
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe semisynthesis of nucleolar human selenoprotein H†
Rebecca Notis
Dardashti
a,
Shay
Laps
 a,
Jacob S.
Gichtin
a and
Norman
Metanis
*abc
a,
Jacob S.
Gichtin
a and
Norman
Metanis
*abc
aThe Institute of Chemistry, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Givat Ram, Jerusalem 9190401, Israel
bCasali Center for Applied Chemistry, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Givat Ram, Jerusalem 9190401, Israel
cThe Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Givat Ram, Jerusalem 9190401, Israel. E-mail: metanis@mail.huji.ac.il
First published on 24th October 2023
Abstract
The human selenoprotein H is the only selenocysteine-containing protein that is located in the cell's nucleolus. In vivo studies have suggested that it plays some role in DNA binding, consumption of reactive oxygen species, and may serve as a safeguard against cancers. However, the protein has never been isolated and, as a result, not yet fully characterized. Here, we used a semi-synthetic approach to obtain the full selenoprotein H with a S43T mutation. Using biolayer interferometry, we also show that the Cys-containing mutant of selenoprotein H is capable of binding DNA with sub-micromolar affinity. Employing state-of-the-art expressed protein ligation (EPL), our devised semi-synthetic approach can be utilized for the production of numerous, hard-to-obtain proteins of biological and therapeutic relevance.
Introduction
Although there are twenty-five known human selenoproteins,1 studies on most have been quite limited. Obtaining sufficient quantities of selenoprotein for structural or enzymatic characterization has eluded scientists for decades,2 as the key residue in these proteins, selenocysteine (Sec, U), is encoded by a stop codon (UGA).3 Although both prokaryotes and eukaryotes have evolved complex mechanisms to override the stop codon during ribosomal translation,3 recombinant methods developed in different labs for E. coli, yeast, or similar expression methods have proven quite inefficient with regards to Sec expression.4 Hence, the majority of studies on human selenoprotein H (SELENOH), to date, have been based on in vivo knockdown or upregulation studies. Obtaining homogenous protein to study its structure and function has not yet been achieved, although its presence has been known for over twenty years.1Chemical (semi-)synthesis of proteins is a powerful approach to provide accesses to uniquely modified or challenging to produce targets for various biochemical studies and applications.5–7 It has provided a route to obtaining selenoproteins, even when recombinant protein expression could not. Expressed protein ligation (EPL), a method explained more in depth later in this introduction, was used to obtain the natural selenoprotein mouse thioreductase 3 (Mtr3), in which the majority of the protein was expressed as an intein fusion, converted to a thioester, and ligated to the C-terminal, Sec-containing tripeptide, which was prepared by SPPS.8 An alternative EPL approach was used to produce human selenoproteins M and W, in which the Cys-containing half of the protein was expressed as an intein fusion, and converted to thioester, while the Sec-containing half of the protein was expressed as selenoprotein in Cys-auxotrophic bacteria.9 Ligation of these segments gave SELENOM and SELENOW as final products.
These two proteins were also produced using entirely synthetic means, where SELENOM and SELENOW were produced from four segments and two segments, respectively.10 SELENOM was produced using the selenazolidine protecting group (Sez) to prevent central segment self-cyclization and unwanted deselenization. Efficacy of the protecting group on the latter was limited, however, and about 10% of the Sez was converted to Ala during deselenization of other Sec residues. This problem was later circumvented in the synthesis of the Trx-fold domain of human selenoprotein F (SELENOF), in which an Fmoc-protected Sez was shown to be completely protected from unwanted deselenization.11 The full-length SELENOF was also synthesized using Sec(Mob) to prevent deselenization.12 In an alternative approach, human selenoprotein K (SELENOK) was synthesized using a selenoester rather than thioester-based ligation, to allow ligation at a challenging Pro-Sec junction.13 Although each synthesis relies on similar techniques (SPPS, thioesterification, NCL), each is truly unique according to the limitations dictated by its protein's particular sequence, and no universal method exists to obtain the entire family of selenoproteins.
Despite gaps in the scientific community's knowledge about its function, enough is known about SELENOH to make it a compelling target for synthesis and study. As with many other selenoproteins,14 SELENOH has a predicted thioredoxin fold (Fig. 1);15–17 it contains a CXXU motif, surrounded by an α-helix and a β-sheet, a classic conserved feature of thioredoxins (traditionally with CXXC).18 As a result, it is predicted that SELENOH, like other thioredoxins, plays a role catalyzing the formation, isomerization or destruction of disulfide bonds in other proteins, although studies on Sec-to-Cys mutants could not confirm thioredoxin activity.15 Additionally, SELENOH's Sec is suspected to play a role in consuming peroxides and perhaps other types of reactive oxygen species (ROS) which can harm the cell. Expressed Sec-to-Cys and Sec-to-Ser mutants of SELENOH showed some activity in consuming peroxides,15 and in vivo studies showed that suppressed SELENOH expression led to peroxide proliferation in the cell,19 while overexpression of SELENOH in cells helped protect against oxidative damage.20 These data suggest that, like many other selenoproteins, SELENOH plays an important role in maintaining redox homeostasis in the cell (Scheme 1).
