DOI:
10.1039/D3SC03194B
(Edge Article)
Chem. Sci., 2023,
14, 9197-9206
Synthetic exploration of electrophilic xanthylation via powerful N-xanthylphthalimides†
Received
23rd June 2023
, Accepted 30th July 2023
First published on 1st August 2023
Abstract
Organic xanthates are broadly applied as synthetic intermediates and bioactive molecules in synthetic chemistry. Electrophilic xanthylation represents a promising approach but has rarely been explored mainly due to the lack of powerful electrophilic reagents. Herein, synthetic exploration of electrophilic xanthylation via powerful N-xanthylphthalimides was investigated. This strategy might provide a new avenue to less-concerned but meaningful electrophilic xanthylation in organic synthesis. With the help of these powerful reagents, electrophilic xanthylation of a wide range of substrates including aryl/alkenyl boronic acids, β-keto esters, 2-oxindole, and alkyl amines, as well as previously inaccessible phenols (first report) was achieved under mild reaction conditions. Notably, this simple electrophilic xanthylation of alkyl amine substrates will occur in the desulfuration reaction, consistent with the previously reported methods. Similarly, xanthamide and thioxanthate groups could also be transformed into desired nucleophiles via this electrophilic reagent strategy. The broad substrate scope, excellent functional group compatibility and late-stage functionalization of bioactive or functional molecules made them very attractive as general reagents which will allow rapid incorporation of SC(S)R (R = OEt, Oalkyl, NEt2 and SEt) into the target molecules.
1 Introduction
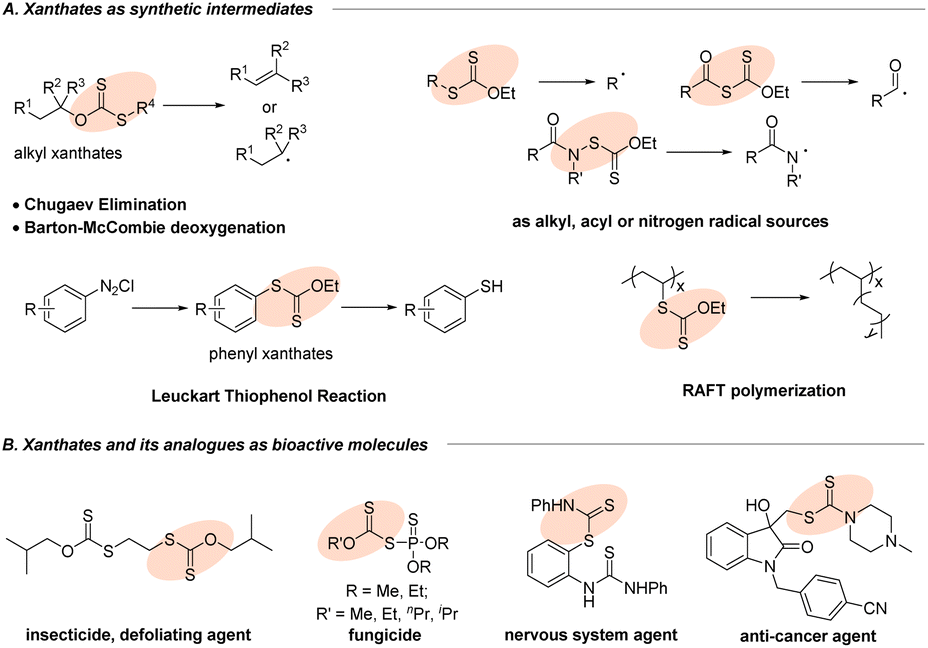
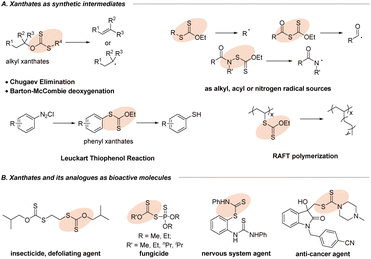
Organic sulfur compounds have become the focus of research interest due to their presence in myriad natural products,1 organic synthesis,2,3 biological processes4 and materials science.5 Xanthates and their analogues, a special class of sulfur-containing compounds with the S–C(S) linkage,6 have gained extensive importance because of their potential applications in the area of synthetic intermediates and bioactive molecules (Fig. 1). As an important functional group in synthetic chemistry, the first encounter with xanthates could generate (Z)-olefins through the famous Chugaev elimination (Fig. 1A)7 at a high temperature, which perhaps serve as key intermediates in the manufacture of viscose from cellulose.8 Besides, the well-developed Barton–McCombie deoxygenation9 serving as a convenient source of alkyl radicals from alcohols10 had a profound impact on the modification of carbohydrates in organic synthesis. Most importantly, organic xanthates could be also utilized as alkyl, acyl or nitrogen radical sources11 for further diverse or multifunctional transformations. Moreover, the extensive application of aryl xanthates is the famous Leuckart thiophenol reaction via nucleophilic xanthylation with diazonium salts (Fig. 1A).12 Furthermore, in the field of materials science, the RAFT polymerization process (Fig. 1A)13 can introduce xanthate groups into long-chain alkyl groups, which can thus avoid the disadvantage of chain termination in traditional polymerization reactions and make the free radical polymerization more controllable, providing a new method for the functional improvement of polymer materials. Additionally, xanthate units and their analogues are present prominently in agrochemicals (Fig. 1B, insecticide and fungicide)14a,b and pharmaceuticals (nervous system agent and anti-cancer agent),14c,d which has attracted increasing interest from both academia and the pharmaceutical industry. Therefore, development of new and valuable strategies for the efficient synthesis of organic xanthates from numerous substrates has been attracting continuous interest from the synthetic community.
 |
| | Fig. 1 Xanthates and its analogues as synthetic intermediates and bioactive molecules. | |
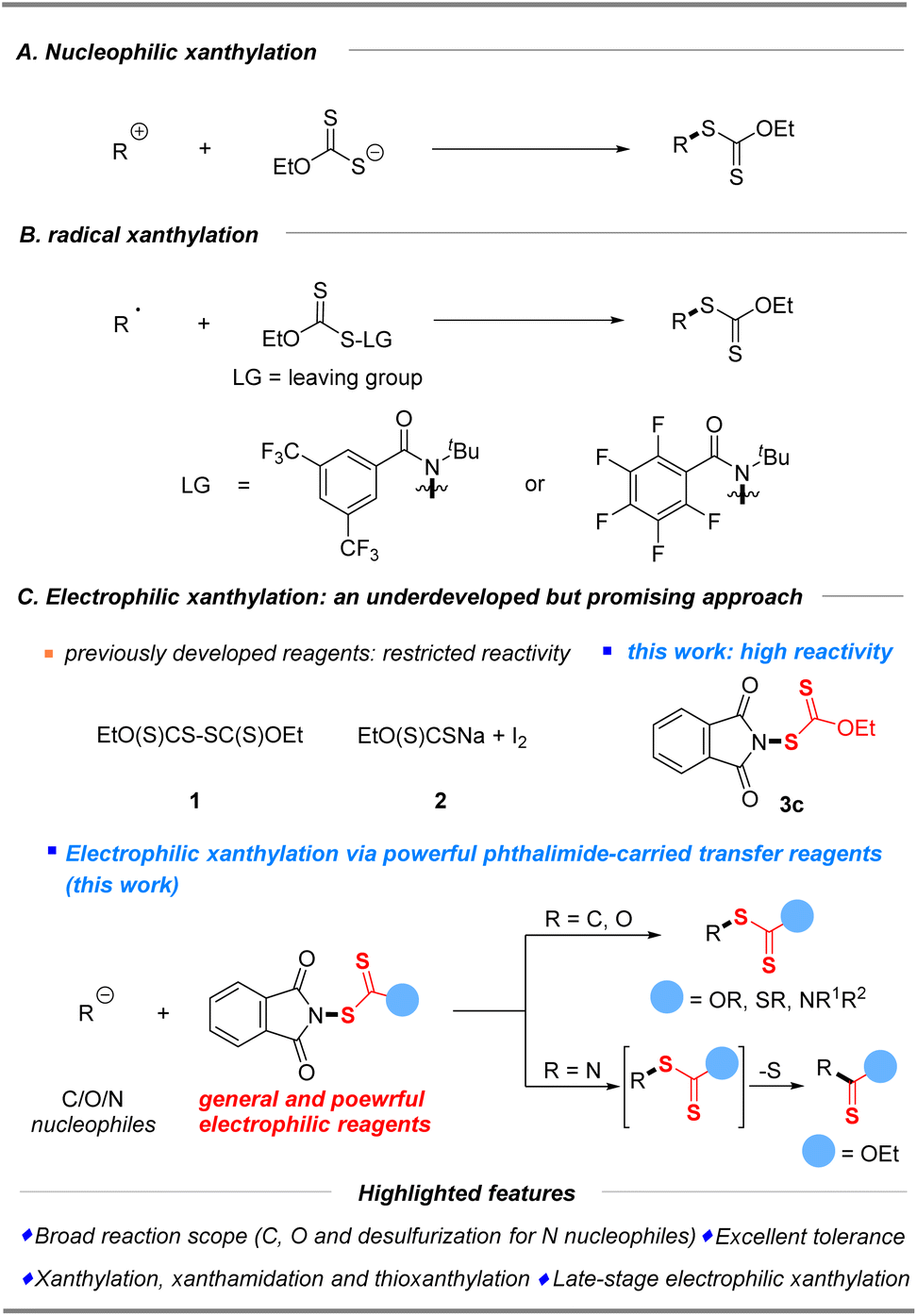
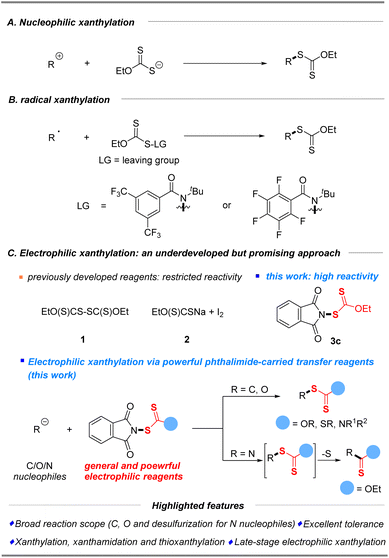
The conventional approach for the construction of xanthylated compounds typically involves metal-free nucleophilic substitution of electrophiles15 or a copper-mediated xanthylation of aryl halides with potassium ethylxanthate,16 well developed by Zard, Sekar, Melchiorre and others (Scheme 1A). As an alternative strategy, recent radical xanthylations using xanthylated synthons as radical acceptors or donor sources have emerged.17–19 In 2022, a facile, efficient, and practical protocol that enables the preparation of aryl xanthates from functionalized dibenzothiophenium salts and sodium ethylxanthogenate via a photoactivated electron donor–acceptor (EDA) complex was first established by Wang and his coworkers.18 Notably, Alexanian19a,b recently reported a direct C–H radical xanthylation or decarboxylative radical xanthylation by distinctive reagent N-ethylxanthylamides under redox-neutral conditions to unlock a wide array of conversions of hydrocarbons (Scheme 1B).
 |
| | Scheme 1 Synthetic strategies for xanthylation. | |
Considering that structurally diverse organic molecules especially lead compounds of drugs are typically electron-rich, electrophilic xanthylation represents an attractive strategy that will be widely applied in the future of drug research and development (R&D), but it has rarely been explored due to the scarcity of general and powerful electrophilic reagents. So far, only two electrophilic reagents (EtO(S)CS–SC(S)OEt (1) and EtO(S)CSNa/I2 (2)) have been reported for electrophilic substitutions of methylene compounds (1) and special amines (2).20 Our further electrophilic attempts disclosed that these previously developed reagents (Scheme 1C, 1–2) have restricted reactivity for common C/N/O nucleophiles (boronic acids, keto esters, alkyl amines, and phenols, please see the ESI† for more details). Therefore, the search for a practical and efficient synthetic auxiliary that can conveniently play the role of xanthyl group transfer, as a general and powerful electrophilic xanthylating reagent, is of high demand and scientific interest. Inspired by the successful development of electrophilic phthalimide-based reagents,21,22 which probably benefits from the cleavage of the weak “N–S” bond, we wished to consider whether N-xanthylphthalimides could serve as broadly applicable reagents that enable electrophilic xanthylation of various nucleophiles. The reactivities of electrophilic xanthyl reagents were then demonstrated by xanthylation of a wide range of substrates, including C and N common nucleophiles as well as the first report of O nucleophiles in good to excellent yields with superb functional group compatibility under mild conditions (Scheme 1C). Notably, this simple electrophilic xanthylation of amine substrates will occur in the desulfuration reaction to give important and valuable thiocarbamates. Especially, the late-stage direct xanthylation of bioactive or functional molecules made this electrophilic xanthyl reagent very attractive to allow rapid incorporation of SC(S)OEt into target electron-rich molecules during the search for new drugs.
2 Results and discussion
2.1. Preparation of electrophilic ethylxanthyl reagents with amide or sulfonamide skeletons
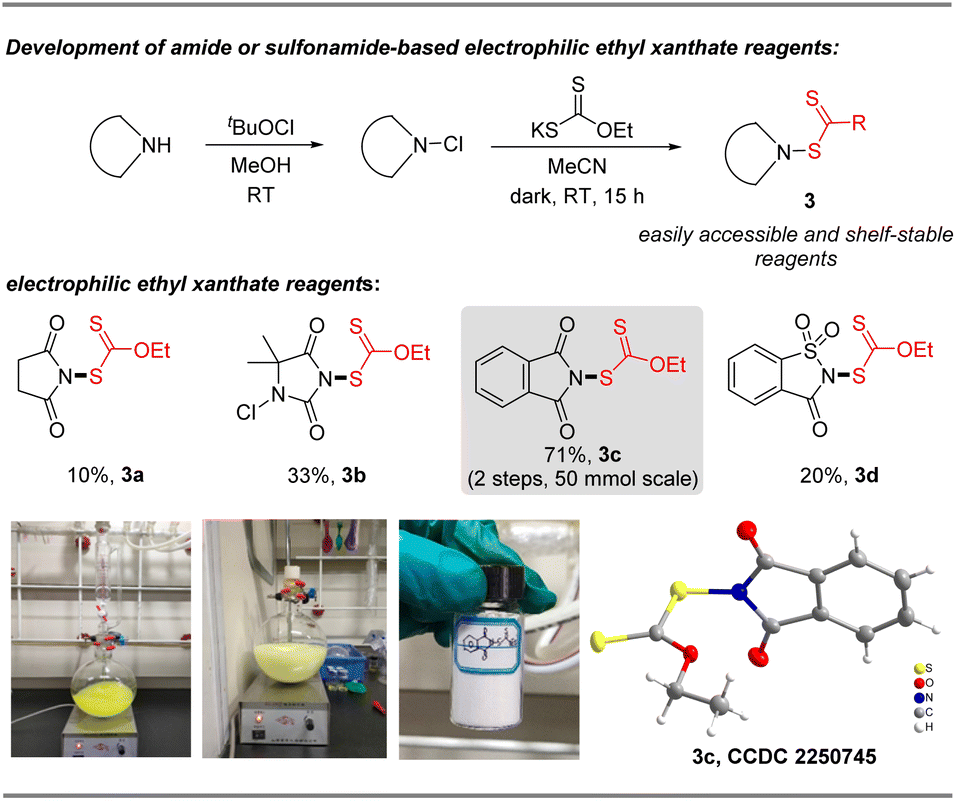
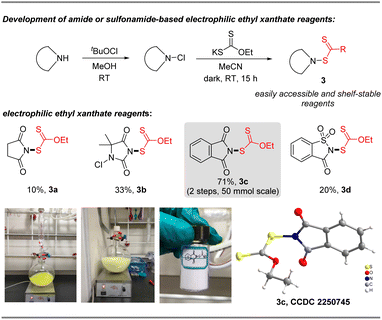
N-Ethylxanthyl phthalimide 3c was originally synthesized in 16% yield by De Greef in 2006,23 but its synthetic applications are unexplored. As an air and moisture-stable solid, it can be simply and rapidly synthesized from commercially available phthalimide and potassium ethylxanthate in 71% yield via a modified synthesis protocol, as shown in Fig. 2. X-ray crystallographic analysis of 3c24 shows that the N (1)–S (1) bond length is 1.683 Å, which is longer than the comparable bonds in a crystal of previously developed N-difluoromethylthiophthalimide and N-trifluoromethylthio succinimide (1.681 Å and 1.676 Å, respectively) (please see the ESI† for more details). These data reveal that reagent 3c is likely to be a potential electrophilic reagent. Similarly, ethylxanthyl reagents based on other amide or sulfonamide skeletons (succinimide 3a, 5,5-dimethylhydantoin 3b, and saccharin 3d) could also be prepared in acceptable yields (10–33%) via this strategy.
 |
| | Fig. 2 Two-step scalable preparation of N-ethylxanthylphthalimide 3c and its analogues. | |
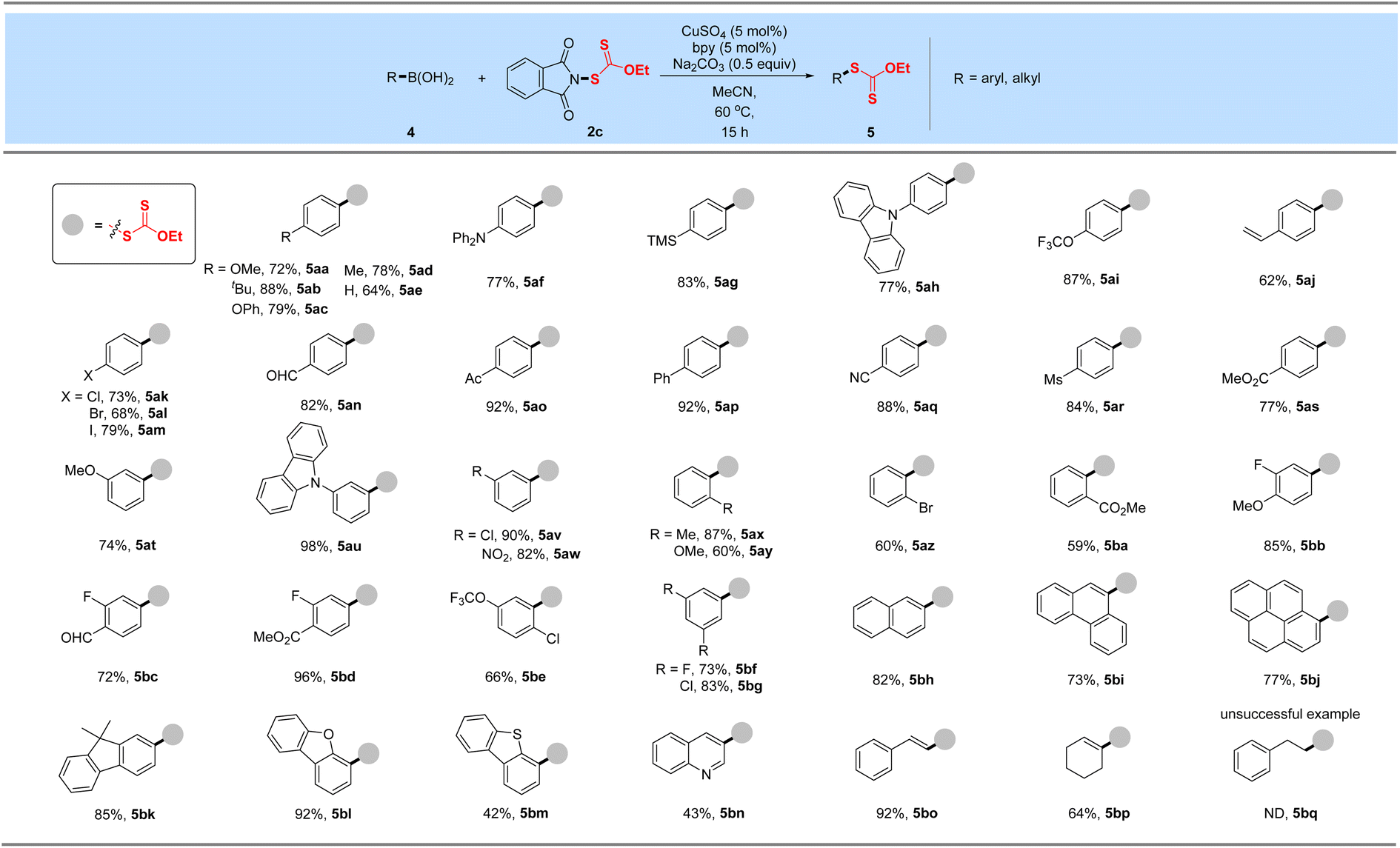
2.2. Xanthylation of aryl/alkenyl boronic acids
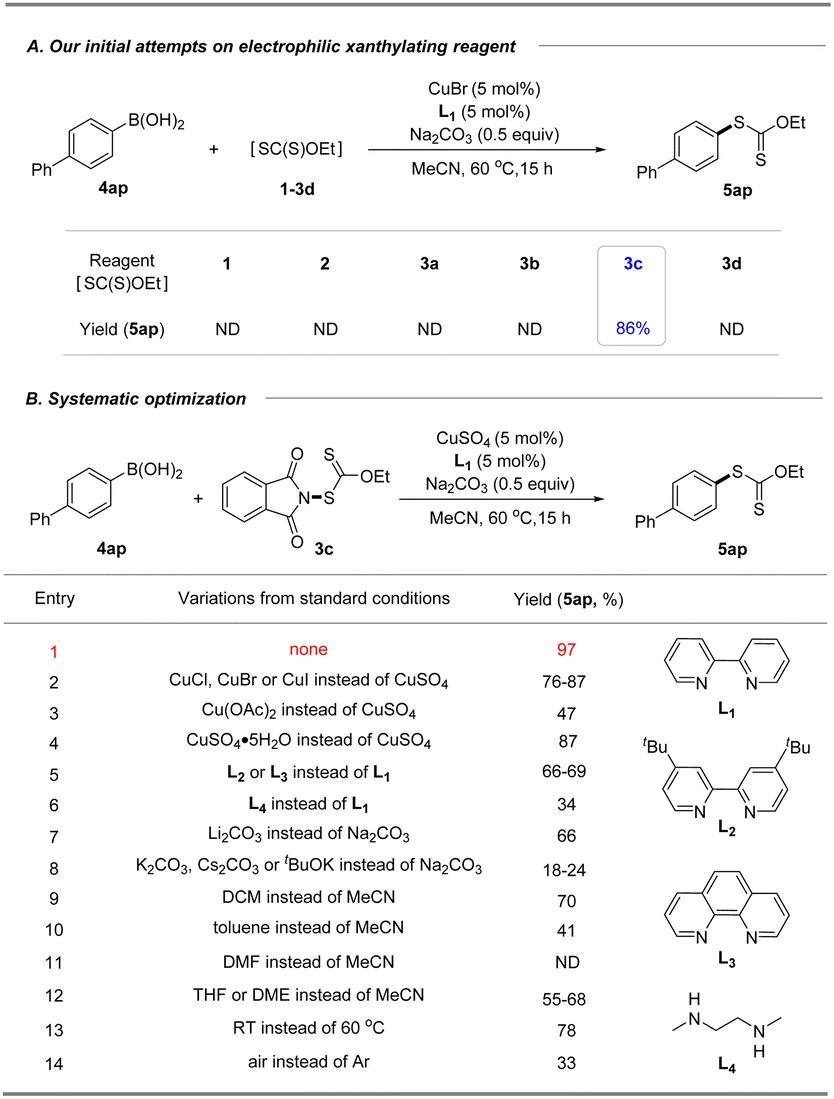
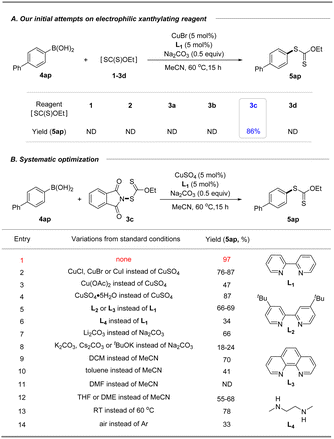
With these xanthyl reagents 3a–3d based on the amide or sulfonamide skeleton in hand, radical aliphatic C–H xanthylations were investigated and failed to give xanthylated hydrocarbons, indicating that the current N-xanthyl reagents, developed by De Greef and our group, are significantly different from Alexanian's N-xanthyl amides.19a,b Thus, we are trying to determine whether they can achieve electrophilic xanthylation that has received little attention but represents a meaningful strategy. Thus, we chose easily available, bench-stable aryl boronic acids as the precursors of aryl nucleophiles to investigate Chan–Lam-type xanthylation with electrophilic reagents 1–3d (Table 1A). The initial attempt had proved that EtO(S)CS–SC(S)OEt (1) or EtO(S)CSNa/I2 (2) has no reactivity for the cross-coupling of 4-biphenyl boronic acid. Further explorations with the mixing of 4ap and N-ethylxanthyl succinimide (3a) or N-ethylxanthyl saccharin (3d) in the presence of CuBr at 60 °C failed to deliver the xanthyl group to the aromatic ring, while unsuccessful conversion also occurred if the commercially available 5,5-dimethylhydantoin auxiliary (3b) was used. Surprisingly, the phthalimide skeleton (3c) was masked as the excellent leaving group in promoting the envisioned electrophilic transformation and could afford the desired ethylxanthyl-substituted 5ap in 86% yield. Subsequently, systematic investigations were conducted to obtain the optimal conditions of the reaction by choosing 3c as the model xanthyl reagent (Table 1B). A range of Cu(I) or Cu(II) catalysts (CuCl, CuBr, CuI and Cu(OAc)2) were screened, but no enhancement in the yield was detected (entries 2–3). Surprisingly, a reaction in the presence of cheap CuSO4 using 2,2′-bipyridine as the ligand in MeCN at 60 °C for 15 h provided a 97% yield of EtOC(S)S-derived product 5ap (entry 1). Notably, this Chan-Lam-type xanthylation can be catalyzed by a hydrated copper catalyst (CuSO4·5H2O), albeit the yield decreases slightly (entry 4). Control experiments showed that simple 2,2′-bipyridine is the most effective ligand for copper catalysts to promote this conversion, while other bidentate nitrogen ligands such as 4,4′-di-tert-butyl-2,2′-bipyridine (L2), o-phenanthroline (L3), and N,N′-dimethyl-1,2-ethanediamine (L4) gave unsatisfactory results (entries 5–6). Considering that the base plays an important role in the transmetalation process of cross coupling,25 a series of strong and weak bases including Li2CO3, Na2CO3, K2CO3, Cs2CO3 and tBuOK were investigated, and Na2CO3 was found to be the optimal base (entries 1, 7–8). Additionally, the effect of solvent on this coupling reaction was also explored (entries 9–12): DCM, toluene, and ether solvent (THF and DME) could afford the target product 5ap in 41–70% yields, while no target product was observed if DMF was employed which might be attributed to the coordination effect between DMF and the copper catalyst. As anticipated, this xanthate coupling reaction can be carried out at room temperature with a slightly reduced yield (78%, entry 13). By contrast, only 33% yield of the desired product was detected when it was conducted in air instead of an argon atmosphere (entry 14).
Table 1 Reaction optimizationa,b
|
Standard conditions: 4-biphenylboronic acid (0.10 mmol), reagent 3c (0.12 mmol, 1.2 equiv.), CuSO4 (5 mol%), bpy (5 mol%) and Na2CO3 (0.5 equiv.) in MeCN (1.0 mL) at 60 °C for 15 h.
Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard.
|
|
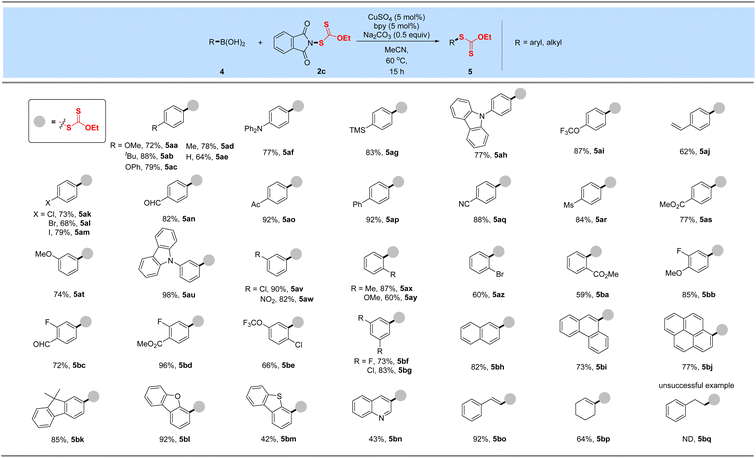
After obtaining the optimal conditions, the boronic acid scope of this Chan–Lam-type coupling was explored in Table 2. Overall, a variety of aryl boronic acids with electron-donating or -withdrawing substituted groups at the para, meta, and ortho-positions proceeded smoothly to deliver a set of corresponding xanthylated products in good to excellent yields (59–98%), which displays excellent reactivity and functional group compatibility over the traditional nucleophilic aromatic xanthylation. Many functional groups including methoxy, tert-butyl, phenoxy, trimethylsilyl, methyl, trifluoromethoxy, alkenyl, chlorine, bromine, iodine, aldehyde, acetyl, ester, methanesulfonyl, cyano and nitro were all tolerated (5aa–5bg). Diphenylamine (5af) and carbazole (5ah, 5au) groups widely used in the field of functional materials can be compatible with this system to afford 77–98% yields of ethyl arylxanthates. This transformation also proceeded smoothly for disubstituted aryl boronic acids bearing methoxy, trifluoromethoxy, fluorine, chlorine, aldehyde or ester substituents and gave desired products in good to excellent yields after simple column chromatography purification (5bb–5bg). These outcomes implied that the vast change in electronic properties of aryl boronic acids did not shut down the occurrence of the reaction. In particular, reserved bromide (5al and 5az), iodine (5am) and unprotected vinyl substituents (5aj) were compatible in this coupling, thus allowing subsequent functional-group orthogonal transformations. Besides, boronic acids from the 9,9-dimethyl-9H-fluorene (5bk) and polybenzo skeleton (naphthalene, phenanthrene or pyrene, 5bh–5bj) were further tested and could be well tolerated in this developed xanthylation, which provide modification methods of these meaningful molecules in fluorescence chemistry. Encouragingly, this simple copper-catalyzed coupling protocol can be applied to structurally diverse heteroaromatic boronic acids such as benzofuran, benzothiophene and quinoline, delivering the desired Chan–Lam-type coupling products in 92%, 42% and 43% yields, respectively (5bl–5bn). Additionally, satisfactory yields could be obtained when styryl boronic acid and cyclohexenyl boronic acid were used as model substrates in this strategy (5bo–5bp). In accordance with the general trend of reactivity, alkyl boronic acid was tested but failed to achieve the desired xanthyl coupling (5bq).
Table 2 Xanthylation of aryl/alkenyl boronic acidsa,b
|
Reaction conditions: aryl or alkenyl boronic acid (4, 0.50 mmol), reagent 3c (0.6 mmol, 1.2 equiv.), CuSO4 (5 mol%), bpy (5 mol%) and Na2CO3 (0.5 equiv.) in MeCN (3.0 mL) at 60 °C for 15 h.
Isolated yields.
|
|
2.3. Xanthylation of other nucleophiles
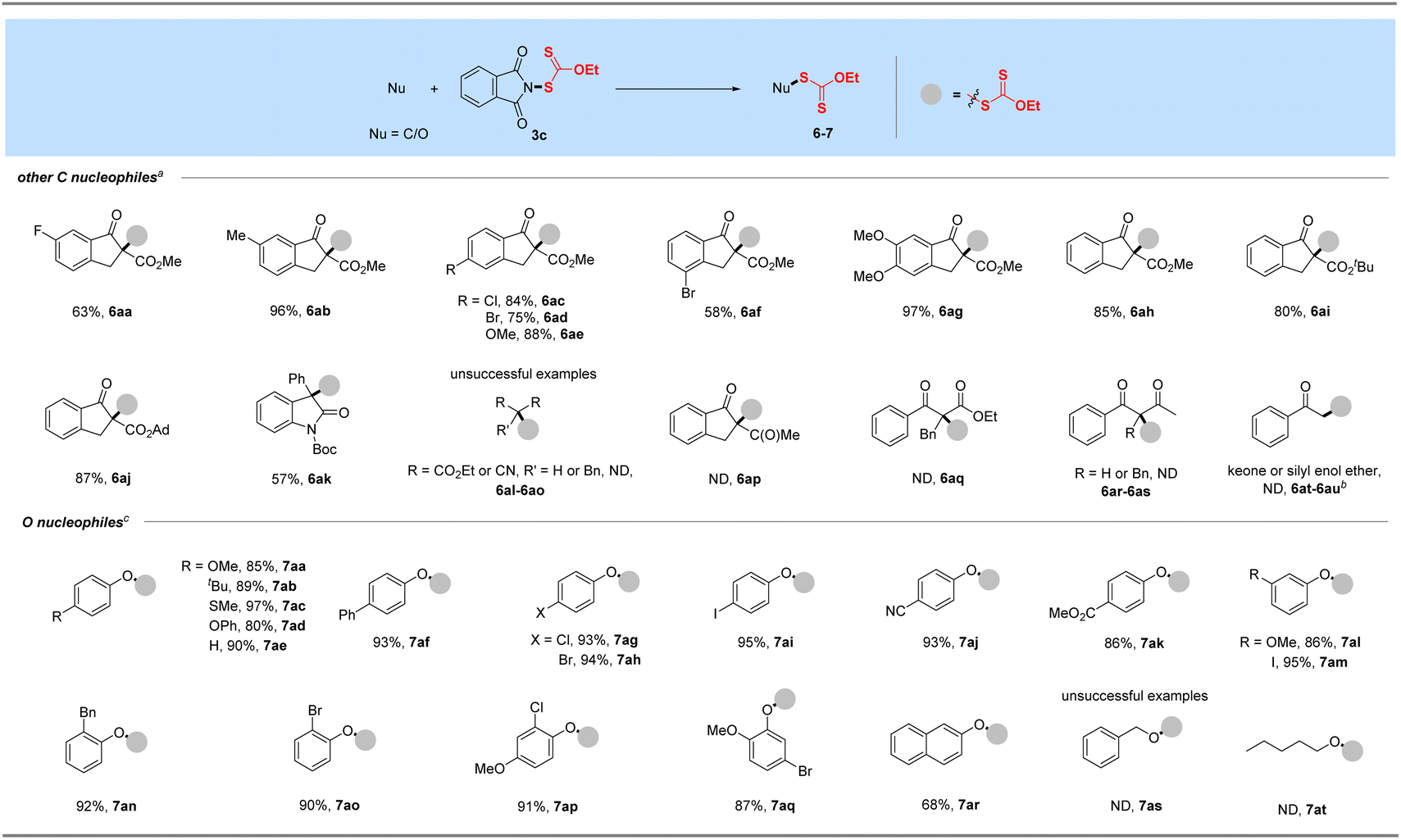
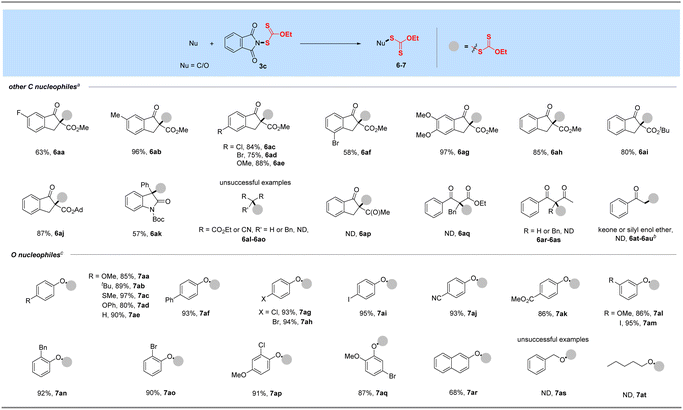
As a supplement to the traditional synthesis of α-xanthylation of 1,3-carbonyl compounds under strong base conditions,20b,c our attempt of N-ethylxanthyl phthalimide 3c with soft nucleophile β-keto esters was conducted under mild conditions to expand the synthesis of Csp3-xanthates, as shown in Table 3. With the assistance of K2CO3, a range of β-keto esters including methyl, tert-butyl and adamantane esters, could be well compatible in this protocol (6aa–6aj). Moreover, this transformation proceeded smoothly to afford the α-xanthyl products from indanone substrates with electron-withdrawing (F, Cl, Br, 6aa, 6ac–6ad, 6af) or electron-donating (Me, OMe, 6ab, 6ae, 6ag) functional groups at different positions of phenyl rings. Notably, the N-Boc-3-phenyl-2-oxindole substrate derived from dye isatin was also tolerated and afforded the xanthylated product in a satisfactory yield for this transformation (6ak). Unfortunately, the reactions of 3c with additional 1,3-dicarbonyls such as cyclic and acyclic diketones, diesters, and dinitrile with or without substituents at the alpha position as well as a ketone or its silyl enol ether failed under our standard or revised conditions.
Table 3 Xanthylation of other C and O nucleophilesa,d
|
Reaction conditions: other C nucleophile (0.50 mmol), reagent 3c (0.6 mmol, 1.2 equiv.) and K2CO3 (1.1 equiv.) in DCE (3.0 mL) at 80 °C for 16 h.
Ketone (1.05 equiv. NaH as the base) or its silyl enol ether (w/or w/o K2CO3 (1.1 equiv.)) was used.
O nucleophile (0.50 mmol), reagent 3c (0.7 mmol, 1.4 equiv.) and Cs2CO3 (1.1 equiv.) in DCE (3.0 mL) at 80 °C for 16 h.
Isolated yields.
|
|
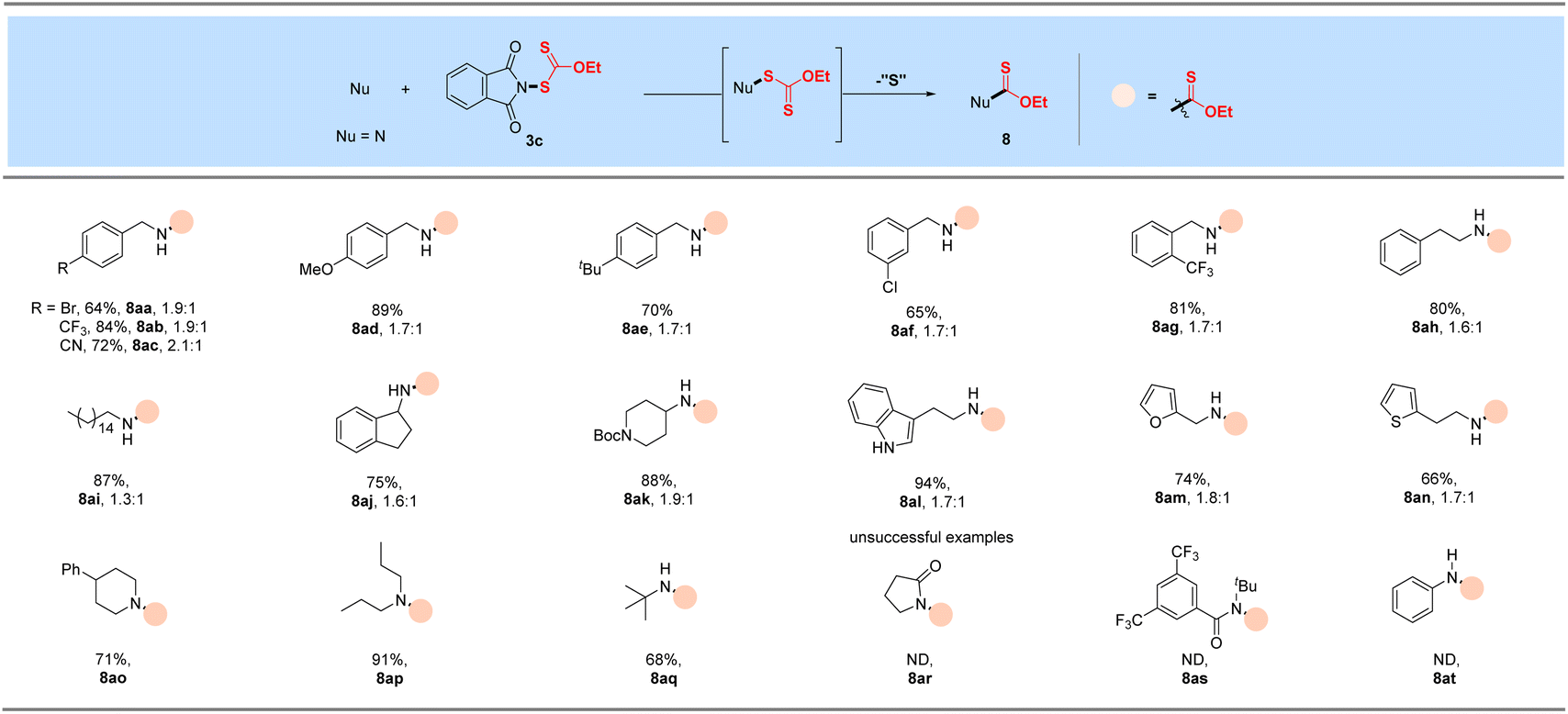
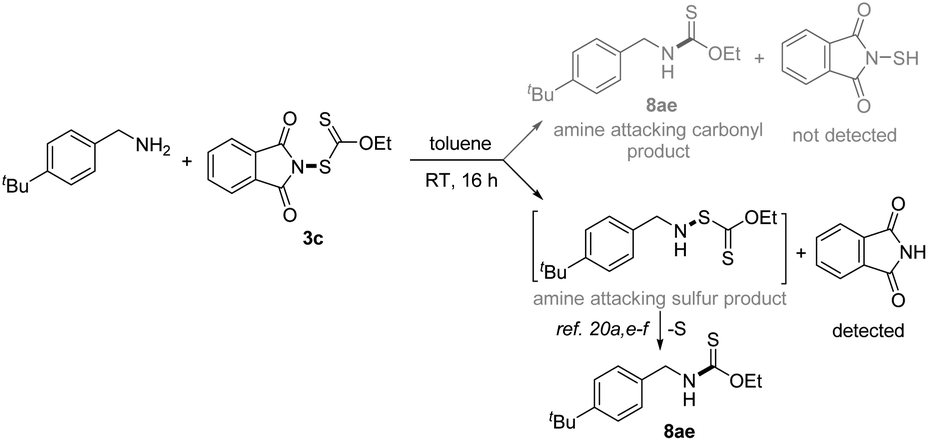
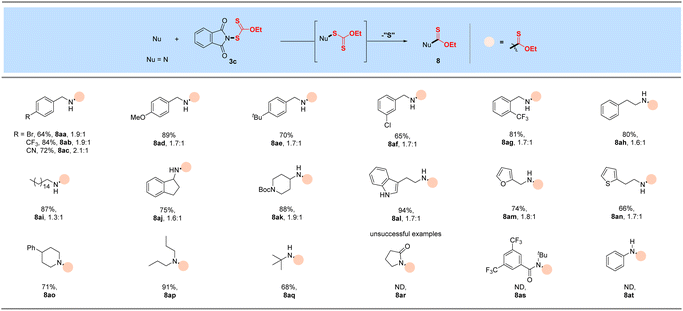
With the successful development of methods for the formation of C-xanthates, we next sought to investigate the electrophilic xanthylation of heteroatomic nucleophiles, which is an attractive yet rarely reported issue in xanthate chemistry. Electrophilic coupling with weakly nucleophilic phenols to obtain O-xanthate products was first reported by us. As summarized in the bottom of Table 3, phenol substrates were smoothly converted to the corresponding O-xanthates in 68–97% yields with the aid of Cs2CO3, bearing multiple functional groups including methoxyl, tert-butyl, methylthio, phenoxy, benzyl, halides, ester and cyano at different positions of phenyl rings (7aa–7ao). Polysubstituted halophenols could successfully deliver the corresponding products in acceptable yields (7ap–7aq). Similarly, 2-naphthol could be also expediently xanthylated under these mild reaction conditions (7ar). However, aliphatic alcohols are not applicable to obtain corresponding O-xanthate products (7as–7at). Encouraged by the superior performance of N-ethylxanthyl phthalimide 3c of phenols, coupling with amine substrates to obtain N-xanthylamines, which could display the potential fungicidal activity,20d was further explored. Surprisingly, it was found that the reaction of alkyl amines with powerful reagent 3c could successfully generate moderate to high yields of structurally diverse thiocarbamates with different rotamers via desulfuration of N-xanthylamines (Table 4), which is indirectly proved by control experiments (Scheme 2, and see the ESI† for more details) and consistent with the occurrence of the desulfurization phenomenon in previous literature.20a,e,f Electron-rich and electron-deficient benzylamines bearing different halogens, trifluoromethyl, cyano, methoxy and tert-butyl were all tolerated in the reaction with 3c to deliver the corresponding thiocarbamate products (8aa–8ag) in high yields. As expected, electrophilic xanthylation of long-chain alkyl amines (phenylethylamine and cetylamine, 8ah–8ai) and even cycloalkyl amines (1-indamine and 4-amino-1-boc-piperidine, 8aj–8ak) can occur smoothly for the desulfuration conversion. Notably, heterocyclic amines including furfurylamine, thiophene-2-ethanamine and tryptamine could be coupled to afford the desired N–C(S)OEt products in this system (8al–8an). In addition to the primary alkyl amines, the reaction of secondary alkyl amines 4-phenylpiperidine and di-n-propylamine with 3c also proceeded in high to excellent yields under standard conditions (8ao–8ap). Additionally, tert-butylamine with a large steric hindrance can also successfully deliver the corresponding target product (8aq). Furthermore, when we extended this system to the substrates of aromatic amines or amide salts from 2-pyrrolidinone or substituted benzamide, the corresponding N-xanthylamides or thiocarbamates cannot be obtained (8ar–8at).
Table 4 Xanthylation of N nucleophilesa,b
|
Reaction conditions: amine or amide salt (0.50 mmol) and reagent 3c (0.6 mmol, 1.2 equiv.) in toluene (3.0 mL) at RT for 16 h.
Isolated yields. The ratio of the thiocarbamate product means the rotamer ratio.
|
|
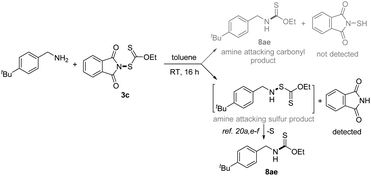 |
| | Scheme 2 Control experiments. | |
2.4. Applications of xanthyl-derived reagents
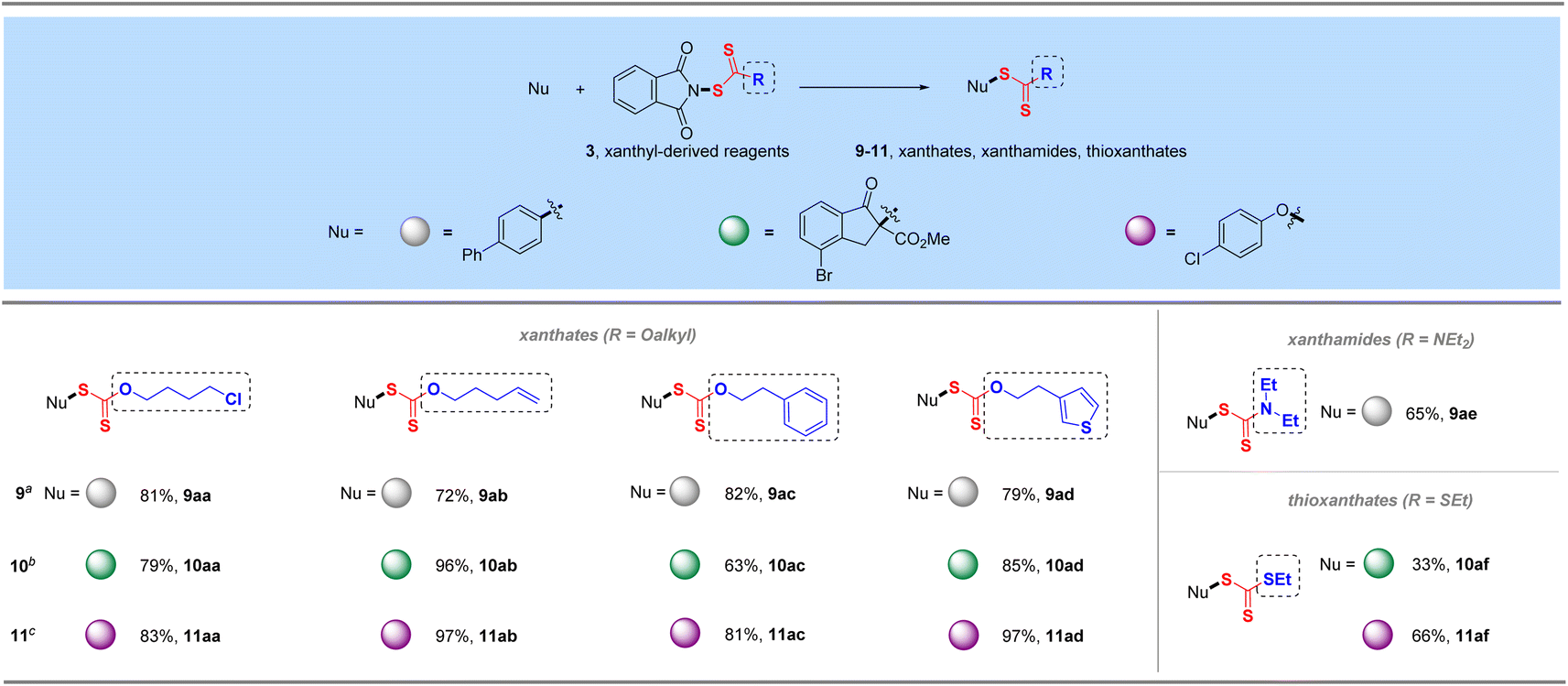
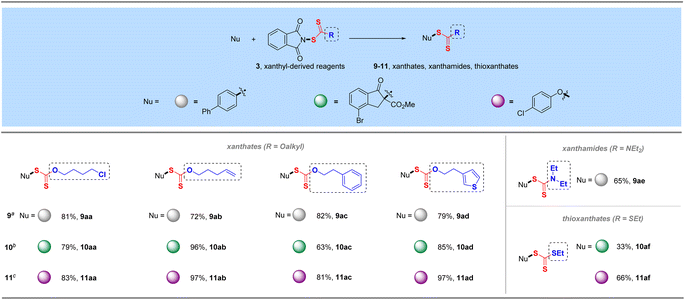
Phthalimide, an efficient amide transfer framework, can also be used in the synthesis of xanthyl-derived reagents, which can also be efficiently coupled with various C and O nucleophiles (4-biphenyl boronic acid, 4-bromoindanone methyl ester and para-chlorophenol) to give manifold xanthate products in excellent yields under the corresponding conditions (9aa–9ad, 10aa–10ad, and 11aa–11ad, Table 5). These diverse xanthyl reagents could tolerate multiple functional groups such as halogen, alkenyl, aromatic ring and thiophene heterocycle during their preparations and synthetic transformations. More specifically, this strategy could also be employed for the transfer of electrophilic xanthamide and thioxanthate groups, incorporating –SC(S)R (R = NEt2 and SEt) into corresponding organic molecules in satisfactory yields (9ae, 10af, 11af), which would provide a novel electrophilic alternative to the traditional synthesis of xanthamide- or thioxanthate-containing compounds.26
Table 5 Applications of xanthyl-derived reagentsa,d
|
Reaction conditions: 4-biphenyl boronic acid (0.50 mmol), reagent 3 (0.6 mmol, 1.2 equiv.), CuSO4 (5 mol%), bpy (5 mol%) and Na2CO3 (0.5 equiv) in MeCN (3.0 mL) at 60 °C for 15 h.
