
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMild and scalable synthesis of phosphonorhodamines†
Joshua L.
Turnbull
a,
Ryan P.
Golden
a,
Brittany R.
Benlian
b,
Katharine M.
Henn
c,
Soren M.
Lipman
 a and
Evan W.
Miller
*abc
a and
Evan W.
Miller
*abc
aDepartment of Chemistry, University of California, Berkeley, CA 94720-1460, USA. E-mail: evanwmiller@berkeley.edu
bDepartment of Molecular & Cell Biology, University of California, Berkeley, CA 94720-1460, USA
cHelen Wills Neuroscience Institute, University of California, Berkeley, CA 94720-1460, USA
First published on 5th October 2023
Abstract
Since their discovery in 1887, rhodamines have become indispensable fluorophores for biological imaging. Recent studies have extensively explored heteroatom substitution at the 10′ position and a variety of substitution patterns on the 3′,6′ nitrogens. Although 3-carboxy- and 3-sulfono-rhodamines were first reported in the 19th century, the 3-phosphono analogues have never been reported. Here, we report a mild, scalable synthetic route to 3-phosphonorhodamines. We explore the substrate scope and investigate mechanistic details of an exogenous acid-free condensation. Tetramethyl-3-phosphonorhodamine (phosTMR) derivatives can be accessed on the 1.5 mmol scale in up to 98% yield (2 steps). phosTMR shows a 12- to 500-fold increase in water solubility relative to 3-carboxy and 3-sulfonorhodamine derivatives and has excellent chemical stability. Additionally, phosphonates allow for chemical derivatization; esterification of phosTMR facilitates intracellular delivery with localization profiles that differ from 3-carboxyrhodamines. The free phosphonate can be incorporated into a molecular wire scaffold to create a phosphonated rhodamine voltage reporter, phosphonoRhoVR. PhosRhoVR 1 can be synthesized in just 6 steps, with an overall yield of 37% to provide >400 mg of material, compared to a 6-step, ∼2% yield for the previously reported RhoVR 1. PhosRhoVR 1 possesses excellent voltage sensitivity (37% ΔF/F) and a 2-fold increase in cellular brightness compared to RhoVR 1.
Introduction
Small molecule fluorophores have revolutionized our ability to visualize complex biological systems.1,2 Biological imaging modalities rely on access to fluorophores with high brightness, stability, and water solubility. Rhodamines, the amino isologues of fluorescein, find widespread use in bioimaging applications. In comparison to fluoresceins, rhodamines are insensitive to pH, exhibit improved photostability, and have tunable excitation and emission across the visible spectrum.3Modification of the structure of rhodamines can produce dramatic changes in the functional properties of the dyes (Scheme 1). Changing the alkylation pattern on the 3′ and 6′ amines tunes absorption and emission wavelengths,4 fused cyclohexanes or 4-membered azetidine rings improve brightness,5 and heteroatom substitution of the 10′ oxygen atom results in a red shift.6–14 Additionally, the delocalized positive charge of rhodamine facilitates cell permeability, making rhodamines attractive scaffolds for intracellular or live-cell imaging applications.15
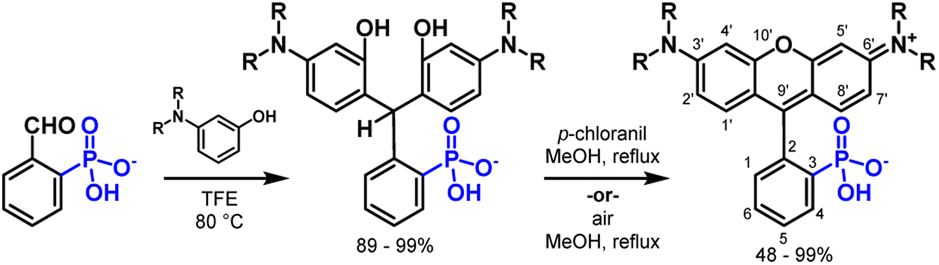 | ||
| Scheme 1 Mild synthesis of phosphonorhodamines. | ||
Substitutions of the rhodamine core have been extensively explored for the modulation of photophysical properties. On the other hand, substitutions at the 3-position of the pendant aryl ring – which include carboxylates, amides, sulfonates, methyl, and substituted methylenes – largely impact subsequent applications of rhodamine fluorophores. We recently disclosed a new class of fluoresceins with 3-phosphonate substitutions. 3-Phosphonofluoresceins exhibit an almost 2-fold improvement in water solubility compared to 3-carboxy analogs,16 and functionalization of the 3-phosphonate with esters improves cellular brightness 70-fold over traditional 3-carboxyfluorescein. Owing to the orthogonality of the xanthene core and the pendant ring, physical changes brought by 3-phosphonate substitution were facilitated without compromising brightness or excitation/emission wavelengths. Expansion of 3-phosphonate substitution to rhodamines may yield additional opportunities in the applications of xanthene fluorophores.
Despite almost 140 years since the first rhodamine was synthesized, synthetic approaches often lack generalizability with respect to varying 3-substitution. In fact, rhodamines are largely constructed by variations of the initially reported Friedel–Crafts condensation.17,18 Phthalic anhydrides are heated with aminophenols in the presence of Brønsted or Lewis acids; however, functionalized phthalides often lead to mixture of regioisomers that are difficult to separate.19 To solve this problem, benzaldehydes can be condensed with aminophenols followed by dehydration and in situ oxidation with exposure to air or use of an organic oxidant such as chloranil.18 The harsh nature of these condensations often results in low yields, functional group incompatibility and troublesome purifications.1,20,21 While the acidic conditions present in all of these reaction schemes increase reactivity of the electrophile, protonation, or Lewis acid complexation, with the aniline decreases the nucleophilicity of aminophenols. An elegant recent example showed that intramolecular activation of the benzaldehyde by an ortho carboxylic acid could furnish rhodamines in high yields without the addition of an acid catalyst.22
Contemporary methods to access rhodamines make use of Pd-catalyzed C–N cross coupling of fluorescein ditriflates with various amines,23 addition of aryl organometallic species into N-alkylated diaminoxanthones,24,25 or addition of dimetallated bisphenyl ethers into electrophiles such as aryl esters or phthalic anhydrides.10,26 While these methods alleviate some of the challenges associated with classic rhodamine condensations, their applications to the synthesis of rhodamines with acidic 3-functionalities (such as phosphonates) are limited.
Here, we report a mild synthesis of 3-phosphonorhodamines in exceptional yields from the condensation of aminophenols and 2-phosphonobenzaldehyde (Scheme 1). 3-Phosphonorhodamines can be synthesized on a 1.5 mmol scale, often without the need for chromatography, alleviating many of the challenges associated with purification of charged fluorophores. The 3-phosphonorhodamines have nearly identical optical properties compared to their classic 3-carboxy- and sulfono-counterparts but possess a >10- to 500-fold increase in water solubility. The tetramethyl-substituted 3-phosphonorhodamine can be applied to diverse cellular imaging contexts. First, esterification of the phosphonate controls cellular localization, and tetramethylphosphonorhodamine shows enhanced cellular brightness compared to the analogous 3-carboxyrhodamine. Next, 3-phosphonorhodamines can be localized to plasma membranes and used for voltage imaging, where they show comparable voltage sensitivity and a 2-fold increase in cellular brightness compared to carboxy-based indicators.
