
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChalcogen-doped, (seco)-hexabenzocoronene-based nanographenes: synthesis, properties, and chalcogen extrusion conversion†
Ranran
Li
a,
Bin
Ma
a,
Shengtao
Li
a,
Chongdao
Lu
 a and
Peng
An
*ab
a and
Peng
An
*ab
aSchool of Chemical Science and Technology, Yunnan University, Kunming 650091, P. R. China. E-mail: anp@ynu.edu.cn
bKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan University, Kunming 650091, P. R. China
First published on 28th July 2023
Abstract
A series of chalcogen-doped nanographenes (NGs) and their oxides are described. Their molecular design is conceptually based on the insertion of different chalcogens into the hexa-peri-hexabenzocoronene (HBC) backbone. All the NGs adopt nonplanar conformations, which would show better solubility compared to planar HBC. Except for the oxygen-doped, saddle-shaped NG, the insertion of large chalcogens like sulfur and selenium leads to a seco-HBC-based, helical geometry. All the three-dimensional structures are unambiguously confirmed by single-crystal X-ray diffractometry. Their photophysical properties including UV-vis absorption, fluorescence, chiroptical, charge distribution, and orbital gaps are investigated experimentally or theoretically. The properties of each structure are significantly affected by the doped chalcogen and its related oxidative state. Notably, upon heating or adding an acid, the selenium-doped NG or its oxide undergoes a selenium extrusion reaction to afford seco-HBC or HBC quantitatively, which can be treated as precursors of hydrocarbon HBCs.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) have fascinated chemists for a long time due to their wide utility in materials science,1 optoelectronic devices,2 biological probes,3 and supramolecular chemistry.4 To exploit three-dimensional aesthetic structures and modulate optoelectronic or photophysical properties, an increasing number of contorted polycyclic aromatic hydrocarbons (PAHs) or nanographenes (NGs) have been developed in the past decades.5 Such nonplanar PAHs or NGs have been constructed with a diversity of shapes, including bowls,6 saddles,7 helixes,8 belts,9 rings,10 and hybrid conformations11 by “bottom-up” strategies. Compared to their planar counterparts, nonplanar PAHs or NGs have shown intriguing properties including high solubility, different aggregation patterns, tunable optoelectronic properties, and specific supramolecular interactions.3a,12 Strategically, the introduction of main-group elements into hydrocarbon PAHs is another powerful method to not only modulate their three-dimensional structures but tune their intrinsic properties including chemical reactivity, electronic energy gap, and photophysical behavior.13As a planar NG unit, hexa-peri-hexabenzocoronene (HBC), a D6h – symmetric planar π scaffold (Fig. 1a), and its derivative have been intensively studied.14 Owing to the nature of its planar structure, HBC generally exhibits low solubility and a high aggregation tendency in solvents, which commonly hampers its application, particularly in solution-based device fabrication.3a,15 Thus, numerous efforts have been made to engineer the structure of HBC. For example, by incorporation of a non-hexagonal ring like a seven-,7a eight-,16 or nine-membered17 ring into the HBC skeleton, saddle-, or helical-, seco-HBC-based (Fig. 1a) structures were revealed, and these novel nonplanar structures exhibit distinct photophysical properties and aromaticity compared to the parent HBC. Recently, by insertion of nitrogen into the HBC backbone in the fjord region, our group reported nitrogen-doped HBC- or seco-HBC-based NGs7b,18 (Fig. 1b), and the optical properties can be mediated by introducing versatile substituents at the peripheral nitrogen. In this context, we focused our attention on the insertion of chalcogens into the HBC backbone. We envisioned that varied chalcogens in the new backbone of NGs would significantly affect the properties of the entire π-system in the following aspects: (1) specific atomic radii of doped chalcogens may lead to diverse three-dimensional structures; (2) distinct electronegativities and multiple oxidation states of chalcogens would be beneficial to sophisticatedly tune the electronic properties; (3) by means of the unique chalcogen-extrusion reaction,19 a specific reactivity toward atom-extrusion conversion would offer dynamic control of ring-reconstruction.
 | ||
| Fig. 1 (a) Structures of planar HBC and partially cyclized seco-HBC. (b) Structures of nitrogen-doped, (seco)-HBC-based NGs. (c) Insertion of chalcogens to design heteroatom-doped HBC or seco-HBCs. | ||
Thus, a series of chalcogen-doped NG analogs were designed and the influences of the doped chalcogens or oxidized chalcogens on the overall synthesis, structural geometry, electronic properties, and chalcogen extrusion reaction of the π-systems are demonstrated (Fig. 1c). Single-crystal X-ray analysis unequivocally reveals the saddle- and saddle–helix-shaped conformations of these novel chalcogen-doped NGs. The photophysical properties as well as the chiroptical properties of the helical, sulfur- and selenium-doped NGs were investigated. Meanwhile, thermal- or acid-induced chalcogen extrusion reactions for these NGs were studied, and the heteroatom-dependent chalcogen-extrusion processes were observed. Particularly, the selenium-doped NGs exhibit controllable, fast, and quantitative selenium extrusion conversion to form HBC under acidic conditions at room temperature. For the first time, the saddle–helix precursors of HBC, with over 100-fold higher solubility than HBC, were identified. The details of these results will be elucidated in this article.
