
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of an autocatalytic reaction cycle in neutral medium for synthesis of life-sustaining sugars†
Hiro
Tabata‡
 a,
Genta
Chikatani‡
a,
Hiroaki
Nishijima
a,
Takashi
Harada
a,
Rika
Miyake
a,
Souichiro
Kato
ab,
Kensuke
Igarashi
b,
Yoshiharu
Mukouyama
ac,
Soichi
Shirai
d,
Minoru
Waki
d,
Yoko
Hase
*ad and
Shuji
Nakanishi
*ae
a,
Genta
Chikatani‡
a,
Hiroaki
Nishijima
a,
Takashi
Harada
a,
Rika
Miyake
a,
Souichiro
Kato
ab,
Kensuke
Igarashi
b,
Yoshiharu
Mukouyama
ac,
Soichi
Shirai
d,
Minoru
Waki
d,
Yoko
Hase
*ad and
Shuji
Nakanishi
*ae
aResearch Center for Solar Energy Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: nakanishi.shuji.es@osaka-u.ac.jp
bBioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 2-17-2-1, Tsukisamu higashi, Toyohira, Sapporo, 062-8517, Japan
cDivision of Science, College of Science and Engineering, Tokyo Denki University, Hatoyama, Saitama 350-0394, Japan
dToyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: y-hase@mosk.tytlabs.co.jp
eInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 9th October 2023
Abstract
Autocatalytic mechanisms in carbon metabolism, such as the Calvin cycle, are responsible for the biological assimilation of CO2 to form organic compounds with complex structures, including sugars. Compounds that form C–C bonds with CO2 are regenerated in these autocatalytic reaction cycles, and the products are concurrently released. The formose reaction in basic aqueous solution has attracted attention as a nonbiological reaction involving an autocatalytic reaction cycle that non-enzymatically synthesizes sugars from the C1 compound formaldehyde. However, formaldehyde and sugars, which are the substrate and products of the formose reaction, respectively, are consumed in Cannizzaro reactions, particularly under basic aqueous conditions, which makes the formose reaction a fragile sugar-production system. Here, we constructed an autocatalytic reaction cycle for sugar synthesis under neutral conditions. We focused on the weak Brønsted basicity of oxometalate anions such as tungstates and molybdates as catalysts, thereby enabling the aldol reaction, retro-aldol reaction, and aldose–ketose transformation, which collectively constitute the autocatalytic reaction cycle. These bases acted on sugar molecules of substrates together with sodium ions of a Lewis acid to promote deprotonation under neutral conditions, which is the initiation step of the reactions forming an autocatalytic cycle, whereas the Cannizzaro reaction was inhibited. The autocatalytic reaction cycle established using this abiotic approach is a robust sugar production system. Furthermore, we found that the synthesized sugars work as energy storage substances that sustain microbial growth despite their absence in nature.
Introduction
Autocatalytic reaction cycles assume an important role in reaction networks that support the central carbon metabolism in living organisms.1–3 For example, in the Calvin cycle that is the starting point of carbon metabolism in plants, CO2 reacts with a five-carbon (C5) molecule, ribulose-1,5-bisphosphate (RuBP), with aldol-like addition by the enzyme of ribulose-1,5-bisphosphate carboxylase (RuBisCO) to generate a six-carbon (C6) reaction intermediate. This C6 intermediate splits into 3-phosphoglyceric acid (3PGA), C3-compound, by C–C bond cleavage (Fig. S1†). Part of the 3PGA is released out of the cycle, while the remaining 3PGAs are used to regenerate the CO2 acceptor, RuBP. In other words, the CO2-accepting RuBP is regenerated in each turnover that the Calvin cycle goes around. Note that the autocatalytic increase in the concentrations of the components of these cycles potentially has the property of self-amplification of the flux of the cycle-forming substances.1 These properties of the autocatalytic cycle are attractive from the perspective of upgrading lower-value chemicals.The formose reaction is well-known to comprise an autocatalytic cycle as proposed by Breslow (Fig. 1a)4 and has attracted much interest as a non-enzymatic sugar synthesis reaction using formaldehyde (HCHO) as a substrate,5–7 which can be synthesized from fossil fuels8 and CO2.9 The Breslow cycle is formed by the consecutive steps of the aldol reaction (r-2 and r-4 in Fig. 1a), retro-aldol reaction (r-6 in Fig. 1a), and Lobry de Bruyn–Van Ekenstein (LBVE) transformation (r-3 and r-5 in Fig. 1a). The retro-aldol reaction, which cleaves the C–C bond of aldotetrose (C4a) to form two molecules of glycolaldehyde (C2a), is the origin of the autocatalytic mechanism in the Breslow cycle. In this reaction, one C2a that can be converted to the enolate of a possible nucleophile for HCHO is regenerated simultaneously with the formation of another C2a molecule as a product. Therefore, nucleophilic addition to HCHO occurs continuously without depletion of nucleophile molecules, using only HCHO as feed.
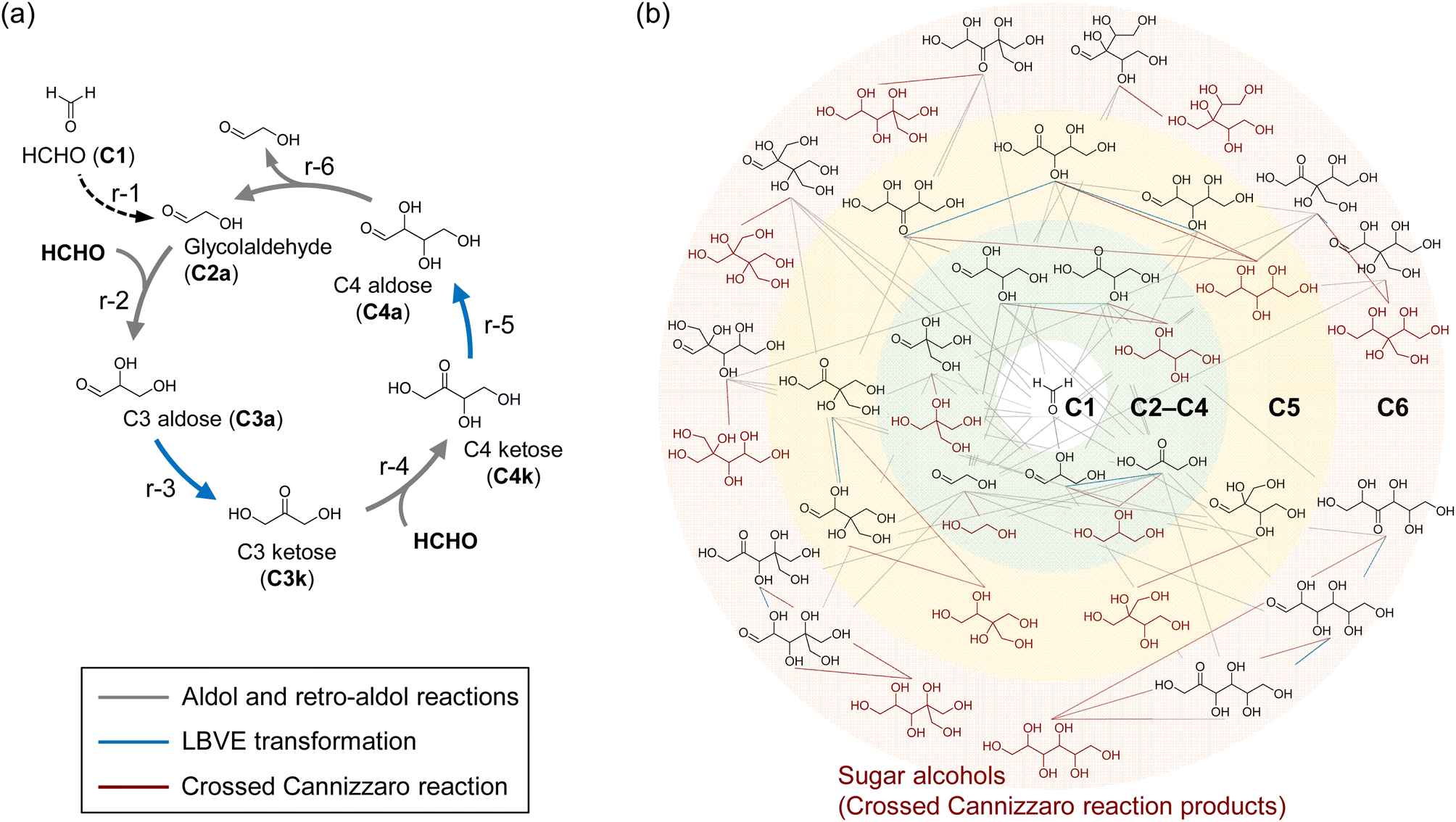 | ||
