted by ∼600 mV with respect to the corresponding reductions of the deprotonated species. The positive change of the potential can be explained by the stabilization effect of the associated proton on the reduced complexes. The calculations also show that the reduction of the singly reduced, monoprotonated complex Cu+-AnQH occurs at a more positive potential than the reduction of the starting complex: Cu+-AnQ (E (Cu+-AnQH/Cu+-AnQH−) = −0.47 V and E (Cu+-AnQ/Cu+-AnQ−) = −0.51 V). This potential inversion has been previously observed by other studies with Ru complexes21,84,85 and has been regarded as an important strategy to achieve photoinduced charge accumulation at mild potentials in photoactive supramolecular assemblies.7
Further protonation of Cu+-AnQH− to form Cu+-AnQH2 is possible at a moderate pKa4 (14.7) for Cu+-AnQH2. This suggests that Cu+-AnQH− is less basic than Cu+-AnQ2−. However, under reaction conditions limited by BIH˙+ concentration, as in the case of the photolysis experiment, any Cu+-AnQH2 that is generated has a propensity to react with Cu+-AnQ2− in a comproportionation reaction to yield Cu+-AnQH−. The extremely high driving force for the comproportionation reaction (Kc2 = 3.7 × 1014) is a reasonable explanation for the observation of Cu+-AnQH− as the major product in photochemical charge accumulation experiment.
The thermodynamic data provide insights into the process which generates the doubly reduced monoprotonated species in the dark. This is related to the disproportionation of the monoprotonated singly reduced Cu+-AnQH into Cu+-AnQ and Cu+-AnQH2. With the pKa and reduction potential E, the equilibrium constant of the disproportionation of Cu+-AnQH is estimated to be 3.2 × 107. The moderate Kd indicates that Cu+-AnQH, derived from the protonation of Cu+-AnQ− by BIH˙+, is highly inclined to undergo disproportionation and affords Cu+-AnQ and Cu+-AnQH2. The subsequent reaction involves proton transfer from Cu+-AnQH2 to Cu+-AnQ2−, producing Cu+-AnQH. Although the disproportionation of the protonated Cu species does not require light, the major pathway for charge accumulation should be photochemical. This is based on the fact that the complete conversion of Cu+-AnQ to Cu+-AnQH− is only realized under illumination (vide supra). Overall, the calculated pKa and E agree well with what we observed in the experimental results which shows the Cu+-AnQH− is the final product.