 | ||
| Fig. 1 (A) SELENOH's predicted structure from AlphaFold overlaid on its sequence; the amino acid of SELENOH contains a critical DNA-binding region at the N-terminus, followed by a hydrophobic region. Proposed ligation sites are underlined; (B) schematic depiction of the protein's predicted structure from AlphaFold. | ||
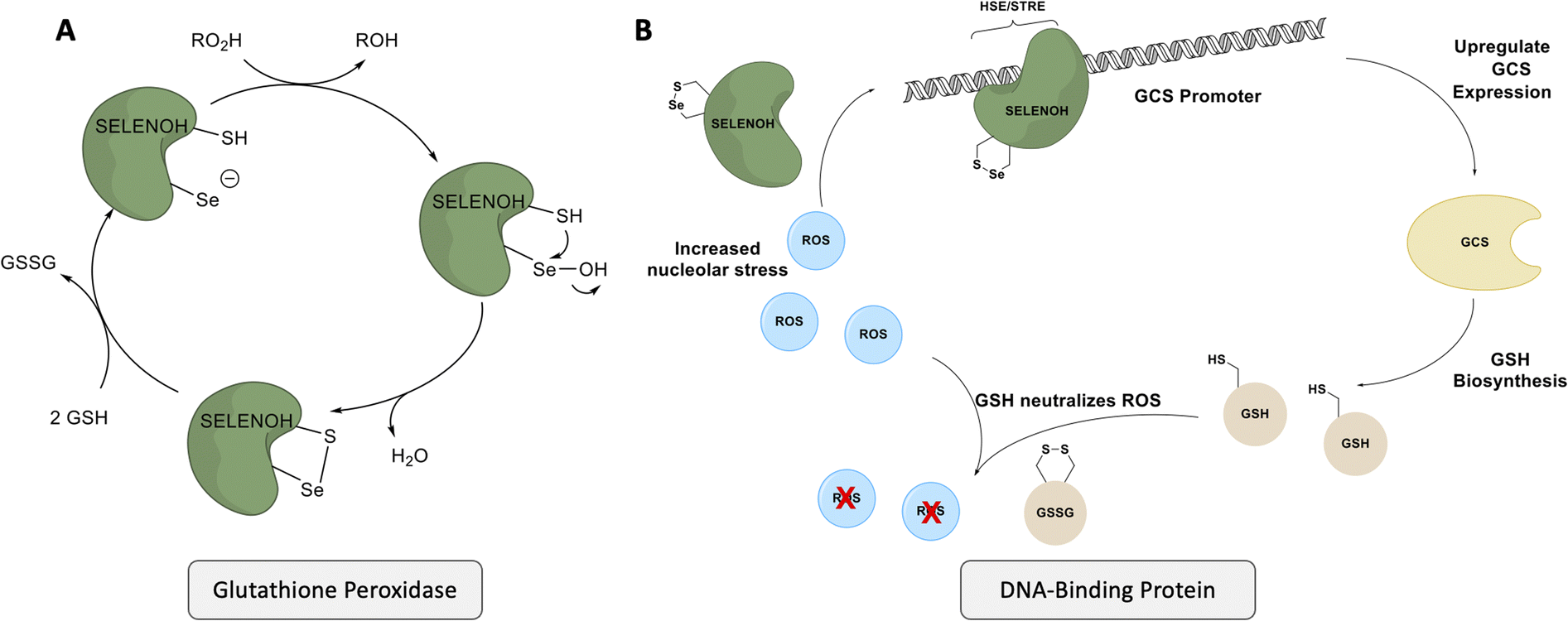 | ||
| Scheme 1 SELENOH's suggested two modes of actions: (A) a glutathione peroxidase or (B) a DNA-binding protein that responds to oxidative stress. | ||
Although there are many shared features between SELENOH and other selenoproteins, SELENOH is unique in its nucleolar localization, and, notably, its ability to bind DNA. In a 2007 study by Berry and coworkers, cells were subjected to oxidative stress and then SELENOH was immunoprecipitated and found to be bound to DNA fragments.20 It is suspected that SELENOH's nuclear localization signal, at its N-terminus, is also the source of its DNA binding ability (Fig. 1). Indeed, this highly positively charged sequence is capable of forming electrostatic interactions with DNA's negatively charged backbone, and its sequence has been compared to an AT-hook,20 a conserved sequence of amino acids known to bind DNA's minor groove. Immunoprecipitated SELENOH-bound DNA fragments closely aligned with heat shock element (HSE) or stress response element (STRE), two sequences of DNA which appear frequently upstream of genes that can respond to cell stress.20 As a result, it was suggested that SELENOH senses an excess of ROS in the cell, and, as a result, binds the DNA promoter upstream of the gene expressing α-GCS. This initiates the expression of the protein, which in turn is responsible for the production of glutathione (GSH). GSH is then released into the cell and consumes the ROS, thereby saving the cell from oxidative stress (Scheme 1B). Computational modeling of the protein has suggested that Sec plays some role in enhancing the binding between the protein and selected DNA sequences,21 but there has not yet been a definitive study of the protein's interactions with DNA.
SELENOH has also recently been shown to be a target for cancer treatment.22,23 Previous biological studies have demonstrated a clear relationship between seph (the zebrafish variant of selenoprotein H), activation of the p53 pathway, and tumor proliferation.24 The antibiotic carrimycin has been known to inhibit some cancers.25,26 Recently, one component of carrimycin, known as ISP I, was isolated and shown to specifically bind and inhibit SELENOH.23 As a direct result of SELENOH inhibition, cancer cells are overrun with ROS, leading them to initiate apoptosis and the growth of certain cancers is thereby stalled.23 Further work to characterize the structural interaction of SELENOH and ISP I as well as understanding the mechanism of degradation of SELENOH have been hampered by the inability to produce significant quantities of pure, isolated SELENOH.
Results and discussion
A three-segment strategy was designed for the synthesis of SELENOH (Fig. 1A and Scheme 2), using two ligation sites at positions 43–44 and 90–91. Dividing the protein into three parts gives segments under 50 amino acids each, which is considered ideal for SPPS, and an Ala was chosen as a ligation site because it is easily replaced with Sec and then converted to Ala following ligation via radical deselenization.27 Our designed segments include the C-terminal SELENOH(91–122), with a temporary A91U substitution to allow for ligation and subsequent deselenization.27,28 Although A91 is located in a predicted β-strand region, SPPS and NCL occur under denaturing conditions and, as such, it should not affect the ligation rates. The middle segment, SELENOH(44–90), would contain a C-terminal thioester precursor and an Fmoc-selenazolidine (Sez) protecting group11 on its natural Sec44, to prevent unwanted cyclization with thioester and prevents conversion to Ala during the deselenization step of A91U.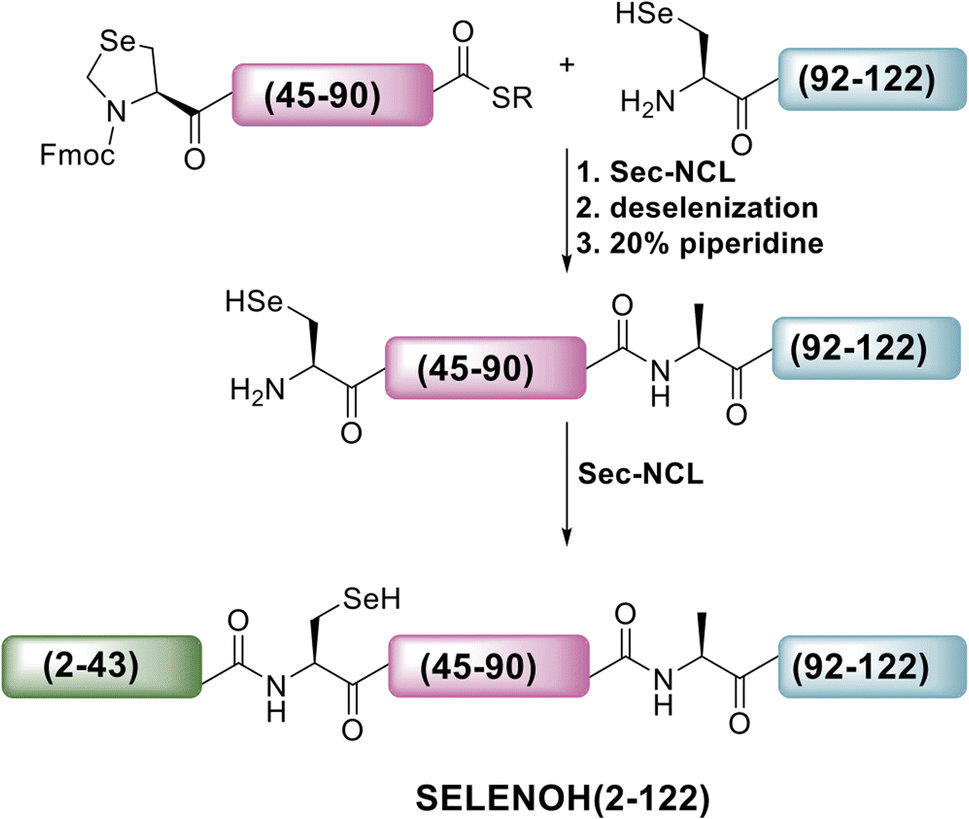 | ||
| Scheme 2 Our initial proposed synthetic route to full SELENOH. | ||
The N-terminal segment would also contain a C-terminal thioester. All peptides would be prepared via SPPS, and thioesters would be prepared from C-terminal hydrazides.29
The middle (Fmoc-SELENOH(44–90)(U44Sez)-COSR) and C-terminal (SELENOH(91–122)(A91U)) segments of the protein were prepared synthetically and assembled in a straightforward fashion. SPPS and purification methods for the individual peptides are discussed in depth in the ESI (Fig. S1 and S2).†
Following purification, SELENOH(44–122) was attained through a one-pot ligation, purification, and selenazolidine opening based on a method previously developed in our group (Fig. 2).11 In short, SELENOH(44–90)-COSR with a N-terminal Sez caging group was ligated with the Sec substituted for Ala91 in SELENOH(91–122)(A91U). Following ligation, deselenization was performed to convert Sec to native Ala91, and the Sez group was then opened to native Sec44. In all, 2.5 mg purified SELENOH(44–122) was obtained from 5.0 mg of the C-terminal and 8.7 mg of the central segment, in 22% yield.
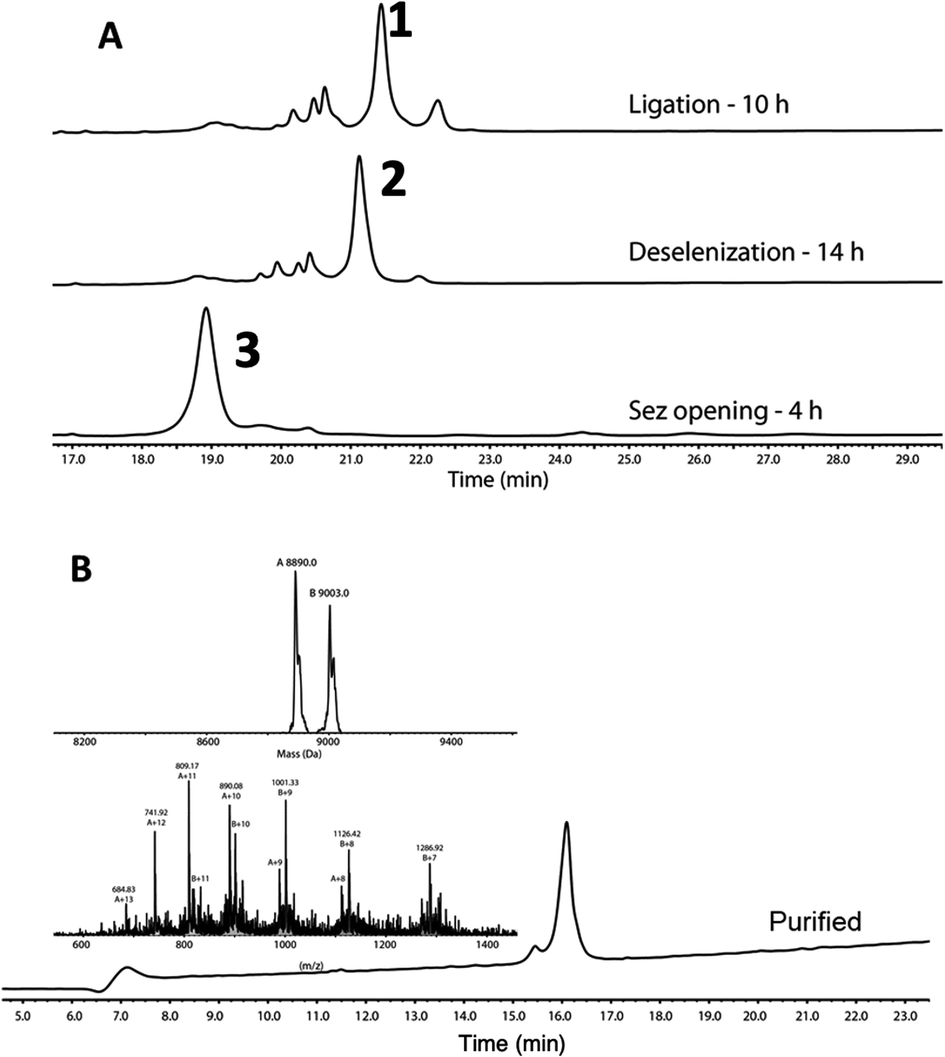 | ||
| Fig. 2 (A) Stacked HPLC chromatograms showing reaction progress of one-pot ligation, deselenization, and Sez opening, monitored on C4 analytical column. (B) HPLC and ESI-MS of purified SELENOH(44–122). Calc. mass 8890 Da, obs. mass 8890.0 Da, 9003.0 Da (+1 TFA salt). | ||
Accessing the N-terminal segment of the protein, SELENOH(2–43), was hindered by many obstacles. A truncated version, SELENOH(19–43) was synthesized in good quality, but the quality of the synthesis suffered greatly after the incorporation of the hydrophobic sequence AAVVAVA at positions 12–18, and a peak on HPLC chromatogram of the crude product could not be identified with the desired mass of the full peptide. Because the sequence contains no prolines and was highly hydrophobic, we theorized that the peptide was undergoing on-resin aggregation.30 Incorporating pseudoproline was not possible, as the segment contained no natural Ser or Thr residues. Attempts to thwart aggregation using chemical means: C-terminal or Cys-linked solubilizing tags,31,32 chaotropic agents such as LiCl during washes,33 Boc synthesis,34 and microwave SPPS were similarly unsuccessful. Further attempts were made to assemble the sequence from two segments, by introducing a Sec at either Ala12 or Ala16. However, in both cases, the ligation product was insoluble and could not be purified, even with addition of a C-terminal solubilizing tag (data not shown). Given these results, we were forced to conclude that SELENOH(2–43) was not attainable via SPPS using the tools that were currently available to peptide chemists, and chose to pursue the segment using protein expression.
Ultimately, SELENOH(2–43) was obtained through expression. The segment was fused to a C-terminal intein (MxeGyrA) for downstream conversion to thioester (Scheme 3). Additionally, with the awareness that this portion of the protein was aggregation-prone, following recent works of Müller, Ramos, and others, we designed an N-terminal SUMO fusion to increase solubility with the intention of removing the SUMO with SUMO- protease Ulp-1 following ligation to the synthetic remainder of the protein.35,36 The protein was expressed in reasonable yields (Fig. S3 and S4,† 57 mg L−1), but the conversion to thioester was unacceptably inefficient (less than 10% after 4 h, with subsequent degradation of protein, Fig. 3). This low conversion rate was attributed to the amino acid at position 43 immediately adjacent to the thiolysis site, Ser. The identity of amino acids at the −1 site from the intein affects rate, a phenomenon that has been previously observed.37 As a result, we chose to mutate Ser43 to Ala or Thr in order to improve thiolysis efficiency (Fig. S3 and S4†). Indeed, following expression, we saw that conversion to thioester via hydrazide38,39 was much faster than the wild-type (fully completed within 4 h, Fig. 3).
 | ||
| Scheme 3 Modified approach to full SELENOH through semi-synthesis. The N-terminal portion of the protein is expressed with N-terminal SUMO to enhance solubility and C-terminal intein to enable downstream thioesterification. | ||
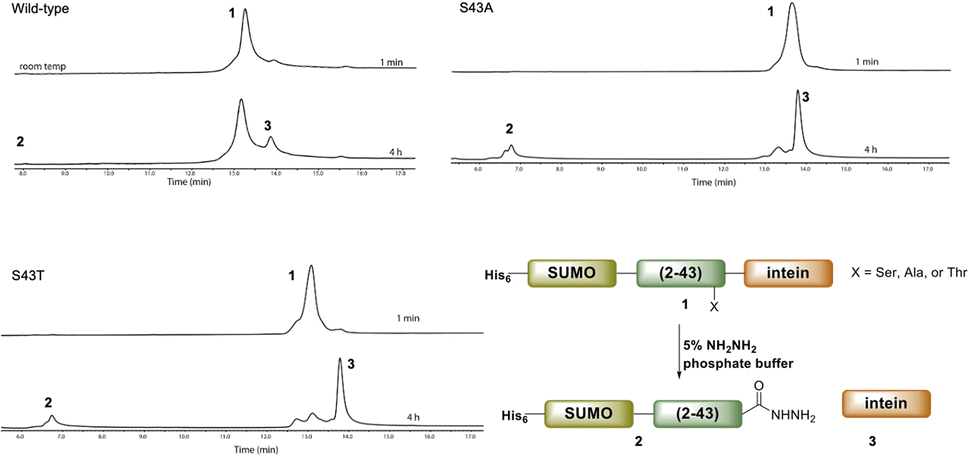 | ||
| Fig. 3 A comparison of hydrazination rates between SUMO-SELENOH(2–43)-intein based on the amino acid at −1 position, Ser43 (wild-type), S43A, or S43T. | ||
The purified SELENOH(44–122) was then ligated with both S43A and S43T variants of SUMO-SELENOH(2–43)-COSR. In both cases, ligation yields could not exceed 50%, as the Cys41 performed internal cyclization with the peptide's C-terminal thioester. This reaction led to trapped cyclization product, in direct competition with the ligation product (Fig. 4). The resulting SUMO-SELENOH variants were refolded and SUMO was removed using the deSUMOylating enzyme ULP-1. Overall yield was approximately 50 μg from the reaction, or about 5 mg L−1 expression (Fig. 5).
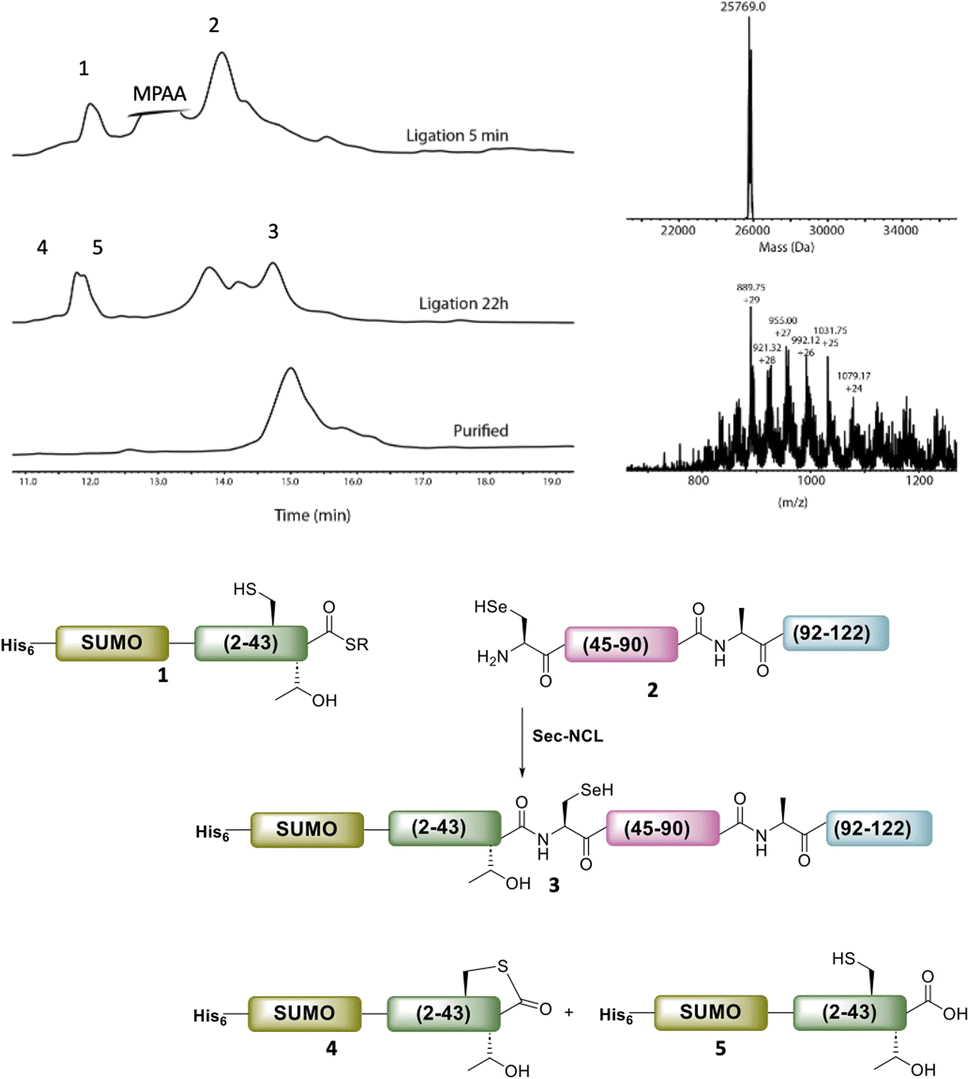 | ||
| Fig. 4 HPLC chromatograms of the ligation of SUMO-SELENOH(2–43)(S43T)-COSR with SELENOH(44–122). Calc. mass for the oxidized form (with selenylsulfide bond) is 25769.2, obs. mass 25769.0 Da. In addition to ligation with 2 to give the desired ligation product 3, the thioester 1 underwent internal cyclization with Cys41 (4) and irreversible hydrolysis (5). | ||
 | ||
| Fig. 5 SDS-PAGE (left) of SUMO removal (scheme on right). SUMO-SELENOH was immobilized on Ni beads, and following SUMO cleavage, the full SELENOH(S43T) protein was eluted from the beads. | ||
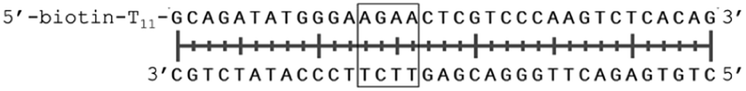
In order to study the protein's DNA binding properties, more material was required. We reasoned that the protein's DNA-binding properties could be studied using a Cys-containing mutant of the protein, one that could easily be produced via bacterial expression. We therefore expressed and purified SELENOH(U44C) (Fig. S6–S9†) and used biolayer interferometry (Fig. S10†) to characterize its interactions with several DNA sequences. Interaction of SELENOH with three different double-stranded DNA segments was explored: STRE (stress-response element) and HSE (heat-shock element) were both prepared based on real sequences found in the GCS promoter, and a scrambled sequence of DNA was included as a control (Table 1).
| Strand name | Sequence |
|---|---|
| HSE dsDNA |
|
| STRE dsDNA |

|
| Scrambled dsDNA |

|
| Scrambled dsDNA (AT poor) |
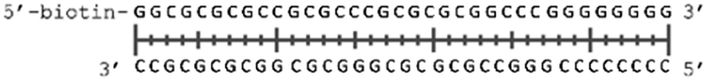
|
We found that all three DNA segments bound the SELENOH protein in the micro- to sub-micromolar range (Table 2 and Fig. S9†), indicating an indisputable affinity between the protein and DNA but one that is not sequence-specific. It is worth noting that the tested scrambled DNA sequence is still somewhat AT-rich, which may be fueling the affinity between the protein and the sequence. Intrigued by this, we sought to examine another, AT-poor scrambled DNA sequence. To our surprise, a somehow similar affinity with Kd ∼1.7 μM has been obtained also in this case (Table 2). However, when we tested the N-terminal segment SELENOH(2–15), no binding to the HSE dsDNA was detected (data not shown), suggesting that the full structured protein is required for efficient DNA binding. Overall, the DNA binding results indicate a more versatile role of SELENOH as was speculated up to date. The power of the precise chemical synthesis introduced above will pave the way to further exploration of SELENOH's biological properties by us and others.
| DNA type | K D (nM) | Error |
|---|---|---|
| HSE | 1200 | ±400 |
| STRE | 1000 | ±200 |
| Scrambled | 700 | ±100 |
| Scrambled AT poor | 1700 | ±800 |
Conclusions
Though they are critical for cell health, over half of the family of human selenoproteins remain uncharacterized, among them SELENOH. Recent findings indicate that this protein is a candidate target in the treatment of cancer; the complex nature of cellular Sec translation has prevented study of SELENOH using recombinant expression. This communication reported the semisynthesis of SELENOH, in which the Sec-containing, C-terminal portion of the protein was assembled from two synthetic segments in a one-pot series of reactions, and a synthesis-resistant, N-terminal peptide was obtained via expression as an S43A or S43T mutant. The semisynthesis approach presented in this study should be useful for the preparation of other hard to access proteins. In addition to the assembly of Sec-containing SELENOH, a Cys-containing mutant was expressed and found to bind various DNA sequences in the micro- or sub-micromolar range in vitro, supporting one proposed function of SELENOH. The examination of various factors affecting the DNA binding properties (e.g. mutations in the N-terminal region of the sequence) is an interesting direction for further investigation, which will be more feasible utilizing our reported synthetic scheme. The synthetic methods honed in this work will provide a route towards full characterization of this unique and therapeutically relevant protein.Data availability
Data supporting this article has been provided as ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. R. N. D., S. L. and J. S. G. performed experimental investigations and data analysis. The project was conceived and supervised by N. M.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Yael Nir-Keren, Dr Tsafi Danieli, and Dr Noa Dekel for their aid in protein expression, and to Dr Hadar Amartely for assistance in protein purification, CD measurements, and BLI binding studies in the Wolfson Center for Applied Structural Biology at HUJI. Thank you to Dar Gliksberg for help in attempts to synthesize SELENOH(2–43) by ligation (results not shown in this paper). Thanks to Dr Hannan Schoffman for help obtaining HR-MS measurements. R. N. D. was supported by the Kaete Klausner PhD scholarship. S. L. is supported by the Emergency Postdoctoral Fellowships for Israeli Researchers in Israel of the Israel Academy of Sciences and Humanities. N. M. acknowledges the financial support of the Israel Science Foundation (1388/22).References
- G. V. Kryukov, S. Castellano, S. V. Novoselov, A. V. Lobanov, O. Zehtab, R. Guigó and V. N. Gladyshev, Science, 2003, 300, 1439–1443 CrossRef CAS PubMed.
- L. Flohé, Biochim. Biophys. Acta, 2009, 1790, 1389–1403 CrossRef PubMed.
- T. C. Stadtman, Annu. Rev. Biochem., 1996, 65, 83–100 CrossRef CAS PubMed.
- R. Mousa, R. Notis Dardashti and N. Metanis, Angew. Chem., Int. Ed., 2017, 56, 15818–15827 CrossRef CAS PubMed.
- Z. Zhao and N. Metanis, Angew. Chem., Int. Ed., 2019, 58, 14610–14614 CrossRef CAS PubMed.
- S. Laps, F. Atamleh, G. Kamnesky, S. Uzi, M. M. Meijler and A. Brik, Angew. Chem., Int. Ed., 2021, 60, 24137–24143 CrossRef CAS PubMed.
- R. Mousa, T. Hidmi, S. Pomyalov, S. Lansky, L. Khouri, D. E. Shalev, G. Shoham and N. Metanis, Commun. Chem., 2021, 4, 1–9 CrossRef PubMed.
- B. Eckenroth, K. Harris, A. A. Turanov, V. N. Gladyshev, R. T. Raines and R. J. Hondal, Biochemistry, 2006, 45, 5158–5170 CrossRef CAS PubMed.
- J. Liu, Q. Chen and S. Rozovsky, J. Am. Chem. Soc., 2017, 139, 3430–3437 CrossRef CAS PubMed.
- L. Dery, P. Sai Reddy, S. Dery, R. Mousa, O. Ktorza, A. Talhami and N. Metanis, Chem. Sci., 2017, 8, 1922–1926 RSC.
- Z. Zhao, R. Mousa and N. Metanis, Chem. –Eur. J., 2022, 28, e202200279 CrossRef CAS PubMed.
- P. Liao, H. Liu and C. He, Chem. Sci., 2022, 13, 6322–6327 RSC.
- N. J. Mitchell, S. S. Kulkarni, B. Chan, L. Radom, J. Payne, N. J. Mitchell, J. Sayers, S. S. Kulkarni, D. Clayton, A. M. Goldys, J. Ripoll-rozada, P. Jose, B. Pereira, B. Chan, L. Radom, P. J. B. Pereira, B. Chan, L. Radom and R. J. Payne, Chem, 2017, 2, 703–715 CAS.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- S. V. Novoselov, G. V. Kryukov, X. M. Xu, B. A. Carlson, D. L. Hatfield and V. N. Gladyshev, J. Biol. Chem., 2007, 282, 11960–11968 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS PubMed.
- P. T. Chivers, K. E. Prehoda and R. T. Raines, Biochemistry, 1997, 36, 4061–4066 CrossRef CAS PubMed.
- R. T. Y. Wu, L. Cao, B. P. C. Chen and W. H. Cheng, J. Biol. Chem., 2014, 289, 34378–34388 CrossRef CAS PubMed.
- J. Panee, Z. R. Stoytcheva, W. Liu and M. J. Berry, J. Biol. Chem., 2007, 282, 23759–23765 CrossRef CAS PubMed.
- S. H. Barage, D. D. Deobagkar and V. B. Baladhye, Amino Acids, 2018, 50, 593–607 CrossRef CAS PubMed.
- L. C. Yu, D. D. Dang, S. Zhuang, S. Chen, Z. Zhuang and J. S. Rosenblum, Cancer Pathog. Therapy, 2023, 1, 111–115 CrossRef PubMed.
- J. Cui, J. Zhou, W. He, J. Ye, T. Westlake, R. Medina, H. Wang, B. L. Thakur, J. Liu, M. Xia, Z. He, F. E. Indig, A. Li, Y. Li, R. J. Weil, M. I. Aladjem, L. Zhong, M. R. Gilbert and Z. Zhuang, J. Exp. Clin. Cancer Res., 2022, 41, 126 CrossRef CAS PubMed.
- A. G. Cox, A. Tsomides, A. J. Kim, D. Saunders, K. L. Hwang, K. J. Evason, J. Heidel, K. K. Brown, M. Yuan, E. C. Lien, B. C. Lee, S. Nissim, B. Dickinson, S. Chhangawala, C. J. Chang, J. M. Asara, Y. Houvras, V. N. Gladyshev and W. Goessling, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5562–E5571 CrossRef CAS PubMed.
- S. Liang, T. Zhao, Z. Zhou, W. Ju, Y. Liu, Y. Tan, D. Zhu, Z. Zhang and L. Zhong, Transl. Oncol., 2021, 14, 101074 CrossRef CAS PubMed.
- Y. Jin, H. X. Zuo, M. Y. Li, Z. H. Zhang, Y. Xing, J. Y. Wang, J. Ma, G. Li, H. Piao, P. Gu and X. Jin, Front. Pharmacol, 2021, 12, 774231 CrossRef CAS PubMed.
- N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed.
- S. Dery, P. S. Reddy, L. Dery, R. Mousa, R. N. Dardashti and N. Metanis, Chem. Sci., 2015, 6, 6207–6212 RSC.
- D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 130, 11808–11813 CrossRef.
- M. Amblard, J.-A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed.
- S. Tsuda, M. Mochizuki, H. Ishiba, K. Yoshizawa-Kumagaye, H. Nishio, S. Oishi and T. Yoshiya, Angew. Chem., Int. Ed., 2018, 130, 2127–2131 CrossRef.
- J.-X. Wang, G.-M. Fang, Y. He, D.-L. Qu, M. Yu, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 2194–2198 CrossRef CAS PubMed.
- J. C. Hendrix, K. J. Halverson, J. T. Jarrett and P. T. Lansbury Jr, J. Org. Chem., 1990, 55, 4517–4518 CrossRef CAS.
- M. Schnölzer, P. Alewood, A. Jones, D. Alewood and S. B. H. Kent, Int. J. Pept. Res. Ther., 2007, 13, 31–44 CrossRef.
- S. Margiola, K. Gerecht and M. M. Müller, Chem. Sci., 2021, 12, 8563–8570 RSC.
- C. Gallagher, F. Burlina, J. Offer and A. Ramos, Sci. Rep., 2017, 7, 14083 CrossRef PubMed.
- M. W. Southworth, K. Amaya, T. C. Evans, M.-Q. Xu and F. B. Perler, BioTechniques, 1999, 27, 110–120 CrossRef CAS PubMed.
- J. Thom, D. Anderson, J. McGregor and G. Cotton, Bioconjugate Chem., 2011, 22, 1017–1020 CrossRef CAS PubMed.
- S. S. Kulkarni, E. E. Watson, J. W. C. Maxwell, G. Niederacher, J. Johansen-Leete, S. Huhmann, S. Mukherjee, A. R. Norman, J. Kriegesmann, C. F. W. Becker and R. J. Payne, Angew. Chem., Int. Ed., 2022, 134, e202200163 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03059h |
| This journal is © The Royal Society of Chemistry 2023 |