| Fig. 1 (a) One of the autocatalytic reaction cycles in the chemical reaction network (CRN) of the formose reaction proposed by Breslow (the Breslow cycle). The dimerization of HCHO (r-1), aldol (r-2, r-4), and retro-aldol reactions (r-6), and LBVE transformation (r-3, r-5) are shown as black dotted line, gray solid lines, and blue solid lines, respectively. (b) The CRN of the formose reaction. Sugar alcohols produced by the crossed Cannizzaro reaction and reaction pathways are shown in brown. The other reaction pathways are also shown as described for (a). | ||
In the formose reaction, continuous reaction pathways form a chemical reaction network (CRN) consisting of a variety of monosaccharides (Fig. 1b). Once C2a is formed, the CRN of the formose reaction proceeds via consecutive reactions, which include C–C bond formation and cleavage through aldol and retro-aldol reactions, respectively, and LBVE transformation, catalyzed by hydroxide ions of a Brønsted base. The dimerization of HCHO (r-1 in Fig. 1a) is thought to be the entry point for the formose reaction,10,11 whereas the CRN of the formose reaction is also formed by addition of small amounts of C2a as an initiator without activating the dimerization pathway. Notably, the Breslow cycle is only part of the CRN of formose reactions that form under basic conditions.12 CRNs catalyzed by hydroxide ions produce a variety of monosaccharides, as mentioned above. In addition, various side reactions, such as the crossed Cannizzaro reaction leading to the degradation of monosaccharides and the consumption of HCHO, and the oxidative degradation of sugars by oxygen,13 comprise a large part of the CRN.10,14–16 Although several studies of catalysts in the formose reaction have been reported,17–21 the selective synthesis of sugars remains a challenge due to the large CRN. To avoid the consumption of HCHO and sugars by these side reactions and form a robust Breslow cycle, it is necessary to proceed with reactions r-2, r-3, r-4, r-5, and r-6 using catalysts other than hydroxide ions.
Here, we applied catalysts in which an acid–base pair act cooperatively, similar to the function of enzymes that activate the formation and cleavage of C–C bonds under neutral conditions.22,55 A combination of experimental and computational chemistry was used to identify candidate catalysts that promote r-2, r-3, r-4, r-5, and r-6 of the Breslow cycle in neutral aqueous solution. Computational chemistry analyses revealed a reaction mechanism in which the oxometalate anions, WO42− and MoO42−, act on sugar molecules as Brønsted bases in the presence of Na+ counter ions. These catalysts form an autocatalytic reaction cycle for sugar synthesis, which has been difficult to construct under neutral conditions. Our approach improved the robustness of the sugar synthesis system due to suppression of the side reactions that proceed under basic conditions. Moreover, sugar products synthesized by tungstate were fed to soil microorganisms, and consumption of sugars and microbial growth were clearly observed. These results indicate the value of the non-enzymatically obtained sugars as carbon resources.
Results and discussion
Selection of candidate catalysts of C–C bond formation in neutral medium
In selecting catalysts to construct the Breslow cycle under neutral conditions, we first focused on the aldol reaction for C–C bond formation. The reaction of r-1, that is, dimerization of HCHO to form C2a, does not proceed under neutral conditions except under severe conditions, such as hydrothermal or reflux reactions.11,23–25 Therefore, we employed C2a as a reaction initiator and selected catalysts based on the following concept.The aldol reaction of HCHO and C2a (r-2), which is a point of entry for HCHO to the Breslow cycle, was chosen as the reaction to evaluate for catalyst selection (Fig. 2a). In the Breslow cycle, Brønsted bases are candidate catalysts to promote r-2 because the rate-limiting step of the aldol reaction is the elimination of hydrogen at the α-position of C2a (i.e., α-H elimination). However, the use of a typical Brønsted base catalyst of hydroxide ions that form pairs with alkali metal and alkaline earth metal cations or ammonium ions, etc., will inevitably increase the pH of the aqueous solution. Therefore, in the present work, we focused on oxometalate anions as Brønsted bases, which are stable under neutral conditions and do not cause an increase in pH.26 The function of oxometalate anions as base catalysts was recently discovered by Kimura et al.,27 and various reactions catalyzed by this category of materials have been reported.28–31 Furthermore, molybdate and its condensates was reported to catalyze epimerization reaction, i.e., the Bilik reaction.32–34 Based on these emerging results, we expected that oxometalate anions such as WO42− and MoO42− could also be used to catalyze aldol reactions, even under neutral conditions. The salts of oxometalate anions and Na+ cation were applied in this study because of their high solubility in water.