Discussion
Reaction pathway of the photochemical charge accumulation
Based on the experimental and computational results, we are now equipped to propose a mechanism of the photochemical charge accumulation of Cu+-AnQ using BIH as a sacrificial electron donor, summarized in Scheme 4. First, the reduction of Cu+-AnQ to Cu+-AnQ− undergoes via thermal-driven mechanism or photodriven mechanism depending on the concentration of BIH in the reaction mixture. Since we observe quite efficient formation of Cu+-AnQ− upon introduction of a large excess of BIH, even prior to illumination, we attribute it to pre-assembly of BIH with the complex resulting in thermal-induced electron transfer generating Cu+-AnQ−. However, as [BIH] decreases, we observe less reduction in the dark and more Cu+-AnQ− is afforded by photodriven process due to the lower propensity of pre-assembly formation under these conditions.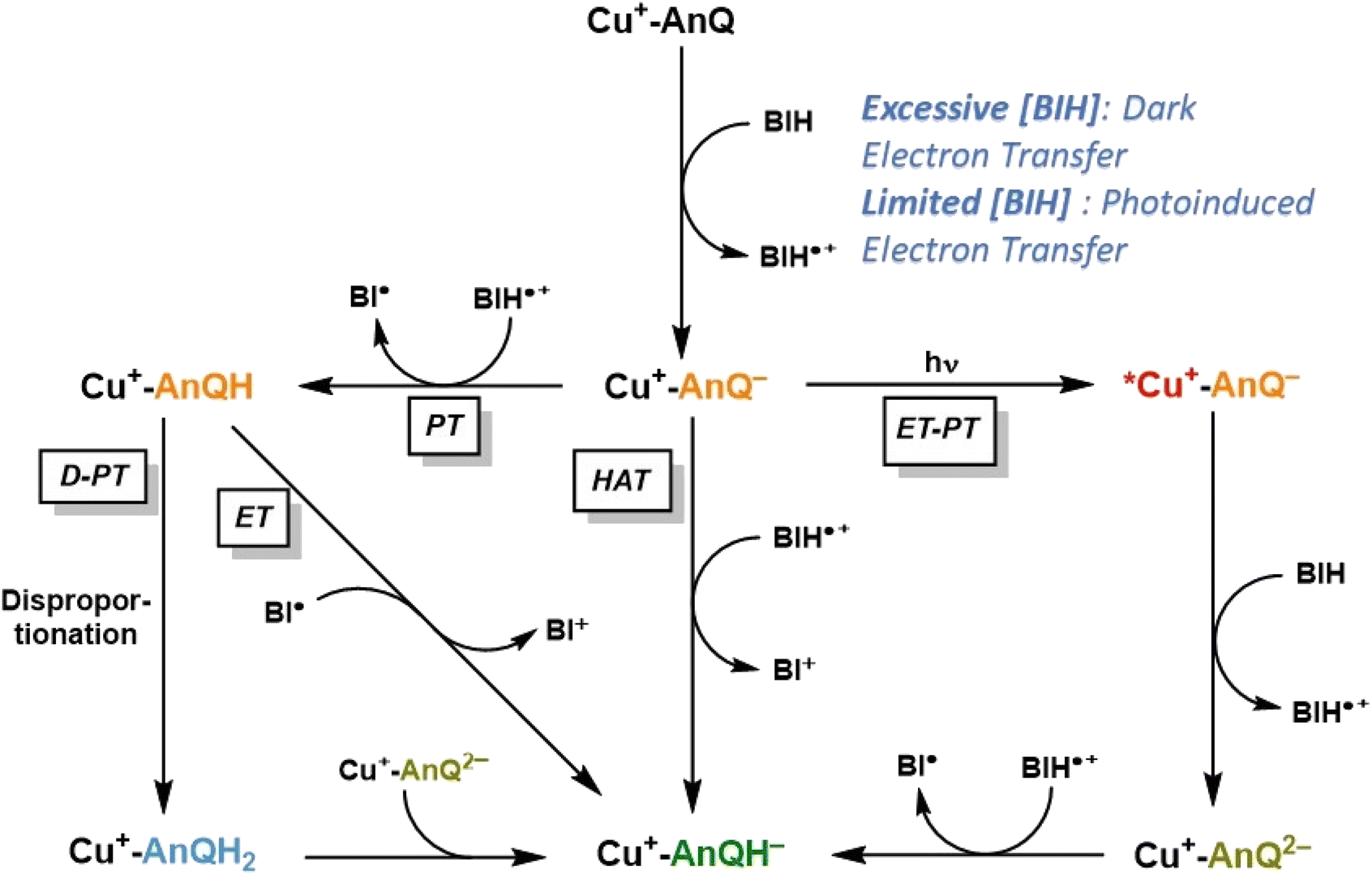 | ||
| Scheme 4 Proposed reaction mechanism of charge accumulation of Cu+-AnQ with BIH. PT = proton transfer; ET = electron transfer; D = disproportionation; HAT = hydrogen atom transfer. | ||
The following steps involve two diverging pathways, either the photoinduced pathway or thermal-induced pathway. The photoinduced pathway is comprised of the photoexcitation of Cu center, intramolecular electron transfer (ET) from Cu*+ to AnQ−, intermolecular electron transfer from BIH to Cu2+ and protonation (PT) of AnQ2− by BIH˙+. We hypothesize this pathway as the major contributing factor to drive charge accumulation reaction to completion. As shown in Fig. 6 and S31,† excitation with 455 nm light accelerates the formation of Cu+-AnQ2−, resulting in a quantitative generation of the doubly reduced species. However, this species is only afforded at very low yield in the dark (Fig. S31†). Although excitation of the copper-based MLCT initiates intramolecular photoinduced electron transfer from the Cu(I) excited state to the AnQ− ligand which generates Cu2+-AnQ2−, we do not observe this intermediate, likely because the Cu2+ center is a very good electron acceptor for BIH (or BIH˙+) which rapidly forms Cu+-AnQ2−. This intermediate is highly basic (pKa3 29.2) and readily accepts an electron from BIH˙+ that is generated from previous electron transfer steps and finally yields Cu+-AnQH−.