Results and discussion
Synthesis of 3-phosphonorhodamines
The key role of acid in the Friedel–Crafts condensations of xanthene fluorophores, such as rhodamines, is to activate the aldehyde and promote nucleophilic attack from electron rich arenes. In the case of 2-carboxybenzaldehyde, the acidic nature of the ortho carboxylate provides intramolecular activation of the aldehyde, increasing electrophilicity.27 The intramolecular activation is evident through slight formation (13%) of the corresponding hemiacetal in alcoholic solvents such as CD3OD (Table S1 and Scheme S1†). Dwight and Levin previously reported a relatively mild, acid-free synthesis of 3-carboxyrhodamines that is mediated by this phenomenon of intramolecular activation.22By analogy to this previous report,22 acidic phosphonates may equip 2-phosphonobenzaldehyde (1) with similar intramolecular aldehyde activation and provide a path to 3-phosphonorhodamines in the absence of exogenous acid. This is the case. Heating of 1 with 3-(dimethylamino)phenol, 2, in 2,2,2-trifluoroethanol (TFE) results in clean precipitation of triarylmethane 9 in an excellent 98% yield. 9 can be isolated by precipitation and undergoes quantitative dehydration and oxidation to the corresponding rhodamine 16 (Scheme 2).
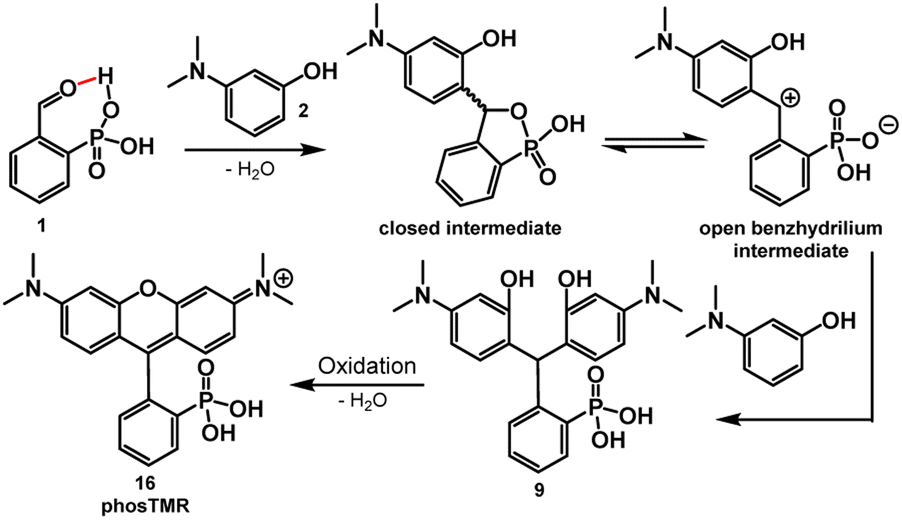 | ||
| Scheme 2 Stepwise formation of phosTMR. | ||
In previous studies, the analogous reaction with 2-carboxybenzaldehyde required TFE to achieve appreciable yields.22 This is likely a result of stabilization of the benzhydrilium intermediate by the high ionizing power, low nucleophilicity, and hydrogen bond donating ability of fluorinated alcohols (such as TFE) compared to hydrocarbon-based alcohols.22,28–30 As in previous studies,22 the reaction between 1 and 2 in TFE produces 9 in high yields (Scheme 3 and Table 1). However, the reaction of 1 and 2 in methanol also produces 9 in moderate yield (up to 58%, Scheme 3 and Table 1). This increased reactivity is likely a result of the greater extent of intramolecular activation of aldehyde 1 compared to 2-carboxybenzaldehyde. NMR studies support this hypothesis: 2-phosphonobenzaldehyde (1) shows a greater propensity to form the corresponding hemiacetal in nucleophilic solvents such as CD3OD (Table S1 and Scheme S1†). Aryl phosphonates generally have pKa1 values that are about 2 pH units lower than the corresponding benzoic acid pKa.31 The low pKa of the phosphonate may also contribute to greater stabilization of the benzhydrilium intermediate.
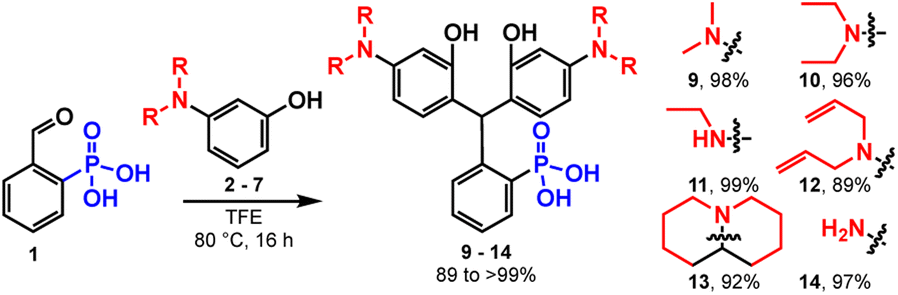 | ||
| Scheme 3 Synthesis of triarylmethane intermediates. | ||
| Aniline | Product | Conditions | Yielda | |
|---|---|---|---|---|
| a Isolated yields. b Performed under a nitrogen atmosphere. | ||||
| 2 |
|
9 | TFE, 80 °C, 16 hb | 98% |
| MeOH, 60 °C, 16 h | 58% | |||
| MeOH, rt, 16 h | 22% | |||
| 3 |
|
10 | TFE, 80 °C, 16 h | 96% |
| MeOH, rt 16 h | 27% | |||
| 4 |
|
11 | TFE, 80 °C, 16 h | 99% |
| 5 |
|
12 | TFE 80 °C, 16 hb | 89% |
| 6 |
|
13 | TFE, 80 °C, 16 hb | 92% |
| MeOH, rt, 16 h | 65% | |||
| 7 |
|
14 | TFE, 80 °C, 16 h | 97% |
To examine the generalizability of this synthetic approach, we exposed aldehyde 1 to a series of anilines, 2 to 7, under the same conditions (TFE, 80 °C, 16 hours) and observe precipitation of triarylmethanes 9 to 14 in excellent yields, ranging from 89 to 99% (Table 1). The exceptional yields of 9 to 14 are likely due in part to the insolubility of the triarylmethanes in alcoholic solvents. Friedel–Crafts condensations are reversible, as evidenced by the degradation of the triarylmethane intermediates in the presence of acid.32 Precipitation from the reaction medium drives reaction progression and contributes to the near quantitative yields. For more soluble intermediates, like 13, performing the reaction under an inert atmosphere improves the yield, by avoiding premature oxidation to the final rhodamine, which would otherwise complicate purification at this stage (Table 1).