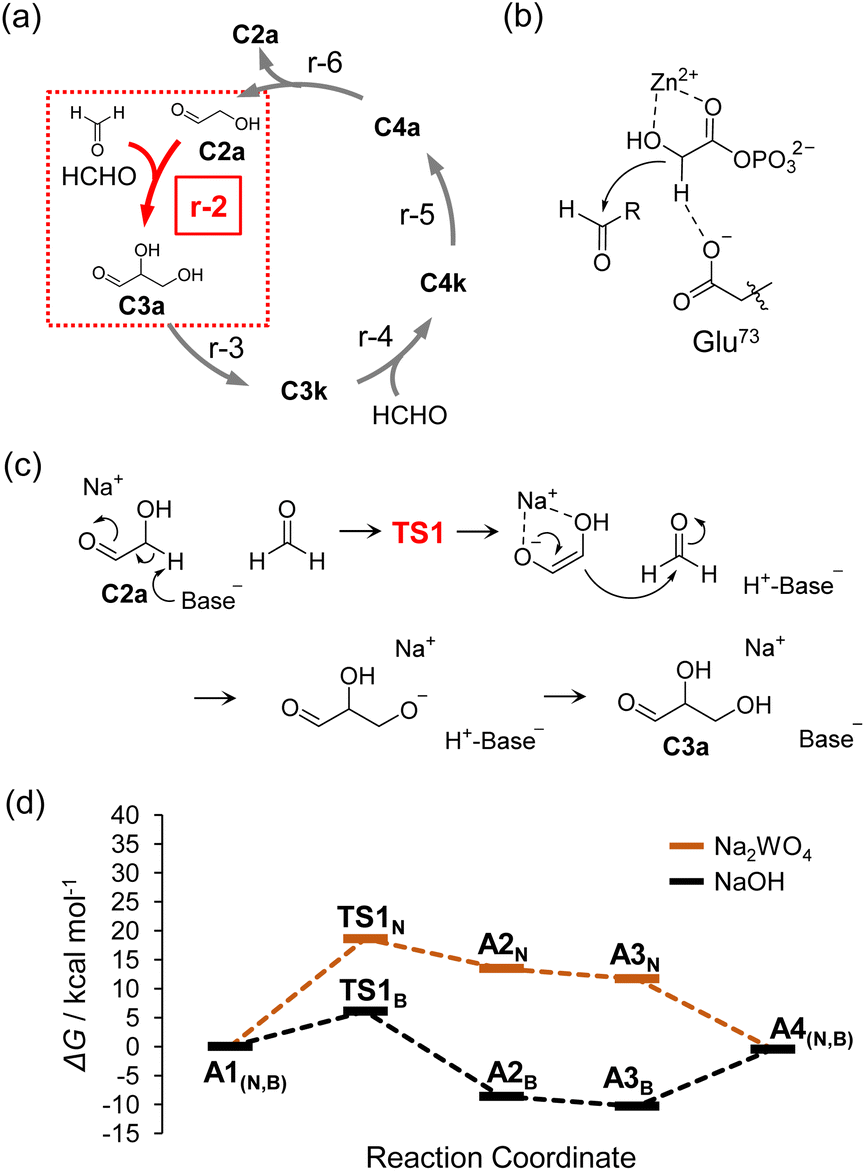 | ||
| Fig. 2 (a) The aldol reaction of HCHO and C2a (r-2, red lines) as an initiation reaction of the Breslow cycle. (b) Cooperative activation of DHAP in class II aldolase. (c) Proposed reaction scheme for r-2 in which a sodium ion and a Brønsted base work concertedly on HCHO and C2a. (d) Calculated free energy diagram for r-2 catalyzed by NaOH and Na2WO4 in water. | ||
Here, we discuss the reaction mechanism catalyzed by oxometalate anion. We used density functional theory (DFT) to evaluate catalytic activities of oxometalate anions. All the calculations in this work were performed using Gaussian 09 programs35 with B3LYP functional.36–39 The basis set used was 6-31++G (d, p)40–44 for C, H, O, and Na, whereas the Lanl2DZ basis set with the effective core potential45–47 was used for W and Mo. The integral equation formalism polarizable continuum model48,49 was used to consider the solvent effect of water. More detailed computational methods are provided in the ESI.†
DFT calculations showed that α-H elimination can proceed via a concerted acid–base action on the sugar molecule, even with a weak base such as oxometalate anion. In the enzymatic reaction mechanism of class II aldolase as shown in Fig. 2b, α-H elimination is promoted by a zinc ion acting as a Lewis acid on the carbonyl group and the oxygen of the OH group at the α-position and simultaneous activity on the hydrogen at the α-position by carboxylate anions, which are weak bases, as in oxometalate anions.22 Referring to class II aldolase, we set up a reaction mechanism in which the C–C bond is formed via transition state (TS1), and the free energy profile of the reaction was evaluated using the DFT calculations (Fig. 2c). WO42− was selected as the Brønsted base and Na+ as the Lewis acid, and the free energy profile of α-H elimination was calculated (Fig. 2d). The results for the case in which OH− was employed as a Brønsted base are shown as comparative data in the same figure; the activation energy of the pathway involving WO42−via TS1N was 18.6 kcal mol−1, which was higher than that of the case involving OH−via TS1B, whereas the reaction can proceed at 80 °C (reaction rates estimated using the Eyring equation are summarized in Table S1†). The optimized structures associated with these calculations are shown in Fig. S2 and S3,† respectively. In addition, the case involving the absence of Na+ was also calculated (Fig. S4†). The estimated activation energy was higher than that of the result involving Na+ (Fig. 2d), indicating that Na+ stabilizes the TS1N structure and enolate anions of C2a. Calculations of the process by which the C–C bond is formed following α-H elimination revealed that the reaction proceeds without a clear TS (Fig. S5†). This series of calculations indicated that C–C bond formation proceeds under neutral conditions due to the simultaneous action of WO42− and Na+. In subsequent experiments, Na2WO4, a salt of WO42− and Na+, was selected as the catalyst to investigate the sugar synthesis reaction using HCHO as a substrate. The main text of the article should appear here with headings as appropriate.
Synthesis of sugars under neutral conditions
The reaction using Na2WO4 catalyst and a small amount of C2a as an initiator with HCHO as feedstock was experimentally found to form a C–C bond under neutral conditions, as suggested by the calculations in Fig. 2d. In this experiment, 0.3 M HCHO solution containing 3 mM C2a, 60 mM Na2WO4 catalyst, and 10% CH3OH (The pH value was 7.82) was prepared. CH3OH was added to inhibit the disproportionation of HCHO (i.e., 2HCHO + H2O → CH3OH + HCOOH), known as the Cannizzaro reaction. The solution was kept at 80 °C, and the decrease in HCHO concentration due to consumption over time was analyzed. A quantitative analysis was performed using high-performance liquid chromatography (HPLC) with a reversed-phase column after modification of the generated sugars according to the 2,4-dinitrophenylhydrazine (DNPH) method (see ESI† for details). Fig. 3a shows that almost all of the HCHO was consumed in approximately 5 h, as indicated by the red line in the figure. The formation of all compounds associated with the Breslow cycle was confirmed, indicating that r-2, r-3, r-4, and r-5 proceeded (Fig. 3b). LVBE transformations (r-3, r-5) via α-H elimination were thought to proceed via Na2WO4 catalysis as well as aldol reactions.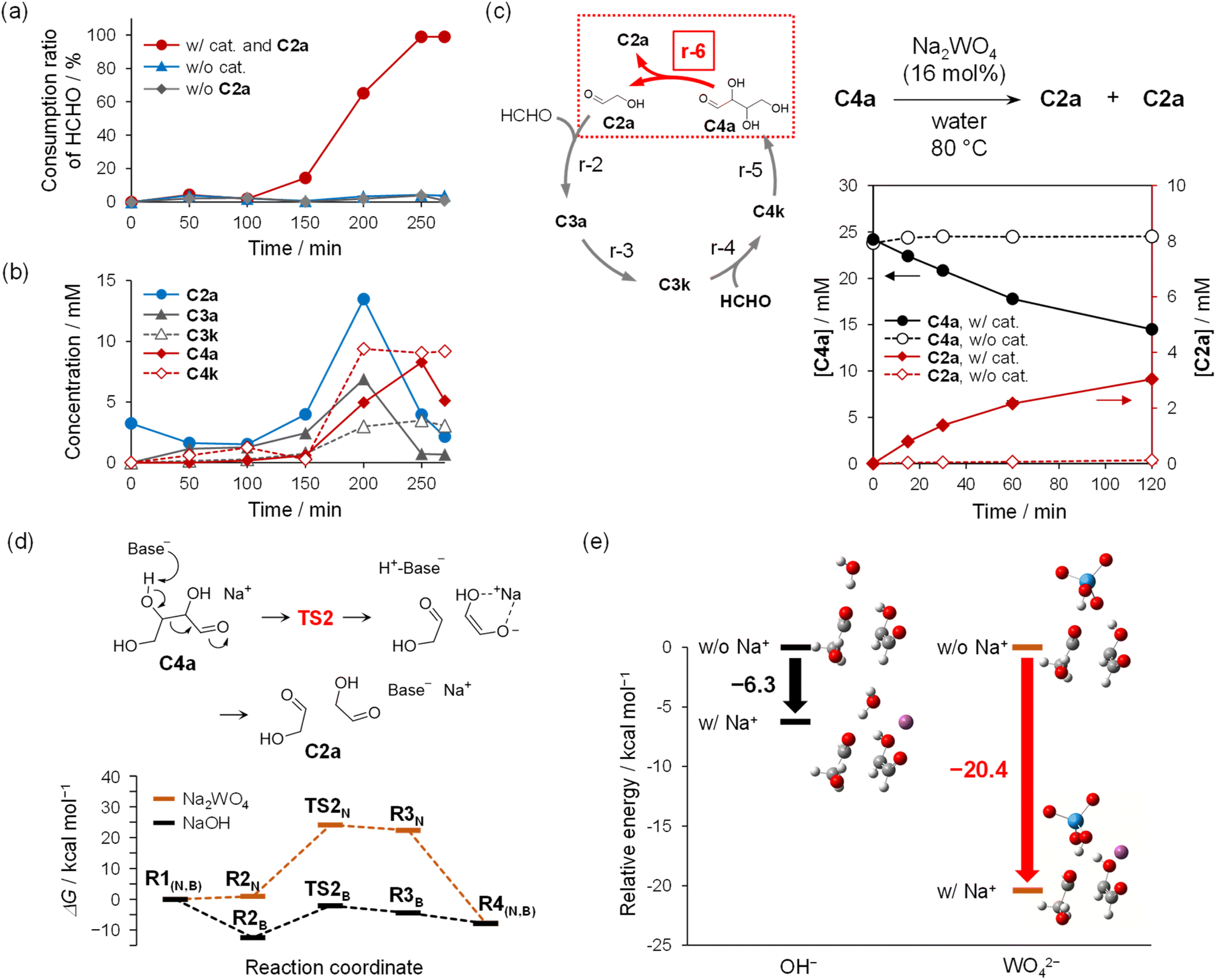 | ||