Results and discussion
Synthesis of chalcogen-doped nanographenes
To obtain a series of chalcogen-doped NGs, we used a universal synthetic route to synthesize NGs with different alkyl side chains for single crystal cultivation (Scheme 1). In brief, the Diels–Alder reaction of dibenzo[b,f]oxepine (DBO) 1 with tetrabromothiophene-S,S-dioxide 2 in toluene followed by oxidative aromatization in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) afforded tetrabrominated aromatics 3 in 75% yield. Subsequently, fourfold Suzuki–Miyaura cross-coupling reactions of compound 3 with (4-ethylphenyl) boronic acid in the presence of 15 mol% PdCl2(CH3CN)2, 30 mol% SPhos and excess K3PO4 then furnished the hexaphenylbenzene 4 in 81% yield. The treatment of compound 4 with DDQ (10.0 equiv.) in the presence of triflic acid (13.0 equiv.) in DCM in a water-ice bath resulted in the fully fused oxa-NG 5O within 5 minutes in 65% yield with the formation of five new C–C bonds. For sulfur- or selenium-doped analogs, thiepine 8 and selenepine 9 were prepared in a two-step reaction with overall 58% and 24% yields respectively: the copper-catalyzed oxidative coupling of (2-formalphenyl) boronic acid with the sulfur or selenium followed by intramolecular McMurry olefin formation. Then Diels–Alder reactions of thiepine 8 and selenepine 9 with compound 2 followed by oxidative aromatization with DDQ furnished compounds 10 and 11 in 90% and 56% yields respectively. Notably, a higher temperature (140 °C in xylene) and longer reaction time are needed for selenepine 9 compared to that for oxepine 1 and thiepine 8 in the Diels–Alder reaction (synthetic details in the ESI†). Then arylations for compounds 10 and 11 were performed using Suzuki–Miyaura cross-coupling conditions in the presence of 15 mol% PdCl2(CH3CN)2, 30 mol% SPhos, excess boronic acid, and K3PO4, affording hexaphenylbenzene 12–15 in 54–78% yields. When compounds 12–15 were subjected to Scholl reactions under the conditions of DDQ/CF3SO3H in DCM at 0 °C, the seco-HBC-based structures 16S, 17S, 18Se, and 19Se were obtained with the formation of four continuous C–C bonds. Instead of the fully cyclized saddle of oxygen-embedded NG, helical structures were formed for sulfur- and selenium-doped NGs presumably due to the large strain caused by atom size increasing. In any attempts of increasing the temperature, adding more equivalent reagents, or using other oxidative cyclization conditions, no fully fivefold cyclization product was observed (Scheme S6†). Meanwhile, the synthesis of tellurium-doped NGs was also investigated (Scheme S4†). In order to construct a tellurium-doped analog, we synthesized dibenzo[b,f]tellurepine 21 from dibromide 20 after failed attempts by using the same synthetic procedure as compounds 6 and 7 from boronic acid. Through two-fold lithium-halide exchange with n-butyl lithium followed by tellurium insertion, compound 20 was successfully transformed to dibenzo[b,f]tellurepine 21 in 58% yield. The mono-tellurium-doped aromatics 21 was unambiguously confirmed by X-ray crystallography analysis.20 Subsequently, we subjected compound 21 to similar Diels–Alder reaction conditions by reacting with tetrabromide 2. Consequently, no tellurium-insertion product 22 was detected by gradually raising the heating temperature. Instead, we observed phenanthrene formed as the dominant product through tellurium extrusion of compound 21 upon heating to 140 °C, and some amount of tetrabromo 23 was observed when the heating temperature raised up to 180 °C. These results indicated that telluride 21 is prone to undergo a tellurium extrusion reaction prior to the Diels–Alder reaction, and compound 23 was most likely formed by [4 + 2] cycloaddition of phenanthrene and compound 2. Structure 23 was also confirmed by X-ray crystallography analysis.20 Hence, the tellurium extrusion process suggested a higher possibility of heteroatom-extrusion by doping a larger-size chalcogen.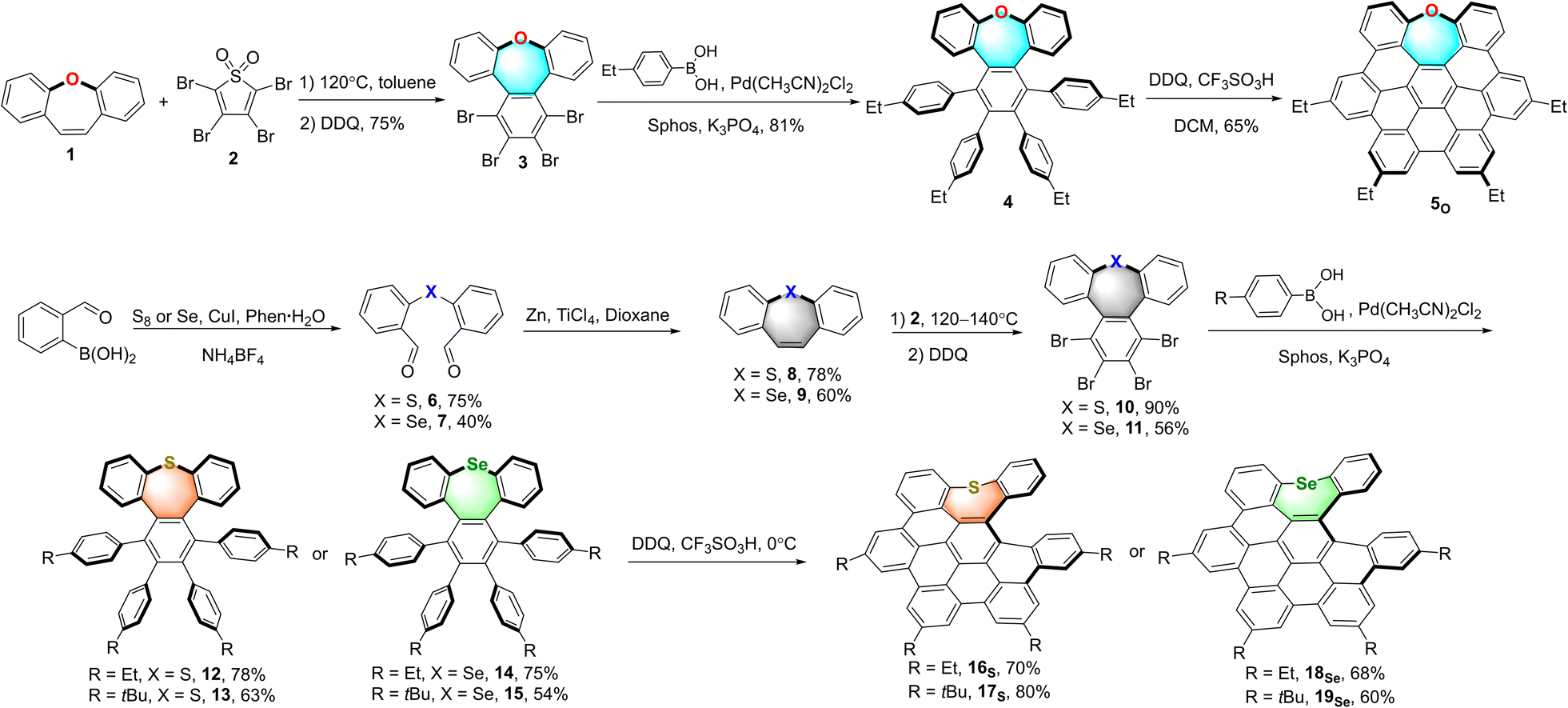 | ||
| Scheme 1 Synthesis of oxygen-doped NG 5O, sulfur-doped NGs 16S, and 17S, and selenium-doped NGs 18Se and 19Se. | ||
With the sulfur- and selenium-doped NGs in hand, their oxidative analogs were prepared (Scheme 2). The selective oxidation of sulfur-NGs 16S and 17S and selenium-NGs 18Se and 19Se was performed by treating them with oxone at 0 °C for 2 hours to afford the corresponding mono-oxides 24SO and 25SO, and 26SeO and 27SeO in nearly quantitative conversion. Meanwhile, the S,S-dioxide 28SO2 was obtained by oxidation of compound 25SO at room temperature in the presence of an excess amount of meta-chloroperoxybenzoic acid (m-CPBA), while 27SeO cannot be oxidized under these conditions. Due to the highly distorted skeletons, all the chalcogen-doped NGs as well as the corresponding oxides show good solubility in organic solvents; therefore, they could be unequivocally confirmed by means of 1H- and 13C NMR spectroscopy (Fig. S29–S50†) and high-resolution mass spectrometry (see the ESI†). Meanwhile, the seco-HBC-type molecules including 16S, 18Se, and 24SO were further characterized by 2D 1H–1H ROESY and COSY (Fig. S1–S3†).