4-Bromoindanone methyl ester (0.50 mmol), reagent 3 (0.6 mmol, 1.2 equiv.) and K2CO3 (1.1 equiv.) in DCE (3.0 mL) at 80 °C for 16 h.
4-Chlorophenol (0.50 mmol), reagent 3 (0.7 mmol, 1.4 equiv.) and Cs2CO3 (1.1 equiv.) in DCE (3.0 mL) at 80 °C for 16 h.
Isolated yields.
|
|
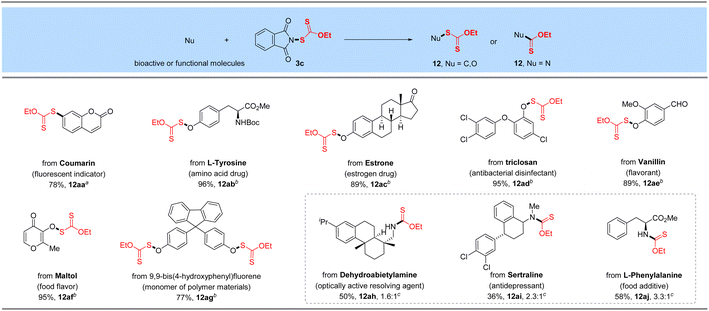
2.5. Late-stage xanthylation of bioactive or functional molecules
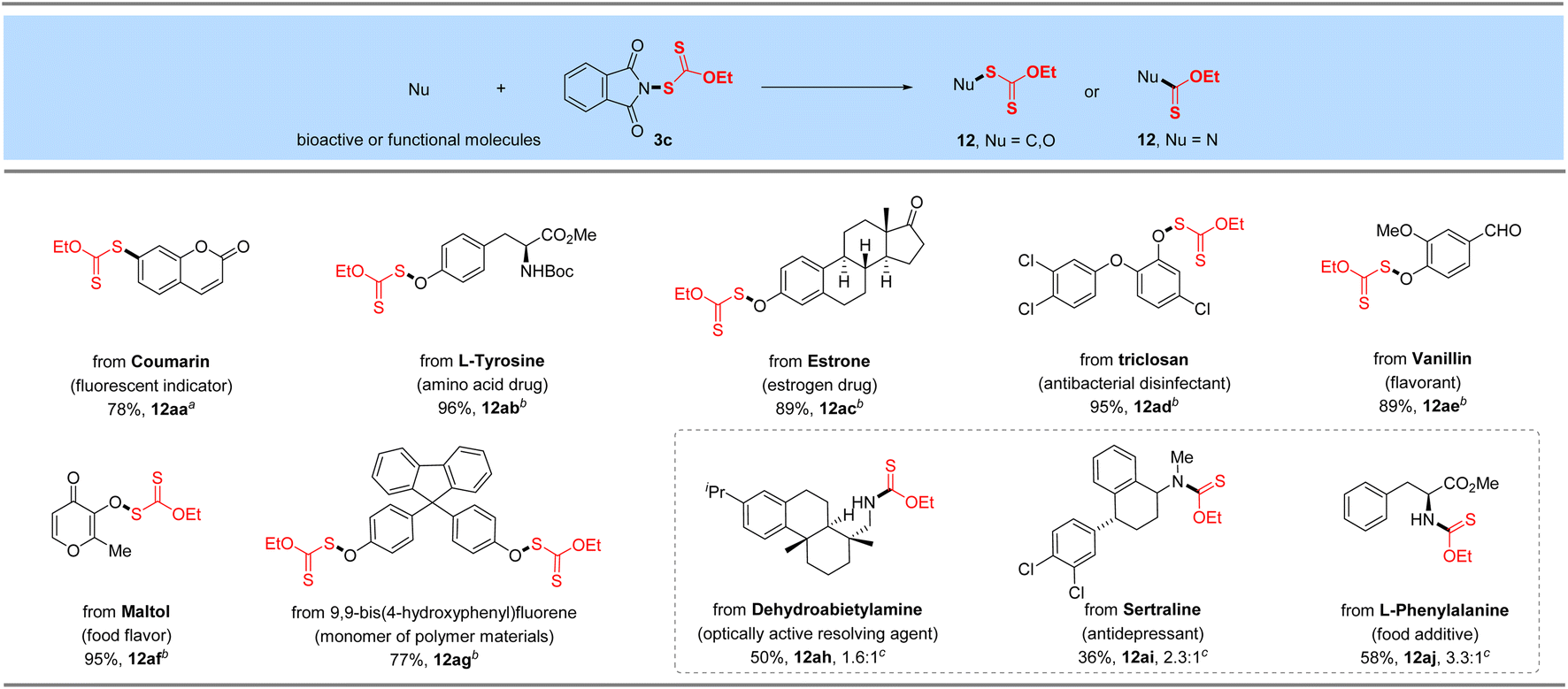
The promising functional group tolerance and high efficiency of this protocol enabled its application to the late-stage xanthylation of structurally complicated bioactive, functional molecules or druglike scaffolds (Table 6). For instance, boronic acid substrate derived from fluorescent indicator 7-hydroxycoumarin reacted smoothly with N-xanthylphthalimide 3c catalyzed by cheap CuSO4 (12aa). Besides, a 96% yield of the target O-xanthate was successfully obtained for the N-protected amino acid drug L-tyrosine (12ab). Moreover, the bioactive phenol substrate estrone drug was tested as a superior candidate under the standard reaction conditions and furnished the O–SC(S)OEt product in 89% yield (12ac). As a broad-spectrum antibacterial disinfectant against Staphylococcus, Escherichia coli and Candida albicans, phenol-derived triclosan could be decorated with the –SC(S)OEt linkage in an excellent yield (95%, 12ad) through our developed electrophilic ethylxanthyl reagent 3c. Likewise, natural or synthetic food flavoring agents such as vanillin and maltol successfully underwent the reaction with 89–95% yields of target O–S coupling products 12ae–12af, which shows good compatibility with activated carbon–carbon double bonds. Finally, 9,9-bis(4-hydroxyphenyl)fluorene as a monomer of polymer materials, was investigated and converted to the corresponding xanthyl product in 77% yield under the optimized conditions (12ag), displaying its potential applications in the field of functional materials. It is noteworthy that dehydroabietylamine (optically active resolving agent) and an antidepressant sertraline were coupled with reagent 3c to yield the corresponding desulfurating thiocarbamates (12ah–12ai) in 36–50% yields. As expected, modification of esterified amino acid L-phenylalanine can also produce the desired N–C(S)OEt product (12aj).
Table 6 Late-stage xanthylation of bioactive or functional moleculesa,d
|
Reaction conditions: aryl boronic acid (0.50 mmol), reagent 3c (0.6 mmol, 1.2 equiv.), CuSO4 (5 mol%), bpy (5 mol%) and Na2CO3 (0.5 equiv.) in MeCN (3.0 mL) at 60 °C for 15 h.
Phenol (0.50 mmol), reagent 3c (0.7 mmol, 1.4 equiv.) and Cs2CO3 (1.1 equiv.) in DCE (3.0 mL) at 80 °C for 16 h.
Alkyl amine (0.50 mmol) and reagent 3c (0.6 mmol, 1.2 equiv.) in toluene (3.0 mL) at RT for 16 h. The ratio of the thiocarbamate product means the rotamer ratio.
Isolated yields.
|
|
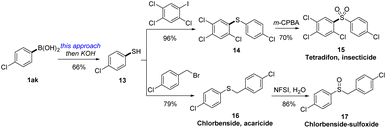
2.6. Synthetic applications
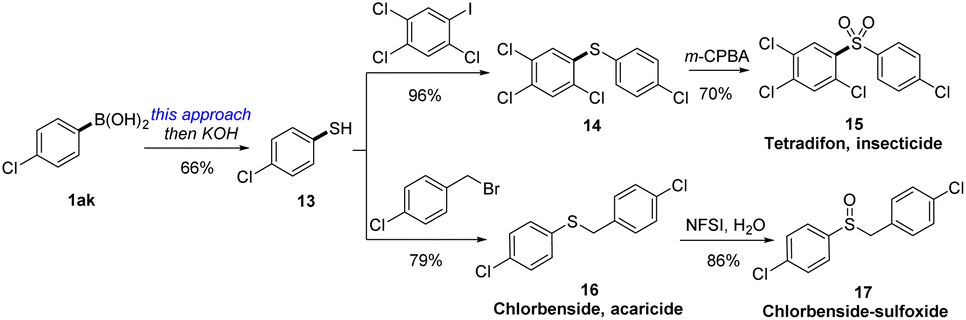
To highlight the practical utility of this protocol in synthesis, we then unlock the synthesis of two important thioether-containing bioactive insecticides (Scheme 3). Thus, treating 4-chlorophenylboric acid with ethyl xanthyl reagent 3c by this electrophilic xanthylation approach and thiolysis under alkaline conditions could generate 4-chlorothiophenol. After cross coupling with 2,4,5-trichloro-1-iodobenzene and excessive oxidation by m-CPBA, a famous insecticide tetradifon (15)27 was synthesized in 44% overall yield. Likewise, we then applied this strategy in the synthesis of an acaricide chlorbenside (16), which could also be selectively converted into the sulfoxide derivative (18) by NFSI via a controllable oxidation.28
 |
| | Scheme 3 Synthesis of bioactive tetradifon and chlorbenside-sulfoxide. | |
3 Conclusions
In summary, a highly desirable and remarkable electrophilic xanthylation via easily accessible and shelf-stable xanthylating reagent N-xanthylphthalimides has been developed. The present protocol might provide a new avenue to the xanthate derivatives in an unmet electrophilic manner in organic synthesis. These xanthylating reagents are effective and powerful for the direct transfer of the –SC(S)OR groups to a rich portfolio of substrates, including Cu-catalyzed Cham–Lam-type xanthylation with organoboronic acids, and electrophilic xanthylation of β-keto esters, 2-oxindole, alkyl amines, as well as previously inaccessible phenols (first report) under metal-free conditions. Notably, this simple electrophilic xanthylation of alkyl amines reported by us also occurs in the desulfuration reaction, providing a novel synthesis method to access valuable O-ethylthiocarbamates. Additionally, we also report the introduction of xanthamide and thioxanthate groups into organic molecules via this electrophilic reagent strategy. Highlighted features of mild conditions, broad substrate scope, and excellent tolerance, as well as late-stage functionalization of biologically active or functional molecules and further application to the synthesis of bioactive drugs made them very attractive as general reagents which will allow rapid incorporation of SC(S)R (R = OEt, Oalkyl, NEt2 and SEt) into densely functionalized drug-like molecules. Expanding the synthetic application of these electrophilic reagents is underway and will be reported shortly.
Data availability
All experimental data in this manuscript are available in the ESI.†
Author contributions
S. W., L. Y., F. L., Y. Z. and D. Z. conceived and designed the experiments. S. W., L. Y., F. L. and Y. Z. performed the experiments and analyzed the data. X.-R. L. performed the single crystal diffraction studies. Q. W., and D. Z. co-wrote the paper. Q. W. assisted in editing the manuscript and approved its contents. D. Z. directed the project. All authors discussed the experimental results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by generous grants from the National Natural Science Foundation of China (21901201 and 32200119), the Natural Science Foundation of Shaanxi Province (2020JQ-571) and the Education Department of Shaanxi Province (20JS146 and 21JK0930). We would like to sincerely thank Professor Xinjun Luan for his help and support to our project.
Notes and references
-
(a) C. Jacob, Nat. Prod. Rep., 2006, 23, 851–863 RSC;
(b) K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS PubMed;
(c) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC.
-
(a) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed;
(b) M. H. Feng, B. Q. Tang, H. L. Steven and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed;
(c) P. Devendar and G. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed;
(d) S. Huang, M. Wang and X. Jiang, Chem. Soc. Rev., 2022, 51, 8351–8377 RSC.
-
(a) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611–620 CrossRef CAS PubMed;
(b) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133–5209 CrossRef CAS PubMed;
(c) Y. Li and M.-H. Xu, Chem. Commun., 2014, 50, 3771–3782 RSC;
(d) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed.
-
(a) G. A. Benavides, G. L. Squadrito, R. W. Mills, H. D. Patel, T. S. Isbell, R. P. Patel, V. M. Darley-Usmar, J. E. Doeller and D. W. Kraus, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17977–17982 CrossRef CAS PubMed;
(b) J. A. Cebollada, P. Kosuri, J. A. R. Pardo and J. M. Fernàndez, Nat. Chem., 2011, 3, 882–887 CrossRef PubMed;
(c) M. Wang and X. Jiang, Topics Curr. Chem., 2018, 376, 1–40 CrossRef CAS PubMed.
-
(a) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed;
(b) X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535–538 CrossRef CAS PubMed;
(c) H. Iino, T. Usui and J. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS PubMed;
(d) D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
-
(a) P. Delduc, C. Tailhan and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1988, 308–310 RSC;
(b) S. Z. Zard, Angew. Chem., Int. Ed., 1997, 36, 672–685 CrossRef.
-
(a) L. Chugaev, Chem. Ber., 1899, 32, 3332–3335 CrossRef;
(b) H. R. Nace, Org. React., 1962, 12, 57–100 CAS;
(c) K. Lee and D. L. Boger, J. Am. Chem. Soc., 2014, 136, 3312–3317 CrossRef CAS PubMed;
(d) D. H. Dethe and B. D. Dherange, J. Org. Chem., 2018, 83, 3392–3396 CrossRef CAS PubMed;
(e) F. S. Karimi, A. Seif and E. Zahedi, ChemistrySelect, 2022, 7, e202202169 CrossRef CAS.
-
S. R. Rao, Xanthates and Related Compounds, Marcel Dekker Inc., New York, 1971, ch. 9 Search PubMed.
-
(a) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 16, 1574–1585 RSC;
(b) D. H. R. Barton, S. I. Parekh and C.-L. Tse, Tetrahedron Lett., 1993, 34, 2733–2736 CrossRef CAS;
(c)
D. H. R. Barton, Half a Century of Free Radical Chemistry, Cambridge University Press. Cambridge, 1993 CrossRef;
(d) H. S. Park, H. Y. Lee and Y. H. Kim, Org. Lett., 2005, 7, 3187–3190 CrossRef CAS PubMed;
(e) H. Ishikawa, G. I. Elliott, J. Velcicky, Y. Choi and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 10596–10612 CrossRef CAS PubMed;
(f) S. Koshino, E. Kwon and Y. Hayashi, Eur. J. Org. Chem., 2018, 5629–5638 CrossRef CAS;
(g) F. W. Friese and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9561–9564 CrossRef CAS PubMed;
(h) M. Konya, K. Shimoyama, S. Arima, T. Fukuda, R. Uchida, H. Tomoda and T. Nagamitsu, Org. Lett., 2020, 22, 5131–5134 CrossRef CAS PubMed.
-
(a) W. Hartwig, Tetrahedron, 1983, 39, 2609–2645 CrossRef CAS;
(b) D. Crich and L. Quintero, Chem. Rev., 1989, 89, 1413–1432 CrossRef CAS;
(c) Q. Huang, J. Michalland and S. Z. Zard, Angew. Chem., Int. Ed., 2019, 58, 16936–16942 CrossRef CAS PubMed.
-
(a) F. Gagosz, C. Moutrille and S. Z. Zard, Org. Lett., 2002, 4, 2707–2709 CrossRef CAS PubMed;
(b) B. Quiclet-Sire, N. D. My Tran and S. Z. Zard, Org. Lett., 2012, 14, 5514–5517 CrossRef CAS PubMed;
(c) L. Debien and S. Z. Zard, J. Am. Chem. Soc., 2013, 135, 3808–3811 CrossRef CAS PubMed;
(d) K. K. K. Goh, S. Kim and S. Z. Zard, Org. Lett., 2013, 15, 4818–4821 CrossRef CAS PubMed;
(e) L. Qin and S. Z. Zard, Org. Lett., 2015, 17, 1577–1580 CrossRef CAS PubMed;
(f) L. Anthore, S. Li, L. V. White and S. Z. Zard, Org. Lett., 2015, 17, 5320–5323 CrossRef CAS PubMed;
(g) B. Quiclet-Sire and S. Z. Zard, Synlett, 2016, 27, 680–701 CrossRef CAS;
(h) C. G. Na and E. J. Alexanian, Angew. Chem., Int. Ed., 2018, 57, 13106–13109 CrossRef CAS PubMed;
(i) A. M. Carestia, D. Ravelli and E. J. Alexanian, Chem. Sci., 2018, 9, 5360–5365 RSC;
(j) S. K. Ayer and J. L. Roizen, J. Org. Chem., 2019, 84, 3508–3523 CrossRef CAS PubMed;
(k) B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed;
(l) D. Spinnato, B. Schweitzer-Chaput, G. Goti, M. Ošeka and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 9485–9490 CrossRef CAS PubMed;
(m) X. Chen and S. Z. Zard, Org. Lett., 2020, 22, 3628–3632 CrossRef CAS PubMed;
(n) E. de Pedro Beato, D. Mazzarella, M. Balletti and P. Melchiorre, Chem. Sci., 2020, 11, 6312–6324 RSC;
(o) V. S. Dorokhov and S. Z. Zard, Org. Lett., 2021, 23, 2164–2168 CrossRef CAS PubMed;
(p) J. Li, J. Li, X. Ji, R. He, Y. Liu, Z. Chen, Y. Huang, Q. Liu and Y. Li, Org. Lett., 2021, 23, 7412–7417 CrossRef CAS PubMed;
(q) V. S. Dorokhov, B. Quiclet-Sire and S. Z. Zard, Org. Lett., 2022, 24, 2878–2882 CrossRef CAS PubMed;
(r) J. Michalland, N. Casaretto and S. Z. Zard, Angew. Chem., Int. Ed., 2022, 61, e202113333 CrossRef CAS;
(s) B. Bieszczad and S. Z. Zard, Org. Lett., 2022, 24, 6973–6977 CrossRef CAS PubMed;
(t) T. Zhou, H. Chen, Y. Liu, H. Wang, Q. Yan, W. Wang and F. Chen, J. Org. Chem., 2022, 87, 15582–15597 CrossRef CAS PubMed.
- R. Leuckart, J. Prakt. Chem., 1890, 41, 179–224 CrossRef CAS.
-
(a) R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977–6980 CrossRef CAS;
(b) G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS;
(c) S. Binauld, L. Delafresnaye, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2014, 47, 3461–3472 CrossRef CAS;
(d) C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2014, 53, 6683–6686 CrossRef CAS PubMed;
(e) S. Allison-Logan, F. Karimi, Y. Sun, T. G. McKenzie, M. D. Nothling, G. Bryant and G. G. Qiao, ACS Macro Lett., 2019, 8, 1291–1295 CrossRef CAS PubMed;
(f) R. Li and Z. An, Angew. Chem., Int. Ed., 2020, 59, 22258–22264 CrossRef CAS;
(g) P. G. Georgiou, H. L. Marton, A. N. Baker, T. R. Congdon, T. F. Whale and M. I. Gibson, J. Am. Chem. Soc., 2021, 143, 7449–7461 CrossRef CAS PubMed;
(h) X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed;
(i) P. Das Karmakar and S. Pal, Polym. Chem., 2022, 13, 1394–1400 RSC;
(j) X. Zhang, Z. Yang, Y. Jiang and S. Liao, J. Am. Chem. Soc., 2022, 144, 679–684 CrossRef CAS PubMed.
-
(a)
R. H. Jones and S. L. Giolito, US Pat. 2743209A, 1956;
(b)
R. J. Venkateswara, V. Yenamandra and R. K. Vijaya, WO Pat. 02079179 A1, 2002;
(c) S. M. Sondhi, R. P. Verma, N. Singhal, R. Shukla, R. Raghubir and M. P. Dubey, Indian Drugs, 1999, 36, 50–54 CAS;
(d) S. Bhandari, A. R. Katore, D. M. Bajaj, P. Sharma, V. Talla and N. Shankaraiah, ChemistrySelect, 2018, 3, 6766–6774 CrossRef CAS.