| Fig. 3 (a) Time course of the consumption ratio of HCHO. The experiment was performed with catalyst (Na2WO4) and C2a as an initiator in water at 80 °C (red line). Results obtained without catalyst (blue line) and without initiator (gray line) are also shown for comparison. Gray and blue lines indicate the average of three experiments, respectively, and the error bars indicate standard errors. (b) Time course of change in concentration of intermediate products (number of carbon atoms: 2–4) obtained under the same experimental conditions shown for the red line in (a). (c) Change in C4a (black line) and C2a (red line) concentrations over time when C4a containing Na2WO4 was heated in water at 80 °C. Results without the addition of a catalyst are shown as dotted lines. Experiments were performed in triplicate, and error bars indicate standard errors. (d) Proposed reaction scheme for r-2 in which a sodium ion and a Brønsted base work concertedly on HCHO and C2a. Calculated free energy diagram for r-6 catalyzed by NaOH (black) or Na2WO4 (brown) in water. (e) Energy of TS2N and TS2B with Na+ ion relative to the case without Na+ ion. | ||
A significant aspect of this experiment should be noted: the consumption curve of HCHO was sigmoidal (red line in Fig. 3a), which indicates that the autocatalytic reaction cycle regenerated compounds with a carbon number ≥2, and these compounds can then be turned to enolates. The red line in Fig. 3a, which depicts the sigmoidal shape of the curve, was not assigned error bars due to variations in the induction period resulting from autocatalysis. A similar trend was confirmed for at least three experiments (see Fig. S6†). Note that when either the Na2WO4 catalyst (the pH value was 8.02) or C2a (the pH value was 8.00) as the initiator was omitted, the reaction did not proceed (blue and gray lines in Fig. 3a and S7†).
Notably, the results shown in Fig. 3a and b suggested that the Na2WO4-catalyzed Breslow cycle was constructed and that C2a was regenerated via the retro-aldol reaction. The concentration of C2a increased from 100 to 200 min (Fig. 3b) relative to the starting concentration, in agreement with the HCHO consumption curve. Simultaneously, the concentrations of the other compounds also tended to increase after 150 min, at which time the rate of HCHO consumption began to rapidly increase. These time-associated trends in concentration were thought to be triggered by the propagation of C2a at r-6 (Fig. 3c).
To verify this hypothesis, the following experiments were performed. An aqueous solution of C4a (24 mM) was heated to 80 °C with 4 mM of Na2WO4 under neutral conditions (pH value was 7.81, Fig. 3c). A decrease in C4a (solid black line) and synchronous increase in C2a (solid red line) were observed with Na2WO4 as the catalyst, but these trends were not observed without a catalyst (pH value was 5.01, dashed lines) and in the solution that is adjusted at pH 7.8. Moreover, the decrease in C4a and the concurrent increase in C2a were more rapid at higher concentrations of the catalyst (Fig. S8†). These results provide clear evidence that Na2WO4 catalyzed r-6. Na2MoO4, another oxometalate sodium salt, gave the same results (Fig. S9†).
The origin of the catalytic activity of oxometalate anions in regard to the retro-aldol reaction was examined using the DFT calculations. The retro-aldol reaction, the reverse of the aldol reaction, is assumed to be initiated when the Brønsted base abstracts the proton from the hydroxy group at the β position of C4a. Assuming simultaneous activity of WO42− and Na+ ions as in the aldol reaction, the change in free energy profile was calculated (Fig. 3d). For comparison, the results calculated for the pair consisting of OH− and Na+ ions are also shown. The optimized structures for these calculations are shown in Fig. S10 and S11,† respectively. The calculated structures suggested that WO42− and Na+ ions act in concert, causing deprotonation of the OH group at the β position, which triggers C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond formation and C–C bond cleavage. The activation energy for this reaction process was estimated to be 24.2 kcal mol−1, which indicates that the reaction could proceed at 80 °C (see Table S1†). The transition state of the retro-aldol reaction in the absence of Na+ cation involvement was calculated to estimate the effect of Na+ cation. The results showed that the Gibbs free energy of the TS was strongly dependent on the presence of Na+ ions; the activation barrier was lowered by more than 20 kcal mol−1 due to the presence of Na+ (Fig. 3e). This result suggested that a Na+ ion stabilizes the TS cooperatively with the Brønsted base by accepting the negative charge of the enolate anion.
O double bond formation and C–C bond cleavage. The activation energy for this reaction process was estimated to be 24.2 kcal mol−1, which indicates that the reaction could proceed at 80 °C (see Table S1†). The transition state of the retro-aldol reaction in the absence of Na+ cation involvement was calculated to estimate the effect of Na+ cation. The results showed that the Gibbs free energy of the TS was strongly dependent on the presence of Na+ ions; the activation barrier was lowered by more than 20 kcal mol−1 due to the presence of Na+ (Fig. 3e). This result suggested that a Na+ ion stabilizes the TS cooperatively with the Brønsted base by accepting the negative charge of the enolate anion.