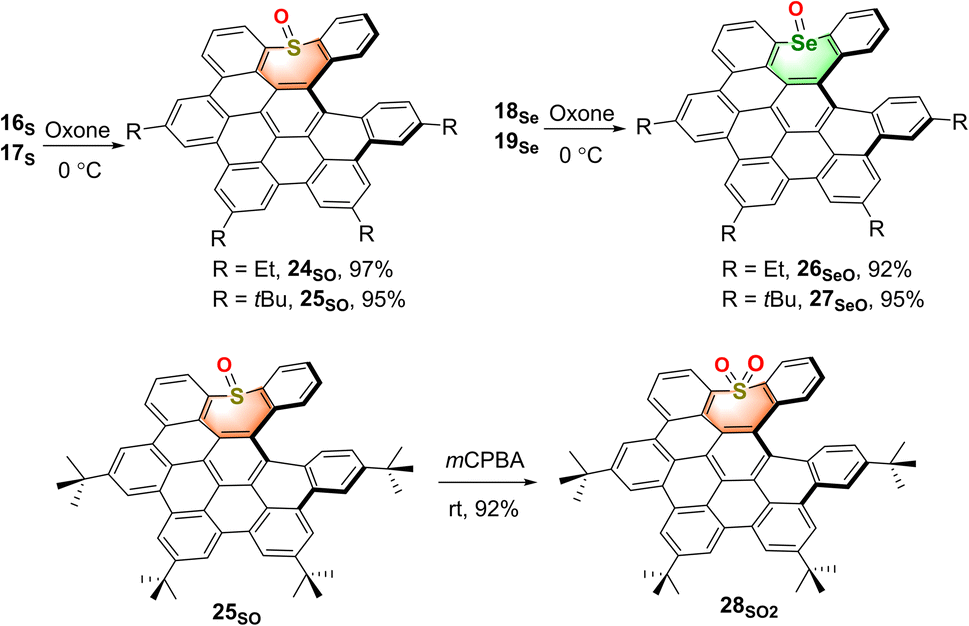 | ||
| Scheme 2 Synthesis of oxidative derivatives of S-, Se-doped NGs. | ||
Structural analysis of NGs
We next try to grow single crystals for all the NGs with different alkyl chains, and as a result, single crystals of 5O, 17S, 18Se, and 24SO suitable for X-ray diffraction analysis were obtained by slowly evaporating their solutions at room temperature (Tables S1 and S2†).20 As shown in Fig. 2a–d, all the molecular backbones are distorted due to the incorporation of a heteroatom-doped, seven-membered ring. All the seven-membered rings exhibit different degrees of antiaromaticity suggested by nucleus-independent chemical shift (NICS)21 by DFT calculations at the B3LYP/6-311+G(d,p) level of theory (Fig. S27†). Positive NICS (0) values between 2.79 and 6.49 were observed for all chalcogen-doped, seven-membered rings. The localized orbital locator (LOL)-π electron distribution calculation indicated that the electrons were localized on the seven hexagonal rings with strong π-conjugation (Fig. S27†). In detail, the X-ray crystallographic analysis of 5O reveals a saddle-shaped, negatively curved structure in which the oxygen atom protrudes from the π surface. Such a distorted conformation resembles that of a previously reported N–Me doped analog.7b The depths of the saddle, defined as the perpendicular distance from the centre of ring A to the line across C14 and C3 is 0.91 A (Fig. 2e), slightly shallower than the N–Me doped analogue. Meanwhile, the structures of 17S, 18Se, and 24SO exhibited saddle–helix geometry with a protruding chalcogen out of the π surface (Fig. 2b–d). The end-to-end dihedral angle of [5]helicene in NG 17S defined as the angle formed by the two benzene rings located at its terminal edges is 71.4°, and the torsion angle (C8–C14–C12–C38) is 34.9° (Fig. 2b). Meanwhile the dihedral angle and torsion angle in NG 18Se are 73.0° and 32.2° (C5–C4–C3–C2) respectively (Fig. 2c). Longer Se1–C1 (1.917 Å) and Se1–C35 (1.920 Å) bonds in 18Se were observed compared to C–S bonds (1.766 and 1.778 Å) in 17S due to the larger atomic radii of selenium. Both helicenes 17S and 18Se exhibited a larger end-to-end dihedral angle and torsion angle than the nitrogen-doped analogs (64.4° and 31.1° respectively)18b due to the increased atom size of sulfur and selenium. As helicenes, the crystallography of both 17S and 18Se, shown in Fig. 2f as a representative example, displays P/M enantiomers in single crystal structures (Fig. S5 and S6†). The S-oxide 24SO was also unequivocally confirmed by its X-ray structure (Fig. 2d). The formation of the SO bond decreased the end-to-end dihedral angle (58° in 24SO) and torsion angle (29.8°, C23–C22–C21–C29). Due to the electronegativity of sulfoxide, the intermolecular hydrogen bond interaction between sulfoxide and dichloromethane was observed in the crystal packing besides the π–π stacking (Fig. S7†). The electron-static potential (ESP) map of 17S, 24SO, and 28SO2 obtained through DFT calculations at the CAM-B3LYP/6-31+(d,p) level of theory (Fig. 2g) clearly showed the influence of different sulfur-oxidation states on the charge of the NG molecules. As expected, the electrons were distributed almost evenly on the surface of sulfur-doped NG 17S, and aggregated at the oxygen once the sulfur was oxidized in 24SO, and 28SO2. The HOMO and LUMO orbitals22 of seco-HBC-based NGs 17S, 19Se, 24SO, 26SeO, and 28SO2 calculated at the PBE0/6-311G(d,p) level do not differ a lot from each other: each HOMO is found to be delocalized on the entire molecule, while the LUMOs are delocalized on the coplanar benzenoid rings (Fig. 2h).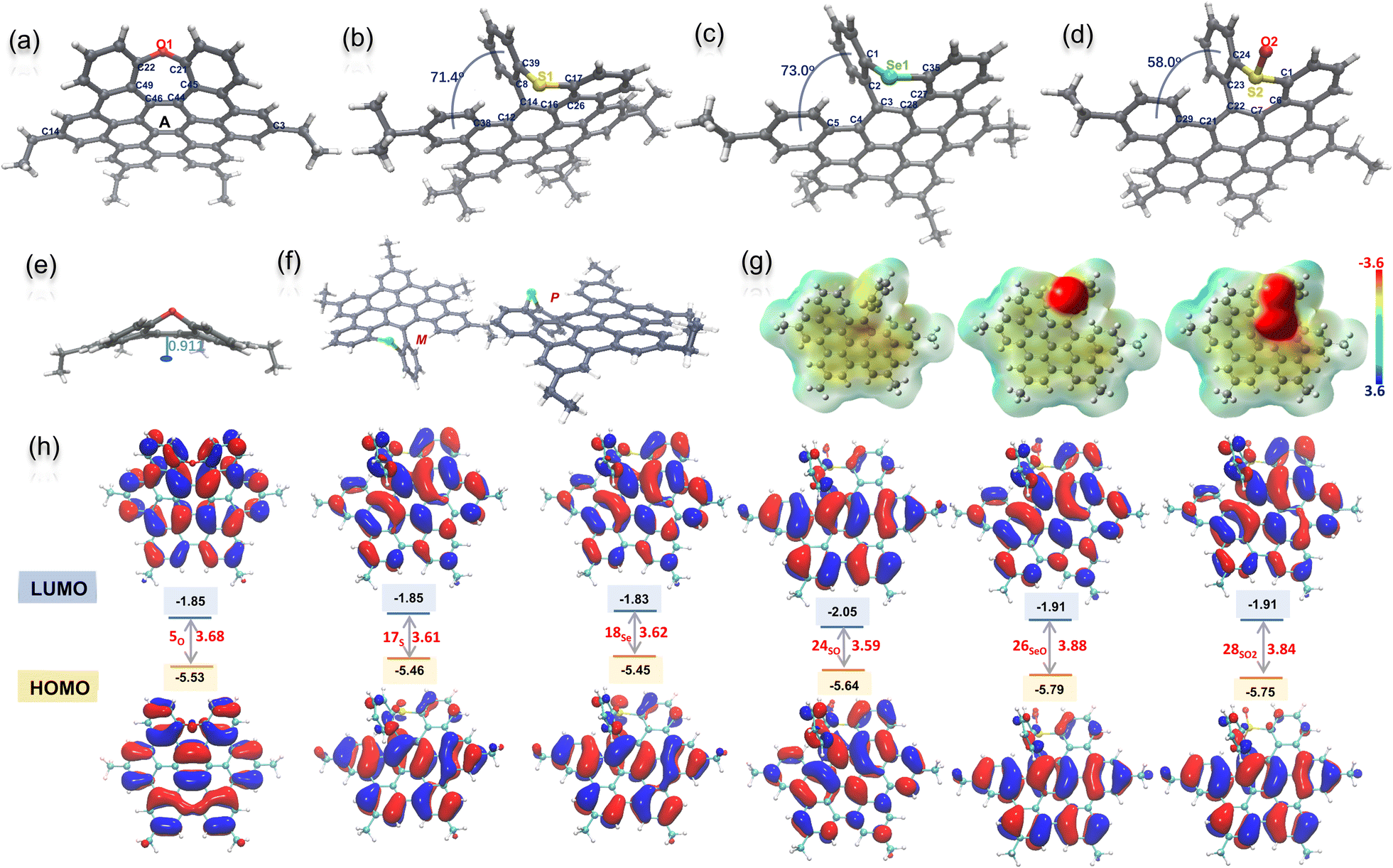 | ||