The thermal-induced pathway includes three possible mechanisms: PT-D-PT, PT-ET, and HAT process (Scheme 4), and we propose that these three pathways are all kinetically slower than the photoinduced pathway. The first two thermal pathways start with a common intermolecular proton transfer (PT) from BIH˙+ to AnQ−. The subsequent steps bifurcate into either electron transfer (ET) from radical BI˙ to Cu+-AnQH, or disproportionation (D) of Cu+-AnQH into Cu+-AnQ and Cu+-AnQH2, and proton transfer (PT) from Cu+-AnQH2 to Cu+-AnQ2−. The rate of these two pathways can be greatly limited by the first PT step owing to the slightly uphill thermodynamics. Considering the pKa of the two individual species, the small ΔpKa between BIH˙+ and Cu+-AnQH− suggests that the proton transfer to Cu+-AnQ− has a small thermodynamic driving force. It has been reported that the rate of deprotonation of BIH˙+ is linearly dependent on the pKa of the conjugate acid of the proton acceptor, and with a small ΔpKa (<2 units) the proton transfer from BIH˙+ to proton acceptor shows a rate constant of 103 M−1 s−1.70 On the other hand, the (photoinduced) electron transfer from BIH to Cu+-AnQ− is estimated to be kinetically efficient based on reported rate constants for photoinduced electron transfer from BIH to similar Cu(I) diimine complexes, on the order of 109 M−1 s−1.86–88 Further support for a fast electron transfer step comes from a closely related example in a molecular copper purpurin chromophore with an anthraquinone moiety that undergoes photoinduced electron transfer with BIH with a quenching rate constant of 9.23 × 109 M−1 s−1.86 Therefore, these studies suggest that the photodriven reduction of Cu+-AnQ− by BIH should outcompete the proton transfer of BIH˙+ to Cu+-AnQ−.
The third possible thermal pathway could proceed through a hydrogen atom transfer (HAT) mechanism which directly transfers a H˙ from BIH˙+ to Cu+-AnQ−, forming Cu+-AnQH−. Such mechanism has been observed in photochemical CO2 reduction involving proton-donating organic sacrificial electron donors89,90 and photoreduction of phenanthrenequinone by alcohols.91 Considering the pKa and E of the Cu species and BIH˙+, the ΔG for the HAT pathway is estimated to be −1.50 eV (Scheme S4†), indicating a viable pathway for the thermal-induced charge accumulation. However, compared with the photoinduced ET-PT process, we deduce that such HAT process could also be kinetically inefficient as indicated by the literature precedents.92,93 Accordingly, the slower kinetics of the thermal-induced pathways than the photoinduced pathway implies that a sequential electron transfer then proton transfer process consisting of the photoinduced reduction of Cu+-AnQ− and the ensuing formation of Cu+-AnQH− is the most viable mechanism for the accumulation of the second charge.
Solar energy storage by the Cu anthraquinone system
An important outcome of understanding how molecular systems can be designed for photochemical charge accumulation is to be able to utilize the system to collect multiple redox equivalents following photoexcitation and perform multi-electron catalysis so that the absorbed energy can be converted into value-added chemicals or fuels. It is, therefore, crucial for the molecular system to not only efficiently accumulate charges but also absorb solar energy with low energy loss. Based on the redox potential and pKa values obtained in the experiment and calculation, we have calculated the ΔG for the following three processes (Scheme 5, see ESI† for calculation details), i.e., single electron transfer (reaction 1), double electron transfer (reaction 2) and PCET process (reaction 3), in order to assess the capacity of energy storage of Cu+-AnQ during photochemical charge accumulation. To simplify this discussion, we only consider photodriven reaction, i.e., assuming the first and second reductions only involve photoexcitation.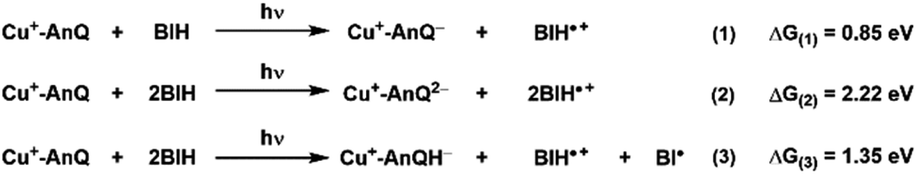 | ||
| Scheme 5 Reactions and the change of free energy of the photoinduced single ET, double ET and PCET processes between Cu+-AnQ and BIH. | ||
For reduction-only processes (reactions 1 and 2), the single electron transfer moves one electron from BIH to Cu complex and stores energy of 0.85 eV in Cu+-AnQ−. A second electron transfer increases the total energy accumulated in Cu+-AnQ2− to 2.22 eV. The energy stored in the Cu-AnQ system is lower than the previously-reported Ru(II) triarylamine anthraquinone pentad complex, which contains similar anthraquinone group as the charge accumulation site.21,22 The Ru pentad system transfers electron from triarylamine to anthraquinone under photochemical conditions and stored substantial amount of energy in the deprotonated doubly reduced species (ΔG = 3.56 eV). We attribute the lower energy storage in our deprotonated species than the Ru pentad complex to the change of the energy level of the electron donors and acceptors. First, the energy of the electron donor (BIH) used with Cu+-AnQ is higher than that of the Ru pentad (the electron donor in Ru pentad is triarylamine with Eox = 0.70 V vs. SCE and the BIH Eox = 0.32 V vs. SCE), making it easier for BIH to donate electrons to the AnQ moiety than triethylamine and thus limits the energy capacity of the system. Second, the extended π-conjugation of the AnQ ligand in Cu+-AnQ decreases the energy level of the electron acceptor, rendering the reduction potential of the AnQ ligand more positive (E (AnQ0/1−) = −0.53 V vs. SCE, E (AnQ−/2−) = −1.05 V vs. SCE) than the reduction potential of anthraquinone in the Ru pentad (E (AQ0/1−) = −0.81 V vs. SCE, E (AQ1−/2−) = −1.31 V vs. SCE). This is ascribed to the fact that the anthraquinone in the Ru pentad complex are linked by an aryl group; the connection between the anthraquinone and the Ru chromophore is not a conjugated π-system. This allows the anthraquinone moiety in the Ru pentad to maintain its electronic properties close to a discrete molecule and preserve higher energetics in the system.
Considering the effect of protonation (reaction 3), however, the difference in potential energy stored between the two systems becomes less remarkable. As shown in Scheme 5, protonation of Cu+-AnQ2− dramatically stabilizes the doubly reduced species, reducing the stored energy to 1.35 eV. Whereas for the Ru pentad, the energy loss resulting from the protonation is about 2 eV, reducing the total stored energy down to 1.51 eV. The lower energy loss observed here for the Cu-AnQ complex can be explained by the fact that the photoproduct of the Cu-AnQ system is monoprotonated due to the low basicity of Cu+-AnQH− (pKa = 14.7 for Cu+-AnQH2), while the final photoproduct of Ru pentad, according to the authors, is a diprotonated species (AQH2, pKa was estimated to be 29.3). This demonstrates a unique strategy to preserve the solar energy accumulated in the molecular system by tuning the basicity and structure of the reduced species to control the protonation state of the product.
Conclusions
In summary, we have described the synthesis and complete characterization of the first photoactive earth-abundant Cu(I) diimine complex, Cu+-AnQ, that is effective for long-lived and stable photochemical charge accumulation. Electrochemical characterization (CV and SEC) confirms that the AnQ ligand can reversibly accept up to three electrons in the π system of the ligand under electrochemical conditions. The singly reduced Cu+-AnQ− and the doubly reduced species Cu+-AnQ2− were also generated by chemical reduction and characterized by NMR, EPR and DFT calculations which uncover that the accumulated electrons of the singly and doubly reduced products reside mainly on the anthraquinone moiety. Regeneration of the Cu+-AnQ complex upon exposure of the reduced products to air demonstrates the reversibility of the charge accumulation process with the Cu system and opens the door for its study on photochemical charge accumulation. Continuous photolysis of Cu+-AnQ in acetonitrile in the presence of BIH as sacrificial electron donor exhibits photodriven reduction of the AnQ ligand by BIH. The structure of the photoproduct is conclusively determined by NMR and X-ray crystallography as the monoprotonated, doubly reduced Cu+-AnQH−. Computational investigation of the thermodynamics of the possible intermediates of the Cu-AnQ system suggests that an ET process generating Cu+-AnQ− and a subsequent ET-PT process is the probable reaction pathway in which the photochemical charge accumulation occurs. The experimental characterization and DFT calculations allow us to estimate that the energy stored in the photochemical charge accumulation by Cu+-AnQH− is 1.34 eV.This work demonstrates that the protonation state and location of a photo- or electrochemically reduced species can be controlled by ligand design and proton stabilization via intramolecular hydrogen bonding interactions. Such control of the protonation state can be a potentially universal strategy to preserve harnessed solar energy in a molecular assembly for photochemical charge accumulation. Furthermore, the ability of Cu+-AnQ to achieve photochemical charge accumulation is reminiscent of the biological electron transfer molecule plastoquinone (PQ) in photosystem II. Like the photosystem converts the solar energy into reduction equivalents that are utilized for biological CO2 reduction, it is interesting to further investigate reactions that exploit the accumulated charges by the Cu anthraquinone complex to achieve solar energy conversion. Ongoing work in our group is focused on time-resolved studies to evaluate how ultrafast kinetics of charge separation influences longer-timescale charge accumulation and protonation events. Additionally, given the exciting potential for quinone compounds for direct electrochemical capture of CO2,94,95 we are pursuing this and related systems in the context of photocatalytic CO2 capture and sequestration.
Data availability
Data are available from the corresponding author upon reasonable request.Author contributions
Conceptualization—Z.-L. X., K. L. M.; investigation—Z.-L. X., N. G., J. N., O. G. P., V. M. L.; formal analysis—Z.-L. X., J. N., O. G. P., V. M. L.; writing original draft—Z.-L. X., K. L. M.; writing, review and editing—Z.-L. X., N. G., J. N., O. G. P., K. D. G., K. L. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through contract no. DE-AC02-06CH11357. We gratefully acknowledge the computing resources provided on Bebop, a high-performance computing cluster operated by the Laboratory Computing Resource Center at Argonne National Laboratory. We thank Dr John Muntean for helpful discussions regarding the NMR experiments.Notes and references
- S. Xu and E. A. Carter, Chem. Rev., 2018, 119, 6631–6669 CrossRef PubMed.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790–6831 RSC.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- T. H. Bürgin and O. S. Wenger, Energy Fuels, 2021, 35, 18848–18856 CrossRef PubMed.
- Y. Pellegrin and F. Odobel, Coord. Chem. Rev., 2011, 255, 2578–2593 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 2002, 92, 435–461 CrossRef.
- J. Barber, Chem. Soc. Rev., 2008, 38, 185–196 RSC.
- V. Krewald, M. Retegan and D. A. Pantazis, Top. Curr. Chem., 2015, 371, 23–48 CrossRef PubMed.
- N. Nelson and C. F. Yocum, Annu. Rev. Plant Biol., 2006, 57, 521–565 CrossRef CAS PubMed.
- P. Heathcote, P. K. Fyfe and M. R. Jones, Trends Biochem. Sci., 2002, 27, 79–87 CrossRef CAS PubMed.
- M. R. Gunner, J. Madeo and Z. Zhu, J. Bioenerg. Biomembr., 2008, 40, 509–519 CrossRef PubMed.
- H. Kurreck and M. Huber, Angew. Chem., Int. Ed., 1995, 34, 849–866 CrossRef CAS.
- J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef CAS PubMed.
- M. E. El-Khouly, E. El-Mohsnawy and S. Fukuzumi, J. Photochem. Photobiol., C, 2017, 31, 36–83 CrossRef CAS.
- R. Konduri, N. R. de Tacconi, A. Krishnan Rajeshwar and F. M. MacDonnell, J. Am. Chem. Soc., 2004, 126, 11621–11629 CrossRef CAS PubMed.
- R. Konduri, H. Ye, F. M. Macdonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed.
- N. R. de Tacconi, R. O. Lezna, R. Konduri, F. Ongeri, K. Rajeshwar and F. M. MacDonnell, Chem.–Eur. J., 2005, 11, 4327–4339 CrossRef CAS PubMed.
- M. Kuss-Petermann, M. Orazietti, M. Neuburger, P. Hamm and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 5225–5232 CrossRef CAS PubMed.
- M. Orazietti, M. Kuss-Petermann, P. Hamm and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 9407–9410 CrossRef CAS PubMed.
- S. Singh, N. R. de Tacconi, N. R. G. Diaz, R. O. Lezna, J. M. Zuñiga, K. Abayan and F. M. MacDonnell, Inorg. Chem., 2011, 50, 9318–9328 CrossRef CAS PubMed.
- J.-F. F. Lefebvre, J. Schindler, P. Traber, Y. Zhang, S. Kupfer, S. Gräfe, I. Baussanne, M. Demeunynck, J.-M. M. Mouesca, S. Gambarelli, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, Chem. Sci., 2018, 9, 4152–4159 RSC.
- N. M. Randell, J. Rendon, M. Demeunynck, P. Bayle, S. Gambarelli, V. Artero, J. Mouesca and M. Chavarot-Kerlidou, Chem.–Eur. J., 2019, 25, 13911–13920 CrossRef CAS PubMed.
- D. Polyansky, D. Cabelli, J. T. Muckerman, E. Fujita, T. Koizumi, T. Fukushima, T. Wada and K. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 4169–4172 CrossRef CAS PubMed.
- T. Fukushima, T. Wada, H. Ohtsu and K. Tanaka, Dalton Trans., 2010, 39, 11526–11534 RSC.
- K. Kobayashi, H. Ohtsu, K. Nozaki, S. Kitagawa and K. Tanaka, Inorg. Chem., 2016, 55, 2076–2084 CrossRef CAS PubMed.
- H. Ohtsu and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 9792–9795 CrossRef CAS PubMed.
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 RSC.
- C. Bozal-Ginesta, C. A. Mesa, A. Eisenschmidt, L. Francàs, R. B. Shankar, D. Antón-García, J. Warnan, J. Willkomm, A. Reynal, E. Reisner and J. R. Durrant, Chem. Sci., 2021, 12, 946–959 RSC.
- M. Schulz, N. Hagmeyer, F. Wehmeyer, G. Lowe, M. Rosenkranz, B. Seidler, A. Popov, C. Streb, J. G. Vos and B. Dietzek, J. Am. Chem. Soc., 2020, 142, 15722–15728 CrossRef CAS PubMed.
- M. Ruthkosky, C. A. Kelly, F. N. Castellano and G. J. Meyer, Coord. Chem. Rev., 1998, 171, 309–322 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201–1219 CrossRef PubMed.
- C. T. Cunningham, K. L. H. Cunningham, J. F. Michalec and D. R. McMillin, Inorg. Chem., 1999, 38, 4388–4392 CrossRef CAS PubMed.
- M. Sandroni, Y. Pellegrin and F. Odobel, C. R. Chim., 2016, 19, 79–93 CrossRef CAS.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2013, 52, 8114–8120 CrossRef CAS PubMed.
- S. Garakyaraghi, C. E. McCusker, S. Khan, P. Koutnik, A. T. Bui and F. N. Castellano, Inorg. Chem., 2018, 57, 2296–2307 CrossRef CAS PubMed.
- M. C. Rosko, E. M. Espinoza, S. Arteta, S. Kromer, J. P. Wheeler and F. N. Castellano, Inorg. Chem., 2023, 62, 3248–3259 CrossRef CAS PubMed.