Identification of sugars produced under Na2WO4
The structures of the major sugars synthesized via the Breslow cycle constructed using Na2WO4 as a catalyst under neutral conditions were identified. Fig. 4b shows an HPLC chromatogram of the sugars in the sample. The sample was collected when approximately 99% of the HCHO had been consumed in a 230 min reaction. Four large peaks (A–D) were observed at retention times of 8–12 min. The results shown in Fig. 3a and b regarding quantification of HCHO and C2–C4 indicated that HCHO, C2a, and C4 ketose (C4k) were abundant in the reaction solution at reaction times around 200 min. The intensity of these peaks increased with the onset of the reaction, reached a maximum, and then decreased as HCHO was consumed (Fig. S12†). Therefore, we extrapolated that the compound corresponding to peaks A–D was formed by the aldol reaction of C4k with HCHO or C2a, and we then performed experiments using C4k and HCHO or C4k and C2a as substrates (Fig. 4a, see ESI† for details). As a result, A and B were obtained as the main products from C4k and HCHO (Fig. 4c), respectively, whereas C and D were obtained as the main products from C4k and C2a (Fig. 4d), respectively. The structures of the products obtained in these experiments were identified by 1H-NMR, 13C-NMR, and mass spectrometry as 1,3,4-trihydroxy-3-(hydroxymethyl)butan-2-one (A), 1,3,4,5-tetrahydroxypentan-2-one (C5 ketose, C5k; B), and stereoisomers of 1,2,4,5,6-pentahydroxyhexan-3-one (3-hexulose, C and D). The sugar analysis HPLC retention times of peaks A, C, and D differed from those of commercially available monosaccharides but were also consistent with the results of the structural analysis of the compounds (see Fig. S13† for the peaks of commercially available sugar samples).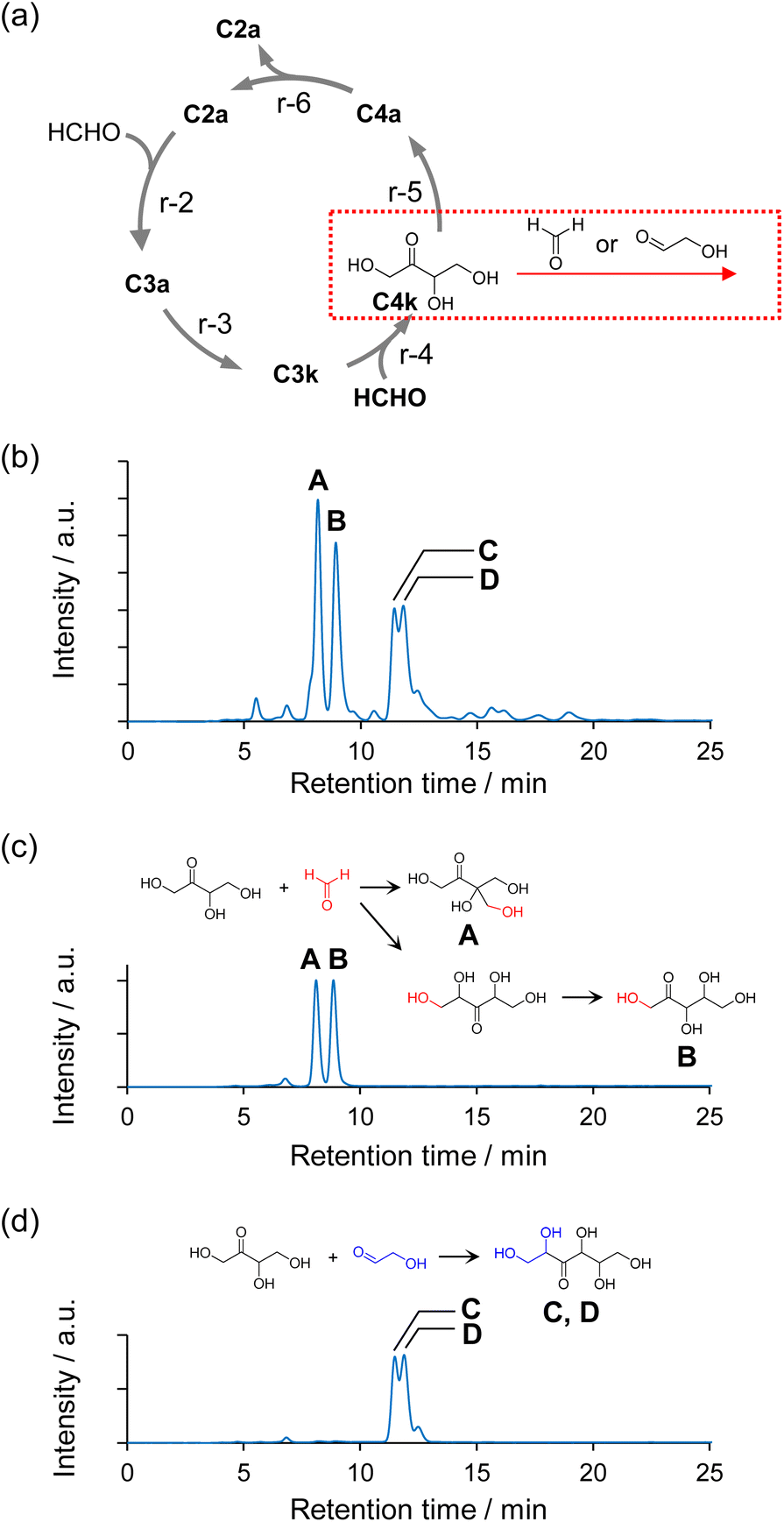 | ||
| Fig. 4 (a) Possible reaction pathways represented by the red arrow that produce C5 and C6 of the main products. (b) Chromatogram of the products obtained in the reaction of HCHO and C2a (formose reaction) with Na2WO4 catalyst, analyzed on a Chromaster® HPLC system. (c) Chromatogram of A and B, which were produced via the reaction of C4k and HCHO, and the chemical structures of A and B, as determined by 1H-NMR, 13C-NMR, and mass spectrometry. (d) Chromatogram of C and D obtained by the reaction of C4k and C2a, and the chemical structures of C and D, determined as described above for A and B. | ||
Results similar to those shown in Fig. 4b were obtained when Na2MoO4 was used as the catalyst (Fig. S14†); however, the reaction driven by the Na2MoO4 catalyst was slower than that driven by Na2WO4. This difference in reaction rate could be explained by the Brønsted basicity of the oxometalate anions, which can be estimated from the charge density of the oxygen atoms in the natural bond orbital (NBO) analysis.50 Fig. S15† shows the calculated NBO charge in the optimized structures of WO42− and MoO42−. The calculated NBO charge densities of the oxygen atoms of WO42− and MoO42− are in the regions −0.934 to −0.935 and −0.857 to −0.859, respectively, which indicates that WO42− exhibits stronger basicity.
Effect of neutral conditions on the robustness of the sugar production system
Under neutral conditions, the robustness of the sugar production system should improve due to suppression of the Cannizzaro reaction and other side reactions that proceed under basic conditions. The robustness of the sugar synthesis system was evaluated by quantifying the amount of sugar produced and the amount of formic acid, a product of the Cannizzaro reaction. For this experiment, 0.3 M HCHO aqueous solution with 3 mM C2a as an initiator and 60 mM catalyst (i.e., Na2WO4 or NaOH) and containing 10 vol% of CH3OH was maintained at 80 °C. The reaction rates differed significantly under neutral and basic conditions; therefore, the experiments were terminated when approximately 99% of the HCHO was consumed. The reaction time at termination was 17 and 230 min under basic and neutral conditions, respectively. The corresponding results obtained under basic conditions analyzed by HPLC for sugars are shown in Fig. S16.†Monosaccharides with carbon numbers 2–6 synthesized under both basic and neutral conditions were comparatively quantified. Monosaccharides with carbon numbers 2–4 were quantified by HPLC, and the compounds were classified as C2–C4 according to carbon number, whereas the phenol–sulfuric acid method, a colorimetric method for sugars using furfural derivatization, was employed to quantify C5 and C6 sugars. The results showed that the quantity of sugar produced under neutral conditions was greater than that produced under basic conditions (Fig. 5a). The detailed quantification results for each compound are shown in Table S2.† Formic acid generated in the reaction was quantified by reversed-phase HPLC. The addition of CH3OH inhibits only the disproportionation of HCHO (Cannizzaro reaction) and not decomposition of the sugar (crossed Cannizzaro reaction). Therefore, quantification of formic acid provides an experimental estimate of the amount of HCHO and sugars consumed in the crossed Cannizaro reaction. This analysis revealed that the amount of formic acid produced under neutral conditions was much lower than that produced under basic conditions (Fig. 5b), indicating that the crossed Cannizzaro reaction was suppressed, as expected. Notably, the formation of formic acid can cause the pH of the reaction solution to decrease below the range suitable for the formose reaction to proceed. Thus, suppression of the formic acid-producing crossed Cannizzaro reaction is also beneficial from the viewpoint of sustained sugar synthesis.