-
(a) L. Degani and R. Fochi, Synthesis, 1978, 365–368 CrossRef;
(b) Y. Lu, C.-M. Li, Z. Wang, J. Chen, M. L. Mohler, W. Li, J. T. Dalton and D. D. Miller, J. Med. Chem., 2011, 54, 4678–4693 CrossRef CAS PubMed;
(c) A. Coluccia, S. Passacantilli, V. Famiglini, M. Sabatino, A. Patsilinakos, R. Ragno, C. Mazzoccoli, L. Sisinni, A. Okuno, O. Takikawa, R. Silvestri and G. La Regina, J. Med. Chem., 2016, 59, 9760–9773 CrossRef CAS PubMed;
(d) Y. Li, X. Luo, Y. Shao and L. Chen, J. Org. Chem., 2018, 83, 8768–8774 CrossRef CAS PubMed;
(e) Z. Zhu, X. Tang, J. Cen, J. Li, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 3767–3770 RSC;
(f) G. Huang, J. Li, J. Li, J. Li, M. Sun, P. Zhou, L. Chen, Y. Huang, S. Jiang and Y. Li, J. Org. Chem., 2020, 85, 13037–13049 CrossRef CAS PubMed;
(g) E. Badali, H. Rahimzadeh, A. Sharifi, A. Habibi and A. Z. Halimehjani, Org. Biomol. Chem., 2020, 18, 4983–4987 RSC;
(h) N. Sundaravelu, A. Nandy and G. Sekar, Org. Lett., 2021, 23, 3115–3119 CrossRef CAS;
(i) X. Zeng and S. Z. Zard, Org. Lett., 2022, 24, 5245–5248 CrossRef CAS PubMed;
(j) J. Li, Y. Liu, Z. Chen, J. Li, X. Ji, L. Chen, Y. Huang, Q. Liu and Y. Li, J. Org. Chem., 2022, 87, 3555–3566 CrossRef CAS PubMed;
(k) A. Nandy and G. Sekar, J. Org. Chem., 2022, 87, 7536–7546 CrossRef CAS PubMed;
(l) S. Mondal, E. M. Di Tommaso and B. Olofsson, Angew. Chem., Int. Ed., 2023, e202216296 CAS;
(m) D. I. Bugaenko, A. A. Volkov, V. V. Andreychev and A. V. Karchava, Org. Lett., 2023, 25, 272–276 CrossRef CAS PubMed.
-
(a) V. K. Akkilagunta and R. R. Kakulapati, J. Org. Chem., 2011, 76, 6819–6824 CrossRef CAS PubMed;
(b) D. J. C. Prasad and G. Sekar, Org. Lett., 2011, 13, 1008–1011 CrossRef CAS PubMed;
(c) S. Sangeetha, P. Muthupandi and G. Sekar, Org. Lett., 2015, 17, 6006–6009 CrossRef CAS PubMed;
(d) S. Sangeetha and G. Sekar, Org. Lett., 2017, 19, 1670–1673 CrossRef CAS;
(e) M. Soleiman-Beigi and Z. Arzehgar, Synlett, 2018, 29, 986–992 CrossRef CAS;
(f) X. Zhao, Q. Li, J. Xu, D. Wang, D. Zhang-Negrerie and Y. Du, Org. Lett., 2018, 20, 5933–5937 CrossRef CAS;
(g) N. Sundaravelu and G. Sekar, Org. Lett., 2019, 21, 6648–6652 CrossRef CAS PubMed;
(h) S. Sangeetha and G. Sekar, Chem. Commun., 2020, 56, 10906–10909 RSC;
(i) N. Sundaravelu, T. Singha, A. Nandy and G. Sekar, Chem. Commun., 2021, 57, 4512–4515 RSC;
(j) Y. Zhang, W. Lai, L. Zhang, X. Gao, G. Qiu and H. Zhou, Org. Biomol. Chem., 2021, 19, 1827–1834 RSC.
-
(a) G. Bouhadir, N. Legrand, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1999, 40, 277–280 CrossRef CAS;
(b) Y. Kwak, R. Nicolaÿ and K. Matyjaszewski, Macromolecules, 2009, 42, 3738–3742 CrossRef CAS;
(c) S. S. Ashirbaev, V. V. Levin, M. I. Struchkova and A. D. Dilman, J. Fluorine Chem., 2016, 191, 143–148 CrossRef CAS;
(d) R. He, Y. Liu, Y. Feng, L. Chen, Y. Huang, F. Xie and Y. Li, Org. Lett., 2022, 24, 3167–3172 CrossRef CAS.
- M. Zhang, B. Wang, Y. Cao, Y. Liu, Z. Wang and Q. Wang, Org. Lett., 2022, 24, 8895–8900 CrossRef CAS PubMed.
-
(a) W. L. Czaplyski, C. G. Na and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 13854–13857 CrossRef CAS PubMed;
(b) C. G. Na, D. Ravelli and E. J. Alexanian, J. Am. Chem. Soc., 2020, 142, 44–49 CrossRef CAS PubMed;
(c) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516–521 CrossRef CAS.
-
(a)
F. A. Bolth, R. D. Chrozier and L. E. Strow, US Pat. 3907854, 1975;
(b) S. Matsui, Bull. Chem. Soc. Jpn., 1987, 60, 1853–1865 CrossRef CAS;
(c) V. Maslak, Ž. Čeković and R. N. Saičić, Synlett, 1998, 1435–1437 CrossRef CAS;
(d) O. V. Bakbardina, I. Y. Pukhnyarskaya, M. A. Gazalieva, S. D. Fazylov and E. M. Makarov, Russ. J. Appl. Chem., 2006, 79, 1726–1728 CrossRef CAS;
(e) M. M. Milosavljević, A. D. Marinković, S. Petrović and M. Sovrlić, Chem. Ind. Chem. Eng. Q., 2009, 15, 257–262 CrossRef;
(f) M. M. Milosavljević, M. Sovrlić, A. D. Marinković and D. D. Milenković, Monatsh. Chem., 2010, 141, 749–755 CrossRef;
(g) P.-E. Dufils, G. David, B. Boutevin, G. Woodward, G. Otter, A. Guinaudeau, S. Mazières and M. Destarac, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1997–2007 CrossRef CAS.
-
(a) M. Li, X.-S. Xue and J.-P. Cheng, Acc. Chem. Res., 2020, 53, 182–197 CrossRef CAS PubMed;
(b) T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856–12859 CrossRef CAS PubMed;
(c) D. Zhu, Y. Gu, L. Lu and Q. Shen, J. Am. Chem. Soc., 2015, 137, 10547–10553 CrossRef CAS PubMed;
(d) S. Munavalli, D. K. Rohrbaugh, D. I. Rossman, F. J. Berg, G. W. Wagner and H. D. Durst, Synth. Commun., 2000, 30, 2847–2854 CrossRef CAS;
(e) F. Shen, P. Zhang, L. Lu and Q. Shen, Org. Lett., 2017, 19, 1032–1035 CrossRef CAS.
-
(a) M. Watanabe, K. Shirai and T. Kumamoto, Chem. Lett., 1975, 8, 855–858 CrossRef;
(b) J. Klose, C. B. Reese and Q. Song, Tetrahedron, 1997, 53, 14411–14416 CrossRef CAS;
(c) S. E. Denmark, D. J. P. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308–15311 CrossRef CAS PubMed;
(d) J. Qiu, D. Wu, P. G. Karmaker, H. Yin and F.-X. Chen, Org. Lett., 2018, 20, 1600–1603 CrossRef CAS PubMed;
(e) W.-C. Gao, Y.-Z. Shang, H.-H. Chang, X. Li, W.-L. Wei, X.-Z. Yu and R. Zhou, Org. Lett., 2019, 21, 6021–6024 CrossRef CAS PubMed;
(f) J. Zou, J. Chen, T. Shi, Y. Hou, F. Cao, Y. Wang, X. Wang, Z. Jia, Q. Zhao and Z. Wang, ACS Catal., 2019, 9, 11426–11430 CrossRef CAS;
(g) Y.-F. Li, Y.-F. Wei, J. Tian, J. Zhang, H.-H. Chang and W.-C. Gao, Org. Lett., 2022, 24, 5736–5740 CrossRef CAS PubMed.
-
M. De Greef, Novel applications of xanthate radical chemistry: masked radical formylation, synthesis of structurally divers spiroketals and synthesis of phosphorus-containing compounds, 2006, 20140701.
- Deposition number 2250745 (for 3c) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- The base plays an important role in the transmetalation process of cross coupling, please see: A. A. C. Braga, N. H. Morgon, G. Ujaque and F. Maseras, J. Am. Chem. Soc., 2005, 127, 9298–9307 CrossRef CAS PubMed.
-
(a) A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, J. Enzyme Inhib., 2001, 16, 55–63 CrossRef CAS PubMed;
(b) A. Krasovskiy, A. Gavryushin and P. Knochel, Synlett, 2005, 2691–2693 CAS;
(c) D. Enders, A. Rembiak and J. X. Liebich, Synthesis, 2011, 281–286 CrossRef CAS;
(d) Z.-B. Dong, X. Liu and C. Bolm, Org. Lett., 2017, 19, 5916–5919 CrossRef CAS PubMed;
(e) M.-T. Zeng, W. Xu, X. Liu, C.-Z. Chang, H. Zhu and Z.-B. Dong, Eur. J. Org. Chem., 2017, 6060–6066 CrossRef CAS;
(f) Z. Wu, M. Lai, S. Zhang, X. Zhong, H. Song and M. Zhao, Eur. J. Org. Chem., 2018, 7033–7036 CrossRef CAS;
(g) W. Xu, F. Gao and Z.-B. Dong, Eur. J. Org. Chem., 2018, 821–828 CrossRef CAS;
(h) J. B. Williamson, C. G. Na, R. R. Johnson III, W. F. M. Daniel, E. J. Alexanian and F. A. Leibfarth, J. Am. Chem. Soc., 2019, 141, 12815–12823 CrossRef CAS PubMed;
(i) Y. Cheng, H.-Y. Peng and Z.-B. Dong, Tetrahedron Lett., 2019, 60, 617–620 CrossRef CAS;
(j) C. Cheng, M. Zhao, M. Lai, K. Zhai, B. Shi, S. Wang, R. Luo, L. Zhang and Z. Wu, Eur. J. Org. Chem., 2019, 2941–2949 CrossRef CAS;
(k) Z.-B. Huang, X.-J. Xia, Z.-H. Huang, L. Xu, X.-Y. Zhang and R.-Y. Tang, Org. Biomol. Chem., 2020, 18, 1369–1376 RSC.
-
J. Swietoslawski, A. Silowiecki, E. Skotnicki, Z. Zerkowski and B. Kurowska, PL Pat. 133539 B1, 1986.
- X. Xu, L. Yan, S. Wang, P. Wang, A.-X. Yang, X. Li, H. Lu and Z.-Y. Cao, Org. Biomol. Chem., 2021, 19, 8691–8695 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2250745. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03194b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a