Some substrates are not compatible with this methodology. Reaction of 1 with resorcinols does not yield the corresponding phosphonofluorescein. Instead, the reaction produces just a single addition of resorcinol to 1. Electron-rich anilines may be required to stabilize the benhzydrilium intermediate and promote a 2nd Friedel–Crafts addition (Scheme 2).
Further, azetidines are not currently compatible. Alcoholic solvents promote cationic ring opening polymerization of azetidines.33 Use of azetidine-substituted aminophenol 8 – which would provide a mild route to phosphono JaneliaFluor intermediate 15 – gives an intractable solid precipitate under our current reaction conditions (Scheme S2†).
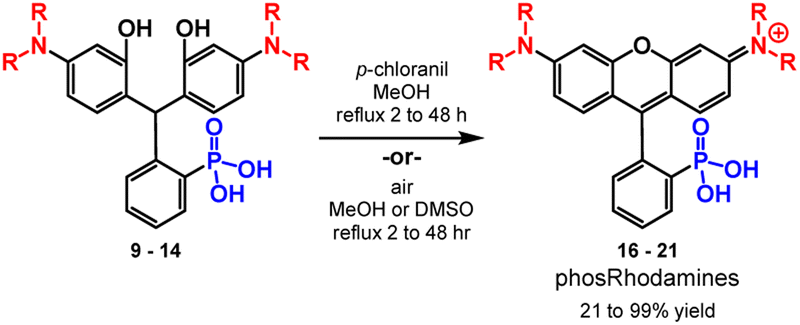
After isolating triarylmethane intermediates 9 to 14 in high yields and purity, we examined conditions for the dehydration and oxidation to the corresponding 3-phosphonorhomdamines 16 to 21 (Scheme 4).
 | ||
| Scheme 4 Synthesis of phosphonorhodamines. | ||
In most cases, reflux in methanol with p-chloranil as an oxidant leads to cyclization and oxidation to the corresponding rhodamine. As dehydration and oxidation to the rhodamines occurs, solubility in methanol increases and so reaction progression can be easily monitored by the dissolution of visible particulates. Reaction times vary from 2 to 48 hours and depend on the volume of methanol used. Upon completion, filtration removes any unoxidized fluorophore and trituration with organic solvent to remove excess chloranil yields rhodamines 16 to 19 in excellent yields (80–99%, Table 2) without need for further purification. Substrates 13 and 14 are sensitive to chloranil oxidation, leading to decomposition. Instead, reflux in methanol or DMSO with exposure to air is sufficient to promote dehydration and oxidation to rhodamines 20 and 21 respectively; however, these methods require purification by reverse phase silica chromatography. The dehydration and oxidation of 14 to phosRho110, 21, is particularly low yielding (21%), owing to the limited solubility of the fluorophore and challenging purification. An alternative synthesis through de-allylation of 19 (Scheme S3†) gives 21 in 71% yield (Table S2†).
| Triarylmethane | Rhodamine | Yielda | Conditions |
|---|---|---|---|
| a Isolated yields. b After reverse phase silica chromatography. c 21 is also accessible by deallylation of 19 in up to 71% yield. | |||
| 9 | 16 phosTMR | 99% | p-Chloranil, MeOH, reflux, 2–48 h |
| 10 | 17 phosRhoB | 80% | p-Chloranil, MeOH, reflux, 2–48 h |
| 11 | 18 phosRho6G | 82% | p-Chloranil, MeOH, reflux, 2–48 h |
| 12 | 19 phosTAR | 93% | p-Chloranil, MeOH, reflux, 2–48 h |
| 13 | 20 phosJulR | 48%b | MeOH, reflux, 48 h |
| 14 | 21 phosRho110 | 21%b,c | DMSO, 100 °C, 12 h |
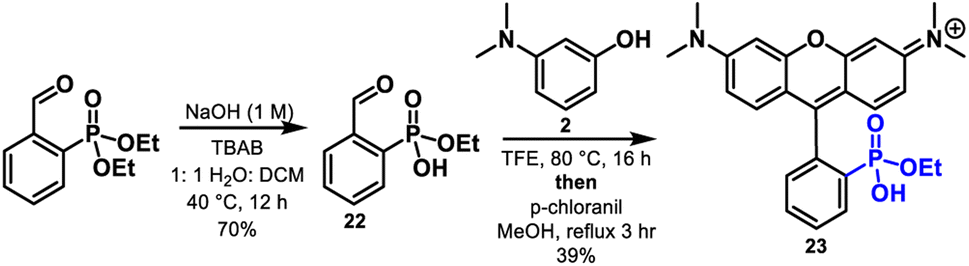
Finally, condensation of aminophenols with phosphonate monoesters are also compatible with this reaction methodology. Condensation of phosphonate monoester 22 with 2, followed by dehydration and chloranil oxidation affords rhodamine 23 in 39% yield with the ethyl ester intact (Scheme 5). Although a reduced yield compared to the 98% for 16, these results show that monoesters possess the ability to facilitate the reaction via intramolecular activation of the aldehyde. Importantly the strongly acidic conditions typically required for these condensations would result in phosphonate ester hydrolysis and thus the direct synthesis of monoester 23 is a testament to the mild nature of this chemistry. The reaction of 2 with the diethylester of 1 gives no reaction in TFE or MeOH.
 | ||
| Scheme 5 Direct synthesis of a functionalized phosTMR. | ||
The conditions used to achieve high yields of phosphonorhodamines do not translate to high yields for 2-carboxy and 2-sulfono rhodamines. While 2-carboxybenzaldehyde also provides intramolecular activation,22,27 reaction with 2 under the same conditions produces carboxyTMR in only 31% yield and requires chromatography (ESI†). This represents more than a 3-fold decrease in the yield compared to phosTMR, 16. The lower pKa of arylphosphonates compared to benzoic acids may account for the differing reactivity. On the other hand, 2-sulfonobenzaldehyde does not produce sulfoTMR under these conditions, suggesting the pKa of the arylsulfonic acid is too low and therefore has no proton to activate the aldehyde. As such, sulfoTMR was synthesized by heating with 2 in neat methanesulfonic acid in just 12% yield (ESI†).