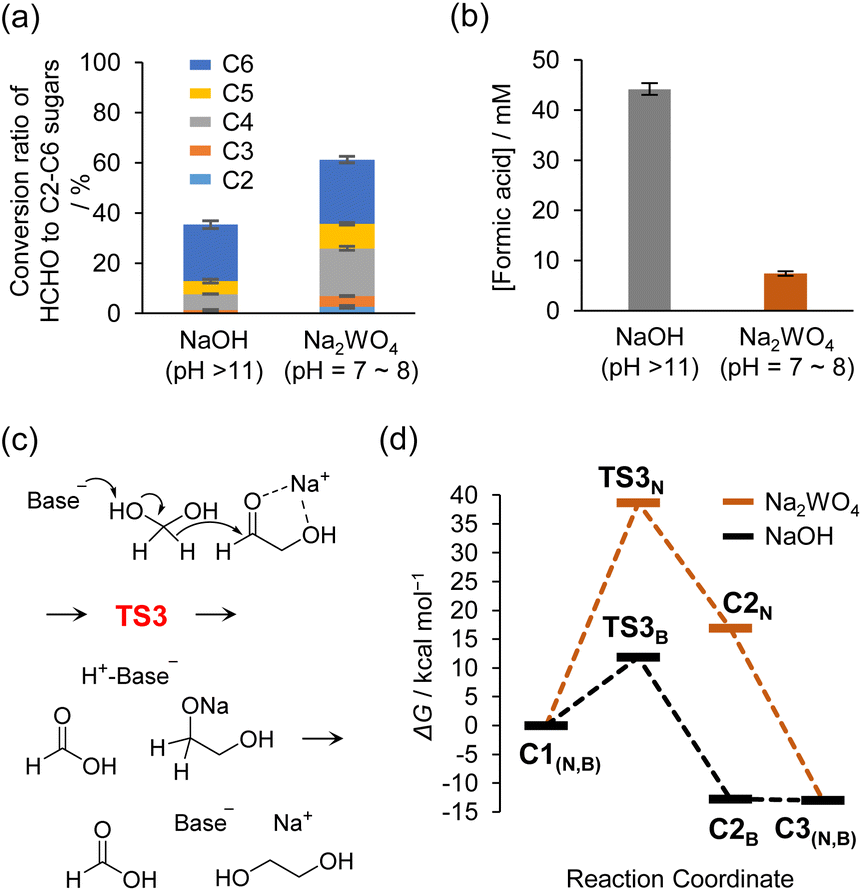 | ||
| Fig. 5 (a) Conversion ratio of HCHO to C2–C6 monosaccharides formed in the formose reactions at 80 °C with NaOH (60 mM, 17 min) and Na2WO4 (60 mM, 230 min) as catalysts. C2–C4 and C5–C6 monosaccharides were quantified by HPLC and phenol–sulfuric acid method, respectively. (b) Concentration of formic acid formed via the formose reaction at 80 °C using NaOH (60 mM, 17 min) or Na2WO4 (60 mM, 230 min) as a catalyst. Formic acid was quantified by HPLC. Experiments shown in (a) and (b) were performed in triplicate, and error bars indicate standard errors. Detailed analytical methods are provided in the ESI.† (c) Proposed reaction scheme for the Cannizzaro reaction of HCHO and C2a. (d) Calculated free energy diagrams for the reaction shown in (c) catalyzed by NaOH or Na2WO4 in water. | ||
The improved selectivity of sugar formation is also corroborated using DFT calculations. The ratios of sugar formation under basic and neutral conditions were consistent with the calculated activation energies of the aldol reaction and the competing crossed Cannizzaro reaction. Fig. 5c and d show schematic illustrations of reaction process and energy diagrams of the crossed Cannizzaro reactions under basic and neutral conditions; the substrates were C2a and HCHO (the optimized structures are shown in Fig. S17 and S18†). The activation energies of the aldol and crossed Cannizzaro reactions catalyzed by OH− were estimated at 6.1 and 11.9 kcal mol−1 as shown in Fig. 2d and 5d, respectively, which indicated that sugar synthesis and degradation proceeded rapidly. In contrast, the activation energies of these reactions catalyzed by oxometalate anions under neutral conditions were estimated at 18.6 and 38.7 kcal mol−1 for the aldol (Fig. 2d) and Cannizzaro reactions (Fig. 5d). In particular, the high activation energy of 38.7 kcal mol−1 for the latter reaction suggested it is unlikely to proceed at 80 °C (estimated reaction rates and half-lives are shown in Table S1†). The reason why tungstates catalyzed aldol and retro-aldol reactions under neutral conditions, whereas the Cannizzaro reaction was suppressed, is explained by the transition state energy and the interaction between the HOMO of the base and the anti-bonding orbital σ* of the substrate (acid) H-A being deprotonated. Details are discussed in Table S3 of the ESI† and in the chapter “Reason for the suppression of the crossed Cannizzaro reaction under neutral conditions”.
Cultivation of microorganisms using chemically synthesized sugars
We verified that the non-enzymatically synthesized sugars could sustain life by using them to cultivate microbes. Soil microbes were collected from forest soil near the National Institute of Advanced Industrial Science and Technology, Hokkaido, Japan. The synthesized sugars fed to the microbial cultures were prepared as follows: 1 M HCHO aqueous solution containing 10 mM C2a and 60 mM Na2WO4 in the presence of 10% CH3OH was maintained at 80 °C for 4 h. After the reaction, catalysts were removed from the solution using an anion exchange resin. It was confirmed by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) that tungsten could be removed to 0.42 mM or less under cultivation conditions. Then, microbial communities generated in three subcultures were analyzed to verify microbial utilization of the synthesized sugars (Fig. 6). Error bars were not assigned in Fig. 6b and c because the distribution of soil bacteria varied in each experiment, although similar experiments were performed at least once more with different flora (Fig. S19†). Fig. 6a shows the results of HPLC analyses of the supernatants of microbial culture medium at 1, 3, and 8 days after cultivation began. The results confirmed that the peak intensity of the sugar products decreased over time. In particular, peak A, assigned to 1,3,4-trihydroxy-3-(hydroxymethyl)butan-2-one, almost disappeared after 8 days. In contrast, it was confirmed that the sugar solution added to the inorganic medium was stable for 8 days and showed no decrease of any peak without the presence of microbes (Fig. S20†). Quantitative analyses of the concentrations of sugars using the phenol–sulfuric acid method revealed that ca. 40% of the C5 and C6 sugars were metabolized by the 8th day of incubation (Fig. 6b). The OD600 value increased as the sugar concentration decreased, indicating growth of the microbial cells (Fig. 6c). Note that there was no decrease of sugar concentration without microbes (gray line in Fig. 6b), and also the increase of OD600 was much smaller without the synthesized sugar (gray line in Fig. 6c). Furthermore, analyses of the microbial consortia indicated that several types of microbes were enriched during incubation with the chemically synthesized sugars (Fig. 6d). Taken together, these data demonstrated that non-enzymatically synthesized sugars can be metabolized and used to sustain microbial life.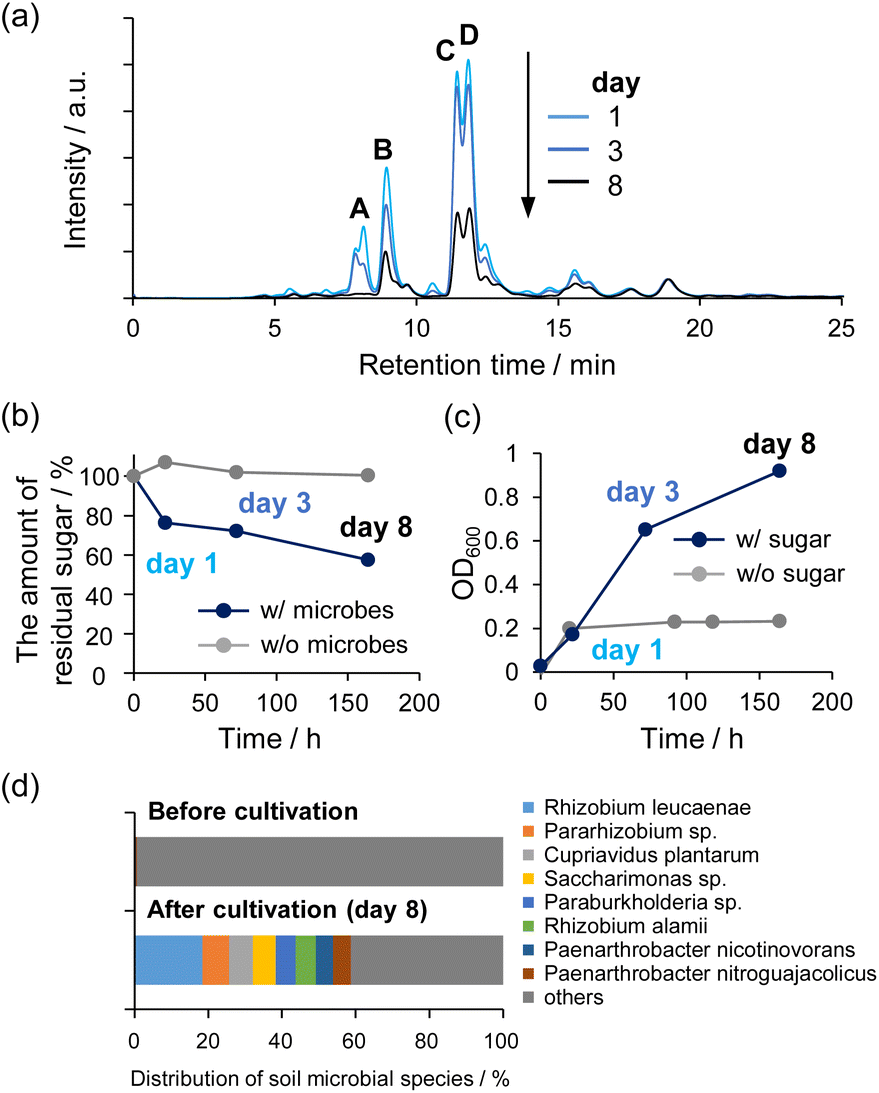 | ||