- M. Ruthkosky, C. A. Kelly, M. C. Zaros and G. J. Meyer, J. Am. Chem. Soc., 1997, 119, 12004–12005 CrossRef CAS.
- M. S. Lazorski, R. H. Gest and C. M. Elliott, J. Am. Chem. Soc., 2012, 134, 17466–17469 CrossRef CAS PubMed.
- A. M. Potocny, B. T. Phelan, E. A. Sprague-Klein, M. W. Mara, D. M. Tiede, L. X. Chen and K. L. Mulfort, Inorg. Chem., 2022, 61, 19119–19133 CrossRef CAS PubMed.
- X. Zhang, M. Cibian, A. Call, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 11263–11273 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- M. Schmittel, C. Michel, S.-X. Liu, D. Schildbach and D. Fenske, Eur. J. Inorg. Chem., 2001, 2001, 1155–1166 CrossRef.
- M. Schmittel and A. Ganz, Chem. Commun., 1997, 999–1000 RSC.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC.
- A. G. Bonn and O. S. Wenger, Phys. Chem. Chem. Phys., 2015, 17, 24001–24010 RSC.
- M. Kuss-Petermann and O. S. Wenger, Chem.–Eur. J., 2017, 23, 10808–10814 CrossRef CAS PubMed.
- L. Gimeno, C. Queffelec, E. Blart and Y. Pellegrin, ACS Omega, 2022, 7, 13112–13119 CrossRef CAS PubMed.
- R. B. López, B. L. Loeb, T. Boussie and T. J. Meyer, Tetrahedron Lett., 1996, 37, 5437–5440 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, J. Chem. Soc., Dalton Trans., 2007, 955–964 RSC.
- M. G. Fraser, H. Van Der Salm, S. A. Cameron, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2013, 52, 2980–2992 CrossRef CAS PubMed.
- J. Habermehl, D. Sorsche, P. Murszat and S. Rau, Eur. J. Inorg. Chem., 2016, 2016, 3423–3428 CrossRef CAS.
- J. Bolger, A. Gourdon, E. Ishow and J.-P. Launay, Inorg. Chem., 1996, 35, 2937–2944 CrossRef CAS.
- S. D. Bergman, D. Reshef, S. Groysman, I. Goldberg and M. Kol, Chem. Commun., 2002, 2, 2374–2375 RSC.
- D. Gut, A. Rudi, J. Kopilov, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2002, 124, 5449–5456 CrossRef CAS PubMed.
- R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS PubMed.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- D. Hayes, L. Kohler, R. G. Hadt, X. Zhang, C. Liu, K. L. Mulfort and L. X. Chen, Chem. Sci., 2018, 9, 860–875 RSC.
- L. Kohler, R. G. Hadt, D. Hayes, L. X. Chen and K. L. Mulfort, Dalton Trans., 2017, 46, 13088–13100 RSC.
- L. Kohler, D. Hayes, J. Hong, T. J. Carter, M. L. Shelby, K. A. Fransted, L. X. Chen and K. L. Mulfort, Dalton Trans., 2016, 45, 9871–9883 RSC.
- Z. L. Xie, X. Liu, A. J. S. Valentine, V. M. Lynch, D. M. Tiede, X. Li and K. L. Mulfort, Angew. Chem., Int. Ed., 2022, 61, e202111764 CrossRef CAS PubMed.
- Y. Sakuragi, B. Zybailov, G. Shen, D. A. Bryant, J. H. Golbeck, B. A. Diner, I. Karygina, Y. Pushkar and D. Stehlik, J. Biol. Chem., 2005, 280, 12371–12381 CrossRef CAS PubMed.
- M. Vuolle and R. Mäkelä, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 51–55 RSC.
- Y. N. Pushkar, I. Karyagina, D. Stehlik, S. Brown and A. Van Der Est, J. Biol. Chem., 2005, 280, 12382–12390 CrossRef CAS PubMed.
- O. Burghaus, M. Plato, M. Rohrer, K. Möbius, F. MacMillan and W. Lubitz, J. Phys. Chem., 1993, 97, 7639–7647 CrossRef CAS.
- R. N. Sampaio, D. C. Grills, D. E. Polyansky, D. J. Szalda and E. Fujita, J. Am. Chem. Soc., 2020, 142, 2413–2428 CrossRef CAS PubMed.
- S. Ilic, A. Alherz, C. B. Musgrave Cde, K. D. Glusac and R. Li, Chem. Commun., 2019, 55, 5583–5586 RSC.
- X. Q. Zhu, M. T. Zhang, A. Yu, C. H. Wang and J. P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- P. J. DeLaive, T. K. Foreman, D. G. Whitten and C. Giannotti, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378–10388 CrossRef CAS PubMed.
- J. M. Aslan, D. J. Boston and F. M. MacDonnell, Chem.–Eur. J., 2015, 21, 17314–17323 CrossRef CAS PubMed.
- J. M. Aslan, M. Yousufuddin, D. J. Boston and F. M. MacDonnell, Inorg. Chim. Acta, 2017, 454, 216–221 CrossRef CAS.
- M. L. Martinez, W. C. Cooper and P. T. Chou, Chem. Phys. Lett., 1992, 193, 151–154 CrossRef CAS.
- P. T. Chou, Y. C. Chen, W. S. Yu, Y. H. Chou, C. Y. Wei and Y. M. Cheng, J. Phys. Chem. A, 2001, 105, 1731–1740 CrossRef CAS.
- S. Hristova, G. Dobrikov, F. S. Kamounah, S. Kawauchi, P. E. Hansen, V. Deneva, D. Nedeltcheva and L. Antonov, RSC Adv., 2015, 5, 102495–102507 RSC.
- R. D. Adams and S. Miao, Inorg. Chem., 2004, 43, 8414–8426 CrossRef CAS PubMed.
- J. G. M. De Carvalho, R. A. Fischer and A. Pöthig, Inorg. Chem., 2021, 60, 4676–4682 CrossRef CAS PubMed.
- A. Babaei, P. A. Connor, A. J. McQuillan and S. Umapathy, J. Chem. Educ., 1997, 74, 1200–1203 CrossRef CAS.
- J. Nomrowski, X. Guo and O. S. Wenger, Chem.–Eur. J., 2018, 24, 14084–14087 CrossRef CAS PubMed.
- J. Nomrowski and O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 5343–5346 CrossRef CAS PubMed.
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Nat. Commun., 2021, 12, 1835 CrossRef CAS PubMed.
- H. Takeda, Y. Monma and O. Ishitani, ACS Catal., 2021, 11, 11973–11984 CrossRef CAS.
- H. Takeda, H. Kamiyama, K. Okamoto, M. Irimajiri, T. Mizutani, K. Koike, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2018, 140, 17241–17254 CrossRef CAS PubMed.
- X. Zheng, M. C. Drummer, H. He, T. M. Rayder, J. Niklas, N. P. Weingartz, I. L. Bolotin, V. Singh, B. V. Kramar, L. X. Chen, J. T. Hupp, O. G. Poluektov, O. K. Farha, P. Zapol and K. D. Glusac, J. Phys. Chem. Lett., 2023, 14, 4334–4341 CrossRef CAS PubMed.
- W. Xie, J. Xu, U. Md Idros, J. Katsuhira, M. Fuki, M. Hayashi, M. Yamanaka, Y. Kobori and R. Matsubara, Nat. Chem., 2023, 15, 794–802 CrossRef CAS PubMed.
- J. Talvitie, I. Alanko, A. Lenarda, N. Durandin, N. Tkachenko, M. Nieger, J. Helaja, J. Talvitie, I. Alanko, A. Lenarda, M. Nieger, J. Helaja, N. Durandin and N. Tkachenko, ChemPhotoChem, 2023, e202300107 CrossRef.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5282–5287 CrossRef CAS PubMed.
- Y. Liu, H. Z. Ye, K. M. Diederichsen, T. Van Voorhis and T. A. Hatton, Nat. Commun., 2020, 11, 1–11 CrossRef CAS PubMed.
- J. M. Barlow and J. Y. Yang, J. Am. Chem. Soc., 2022, 144, 14161–14169 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 22780292278030. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03428c |
| This journal is © The Royal Society of Chemistry 2023 |