Spectroscopic characterization of 3-phosphonorhodamines
The spectroscopic properties of phosTMR, 16 and the ethyl ester analog phosTMR·OEt, 23 are similar compared to sulfo- and carboxyTMR (Fig. 1a–d and Table 3). Compound 16, phosTMR, displays a small hypsochromic shift (546/564 nm), relative to carboxyTMR (549/569 nm) in phosphate buffered saline (PBS, pH 7.4). On the other hand, phosTMR·OEt (23) has a slight bathochromic shift in absorbance and identical emission (551/569 nm) to carboxyTMR. SulfoTMR absorbs at 556 nm and emits at 574 nm. 3-Substituents have little effect on the spectral profiles of tetramethylrhodamines. The small wavelength shifts (∼10 nm) are explained by inductive differences of the pendant ring that arise from these substitutions. Extinction coefficients, Stokes shifts and quantum yields all display little variance in response to 3-substitution owing to the maintained orthogonality of the xanthene chromophore and pendant rings (Table 3).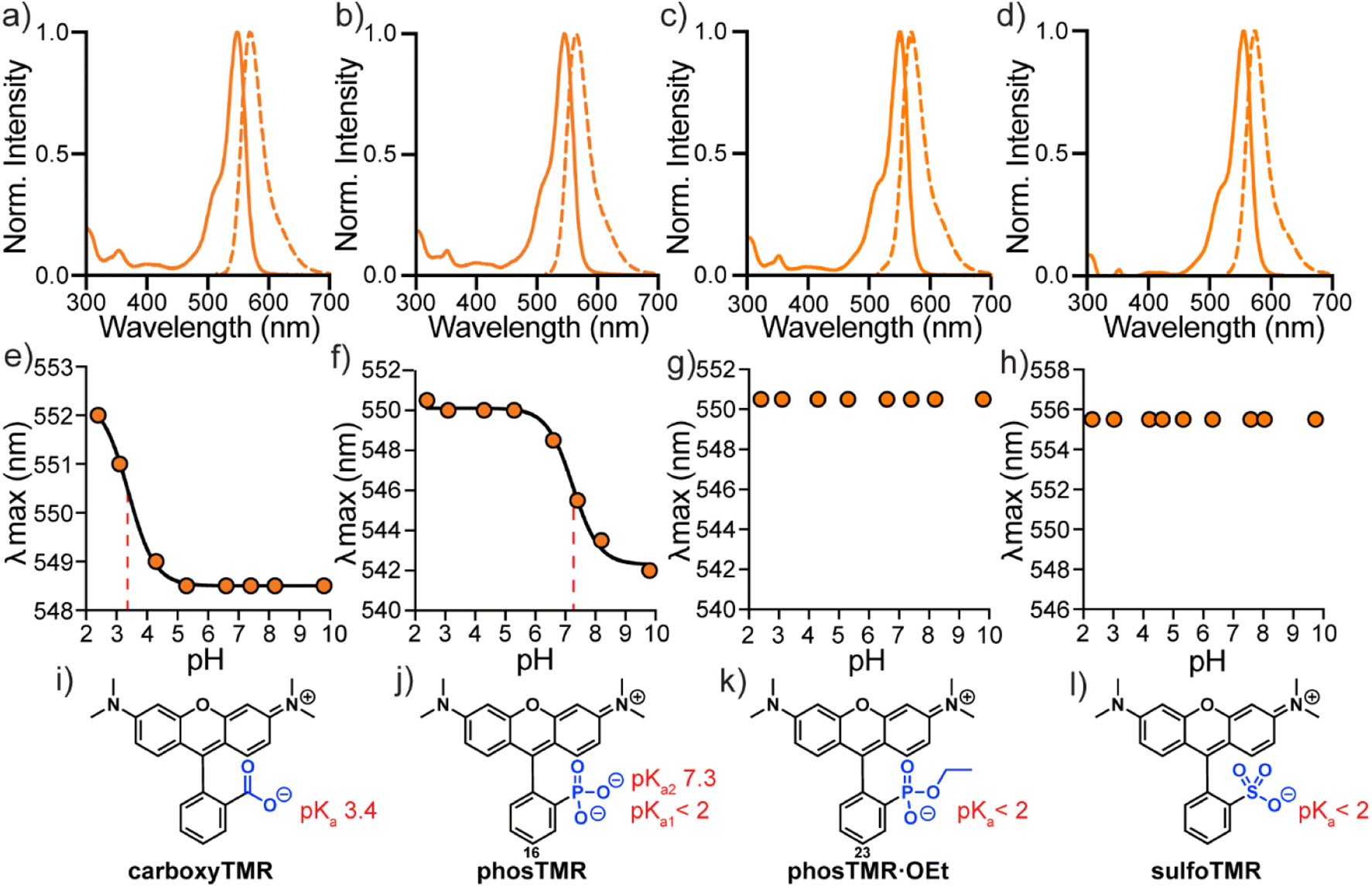 | ||
| Fig. 1 Spectroscopic characterization of tetramethylrhodamines. Normalized absorbance and fluorescence spectra (a–d) in PBS, plots of λmaxvs. pH (e–h) and corresponding chemical structures (i–l) for carboxyTMR (a, e and i), phosTMR (b, f and j), phosTMR·OEt (c, g and k) and sulfoTMR (d, h and l). pH titrations were performed in 10 mM buffered solutions (see ESI†) containing 150 mM NaCl ranging from pH 2.3 to 9.8 at a final dye concentration of 2 μM. Where applicable, titration curves were fit to sigmoidal dose response curves (solid black) to enable pKa determination (dashed red). Determined pKa values are reported next to chemical structures (i–l). | ||
| Rhodamine | λ max /nm | λ em /nm | ε , /M−1 cm−1 | Φ | Solubilityc (mM) |
|---|---|---|---|---|---|
| a Measured in PBS. b At maximum absorption. c Measured in PBS, values are mean ± standard deviation. | |||||
| carboxyTMR | 549 | 569 | 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.45 | 0.9 ± 0.02 |
| sulfoTMR | 556 | 574 | 73000 |
0.46 | 0.02 ± 0.002 |
| phosTMR, 16 | 546 | 564 | 70000 |
0.52 | 12 ± 0.6 |
| phosRhoB, 17 | 553 | 570 | 75000 |
0.38 | — |
| phosRho6G, 18 | 520 | 539 | 76000 |
0.95 | — |
| phosTAR, 19 | 540 | 559 | 53000 |
0.68 | — |
| phosJulR, 20 | 573 | 591 | 91000 |
0.99 | — |
| phosRho110, 21 | 495 | 515 | 76000 |
0.98 | — |
| phosTMR·OEt, 23 | 551 | 569 | 78000 |
0.51 | — |
3-phosphonorhodamine 16 has substantially improved water solubility (12 mM, PBS) compared to sulfono- and carboxyTMR (Fig. S1†). 16 is ∼500× more soluble than sulfonoTMR (0.02 mM) and ∼12× more soluble than carboxyTMR (0.9 mM) (Table 3). This is a larger improvement in solubility than phosphonofluorescein, which is ∼2× more soluble than the related carboxyfluorescein.16 While fluorescein solubility correlates well with the 3-functionality pKa, relative solubility of rhodamines appear more nuanced. In the absence of the negatively charged phenolate of fluoresceins, 3-substituents of rhodamines seem to have a greater impact on relative solubility. Improved water solubility will be a boon for applications where removal of residual fluorophore is important to improve contrast, for example, in vitro protein labelling or next generation sequencing.34–36 3-Phosphonorhodamine 16 shows exceptional stability compared to carboxyTMR. After 1 year of storage as a solid powder, under identical conditions, (at room temperature, 15–22 °C, in the dark), carboxyTMR decomposed into a complex mixture of compounds, including a substantial amount of aniline demethylation (Fig. S2†). However, phosTMR showed no signs of degradation (Fig. S3†).
The absorbance intensity of 3-phosphonorhodamines 16, 17, 18, 19, 20, 21, and 23 at their respective λmax values are all insensitive to pH changes between pH 2 and 10 (Fig. S4†). The λmax values of carboxyTMR and phosTMR shift by 3.5 and 8 nm, respectively, as pH decreases (Fig. 1e and f). Protonation of the 3-substituent results in more electron-withdrawing inductive character of the pendant ring – giving a red-shift in absorbance, as predicted by the Dewar–Knot rules.37,38 Fitting the value of λmax to non-linear sigmoidal curves reveals a 3-carboxylate pKa of 3.4 and 3-phosphonate pKa2 of 7.3. Neither phosTMR·OEt nor sulfoTMR show a shift in absorbance wavelength λmax across this pH range, indicating their pKa values are below 2 (Fig. 1g and h).
All new 3-phosphonorhodamines, 16–21, 23 (Table 3), show characteristic rhodamine absorbance and emission spectra (Fig. S4†). As expected, changing the alkylation pattern of the anilines results in spectral tuning of the absorption and emission wavelengths from 495/515 nm (21) to 573/591 nm (20). The absorption intensity of all 3-phosphonorhodamines is insensitive to pH (between 2 and 10) and phosphonate pKa2 values range from 7.2 to 7.6 (Fig. S4a–r†). The photostabilities of 16, carboxyTMR, and sulfoTMR are indistinguishable from one another (Fig. S4v–x†).
Tunable cellular localization of phosTMRs
Biologically labile acetoxy methyl (AM) esters are commonly used to mask anionic functionalities, such as phosphonates, to facilitate cell permeability.39–41 The AM ester masking strategy can deliver 3-phosphonofluoresceins into cells, where hydrolysis by intracellular esterases releases the phosphonic acid, trapping the fluorophore in the cytosol.16 We hypothesized that a similar AM ester masking strategy could be exploited to deliver phosTMRs into cells.Treatment of phosTMR 16 with either ethyl bromide or bromomethyl acetate with the presence of Ag(I) in MeCN yields esterified phosTMRs 24 and 25 in 22% and 25% yield respectively (Scheme 6). Acetoxymethyl esters are prone to hydrolysis by cellular esterases, and upon in vitro incubation with porcine liver esterase (PLE), both carboxyTMR AM and phosTMR AM, 25, show complete hydrolysis to the corresponding carboxyTMR and phosTMR (Fig. S5 and S6†). In the absence of PLE, carboxyTMR AM shows low levels of hydrolysis, while phosTMR AM 25 shows about ∼50% hydrolysis to the phosphonate monoAM ester (Fig. S5 and S6†). Diethyl ester phosTMR·2OEt 24 is resistant to hydrolysis in the presence or absence of PLE (Fig. S7†).
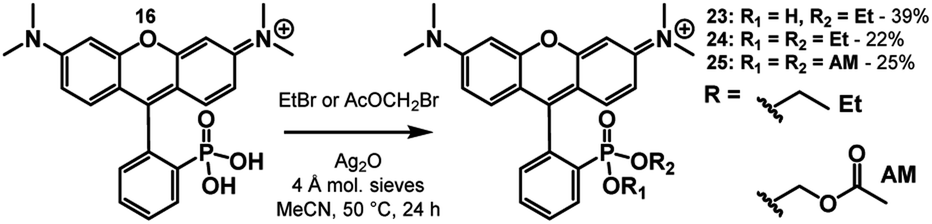 | ||
| Scheme 6 Synthesis of phosTMR diesters 24 and 25. | ||
The identity of the phosphonate ester profoundly affects cellular localization of phosphonorhodamines. Both phosTMR 16 and monoester phosTMR·OEt 23 show negligible cellular fluorescence, since the doubly- and singly-ionized phosphonates preclude entry into cells (Fig. 2a and b). However, diesters 24 (ethyl ester) and 25 (AM ester) exhibit strong cellular fluorescence. phosTMR·2OEt 24 exhibits heterogeneous cellular localization (Fig. 2c), which appears to co-localize to mitochondria (Fig. S8†), as expected for rhodamine esters.15
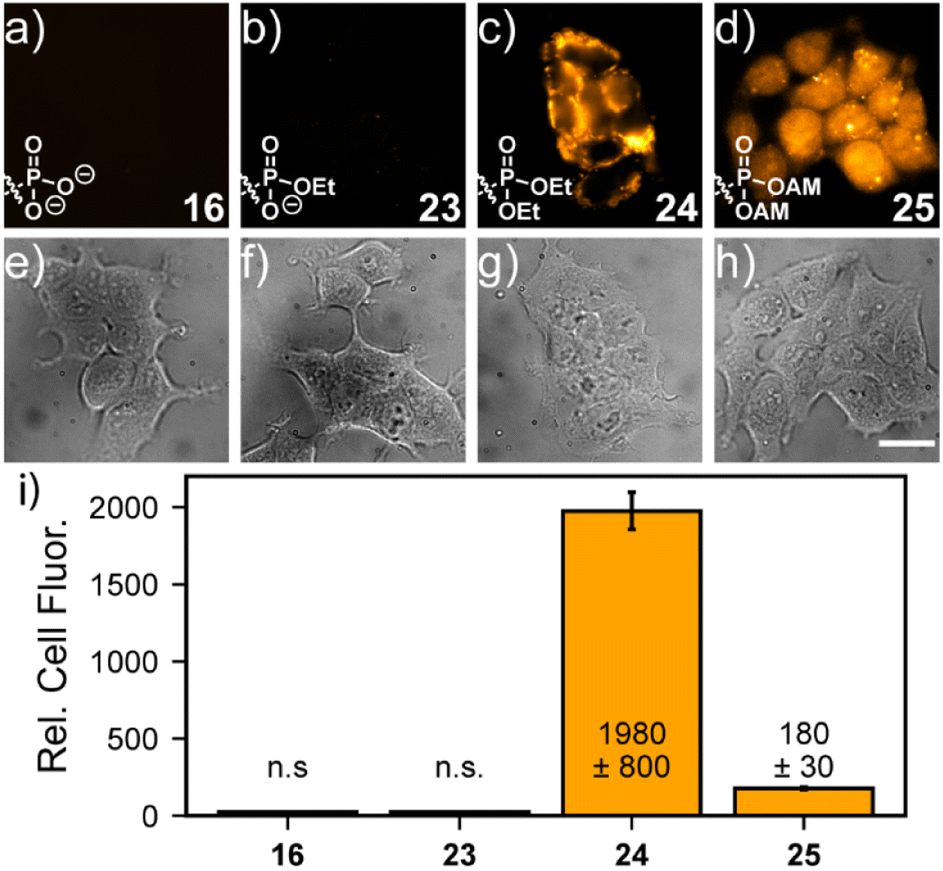 | ||
| Fig. 2 Tunable localization of 3-phosphonoTMRs. Widefield fluorescence (a–d) and transmitted light (e–h) images of HEK293T cells stained with 500 nM dyes 16 (a and e), 23 (b and f), 24 (c and g), or 25 (d and h) in HBSS for 20 min at 37 °C. Quantification (i) of cellular fluorescence. Values are mean fluorescence intensity ± S.E.M. for n = 21, 32, 45, and 19 regions of interest (ROI) for each dye. Each ROI contained between 2 and 20 individual cells. “n.s.” means that the cellular fluorescence values were not above background fluorescence. Coverslips were placed into fresh HBSS prior to imaging. Scale bar is 20 μm. | ||
On the other hand, the more labile phosTMR AM 25 displays more uniform cytosolic localization compared to either phosTMR·2OEt 24 (Fig. 2d and S8e–h†) or carboxyTMR AM (Fig. S9 and S10†). This is different from the behavior of the diethyl ester of phosTMR 24 and different from the behavior of the AM ester of traditional carboxyTMR, which localize to mitochondria (Fig. S9†). The differential cellular localization of 25 may stem from intracellular AM hydrolysis to yield phosTMR with a net negative charge (pKa2 7.3); the greater charge density of phosTMR vs. carboxyTMR (net negative vs. net neutral at pH 7.4) likely precludes localization to the mitochondria.
phosTMR AM 25 also displays excellent cellular retention (Fig. S10a–c, g†). Serial washing of stained cells showed no decrease in the fluorescence intensity of 25, up to 36 min after loading. On the other hand, after 3 washes carboxyTMR AM shows a 40% decrease in mitochondrial fluorescence intensity (Fig. S10d–f, g†), suggesting the additional negative charge provided by a 3-phosphonate enhances cellular retention.
Voltage sensing with phosphonorhodamines
The high water solubility, tunable cellular localization, and persistent anionic charge at physiological pH make phosTMR well-suited for voltage imaging applications, where retention of fluorophores on the extracellular surface of the plasma membrane is a critical feature. Rhodamine Voltage Reporters (RhoVRs) are a class of voltage sensing indicators that exhibit absorption and emission profiles in the green to orange regions of the visible spectrum. While voltage sensitive fluorophores typically rely on 3-sarcosine (RhoVR 1)42 or 3-sulfonate (sRhoVR 1)43 functionalities for membrane anchoring, expansion to a 3-phosphonate (phosRhoVRs) would allow us to take advantage of the scalability of the acid-free condensation chemistry.Reaction of 5-bromo-2-phosphonobenzaldehyde 27 with 3-dimethylaminophenol in TFE, results in precipitation of triarylmethane 28 in quantitative yield (Fig. 3a). Dehydration and oxidation with chloranil gives conversion to the corresponding 6-bromo-3-phosphonotetramethyl rhodamine. This is a large increase the in yield of the condensation to 29 (>99%) relative to the 3-carboxy (57%) and 3-sulfono (44%) analogs.42,43 This reaction can be performed on at least a 1.5 mmol scale to furnish hundreds of milligrams of pure material without column chromatography.
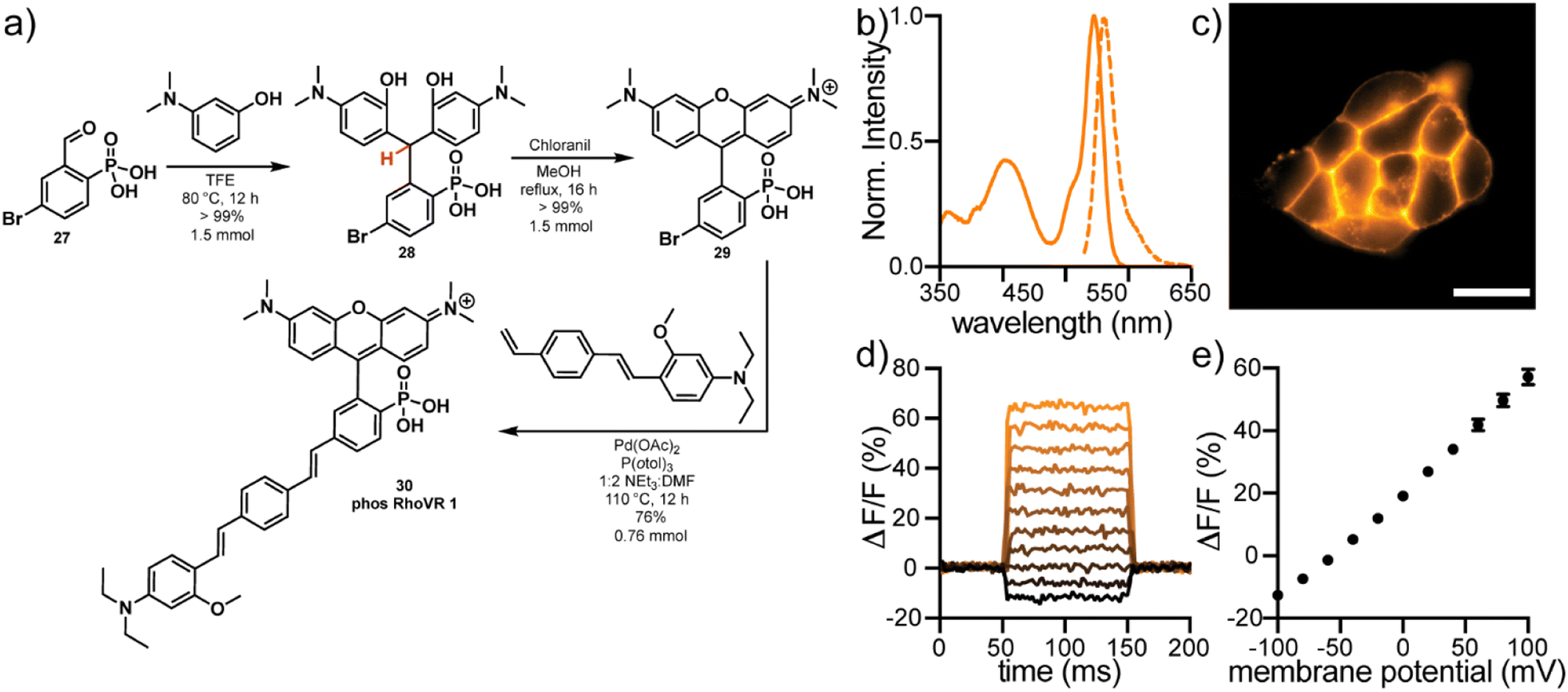 | ||
| Fig. 3 Synthesis and characterization of phosRhoVR 1. (a) Synthesis of phosRhoVR 1, 30 from aldehyde 27. (b) Normalized absorbance (solid line) and fluorescence emission (dashed line) spectra of phosRhoVR 1 in ethanol. (c) HEK cells stained with 500 nM phosRhoVR 1. Scale bar is 20 μm. (d) Plot of the fractional change in fluorescence of phosRhoVR 1 vs. time for 100 ms hyper- and depolarizing steps (±100 mV in 20 mV increments) for single HEK cells under whole-cell voltage-clamp mode. (e) Plot of ΔF/F vs. final membrane potential, revealing a voltage sensitivity of approximately 37% per 100 mV. Error bars are ±SEM for n = 6 cells. If not visible, error bars are smaller than the marker. | ||
Heck coupling of 29 to a phenylenevinylene molecular wire produces phosRhoVR 1, 30, as a crude triethylamine salt which can be purified by reverse phase silica chromatography in 76% yield (0.76 mmol, 432 mg, Fig. 3a). In the comparable synthesis of the 3-amide analog, RhoVR 1, Heck coupling to the analogous 3-carboxytetramethylrhodamine is followed by a HATU coupling with N-Boc-sarcosine and TFA deprotection, resulting in a substantially lower 9% yield over 3 steps.42 The streamlined and higher yielding synthesis of phosRhoVRs not only alleviates the time-consuming bottleneck in voltage reporter synthesis but also enables access to RhoVRs in much greater quantities (hundreds vs. tens of milligrams at a time).
phosRhoVR 1 displays characteristic absorption and emission profiles of 3-phosphono-tetramethylrhodamine, centered at 545 and 562 nm, respectively, with the addition of an absorption band at 402 nm from the molecular wire (Fig. 3b). In live HEK cells, fluorescence is localized primarily to the plasma membranes (Fig. 3c). The negatively charged phosphonate is sufficient to prevent internalization despite the positive charge of the rhodamine. phosRhoVR 1 shows a nearly 2-fold improvement in cellular brightness compared to RhoVR 1, when loaded under identical conditions (500 nM, Fig. S11†). Patch clamp electrophysiology coupled with fluorescence microscopy reveals a voltage sensitivity of 37% ΔF/F per 100 mV for phosRhoVR 1 (Fig. 3d and e), which is comparable but lower than the reported 47% for RhoVR 1 and 44% for sRhoVR 1.42,43 However, given the improved cellular brightness, phosRhoVR 1 provides a nearly 2-fold increase in signal-to-noise for detecting membrane potential changes. PhosRhoVR 1 shows retention on the plasma membrane similar to RhoVR 1 (Fig. S12†). Both RhoVR 1 and phosRhoVR 1 are well-retained on the plasma membrane compared to di-4-ANEPPS, which shows some internalization after 30 minutes (Fig. S12†).
Conclusions
In summary, we report the synthesis of rhodamine fluorophores bearing 3-phosphonates via relatively mild chemistry. This chemistry relies on intramolecular aldehyde activation provided by the phosphonic acid functionality, negating the need for exogenous acid. Phosphonate substitution has little effect on photophysical properties of rhodamines but provides a 12× to 500× increase in water solubility compared to 3-carboxyTMR or 3-sulfonoTMR. PhosphonoTMRs are suitable for live-cell imaging. Intracellular delivery and retention of phosphonorhodamines can be controlled by esterification, and AM esters of phosphonorhodamines show differential localization, increased cell brightness, and improved retention compared to traditional carboxy analogs.Additionally, 3-phosphonate functionality is readily incorporated into voltage sensing scaffolds. Fluorescent voltage reporters with a 3-phosphono group possess excellent voltage sensitivity, 2× improved cellular brightness, and can be synthesized in hundreds of milligram quantities. Future work will expand 3-phosphonate substitution to the entire family of xanthene fluorophores, including rhodols and other 10′ heteroatom substitutions.
Data availability
Data is included in ESI.†Author contributions
JLT: conceptualization, investigation, formal analysis, visualization, original draft preparation, and review & editing, RPG: investigation, formal analysis, and review & editing, BRB: investigation, formal analysis, and review & editing, KMH: investigation, formal analysis, and review, SML: investigation, formal analysis, and review, EWM: conceptualization, funding acquisition, formal analysis, supervision, visualization, original draft preparation, and review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. W. M. acknowledges support from the Camille Dreyfus Teacher-Scholar Fellowship and the National Institute for General Medical Sciences (R35GM119855). B. R. D. was supported, in part, by a training grant from NIGMS (T32GM666098). S. M. L. was supported by a Graduate Research Fellowship from the National Science Foundation (DGE 2146752).Notes and references
- L. D. Lavis and R. T. Raines, Bright building blocks for chemical biology, ACS Chem. Biol., 2014, 9, 855–866 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, Bright ideas for chemical biology, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Synthesis and applications of rhodamine derivatives as fluorescent probes, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- T. Karstens and K. Kobs, Rhodamine B and rhodamine 101 as reference substances for fluorescence quantum yield measurements, J. Phys. Chem., 2002, 84, 1871–1872 CrossRef.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, A general method to improve fluorophores for live-cell and single-molecule microscopy, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, Carbofluoresceins and carborhodamines as scaffolds for high-contrast fluorogenic probes, ACS Chem. Biol., 2013, 8, 1303–1310 CrossRef CAS PubMed.
- J. B. Grimm, T. D. Gruber, G. Ortiz, T. A. Brown and L. D. Lavis, Virginia Orange: A Versatile, Red-Shifted Fluorescein Scaffold for Single- And Dual-Input Fluorogenic Probes, Bioconjugate Chem., 2016, 27, 474–480 CrossRef CAS PubMed.
- M. Fu, Y. Xiao, X. Qian, D. Zhao and Y. Xu, A design concept of long-wavelength fluorescent analogs of rhodamine dyes: replacement of oxygen with silicon atom, Chem. Commun., 2008, 1780–1782 RSC.
- K. Hirabayashi, K. Hanaoka, T. Takayanagi, Y. Toki, T. Egawa, M. Kamiya, T. Komatsu, T. Ueno, T. Terai, K. Yoshida, M. Uchiyama, T. Nagano and Y. Urano, Analysis of Chemical Equilibrium of Silicon-Substituted Fluorescein and Its Application to Develop a Scaffold for Red Fluorescent Probes, Anal. Chem., 2015, 87, 9061–9069 CrossRef CAS PubMed.
- J. B. Grimm, T. A. Brown, A. N. Tkachuk and L. D. Lavis, General Synthetic Method for Si-Fluoresceins and Si-Rhodamines, ACS Cent. Sci., 2017, 3, 975–985 CrossRef CAS PubMed.
- T. Egawa, Y. Koide, K. Hanaoka, T. Komatsu, T. Terai and T. Nagano, Development of a fluorescein analogue, TokyoMagenta, as a novel scaffold for fluorescence probes in red region, Chem. Commun., 2011, 47, 4162–4164 RSC.
- X. Chai, X. Cui, B. Wang, F. Yang, Y. Cai, Q. Wu and T. Wang, Near-Infrared Phosphorus-Substituted Rhodamine with Emission Wavelength above 700 nm for Bioimaging, Chem.–Eur. J., 2015, 21, 16754–16758 CrossRef CAS PubMed.
- X. Zhou, R. Lai, J. R. Beck, H. Li and C. I. Stains, Nebraska Red: a phosphinate-based near-infrared fluorophore scaffold for chemical biology applications, Chem. Commun., 2016, 52, 12290–12293 RSC.
- J. Liu, Y. Q. Sun, H. Zhang, H. Shi, Y. Shi and W. Guo, Sulfone-rhodamines: a new class of near-infrared fluorescent dyes for bioimaging, ACS Appl. Mater. Interfaces, 2016, 8, 22953–22962 CrossRef CAS PubMed.
- L. V. Johnson, M. L. Walsh and L. B. Chen, Localization of mitochondria in living cells with rhodamine 123, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 990–994 CrossRef CAS PubMed.
- J. L. Turnbull, B. R. Benlian, R. P. Golden and E. W. Miller, Phosphonofluoresceins: Synthesis, Spectroscopy, and Applications, J. Am. Chem. Soc., 2021, 143, 6194–6201 CrossRef CAS PubMed.
- M. Ceresole, Verfahren zur Darstellung von Farbstoffen aus der Gruppe des Meta-amidophenol-Phtaleïns, Ger. Pat., 44002, 1887 Search PubMed.
- T. Sandmeyer, Red dye, US Pat., 573299, 1896 Search PubMed.
- M. Fu, X. Zhang, J. Wang, H. Chen and Y. Gao, Progress of Synthesis and Separation of Regioisomerically Pure 5(6)-Substituted Rhodamine, Curr. Org. Chem., 2016, 20, 1584–1590 CrossRef CAS.
- G. Mudd, I. P. Pi, N. Fethers, P. G. Dodd, O. R. Barbeau and M. Auer, A general synthetic route to isomerically pure functionalized rhodamine dyes, Methods Appl. Fluoresc., 2015, 3, 045002 CrossRef PubMed.
- P. E. Deal, R. U. Kulkarni, S. H. Al-Abdullatif and E. W. Miller, Isomerically pure tetramethylrhodamine voltage reporters, J. Am. Chem. Soc., 2016, 138, 9085–9088 CrossRef CAS PubMed.
- S. J. Dwight and S. Levin, Scalable Regioselective Synthesis of Rhodamine Dyes, Org. Lett., 2016, 18, 5316–5319 CrossRef CAS PubMed.
- J. B. Grimm and L. D. Lavis, Synthesis of Rhodamines from Fluoresceins Using Pd-Catalyzed C–N Cross-Coupling, Org. Lett., 2011, 13, 6354–6357 CrossRef CAS PubMed.
- L. Wu and K. Burgess, Synthesis and Spectroscopic Properties of Rosamines with Cyclic Amine Substituents, J. Org. Chem., 2008, 73, 8711–8718 CrossRef CAS PubMed.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z.-G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed.
- C. Fischer and C. Sparr, Direct Transformation of Esters into Heterocyclic Fluorophores, Angew. Chem., Int. Ed., 2018, 57, 2436–2440 CrossRef CAS PubMed.
- J. Kagan, The Structure of Phthalaldehydic Acid, J. Org. Chem., 1967, 32, 4060–4062 CrossRef CAS.
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, Unveiling the ‘booster effect’ of fluorinated alcohol solvents: aggregation-induced conformational changes and cooperatively enhanced H-bonding, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed.
- J. P. Bégué, D. Bonnet-Delpon and B. Crousse, Fluorinated Alcohols: A New Medium for Selective and Clean Reaction, Synlett, 2004, 2004, 18–29 Search PubMed.
- S. Minegishi, S. Kobayashi and H. Mayr, Solvent Nucleophilicity, J. Am. Chem. Soc., 2004, 126, 5174–5181 CrossRef CAS PubMed.
- R. D. Franz, Comparisons of pKa and logP values of some carboxylic and phosphonic acids: synthesis and measurement, AAPS PharmSci, 2001, 3, E10 CrossRef CAS PubMed.
- T. Iwata, R. Kawano, T. Fukami and M. Shindo, Retro-Friedel-Crafts-Type Acidic Ring-Opening of Triptycenes: A New Synthetic Approach to Acenes, Chem.–Eur. J., 2022, 28, e202104160 CrossRef CAS PubMed.
- E. J. Goethals, E. H. Schacht, Y. E. Bogaert, S. I. Ali and Y. Tezuka, The polymerization of azetidines and azetidine derivatives, Polym. J., 1980, 12, 571–581 CrossRef CAS.
- R. Bandichhor, A. D. Petrescu, A. Vespa, A. B. Kier, F. Schroeder and K. Burgess, Synthesis of a new water-soluble rhodamine derivative and application to protein labeling and intracellular imaging, Bioconjugate Chem., 2006, 17, 1219–1225 CrossRef CAS PubMed.
- J. B. Grimm and L. D. Lavis, Caveat fluorophore: an insiders' guide to small-molecule fluorescent labels, Nat. Methods, 2022, 19, 149–158 CrossRef CAS PubMed.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao, W. Y. Leung and R. P. Haugland, Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates, J. Histochem. Cytochem., 1999, 47, 1179–1188 CrossRef CAS PubMed.
- M. J. S. Dewar, 478. Colour and constitution. Part I. Basic dyes, J. Chem. Soc., 1950, 2329–2334 RSC.
- E. B. Knott, 227. The colour of organic compounds. Part I. A general colour rule, J. Chem. Soc., 1951, 1024–1028 RSC.
- R. Y. Tsien, A non-disruptive technique for loading calcium buffers and indicators into cells, Nature, 1981, 290, 527–528 CrossRef CAS PubMed.
- L. D. Lavis, T.-Y. Chao and R. T. Raines, Synthesis and utility of fluorogenic acetoxymethyl ethers, Chem. Sci., 2011, 2, 521–530 RSC.
- C. Schultz, M. Vajanaphanich, A. T. Harootunian, P. J. Sammak, K. E. Barrett and R. Y. Tsien, Acetoxymethyl esters of phosphates, enhancement of the permeability and potency of cAMP, J. Biol. Chem., 1993, 268, 6316–6322 CrossRef CAS PubMed.
- P. E. Deal, R. U. Kulkarni, S. H. Al-Abdullatif and E. W. Miller, Isomerically Pure Tetramethylrhodamine Voltage Reporters, J. Am. Chem. Soc., 2016, 138, 29 CrossRef PubMed.
- R. U. Kulkarni, M. Vandenberghe, M. Thunemann, F. James, O. A. Andreassen, S. Djurovic, A. Devor and E. W. Miller, In Vivo Two-Photon Voltage Imaging with Sulfonated Rhodamine Dyes, ACS Cent. Sci., 2018, 4, 1371–1378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02590j |
| This journal is © The Royal Society of Chemistry 2023 |