| Fig. 6 (a) HPLC chromatograms of the supernatant of microbial culture medium at 1, 3, and 8 days after cultivation was started. (b) and (c) Time course of total residual sugar concentration (C5 and C6) (b) and OD600 (c) after cultivation under the same conditions described for (a). Results obtained without microbes (b) and without sugar (c) are also shown for comparison (gray lines). (d) Analysis of microbial consortia before and after cultivation. Each color represents the microbial species that were included in the consortia. Error bars were not assigned because the distribution of soil bacteria varied for each experiment, although similar results were obtained at least 2 times using different flora, as shown in Fig. S19.† | ||
Conclusions
In this study, an autocatalytic reaction cycle for the non-enzymatic production of sugars using HCHO as a substrate under neutral conditions was successfully constructed. In the presence of an oxometalate catalyst and a small amount of initiator, C2a, the time course curve of HCHO consumption exhibited a sigmoidal shape. In addition, the concentration of C2a increased in the middle of the reaction, becoming higher than the concentration in the initial state. These results proved that the oxometalate-catalyzed sugar formation reaction is based on an autocatalytic reaction cycle involving the regeneration of C2a. The consumption of HCHO and degradation of sugar in the products via the crossed Cannizzaro reaction were significantly suppressed under neutral conditions. Moreover, maintaining the pH in the neutral region resulted in an increase in the robustness of sugar formation. It was also shown that the chemically synthesized sugars in this neutral solution were metabolized by microbial cells and supported their growth, even though the products included branched sugars that are not produced naturally. Given growing concerns regarding the sustainability of natural sugar production, we anticipate that the system for non-enzymatic production of life-sustaining sugars shown in this work will lead to innovations in bioproduction technology, which to date has used only natural sugars as substrates.Formose reactions have attracted considerable attention in terms of the abiotic production of sugars. HCHO, the substrate for the formose reaction, can be synthesized reductively by CO2 electrolysis.51,52 Therefore, there is a need to design sugar synthesis reactions with a view to achieving one-pot sugar synthesis from CO2.7 Previous studies of the formose reaction have explored the use of various catalysts that function under basic conditions to improve the selectivity of sugar production. However, the inevitable neutralization of the reaction solution by an electrolyte containing CO2 must be considered when constructing a sugar synthesis reaction cycle applicable to an integrated system of CO2 electrolysis and sugar synthesis.53,54 The knowledge generated by this study regarding the reaction mechanism of Lewis acid and weak Brønsted base pairs working cooperatively under neutral conditions will be an important guideline for the design of a general reaction system for sugar synthesis from CO2.
Data availability
Experimental and computational data and detailed procedures are included in the ESI.†Author contributions
S. N. and Y. H. conceived the idea and supervised the project. H. T., G. C., H. N., Y. M. and M. W. performed the chemical experiments, analyzed the data. S. N. and H. T. wrote the first draft of the manuscript. T. H. and R. M. performed the HPLC characterization. G. C., S. S. and Y. H. performed the computational studies. S. K. and K. I. performed biological experiments. All the authors contributed to the discussion of the results and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Kakenhi Grants-in-Aid (No. 19K22232 and 22J10537) and JST-Mirai Program Grant No. JPMJMI22E5, Japan. The authors would like to express appreciation to Ms. Ai Miura for assistance with the cultivation of soil microbes.Notes and references
- R. Braakman and E. Smith, The compositional and evolutionary logic of metabolism, Phys. Biol., 2013, 10, 011001 CrossRef CAS PubMed.
- F. L. Sousa, W. Hordijk, M. Steel and W. F. Martin, Autocatalytic sets in E. coli metabolism, J. Syst. Chem., 2015, 6, 1–21 CrossRef CAS PubMed.
- S. A. Marakushev and O. V. Belonogova, Chemical Basis of Carbon Fixation Autotrophic Paleometabolism, Biol. Bull. Russ. Acad. Sci., 2021, 48, 519–529 CrossRef CAS.
- R. Breslow, On the mechanism of the formose reaction, Tetrahedron Lett., 1959, 1, 22–26 CrossRef.
- J. B. García Martínez, K. A. Alvarado, X. Christodoulou and D. C. Denkenberger, Chemical synthesis of food from CO2 for space missions and food resilience, J. CO2 Util., 2021, 53, 101726 CrossRef.
- F. Dinger and U. Platt, Towards an Artificial Carbohydrates Supply on Earth, Front. Sustain. Food Syst., 2020, 4, 90 CrossRef.
- S. Cestellos-Blanco, S. Louisia, M. B. Ross, Y. Li, N. E. Soland, T. C. Detomasi, J. N. Cestellos Spradlin, D. K. Nomura and P. Yang, Toward abiotic sugar synthesis from CO2 electrolysis, Joule, 2022, 6, 2304–2323 CrossRef CAS.
- L. E. Heim, H. Konnerth and M. H. G. Prechtl, Future perspectives for formaldehyde: pathways for reductive synthesis and energy storage, Green Chem., 2016, 19, 2347–2355 RSC.
- M. Rauch, Z. Strater and G. Parkin, Selective Conversion of Carbon Dioxide to Formaldehyde via a Bis(silyl)acetal: Incorporation of Isotopically Labeled C1 Moieties Derived from Carbon Dioxide into Organic Molecules, J. Am. Chem. Soc., 2019, 141, 17754–17762 CrossRef CAS PubMed.