| Fig. 2 X-ray crystallographic structures of 5O (a), 17S (b), 18Se (c), and 24SO (d) with 50% probability of thermal ellipsoids. Side view of the saddle-shaped NG 5O with the saddle-depth marked (e) and a pair of enantiomers shown in the single crystal 18Se (f). Electrostatic potential (ESP) maps and the energy colour bar (a.u.) of S-, SO-, and SO2-doped NGs (g), in which the alkyl chains were replaced by methyl groups. TD-DFT calculation of HOMO and LUMO orbitals with energies (eV) of 5O, 17S, 18Se, 24SO, 26SeO, and 28SO2 at the PBE0/6-311G(d,p) level of theory (h). The alkyl chains were replaced by methyl groups. | ||
Photophysical and electrochemical properties
The optical properties of chalcogen-doped NGs were investigated by UV-vis absorption and fluorescence spectroscopy. The UV-vis spectra of 5O, 17S, 19Se, 25SO, and 27SeO in DCM show that the absorption maxima are nearly identical (351–353 nm) together with multiple shoulder peaks, while sulfone 28SO2 exhibits a red-shifted absorption band with the maximum peak at 360 nm (Fig. 3a). The weak absorption peaks of 5O, 24SO, and 26SeO with ethyl substituents at approximately 450 nm were observed rather than the tert-butyl substituted analogs probably due to the formation of an exciplex by intermolecular π–π interaction (Fig. S8†). To obtain insights into the origin of the absorptions, we conducted time-dependent density-functional theory (TD-DFT) (Fig. S28, Tables S10–S15†). The DFT calculations indicated that the lower-energy bands at around 430 and 400 nm are mainly contributed by the HOMO–LUMO transition. The high-energy bands at approximately 375 and 350 nm can be assigned to either HOMO–LUMO+1 or HOMO−1–LUMO and HOMO−1–LUMO+1 respectively, which exhibit larger oscillator strength. The fluorescence maximum of these NGs is similarly located at around 475 nm with shoulder emission peaks for 5O, and a bathochromic-shifted peak at 490 nm for 28SO2 was observed (Fig. 3b). These compounds exhibit a large Stokes shift of 120 nm (over 7200 cm−1). Meanwhile, these chalcogen-doped NGs exhibit fluorescence intensity that depends on the doped heteroatoms and their oxidative state. With a similar helical conformation, 17S doped with sulfur exhibited 10-fold higher fluorescence intensity and nearly 20-fold higher fluorescence quantum yield (Table S3†) than the selenium-doped analog 19Se. The fluorescence efficiency of NGs can be significantly enhanced by changing the doping heteroatoms S or Se to S-oxide or Se-oxide (17Svs.25SO; 19Sevs.26SeO, Fig. 3b, Table S3†). As a result, the S-oxide 25SO was highly fluorescent with the highest fluorescence quantum yield of 0.30, which is significantly higher than that of the corresponding HBC 30HBC and seco-HBC 29seco-HBC (Table S3†). Notably, the difference in the magnitude of the fluorescence decay was observed by doping different heteroatoms. As shown in Fig. 3c, the oxygen- and sulfur-doped candidates 5O, 17S, 25SO, and 28SO2 exhibit very close decay rates with fluorescence lifetimes over 17 ns, whereas, for selenium-doped compounds 19Se and 27SeO, the fluorescence lifetimes are less than 4 ns. Electrochemical properties were investigated by cyclic voltammetry in CH2Cl2 in the presence of Bu4NPF6 as a supporting electrolyte (Fig. S11†). | ||
| Fig. 3 Photophysical and chiroptical properties of chalcogen-doped NGs. (a) UV-vis absorption spectra, (b) fluorescence emission spectra, and (c) fluorescence decay profiles of 5O, 17S, 19Se, 25SO, 27SeO, and 28SO2. The spectra were collected in DCM solution with 5 μM concentration and λexc. = 350 nm for fluorescence excitation. (d) CD spectra of enantiomeric 17S-P and 17S-M, enantiomeric 19Se-P and 19Se-M, and enantiomeric 25SO-P and 25SO-M in hexane. (e) CPL and direct current (DC) spectra of 17S-P/17S-M and 25SO-P/25SO-M in dichloromethane (λexc. = 350 nm, the measurement was carried out in air). | ||
Notably, the redox properties can be modulated by controlling the oxidative states of the doped chalcogens. The oxidized structures 25SO, 27SeO, and 28SO2 show more oxidation waves than their reduced analogs 17S and 19Se.
Chiroptical properties and determination of the racemization barrier
Helicene-based NGs have triggered much interest in the past decades due to their intrinsic chirality.23 We attempt to resolve the two sets of enantiomers for both 17S and 19Se and study their chiroptical properties. The enantiomers of 17S and 19Se were successfully separated by chiral high-performance liquid chromatography (HPLC) at 25–30 °C with ethanol as the mobile phase (Fig. S12 and S13†). The electronic circular dichroism (ECD) spectra of the enantiomers of 17S and 19Se in hexane demonstrated a perfect mirror image relationship with multiple opposite Cotton effects in the UV/vis region from 200 to 450 nm (Fig. 3d). Due to the homologous chiral structures, similar profiles of enantiomers from 17S and 19Se were displayed. Each enantiomer of 17S or 19Se was confirmed by comparing each ECD spectrum with the calculated spectrum (Fig. S26†). Since sulfoxide 25SO shows high emission efficiency, we prepared the optically pure enantiomers 25SO-P/25SO-M by oxidation of enantiomers 17S-P and 17S-M (Fig. S14†), and the ECD spectra of 25SO-P/25SO-M indicated a perfect enantiomer relationship (Fig. 3d). Next, we carried out circularly polarized luminescence (CPL) measurements for each enantiomer. The CPL spectra of 17S-P/17S-M and 25SO-P/25SO-M exhibited a mirror-imaging relationship in dichloromethane under ambient conditions (Fig. 3e), which suggested that 17S and 25SO show CPL activity.The luminescence dissymmetry ratio (glum), evaluating the degree of CPL, was determined to be 1.1 × 10−3 and 5.6 × 10−4 for 17S and 25SO respectively at 480 nm (Fig. S15†). Due to the low fluorescence quantum yield (ΦF < 0.01), the CPL of 19Se-P and 19Se-M was feeble to be detected. Meanwhile, due to the high extinction coefficient and ΦF, the enantiomers of 17S and 25SO exhibit BCPL up to 13 M−1 cm−1 and 12 M−1 cm−1 respectively. The successful resolution of two enantiomers of 17S or 19Se indicated relatively high enantiomerization barriers for both helicenes.
To experientially determine the isomerization barrier (ΔG‡), thermal racemization of optically pure 17S-P was performed followed by chiral HPLC analysis (Fig. S16†). HPLC traces of the samples after heating at 100 °C exhibited a clean transformation from the 17S-P to the 17S-M isomer. The half-life (τ1/2) for loss of enantiomeric excess was determined to be 5.5 hours at 100 °C. It gives a racemization barrier ΔG‡ (373 K) of 29.6 kcal mol−1 using the Eyring equation, which demonstrated a relatively stable helicene enantiomer and comparable with reported [5]helicene containing NGs.24 In addition, thermal racemization of optically pure 19Se-P was performed similarly. However, 19Se-P stayed stable after heating at 100 °C for hours, which suggested a higher isomerization barrier of 19Se than that of 17S. Interestingly, the selenium-extrusion reaction was observed together with enantiomerization when the heating temperature was raised to 130 °C (Fig. S17†). Nevertheless, excluding other chemical reactivity, the racemization barrier ΔG‡ of 19Se was evaluated to be 31.2 kcal mol−1 at 403 K.
Chalcogen-extrusion processes
After getting a hint from the observation of the selenium-extrusion process during the synthesis, we then investigated the chalcogen-extrusion reactions of these heteroatom-doped NGs. Several organosulfur compounds are susceptible to undergoing photo- or thermal-induced sulfur extrusion reactions.25 Initially, we examined whether the extrusion reactions could be triggered by UV light. Each compound was treated in DCM solution under an atmosphere of N2, followed by photoirradiation with 254 or 365 nm UV light. Unfortunately, no distinct change was observed for all the NGs upon irradiation for a short period of time (<10 min) and compounds gradually degraded over a long time. Alternatively, we performed the thermal chalcogen extrusion for compounds 5O, 17S, 19Se, 25SO, 27SeO, and 28SO2 (Fig. 4a). The experiments were conducted by heating milligrams of solid of each sample in an oven for a specific time, and monitoring the changes in the absorption spectra after re-dissolving the preheated sample in DCM. To our delight, for selenium-doped candidates, 19Se and 27SeO, identical de-selenium products were detected by observing the same absorption profiles formed after heating at 200 °C for only 5 min. These new species were confirmed as selenium-extrusion product 29seco-HBC in comparison with the UV-absorption of standard product 29seco-HBC (Fig. 4be). The HPLC analyses of the brown samples formed upon heating (Fig. 4b, inset) indicated a quantitative formation of 29seco-HBC (Fig. S20 and S21†). In contrast, sulfur 17S and sulfone 28SO2 as well as the oxygen-doped saddle 5O showed no change in the absorption spectra upon heating up to 250 °C for 2 hours, which suggests relatively thermally stable structures (Fig. 4c, f, and S18†). Meanwhile, sulfoxide 25SO exhibited an inert sulfur extrusion process by heating over 250 °C compared to selenium-doped analogs (Fig. 4d and S19†). These results suggested the accessibility and rate of chalcogen extrusion reaction are highly dependent on the sort and oxidative state of the embedded chalcogens.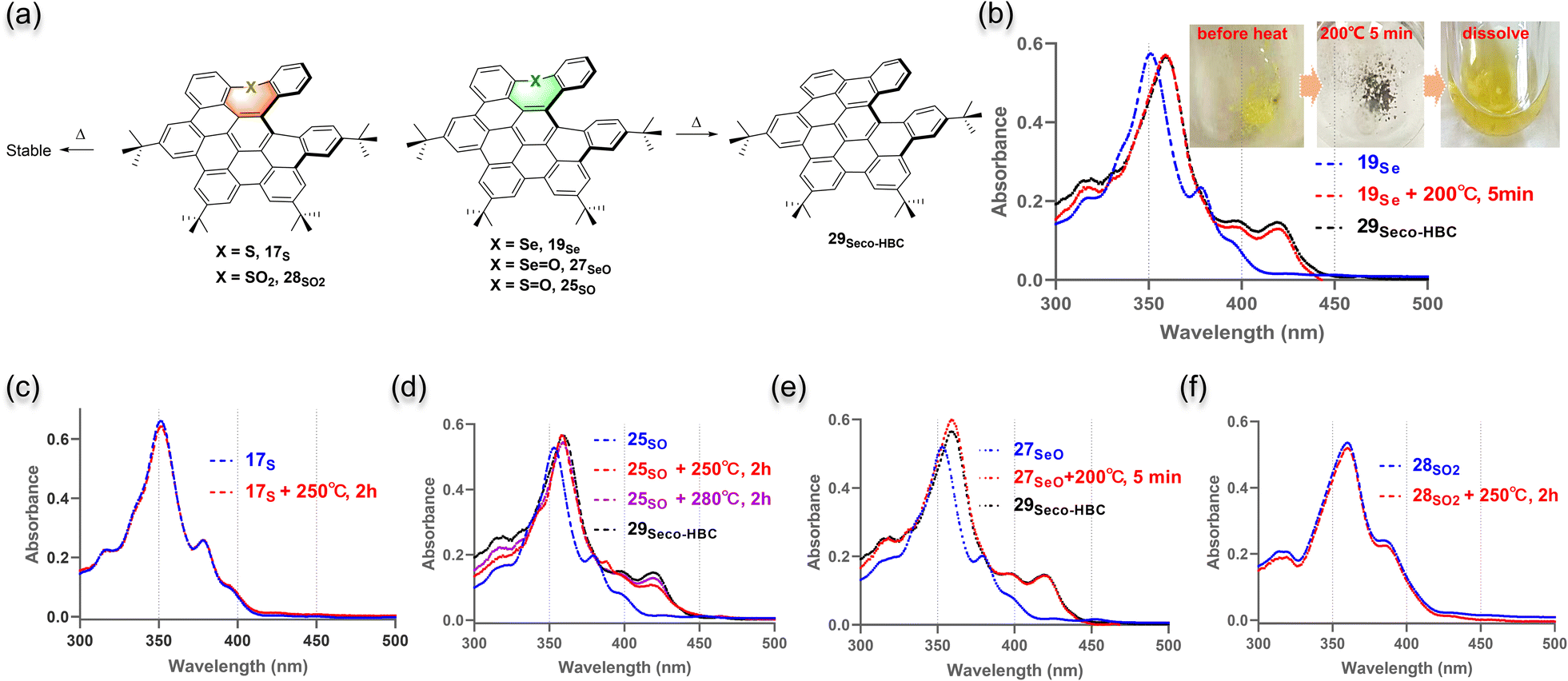 | ||
| Fig. 4 (a) Chalcogen extrusion reactions of S-, Se-doped NGs. UV-vis absorption spectra of 19Se (b), 17S (c), 25SO (d), 27SeO (e), and 28SO2 (f) before and after heating. The spectra were collected by dissolving the solid (before and after heating) in DCM and the heating time of each compound was shown in the corresponding spectrum. For comparison, the UV-vis spectra of the chalcogen-extrusion product 29seco-HBC were shown together with 19Se (b), 25SO (d), and 27SeO (e). The inset in (b) shows the photographic images of the 19Se solid before and after heating, and the dissolved brown heated solid in DCM. | ||
The HBC reveals wide application in materials science but is detrimental to the fabrication of material devices due to aggregation-caused low solubility. Apparently, having a soluble precursor of HBC would significantly facilitate the material fabrication of HBC. We envisioned whether HBC structures could be obtained by spontaneous oxidative cyclodehydrogenation of the chalcogen-extrusion products. By screening commonly used acids like acetic acid, HCl, and trifluoroacetic acid, it was found that selenium oxide 27SeO could slowly convert to 30HBC in a trifluoroacetic acid/DCM (1/2, v/v) solution (Fig. 5a) and the formation of target product 30HBC was unequivocally confirmed by X-ray crystallography (Fig. 5b, Scheme S5†).20 As monitored by UV-vis absorption (Fig. 5c), through selenium extrusion and spontaneous oxidation in air, the characteristic peak of 27SeO at 353 nm gradually shifted to 358 nm over 20 hours in the trifluoroacetic acid/DCM (1/2, v/v) solution, and the absorption spectrum after a 20 hour reaction was almost identical to that of 30HBC, whereas the other NGs were stable under these conditions (Fig. S23†). Together with the HPLC analyses (Fig. S22†), a nearly quantitative conversion was obtained for 27SeO. Moreover, this transformation could be accelerated by the addition of stronger acid methanesulfonic acid instead of trifluoroacetic acid (Fig. S24†). To evaluate the difference in soluble properties between HBC and this precursor, we measured the solubility of 27SeO and 30HBC in dichloromethane and ethanol (Fig. S25†). The precursor 27SeO exhibits more than 100-fold higher solubility in either dichloromethane or polar solvent ethanol.
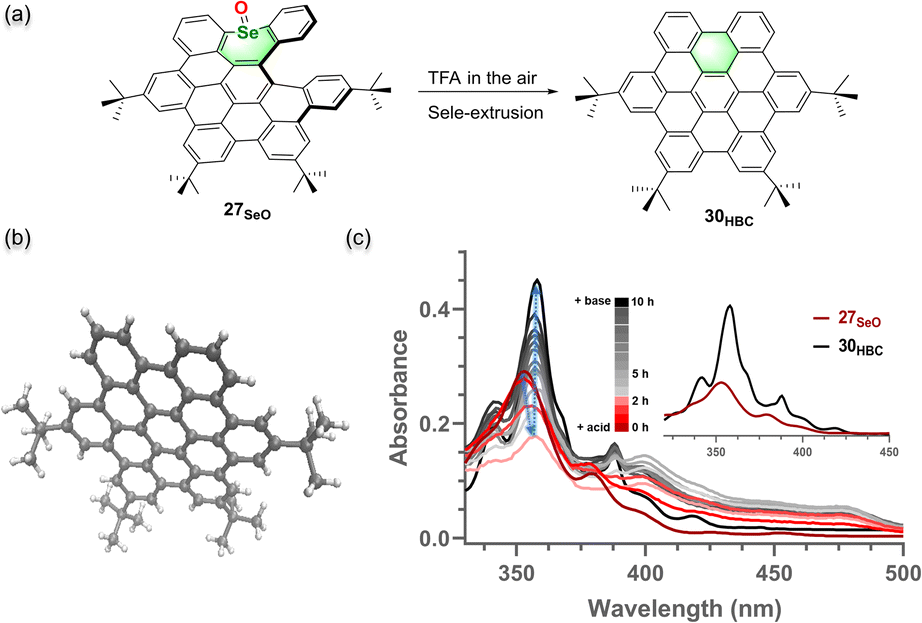 | ||
| Fig. 5 (a) Acid-catalyzed HBC formation from HBC precursor selenium-doped NG 27SeO. (b) X-ray crystallographic structure of 30HBC with 50% probability of thermal ellipsoids. (c) Changes in the UV-vis absorption spectra of 27SeO upon dissolving it in trifluoroacetic acid/DCM (1/2, v/v) with 3.3 μM concentration under ambient conditions. | ||
Conclusions
We have developed a new class of chalcogen-doped NGs, in which a chalcogen is conceptually embedded into the HBC or seco-HBC backbone in the fjord region. As a eesult of the size of chalcogens, two types of structures were obtained toward the final step of the oxidative ring closure reaction: the oxygen-doped saddle and the sulfur- or selenium-doped helix. The saddle and helical three-dimensional structures of the chalcogen-doped NGs and the sulfur-oxide were revealed by single-crystal X-ray diffraction analysis, and both P- and M-enantiomers of helicene enantiomers cocrystallize in a single crystal. Enantiomers of sulfur- and selenium-doped helical NGs were isolated by chiral HPLC. The chiroptical properties including ECD and CPL were investigated and the sulfur-embedded NG exhibited CPL activity with a glum of 1.1 × 10−3 at 480 nm and BCPL of 13 M−1 cm−1. The racemic barriers of S-NG and Se-NG were determined to be 29.6 and 31.2 kcal mol−1 respectively by HPLC-based thermal isomerization. The experimental and computational study including UV-vis absorption, fluorescence emission, efficiency, decay, HOMO–LUMO gaps, and charge distribution indicated that the chalcogen and its oxidative state in the NG significantly affect the electronic properties. The stability measurements of these NGs upon heating suggested that the Se-doped NG and its oxide undergo quantitative selenium-extrusion reactions, which offers dynamic control of the structural reconstruction. Notably, the highly distorted helical Se-oxide NG can transform to planar HBC under acidic conditions. The planar HBC generally exhibits low solubility and a high aggregation tendency in solvents. This HBS precursor would significantly facilitate the application of HBC in material fabrication.Data availability
The full experimental details, synthetic procedures, characterization data, HPLC traces, UV-vis, fluorescence, IR and NMR spectra, computational details, and supplementary discussions associated with this article are provided in the ESI.†Author contributions
Ranran Li performed most of the synthesis work and property studies. Bin Ma started the initial synthesis of compound 5O. Shengtao Li helped with NMR, HPLC, and photophysical property measurements. P. An performed the DFT calculation and conceived the concept. P. An and C. Lu prepared and revised the manuscript. All the authors analyzed and interpreted the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the National Natural Science Foundation of China (21901226 and 22061046). We thank the advanced analysis and measurement center of Yunnan University for assistance with instrumentation. Particularly, we thank Dr Jie Zhou from the Advanced Analysis and Measurement Center of Yunnan University for the X-ray analysis.Notes and references
- (a) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed; (b) K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed; (c) L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J.-L. Brédas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500–509 CrossRef CAS PubMed; (d) X.-Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- (a) Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed; (b) N. Bachar, L. Liberman, F. Muallem, X. Feng, K. Müllen and H. Haick, ACS Appl. Mater. Interfaces, 2013, 5, 11641–11653 CrossRef CAS PubMed; (c) V. B. R. Pedersen, S. K. Pedersen, Z. Jin, N. Kofod, B. W. Laursen, G. V. Baryshnikov, C. Nuckolls and M. Pittelkow, Angew. Chem., Int. Ed., 2022, 61, e202212293 CAS.
- (a) H.-A. Lin, Y. Sato, Y. Segawa, T. Nishihara, N. Sugimoto, L. T. Scott, T. Higashiyama and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 2874–2878 CrossRef CAS PubMed; (b) Q.-Q. Li, Y. Hamamoto, G. Kwek, B. Xing, Y. Li and S. Ito, Angew. Chem., Int. Ed., 2022, 61, e202112638 CAS; (c) Y.-F. Wu, S.-W. Ying, L.-Y. Su, J.-J. Du, L. Zhang, B.-W. Chen, H.-R. Tian, H. Xu, M.-L. Zhang, X. Yan, Q. Zhang, S.-Y. Xie and L.-S. Zheng, J. Am. Chem. Soc., 2022, 144, 10736–10742 CrossRef CAS PubMed.
- (a) T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS PubMed; (b) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (c) W. Pisula, X. Feng and K. Müllen, Chem. Mater., 2011, 23, 554–567 CrossRef CAS; (d) D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed; (e) N. Zhang, L. Yang, W. Li, J. Zhu, K. Chi, D. Chang, Y. Qiao, T. Wang, Y. Zhao, X. Lu and Y. Liu, J. Am. Chem. Soc., 2022, 144, 21521–21529 CrossRef CAS PubMed; (f) L. Chai, Y.-Y. Ju, J.-F. Xing, X.-H. Ma, X.-J. Zhao and Y.-Z. Tan, Angew. Chem., Int. Ed., 2022, 61, e202210268 CAS; (g) S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- (a) M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC; (b) S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed; (c) Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS; (d) M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef CAS PubMed; (e) I. R. Márquez, S. Castro-Fernández, A. Millán and A. G. Campaňa, Chem. Commun., 2018, 54, 6705–6718 RSC; (f) S. R. Peurifoy, T. J. Sisto, F. Ng, M. L. Steigerwald, R. Chen and C. Nuckolls, Chem. Rec., 2019, 19, 1050–1061 CrossRef CAS PubMed; (g) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed; (h) R. A. Pascal, Chem. Rev., 2006, 106, 4809–4819 CrossRef CAS PubMed; (i) A. Bedi and O. Godron, Acc. Chem. Res., 2019, 52, 2482–2490 CrossRef CAS PubMed; (j) M. C. Stuparu, Acc. Chem. Res., 2021, 54, 2858–2870 CrossRef CAS PubMed; (k) J. Wang, X. Zhang, H. Jia, S. Wang and P. Du, Acc. Chem. Res., 2021, 54, 4178–4190 CrossRef CAS PubMed; (l) M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- (a) M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Angew. Chem., Int. Ed., 2021, 60, 14998–15005 CrossRef CAS PubMed; (b) H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed.
- (a) J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796–13803 CrossRef CAS PubMed; (b) P. An, R. Li, B. Ma, R.-Y. He, Y.-K. Zhang, M.-J. Xiao and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24478–24483 CrossRef CAS PubMed; (c) T. Kirschbaum, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2020, 59, 270–274 CrossRef CAS PubMed; (d) T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2020, 26, 14560–14564 CrossRef CAS PubMed; (e) I. R. Márquez, N. Fuentes, C. M. Cruz, V. Puente-Muñoz, L. Sotorrios, M. L. Marcos, D. Choquesillo-Lazarte, B. Biel, L. Crovetto, E. Gómez-Bengoa, M. T. González, R. Martin, J. M. Cuerva and A. G. Campaña, Chem. Sci., 2017, 8, 1068–1074 RSC.
- (a) N. Ogawa, Y. Yamaoka, H. Takikawa, K.-I. Yamada and K. Takasu, J. Am. Chem. Soc., 2020, 142, 13322–13327 CrossRef CAS PubMed; (b) M. M. Martin, F. Hampel and N. Jux, Chem.–Eur. J., 2020, 26, 10210–10212 CrossRef CAS PubMed; (c) J. Ma, Y. Fu, E. Dmitrieva, F. Liu, H. Komber, F. Hennersdorf, A. A. Popov, J. J. Weigand, J. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 5637–5642 CrossRef CAS PubMed; (d) X.-Y. Chen, J.-K. Li and X.-Y. Wang, Chinese J. Org. Chem., 2021, 41, 4105–4137 CrossRef CAS.
- (a) K. Y. Cheung, S. Gui, C. Deng, H. Liang, Z. Xia, Z. Liu, L. Chi and Q. Miao, Chem, 2019, 5, 838–847 CrossRef CAS; (b) S. Wang, J. Yuan, J. Xie, Z. Lu, L. Jiang, Y. Mu, Y. Huo, Y. Tsuchido and K. Zhu, Angew. Chem., Int. Ed., 2021, 60, 18443–18447 CrossRef CAS PubMed.
- M. Hermann, D. Wassy and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 15743–15766 CrossRef CAS PubMed.
- (a) A. Jolly, D. Miao, M. Daigle and J.-F. Morin, Angew. Chem., Int. Ed., 2020, 59, 4624–4633 CrossRef CAS PubMed; (b) J. M. Fernández-García, P. J. Evans, S. M. Rivero, I. Fernández, D. García-Fresnadillo, J. Perles, J. Casado and N. Martín, J. Am. Chem. Soc., 2018, 140, 17188–17196 CrossRef PubMed; (c) T. Fujikawa, D. V. Preda, Y. Segawa, K. Itami and L. T. Scott, Org. Lett., 2016, 18, 3992–3995 CrossRef CAS PubMed; (d) R. Li, B. Ma, R.-Y. He, B. Zhang, Y.-K. Zhang, S.-Y. Feng and P. An, Chem.–Asian J., 2022, 17, e202101365 CAS.
- (a) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed; (b) K. Kato, K. Takaba, S. Maki-Yonekura, N. Mitoma, Y. Nakanashi, T. Nashihara, T. Hata-Keyama, T. Kamada, Y. Hijikata, J. Pirillo, L. T. Scott, K. Yonekura, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2021, 143, 5465–5469 CrossRef CAS PubMed; (c) J. Wang, F. Gordillo Gámez, J. Marín-Beloqui, A. Diaz-Andres, X. Miao, D. Casanova, J. Casado and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202217124 CAS.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (c) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed; (d) V. Barát and M. C. Stuparu, Chem.–Asian J., 2021, 16, 20–29 CrossRef PubMed; (e) V. Barát and M. C. Stuparu, Chem.–Eur. J., 2020, 26, 15135–15139 CrossRef PubMed; (f) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed; (g) Y. Guo, C. Chen and X.-Y. Wang, Chin. J. Chem., 2023, 41, 1355–1373 CrossRef CAS.
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (b) K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed; (c) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed; (d) H. Hölzel, P. Haines, R. Kaur, D. Lungerich, N. JUX and D. M. Guldi, J. Am. Chem. Soc., 2022, 144, 8977–8986 CrossRef PubMed.
- (a) J. Wu, A. Fechtenkötter, J. Gauss, M. D. Watson, M. Kastler, C. Fechtenkötter, M. Wagner and K. Müllen, J. Am. Chem. Soc., 2004, 126, 11311–11321 CrossRef CAS PubMed; (b) J. Wu, J. Li, U. Kolb and K. Müllen, Chem. Commun., 2006, 48–50 RSC.
- M. A. Medel, R. Tapia, V. Blanco, D. Miguel, S. P. Morcillo and A. G. Campaña, Angew. Chem., Int. Ed., 2021, 60, 6094–6100 CrossRef CAS PubMed.
- M. A. Medel, C. M. Cruz, D. Miguel, V. Blanco, S. P. Morcillo and A. G. Campaña, Angew. Chem., Int. Ed., 2021, 60, 22051–22056 CrossRef CAS PubMed.
- (a) B. Ma, M. Xiao, P. Liu and P. An, Synlett, 2022, 33, 409–414 CrossRef CAS; (b) B. Zhang, L. Ruan, Y.-K. Zhang, H. Zhang, R. Li and P. An, Org. Lett., 2023, 25, 732–737 CrossRef CAS PubMed.
- F. S. Guziec Jr and L. J. Sanfilippo, Tetrahedron, 1988, 44, 6241–6285 CrossRef.
- Deposition numbers 2238626–2238632 for compounds 5O, 21, 23, 24SO, 17S, 18Se, and 30HBC contain the supplementary crystallographic data for this paper.†.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- (a) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed; (b) C. Li, Y. Yang and Q. Miao, Chem.–Asian J., 2018, 13, 884–894 CrossRef CAS PubMed; (c) J. M. Fernández-García, P. J. Evans, S. Filippone, M. Á. Herranz and N. Martín, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef PubMed; (d) T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed; (e) Y. Wu, L. Zhang, Q. Zhang, S.-Y. Xie and L.-S. Zheng, Org. Chem. Front., 2022, 9, 4726–4743 RSC; (f) P. Karak and J. Choudhury, Chem. Sci., 2022, 13, 11163–11173 RSC; (g) G. R. Kiel, H. M. Bergman, A. E. Samkian, N. J. Schuster, R. C. Handford, A. J. Rothenberger, R. Gómez-Bombarelli, C. Nuckolls and T. D. Tillet, J. Am. Chem. Soc., 2022, 144, 23421–23427 CrossRef CAS PubMed; (h) Y.-F. Wu, S.-W. Ying, S.-D. Liao, L. Zhang, J.-J. Du, B.-W. Chen, H.-R. Tian, F.-F. Xie, H. Xu, S.-L. Deng, Q. Zhang, S.-Y. Xie and L. S. Zheng, Angew. Chem., Int. Ed., 2022, 61, e202204334 CAS; (i) J.-K. Li, X.-Y. Chen, W.-L. Zhao, Y.-L. Guo, Y. Zhang, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao, M. Li, C.-F. Chen and X.-Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215367 CAS.
- J. M. Fernández-Garcıía, P. Izquierdo-García, M. Buendía, S. Filip-pone and N. Martín, Chem. Commun., 2022, 58, 2634–2645 RSC.
- (a) V. Boekelheide, I. D. Reingold and M. Tuttle, J. Chem. Soc., Chem. Commun., 1973, 406–407 RSC; (b) R. S. Givens, R. J. Olsen and P. L. Wylie, J. Org. Chem., 1979, 44, 1608–1613 CrossRef CAS; (c) A. Wakamiya, T. Nishinaga and K. Komatsu, J. Am. Chem. Soc., 2002, 124, 15038–15050 CrossRef CAS PubMed; (d) T. Okamoto, K. Kudoh, A. Wakamiya and S. Yamaguchi, Org. Lett., 2005, 7, 5301–5304 CrossRef CAS PubMed; (e) H. Kamiya, T. Kondo, T. Sakida, S. Yamaguchi and H. Shinokubo, Chem.–Eur. J., 2012, 18, 16129–16135 CrossRef CAS PubMed; (f) P. R. Christensen, B. O. Patrick, E. Caron and M. O. Wolf, Angew. Chem., Int. Ed., 2013, 52, 12946–12950 CrossRef CAS PubMed; (g) S. Hayakawa, K. Matsuo, H. Yamada, N. Fukui and H. Shinokubo, J. Am. Chem. Soc., 2020, 142, 11663–11668 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2238626–2238632. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02595k |
| This journal is © The Royal Society of Chemistry 2023 |