- I. V. Delidovich, A. N. Simonov, O. P. Taran and V. N. Parmon, Catalytic formation of monosaccharides: from the formose reaction towards selective synthesis, ChemSusChem, 2014, 7, 1833–1846 CrossRef CAS PubMed.
- C. E. Harman, J. F. Kasting and E. T. Wolf, Atmospheric production of glycolaldehyde under hazy prebiotic conditions, Origins Life Evol. Biospheres, 2013, 43, 77–98 CrossRef CAS PubMed.
- P. van Duppen, E. Daines, W. E. Robinson and W. T. S. Huck, Dynamic Environmental Conditions Affect the Composition of a Model Prebiotic Reaction Network, J. Am. Chem. Soc., 2023, 145, 7559–7568 CrossRef CAS PubMed.
- A. N. Simonov, O. P. Pestunova, L. G. Matvienko and V. N. Parmon, 13C NMR studies of isomerization of D-glucose in an aqueous solution of Ca(OH)2. The effect of molecular oxygen, Russ. Chem. Bull., 2005, 54, 1967–1972 CrossRef CAS.
- W. E. Robinson, E. Daines, P. van Duppen, T. de Jong and W. T. S. Huck, Environmental conditions drive self-organization of reaction pathways in a prebiotic reaction network, Nat. Chem., 2022, 14, 623–631 CrossRef CAS PubMed.
- A. Omran, C. Menor-Salvan, G. Springsteen and M. Pasek, The Messy Alkaline Formose Reaction and Its Link to Metabolism, Life, 2020, 10, 125 CrossRef CAS PubMed.
- M. Haas, S. Lamour and O. Trapp, Development of an advanced derivatization protocol for the unambiguous identification of monosaccharides in complex mixtures by gas and liquid chromatography, J. Chromatogr. A, 2018, 1568, 160–167 CrossRef CAS PubMed.
- Y. Furukawa, M. Horiuchi and T. Kakegawa, Selective stabilization of ribose by borate, Origins Life Evol. Biospheres, 2013, 43, 353–361 CrossRef PubMed.
- M. Haas, S. Lamour, S. B. Christ and O. Trapp, Mineral-mediated carbohydrate synthesis by mechanical forces in a primordial geochemical setting, Commun. Chem., 2020, 3, 140 CrossRef CAS PubMed.
- S. Pallmann, J. Šteflová, M. Haas, S. Lamour, A. Henß and O. Trapp, Schreibersite: an effective catalyst in the formose reaction network, New J. Phys., 2018, 20, 055003 CrossRef.
- E. Camprubi, S. A. Harrison, S. F. Jordan, J. Bonnel, S. Pinna and N. Lane, Do Soluble Phosphates Direct the Formose Reaction towards Pentose Sugars?, Astrobiology, 2022, 22, 981–991 CrossRef CAS PubMed.
- A. W. Schwartz and R. M. de Graaf, The prebiotic synthesis of carbohydrates: a reassessment, J. Mol. Evol., 1993, 36, 101–106 CrossRef CAS.
- T. D. Machajewski and C.-H. Wong, The Catalytic Asymmetric Aldol Reaction, Angew. Chem., Int. Ed., 2000, 39, 1352–1375 CrossRef CAS PubMed.
- D. Kopetzki and M. Antonietti, Hydrothermal formose reaction, New J. Chem., 2011, 35, 1787–1794 RSC.
- N. W. Gabel and C. Ponnamperuma, Model for origin of monosaccharides, Nature, 1967, 216, 453–455 CrossRef CAS PubMed.
- S. Civis, R. Szabla, B. M. Szyja, D. Smykowski, O. Ivanek, A. Knizek, P. Kubelik, J. Sponer, M. Ferus and J. E. Sponer, TiO2-catalyzed synthesis of sugars from formaldehyde in extraterrestrial impacts on the early Earth, Sci. Rep., 2016, 6, 23199 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Polyoxometalates in solution: speciation under spotlight, Chem. Soc. Rev., 2020, 49, 7568–7601 RSC.
- T. Kimura, K. Kamata and N. Mizuno, A bifunctional tungstate catalyst for chemical fixation of CO2 at atmospheric pressure, Angew. Chem., Int. Ed. Engl., 2012, 51, 6700–6703 CrossRef CAS PubMed.
- T. Kimura, H. Sunaba, K. Kamata and N. Mizuno, Efficient [WO4]2--catalyzed chemical fixation of carbon dioxide with 2-aminobenzonitriles to quinazoline-2,4(1H,3H)-diones, Inorg. Chem., 2012, 51, 13001–13008 CrossRef CAS PubMed.
- K. Kamata, T. Kimura, H. Sunaba and N. Mizuno, Scope of chemical fixation of carbon dioxide catalyzed by a bifunctional monomeric tungstate, Catal. Today, 2014, 226, 160–166 CrossRef CAS.
- K. Sugahara, T. Kimura, K. Kamata, K. Yamaguchi and N. Mizuno, A highly negatively charged γ-Keggin germanodecatungstate efficient for Knoevenagel condensation, Chem. Commun., 2012, 48, 8422–8424 RSC.
- K. Sugahara, N. Satake, K. Kamata, T. Nakajima and N. Mizuno, A basic germanodecatungstate with a -7 charge: efficient chemoselective acylation of primary alcohols, Angew. Chem., Int. Ed., 2014, 53, 13248–13252 CrossRef CAS PubMed.
- M. L. Hayes, N. J. Pennings, A. S. Serianni and R. Barker, Epimerization of aldoses by molybdate involving a novel rearrangement of the carbon skeleton, J. Am. Chem. Soc., 1982, 104, 6764–6769 CrossRef CAS.
- H. J. Kim, A. Ricardo, H. I. Illangkoon, M. J. Kim, M. A. Carrigan, F. Frye and S. A. Benner, Synthesis of carbohydrates in mineral-guided prebiotic cycles, J. Am. Chem. Soc., 2011, 133, 9457–9468 CrossRef CAS PubMed.
- M. Rellán-Piñeiro, M. Garcia-Ratés and N. López, A mechanism for the selective epimerization of the glucose mannose pair by Mo-based compounds: towards catalyst optimization, Green Chem., 2017, 19, 5932–5939 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision E.01, 2009 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. Hayashi, S. Yamazoe, K. Koyasu and T. Tsukuda, Application of group V polyoxometalate as an efficient base catalyst: a case study of decaniobate clusters, RSC Adv., 2016, 6, 16239–16242 RSC.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, High-yield electrochemical production of formaldehyde from CO2 and seawater, Angew. Chem., Int. Ed. Engl., 2014, 53, 871–874 CrossRef CAS PubMed.
- Y. Y. Birdja and M. T. Koper, The Importance of Cannizzaro-Type Reactions during Electrocatalytic Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2017, 139, 2030–2034 CrossRef CAS PubMed.
- H. Zhong, K. Fujii, Y. Nakano and F. Jin, Effect of CO2 Bubbling into Aqueous Solutions Used for Electrochemical Reduction of CO2 for Energy Conversion and Storage, J. Phys. Chem. C, 2014, 119, 55–61 CrossRef.
- N. Yoshihara, M. Arita and M. Noda, Electrolyte Dependence for the Electrochemical CO2 Reduction Activity on Cu(111) Electrodes, Chem. Lett., 2017, 46, 125–127 CrossRef CAS.
- B. L. Horecker, O. Tsolas and C. Y. Lai, 6 Aldolases, The Enzymes, ed. P. D. Boyer, Academic Press, 1972, vol. 7, pp. 213–258 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03377e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |