
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplying green chemistry principles to iron catalysis: mild and selective domino synthesis of pyrroles from nitroarenes†
Johannes
Fessler
 ,
Kathrin
Junge
* and
Matthias
Beller
*
,
Kathrin
Junge
* and
Matthias
Beller
*
Leibniz-Institut für Katalyse e.V. (LIKAT), Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Kathrin.Junge@catalysis.de; Matthias.Beller@catalysis.de
First published on 9th August 2023
Abstract
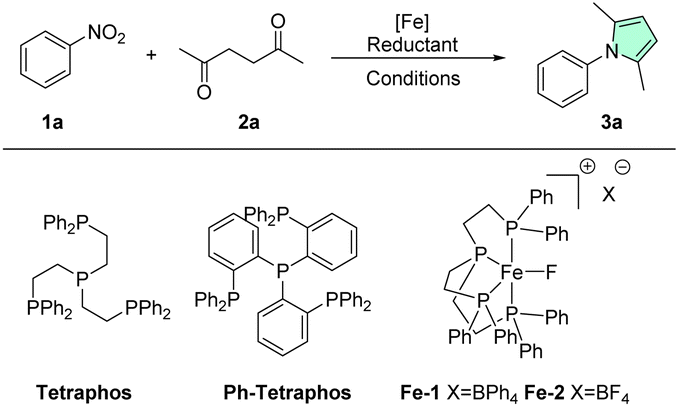
An efficient and general cascade synthesis of pyrroles from nitroarenes using an acid-tolerant homogeneous iron catalyst is presented. Initial (transfer) hydrogenation using the commercially available iron–Tetraphos catalyst is followed by acid catalysed Paal–Knorr condensation. Both formic acid and molecular hydrogen can be used as green reductants in this process. Particularly, under transfer hydrogenation conditions, the homogeneous catalyst shows remarkable reactivity at low temperatures, high functional group tolerance and excellent chemoselectivity transforming a wide variety of substrates. Compared to classical heterogeneous catalysts, this system presents complementing reactivity, showing none of the typical side reactions such as dehalogenation, debenzylation, arene or olefin hydrogenation. It thereby enhances the chemical toolbox in terms of orthogonal reactivity. The methodology was successfully applied to the late-stage modification of multi-functional drug(-like) molecules as well as to the one-pot synthesis of the bioactive agent BM-635.
Introduction
25 years ago Paul Anastas and John Warner formulated the so-called green chemistry principles, in order to lead chemical development and production into a more sustainable future.1 One of these 12 principles, catalysis, can make processes less energy intensive, faster, more selective and reduce the amount of waste compared to e.g. stoichiometric reagents. Within the realm of transition metal catalysis, traditionally emphasis was placed on precious metal catalysts that are in general expensive, toxic, and only present in the Earth's crust in parts per billion (ppb). Iron, however, is the 4th most abundant element in the Earth's crust and the most abundant transition metal.2 Thus, many iron-based compounds are economical, widely available and generally accepted to have lower toxicity compared to other transition metals.3 While heterogeneous iron catalysis has been around for over a century, giving us for example the Haber–Bosch process for the indispensable mass-production of ammonia,4,5 homogeneous iron catalysis has been largely neglected by synthetic chemists and only been on the rise for around the last two decades.6–9 A field that has flourished incredibly over the last years is the application of iron and its neighboring 3d-metals as catalysts for (transfer) hydrogenations.10–22 Combining these sustainable reductive transformations with another “green” topic of increasing interest, cascade reactions (also called tandem or domino reactions), can be highly favorable since it reduces synthetic steps, which in turn can lead to further savings in time, labor, resources and generated waste.23–27 As an example, earth-abundant catalysis has been applied to significantly improve the synthesis of N-heterocycles, which are of importance for many life science applications.28–40Here, pyrroles in particular are interesting targets due to their ubiquitous presence in pharmaceuticals (atorvastatin), agrochemicals (fludioxonil), natural products (porphyrin), dyes (bodipy), materials (polypyrrole) and other fine chemicals.41–44 Traditionally, pyrroles are prepared though the well-established Hantzsch, Knorr, or Paal–Knorr condensation reactions. Especially, the latter two approaches are still of significant interest to sustainable/process chemists today, due to their great atom-economy, high yields, easily available starting materials and benign side products (water).45–48 Therefore, N-aryl pyrroles are commonly made via Paal–Knorr reaction of 1,4-dicarbonyl compounds with anilines, which in turn are typically obtained from the reduction of nitroarenes (Scheme 1). Since nitroarenes are widely available, it is appealing to devise a direct cascade process from nitroarenes to N-aryl pyrroles. While there is some precedent for reductive cascade reactions of nitro-compounds for the synthesis of heterocycles, they mostly rely on precious metals such as Rh, Ru, Pt and Pd.49–52 Recently, a handful of groups have developed nitro reduction cascades using earth-abundant metals such as cobalt and iron.53–55 In 2017, Zhang et al. showed for the first time the cascade synthesis of pyrroles from nitroarenes using a heterogeneous cobalt catalyst and formic acid (FA) as a green reductant.56 This methodology was extended by our group in 2020 to include molecular hydrogen and CO/H2O mixtures as possible reductants.57 Highlighting the current scientific interest, very recently the first heterogeneous iron catalyst for this transformation, using reductive base/formic acid mixtures, was reported by Lin et al.58 All the above-mentioned methodologies have in common that they apply heterogeneous catalysts and more importantly they require relatively harsh reaction conditions (100 to 120 °C).
 | ||
| Scheme 1 State of the art in catalytic pyrrole synthesis from nitroarenes using earth-abundant (transfer) hydrogenation catalysts. | ||
Results and discussion
Inspired by these earlier reports, we wondered if a more active homogeneous iron system could be developed that works under milder conditions and without additives. Furthermore, it should be stable in the presence of water and acidic conditions, which is non-trivial for homogeneous 3d metal catalysts, that tend to be more sensitive than their heavier 4/5d analogs.6–9 With our previous expertise in iron catalysis, our first candidate of choice was the robust Fe–Tetraphos system.59–66 Among other interesting transformations such as carbon dioxide hydrogenation or anti-Markovnikov epoxide hydrogenation, this catalytic system is known for the selective reduction of nitro groups, which represents the first step of our cascade, using both hydrogen and formic acid as reductants.67–70 Considering that formic acid is also a potentially suitable acid catalyst for the second step of the cascade, the Paal–Knorr reaction, its dual role could be particularly desirable. With this idea in mind, we started our investigations, selecting nitrobenzene and hexane-2,5-dione as our benchmark substrates. To our delight, the standard conditions for transfer hydrogenation of nitro arenes (40 °C in EtOH for 2 h) delivered full conversion of nitrobenzene and nearly quantitative yield of the desired phenyl dimethyl pyrrole 3a (Table 1, entry 1). Compared to the previous methods by the groups of Lin and Beller no additional base was necessary and an excellent chemoselectivity (i.e. no reduction of the 1,4-diketone) was observed.57,58 Further optimization (for highlights see Table 1, full screening see ESI†) revealed that the reaction could also be conducted efficiently at room temperature when slightly extending the reaction time to 5 h (entry 5). In comparison to the previously reported heterogeneous systems, comparable or even improved yields are obtained at a quarter of the temperature and less than half the reaction time, clearly highlighting the potential of this novel catalytic process. Control reactions showed that without ligand, metal, or FA no reactivity was observed (entries 2–4). Aside from their effect on reactivity, solvents also have a large effect on the sustainability of a process, accounting for the majority of generated waste and around 60% of the energy-usage of chemical production processes.71–76 Accordingly, different solvents were evaluated: alcohols generally showing the most promise, while ethers and carbonates gave decreased yields (entries 9–12). Although water was well tolerated in the reactions, indicated by the good reactivity of the iron tetrafluoroborate hexahydrate and the general reactivity of the reaction (generating 4 equiv. H2O), when water was used as the sole solvent no reactivity was observed (entry 13), possibly due to solubility issues.|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Ligand | Solvent | Reductant | Yieldb |
| a General reaction conditions: 0.5 mmol 1a, 0.6 mmol 2a, 0.025 mmol Fe-source, 0.026 mmol ligand, 2.25 mmol FA, 2 mL solvent, 25 °C, 5 h. b Determined by GC-FID using n-hexadecane as internal standard. c 40 °C, 2 h. d 0.01 mmol Fe-source, 0.01 mmol ligand, 20 μL TFA, 20 bar, 120 °C, 2 h. e 0.01 mmol Fe-source, 0.01 mmol ligand, 20 μL TFA, 20 bar, 100 °C, 6 h. | |||||
| 1c | Fe(BF4)2·6H2O | Tetraphos | EtOH | HCO2H | 99% |
| 2 | Fe(BF4)2·6H2O | — | EtOH | HCO2H | 0% |
| 3 | — | Tetraphos | EtOH | HCO2H | 0% |
| 4 | Fe(BF4)2·6H2O | Tetraphos | EtOH | — | 0% |
| 5 | Fe(BF4)2·6H2O | Tetraphos | EtOH | HCO2H | 97% |
| 6 | Fe(OTf)2 | Tetraphos | EtOH | HCO2H | 99% |
| 7 | Fe-1, [Fe(Tetraphos)F][BPh4] | EtOH | HCO2H | 24% | |
| 8 | Fe-2, [Fe(Tetraphos)F][BF4] | EtOH | HCO2H | 87% | |
| 9 | Fe(BF4)2·6H2O | Tetraphos | iPrOH | HCO2H | 89% |
| 10 | Fe(BF4)2·6H2O | Tetraphos | THF | HCO2H | 40% |
| 11 | Fe(BF4)2·6H2O | Tetraphos | Dioxane | HCO2H | 25% |
| 12 | Fe(BF4)2·6H2O | Tetraphos | DMC | HCO2H | 47% |
| 13 | Fe(BF4)2·6H2O | Tetraphos | H2O | HCO2H | 0% |
| 14d | Fe(BF4)2·6H2O | Tetraphos | THF | H2 | 16% |
| 15d | Fe(BF4)2·6H2O | Ph–Tetraphos | THF | H2 | 96% |
| 16e | Fe(BF4)2·6H2O | Ph–Tetraphos | THF | H2 | 99% |
In the end, it was decided to proceed with ethanol as the solvent, since it gave the best performance and is considered a relatively green solvent, being biodegradable, low-toxic, and mass-produced from biorenewables.71–78 Alternative iron sources and molecularly defined catalysts were also tested (entries 6–8). While iron triflate gave equally excellent yields, the commercial Fe-1, containing the tetraphenylborate anion, showed significantly reduced yield. On the other hand, the air-stable tetrafluoroborate analog Fe-2 gave very good yield. However, since the in situ prepared catalyst performed the best, it was used for further reactions. Lastly, also hydrogen gas was successfully tested as a reductant for this benchmark reaction (entries 14–16). It is worth noting that to the best of our knowledge, this is the first example of an iron-based catalyst (homogeneous or heterogeneous) directly using molecular hydrogen for this cascade transformation. Substantially increased hydrogenation reactivity was observed when switching from the alkyl bridged Tetraphos ligand used for transfer hydrogenation to the more rigid and less donating phenyl-bridged variant (Ph–Tetraphos). However, more forcing reaction conditions needed to be applied (20 bar, 100–120 °C, trifluoroacetic acid (TFA) additive) as well as a more complex autoclave setup. When applying these harsh hydrogenation conditions in the upcoming substrate scope, the majority of tested examples showed decreased yields. Accordingly, we continued our investigations under the milder transfer hydrogenation conditions.
To test the general applicability of our methodology and the likely improved functional group tolerance, a substrate scope was prepared using our optimized protocol (Schemes 2 and 3). Since pyrroles are commonplace in pharmaceuticals and agrochemicals which are often complex, multifunctionalized molecules, particular focus was placed on a diversity of functional groups prevalent in bioactive molecules. Starting with the unfunctionalized benchmark substrates, phenylpyrrole 3a was isolated in excellent yield of 91%. Next, to assess the influence of steric factors on the reaction, 2-nitrocumene with a bulky ortho isopropyl substituent was tested providing good yields of the corresponding pyrrole 3b. Nitroarenes bearing the common halide substituents (fluorine 1c, chlorine 1d, bromine 1e, iodine 1f) were also converted without issues. Reductive dehalogenation, which is a side reaction frequently occurring with precious metal catalysts was never observed in our methodology.79 Similarly, the trifluoromethyl variant 1g gave 3g in good yield, illustrating nicely that electron poor nitroarenes are well suited substrates. Further, remarkable chemoselectivity was observed, when converting 4-nitrostyrene 1h, showing exclusively nitro reduction and leaving the alkene moiety intact. Additionally, it should be highlighted that polymerization of the starting material 1h or the styrene product 3h was also not encountered using such mild conditions. Nitroarenes bearing electron donating groups such as ethers (1j), thioethers (1i) and hydroxy groups (1k) were also smoothly converted to the respective pyrroles. Often sulfur containing compounds act as catalyst poisons for transition metal catalysts.80–82 For instance, 4-nitrothioanisole 1i specifically was shown to poison Pd/C in hydrogenations.83 However, when 1i was tested under our standard reaction conditions, it provided near quantitative isolated yield (96%) of the corresponding pyrrole 3i. 4-Nitrophenol 1k on the other hand is quite acidic (pKa(H2O) = 7.15), and therefore can become a problem in hydrogenations conducted under neutral or basic conditions, but again the desired product was obtained smoothly.84 Boronic acids and pinacol boronic esters are frequently used building blocks in the fine chemical industry. Noteworthily, boronic acid 1m and its pinacol boronate 1l gave the corresponding pyrroles in decent yields. Protodeboronation, a common side-reaction of organoboron compounds, was not observed in either case.85,86 Nitroarenes containing carbonyl groups such as ketone 1n, ester 1o, and amide 1p could also be converted chemoselectively. Primary amides are widely abundant not only in amino acids, vitamins, cofactors etc. but also in pharmaceuticals. However, their two protic hydrogens and coordination ability can pose a challenge for transition-metal catalyzed transformations and thus they are oftentimes underrepresented in the substrate scope evaluation of a given synthetic methodology. Remarkably, primary amide 1p was well tolerated by our catalytic system. Additionally, it should be noted that no pyrrole formation on the amide nitrogen was detected.
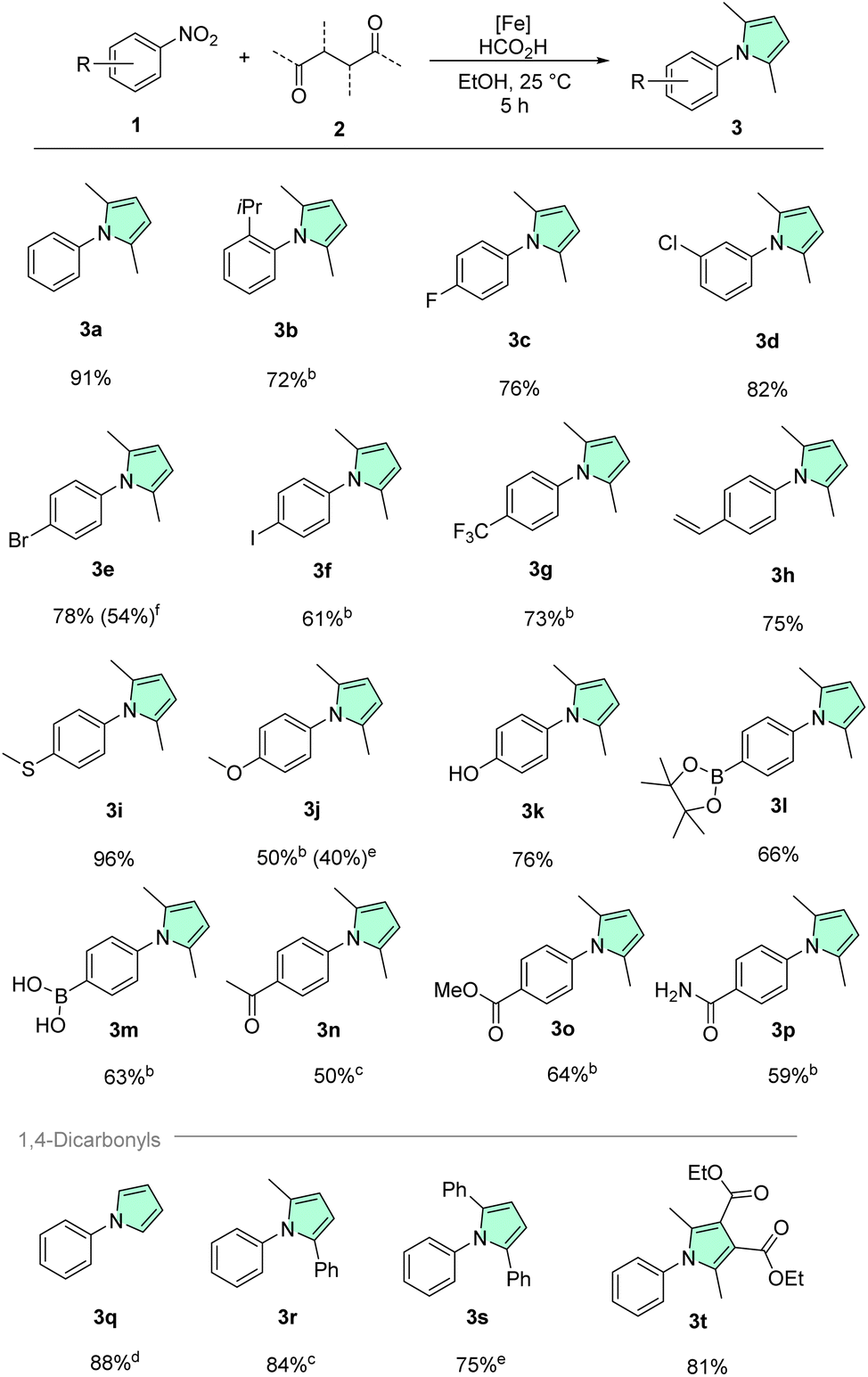 | ||
| Scheme 2 Scope of nitroarenes and examples of 1,4-dicarbonyl compounds.a aGeneral reaction conditions: 0.5 mmol 1, 0.6 mmol 2, 0.025 mmol Fe(BF4)2·6H2O, 0.025 mmol Tetraphos, 2.25 mmol FA, 2 mL EtOH, 25 °C, 5 h. Isolated yields are shown. b24 h. c40 °C, 24 h. d24 h, from 2,5-dimethoxytetrahydrofuran, after 5 h addition of 1.5 mL aq. 50% FA. e0.01 mmol Fe(BF4)2·6H2O, 0.01 mmol Ph–Tetraphos, 20 μL TFA, 1.5 mL THF, 20 bar H2, 120 °C, 20 h. f0.015 mmol Fe(BF4)2·6H2O, 0.015 mmol Ph–Tetraphos, 20 μL TFA, 1.5 mL THF, 20 bar H2, 120 °C, 20 h. | ||
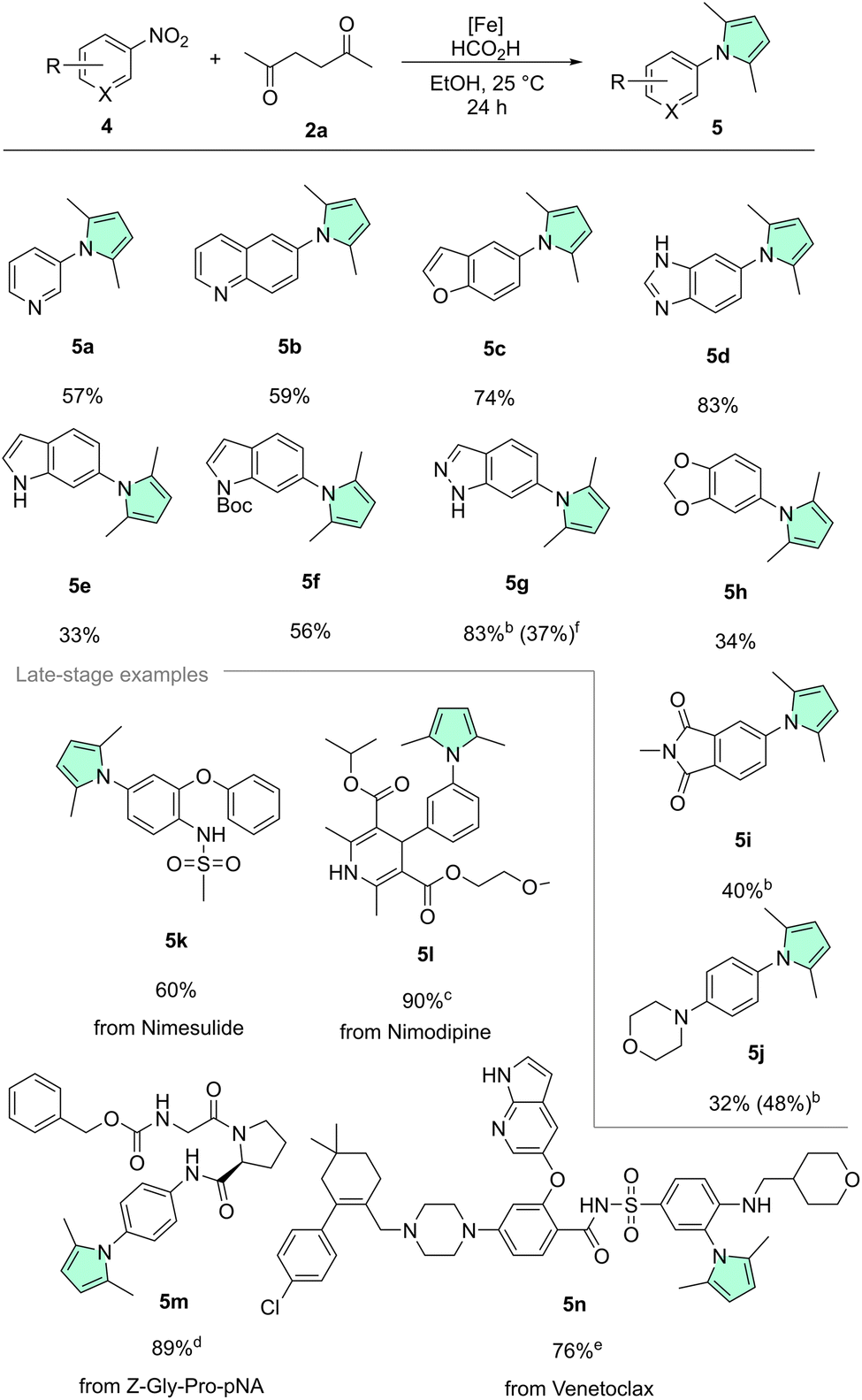 | ||
Scheme 3 Scope of heterocyclic nitro compounds and late-stage modifications.a aGeneral reaction conditions: 0.5 mmol 4, 0.6 mmol 2a, 0.025 mmol Fe(BF4)2·6H2O, 0.026 mmol Tetraphos, 2.25 mmol FA, 2 mL EtOH, 25 °C, 24 h. Isolated yields are shown. b40 °C. c0.25 mmol scale, 5 h. d0.25 mmol scale. e0.25 mmol scale, solvent 2 mL (EtOH/toluene, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). f0.01 mmol Fe(BF4)2·6H2O, 0.01 mmol Ph–Tetraphos, 20 μL TFA, 1.5 mL THF, 20 bar H2, 120 °C, 20 h. :1). f0.01 mmol Fe(BF4)2·6H2O, 0.01 mmol Ph–Tetraphos, 20 μL TFA, 1.5 mL THF, 20 bar H2, 120 °C, 20 h. | ||
Most of the previous reports related to the transformation discussed herein showed little variation in 1,4-dicarbonyl starting materials, we therefore wanted to demonstrate that our protocol also allows to convert different and more challenging diketones efficiently.56–58 Starting from 2,5-dimethoxy tetrahydrofuran, unsubstituted phenyl pyrrole 3q was isolated in 88% yield. Phenyl methyl pyrrole 3r was obtained in very good yield from the corresponding diketone. Even the challenging dibenzoylethane 2s could be converted with 1a to form 1,2,5-triphenyl pyrrole 3s in good yield when using our hydrogenation conditions. Notably, other reported catalysts only showed minimal product formation using this bulky diketone.57 Finally, also a persubstituted pyrrole 3t could be synthesized under mild conditions starting from 2,3,4,5-tetrasubstituted diketone 2t.
Since further heterocycles are often prevalent in bioactive chemicals alongside pyrroles, next we extended the scope to several interesting nitro-containing heterocyclic substrates (Scheme 3, top). Basic nitrogen atoms that could potentially coordinate and thereby poison a catalyst were tolerated very well, as illustrated by 3-nitropyridine 4a, 6-nitroquinoline 4b, and 5-nitrobenzimidazole 4d. Benzofuran derivative 5c was also isolated in a good yield (74%). Surprisingly, poor conversion and yield were obtained with unprotected nitro indole 4e. We believe that the highly electron-rich character of the indole system renders the nitro group of 4e less electrophilic, thus slowing down the reduction. Similar trends were observed for the electron-rich benzodioxole 4h and the morpholino derivative 4j. To overcome this limitation, Boc-protection of the indole nitrogen, to remove some electron density from the heteroarene core, was tried. Indeed, this approach led to a significantly improved yield of the N-protected product 5f. Boc-deprotection was only a minor pathway due to the mild conditions. Another way to tackle the reduced reactivity, in the case of 4j, was simply to slightly increase the reaction temperature to 40 °C, which was associated with a noticeable improvement of both conversion and product yield. Corroborating our theory of electron density being the culprit, related but less electron-rich indazole 4g provided very good yield. Finally, imide 4i was also transformed chemoselectively. Once again, 4i showed no signs of overreduction.
In addition to examining functional group tolerance individually, it was also important for us to test several multi-functionalized, drug(-like) substrates (Scheme 3, bottom). COX-2 inhibitor Nimesulide, notable for its acidic sulfonamide (pKa(MeOH) = 6.46) moiety, was transformed to 5k without problems.87 The calcium channel blocker Nimodipine gave excellent yield, when converted to 5l with our protocol. Remarkably, the prolyl endopeptidase probe Z-Gly-Pro-pNA was also converted in high yield, giving pyrrole-substituted dipeptide 5m. Interestingly, the Cbz-group which would traditionally be cleaved via hydrogenolysis using heterogeneous catalysts such as Pd/C was left intact.88–92 Outstandingly, our methodology was even applicable to the highly-functionalized Bcl-2 inhibitor Venetoclax 4n, giving 76% yield of the corresponding pyrrole product 5n, illustrating once more the striking functional group tolerance of our system.
Aside from derivatizing existing drug molecules, it is intriguing to showcase the advantages of this novel methodology for the synthesis of existing bioactive agents. Therefore, we selected BM-635, a pyrrole-based antitubercular agent, as a synthetic target (Scheme 4).93,94 This compound is typically synthesized through a stepwise process centered on the Paal–Knorr reaction of 4-fluoroaniline 6 and 1,4-diketone 2c.956 is itself traditionally synthesized by reduction of 4-fluoronitrobenzene 1c.96–98 Finally, the resulting Paal–Knorr pyrrole 7 is treated with formaldehyde, morpholine (8) and acetic acid following a Mannich protocol to give BM-635 over 3 steps.95 Initially, we tested our cascade methodology as a replacement for the first two steps (see ESI†). The 1,2,5-substituted pyrrole 7 was isolated with a good yield (73%), and could be further converted to BM-635 according to the literature method.93 However, since the final Mannich reaction is run under acidic conditions similar to our methodology, we hypothesized that the cascade reaction could potentially be expanded to include the third step of the sequence. Accordingly, we performed a one-pot, three-step reaction, where formaldehyde and morpholine were added to the reaction mixture after the complete conversion of 1c. Using this novel multicomponent reaction approach, the practical one-pot synthesis of BM-635 was achieved in 32% overall yield. This corresponds to a good average yield per step of 68%. The one-pot synthesis was also successfully scaled-up to gram-scale producing 1.27 g of the active ingredient in a single batch. We believe this reduction/Paal–Knorr/Mannich cascade conveniently illustrates the synthetic capabilities of our new method, quickly building up molecular complexity in a one-pot fashion. Furthermore, we consider this transformation a useful tool for combinatorial chemistry and drug discovery to potentially generate novel compound libraries from broadly available building blocks.99,100
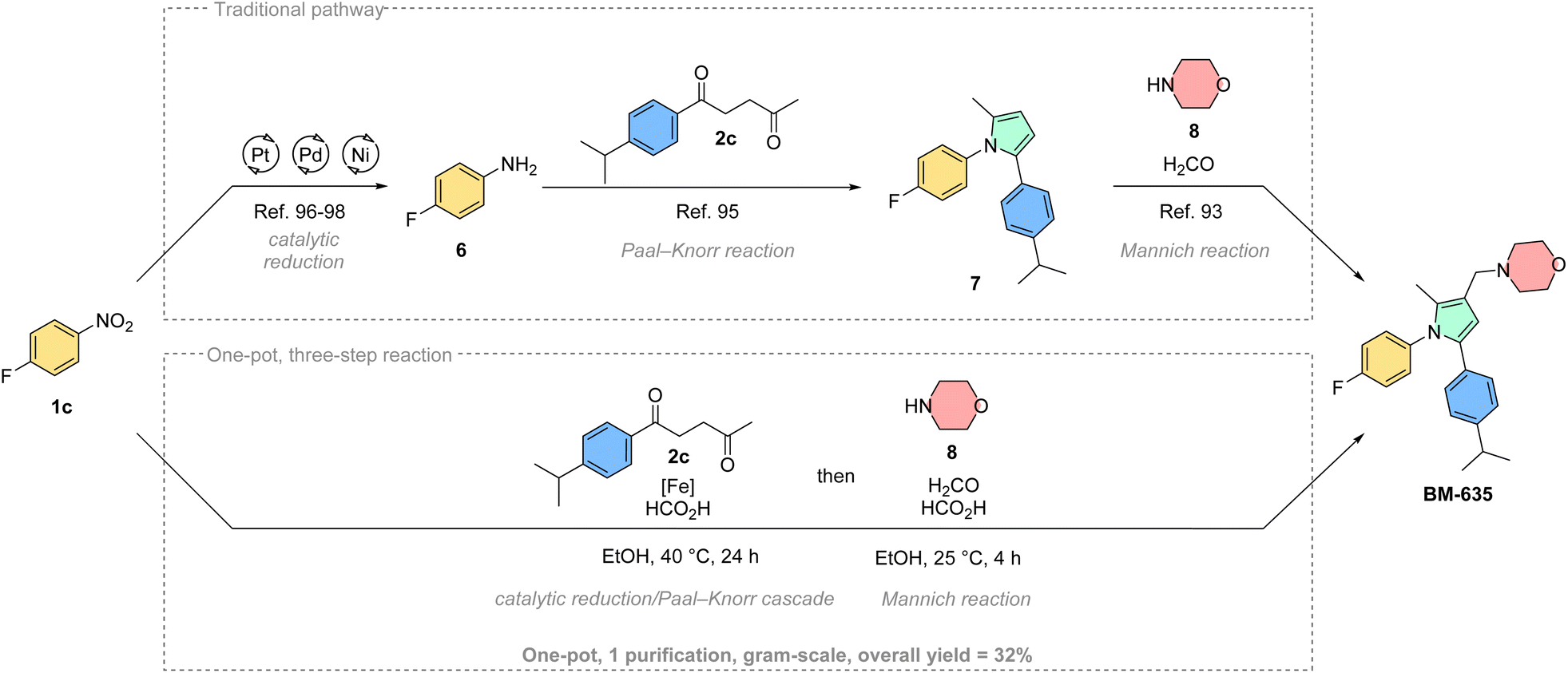 | ||
| Scheme 4 Synthetic application of the Fe-catalysed domino pyrrole synthesis: traditional route to BM-635 starting from 1c (top), scaled-up one-pot, three-step synthesis of BM-635 (bottom).93–98 | ||
Conclusions
To summarize, we have developed a cascade synthesis of pyrroles from nitroarenes for the first time using a commercially available homogeneous iron catalyst. This catalyst can be easily prepared in situ or used as the molecularly-defined complex. Both formic acid and molecular hydrogen can be used as green reductants in the (transfer) hydrogenation/Paal–Knorr reaction sequence. Particularly, under transfer hydrogenation conditions, the homogeneous catalyst showed remarkable reactivity at low temperatures, exceptional functional group tolerance and excellent chemoselectivity using a wide variety of substrates. This system offers a complementing reactivity to classical heterogeneous catalysts, exhibiting none of the typical side reactions such as dehalogenation, debenzylation, ketone, arene or olefin hydrogenation. The methodology thereby enhances the chemical toolbox in terms of orthogonal reactivity. Finally, the advantages of this novel protocol are highlighted by the successful late-stage modification of multi-functionalized drug(-like) molecules as well as the one-pot synthesis of the bioactive agent BM-635 on gram-scale. Further investigations concerning a more efficient use of hydrogen gas for this cascade protocol are currently ongoing in our laboratories.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
J. F. conceptualized the work, performed the investigations and the writing – original draft. K. J. and M. B. provided resources and supervision. All authors contributed to the final version of the manuscript (writing – review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the analytical and technical staff of the Leibniz Institute for Catalysis (LIKAT) for their service. Furthermore, the authors are grateful to Dr Pavel Ryabchuk (now Janssen Pharmaceuticals) for valuable discussions and to Dr Florian Bourriquen (now dsm-firmenich) for assistance during the preparation of the manuscript. Lastly, the State of Mecklenburg–Western Pomerania is acknowledged for generous financial support.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- P. A. Frey and G. H. Reed, ACS Chem. Biol., 2012, 7, 1477–1481 CrossRef CAS PubMed.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- BASF AG, Ger. Pat., DE235421C, 1908 Search PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- A. Guðmundsson and J.-E. Bäckvall, Molecules, 2020, 25, 1349 CrossRef PubMed.
- S. Gaillard and J.-L. Renaud, ChemSusChem, 2008, 1, 505–509 CrossRef CAS PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- G. Bauer and K. A. Kirchner, Angew. Chem., Int. Ed., 2011, 50, 5798–5800 CrossRef CAS PubMed.
- A. Quintard and J. Rodriguez, Angew. Chem., Int. Ed., 2014, 53, 4044–4055 CrossRef CAS PubMed.
- R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed.
- B. Singh, M. B. Gawande, A. D. Kute, R. S. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zbořil, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS PubMed.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131–163 CrossRef.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed.
- J. Zhou, Chem.–Asian J., 2010, 5, 422–434 CrossRef CAS PubMed.
- R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2016, 138, 15543–15546 CrossRef CAS PubMed.
- T. Schwob and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 15175–15179 CrossRef CAS PubMed.
- N. Deibl and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 1663–1666 CrossRef CAS PubMed.
- R. Sreedevi, S. Saranya, K. R. Rohit and G. Anilkumar, Adv. Synth. Catal., 2019, 361, 2236–2249 CrossRef CAS.
- J. C. Borghs, L. M. Azofra, T. Biberger, O. Linnenberg, L. Cavallo, M. Rueping and O. El-Sepelgy, ChemSusChem, 2019, 12, 3083–3088 CrossRef CAS PubMed.
- K. Li, L. Wei, M. Sun, B. Li, M. Liu and C. Li, Angew. Chem., Int. Ed., 2021, 60, 20204–20209 CrossRef CAS PubMed.
- M. Sohail, M. Bilal, T. Maqbool, N. Rasool, M. Ammar, S. Mahmood, A. Malik, M. Zubair and G. A. Ashraf, Arabian J. Chem., 2022, 15, 104095 CrossRef CAS.
- S. Chun, J. Ahn, R. R. Putta, S. B. Lee, D.-C. Oh and S. Hong, J. Org. Chem., 2020, 85, 15314–15324 CrossRef CAS PubMed.
- R. R. Putta, S. Chun, S. H. Choi, S. B. Lee, D.-C. Oh and S. Hong, J. Org. Chem., 2020, 85, 15396–15405 CrossRef CAS PubMed.
- R. R. Putta, S. Chun, S. B. Lee, J. Hong, D.-C. Oh and S. Hong, RSC Adv., 2021, 11, 18225–18230 RSC.
- J. Wu and C. Darcel, J. Org. Chem., 2021, 86, 1023–1036 CrossRef CAS PubMed.
- S. Das, S. Mallick and S. De Sarkar, J. Org. Chem., 2019, 84, 12111–12119 CrossRef CAS PubMed.
- P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 14373–14377 CrossRef CAS PubMed.
- T. Yan and K. Barta, ChemSusChem, 2016, 9, 2321–2325 CrossRef CAS PubMed.
- F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 7261–7265 CrossRef CAS PubMed.
- B. Emayavaramban, M. Sen and B. Sundararaju, Org. Lett., 2017, 19, 6–9 CrossRef CAS PubMed.
- C. Paal, Chem. Ber., 1884, 17, 2756–2767 CrossRef.
- L. Knorr, Chem. Ber., 1884, 17, 2863–2870 CrossRef.
- A. Hantzsch, Chem. Ber., 1890, 23, 1474–1476 CrossRef.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, RSC Adv., 2021, 11, 13585–13601 RSC.
- F. G. Cirujano, A. Leyva-Pérez, A. Corma and F. X. Llabrés i Xamena, ChemCatChem, 2013, 5, 538–549 CrossRef CAS.
- S. Menuel, E. Bertaut, E. Monflier and F. Hapiot, Dalton Trans., 2015, 44, 13504–13512 RSC.
- M. A. Fouad, F. Ferretti, D. Formenti, F. Milani and F. Ragaini, Eur. J. Org. Chem., 2021, 2021, 4876–4894 CrossRef.
- T. Wagener, M. Pierau, A. Heusler and F. Glorius, Adv. Synth. Catal., 2022, 364, 3366–3371 CrossRef CAS PubMed.
- C. Bäumler and R. Kempe, Chem.–Eur. J., 2018, 24, 8989–8993 CrossRef PubMed.
- T. Schwob, M. Ade and R. Kempe, ChemSusChem, 2019, 12, 3013–3017 CrossRef CAS PubMed.
- H. Song, Z. Yang, C.-H. Tung and W. Wang, ACS Catal., 2020, 10, 276–281 CrossRef CAS.
- Z. Gong, Y. Lei, P. Zhou and Z. Zhang, New J. Chem., 2017, 41, 10613–10618 RSC.
- P. Ryabchuk, T. Leischner, C. Kreyenschulte, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 18679–18685 CrossRef CAS PubMed.
- Y. Lin, F. Wang, E. Ren, F. Zhu, Q. Zhang and G.-P. Lu, J. Catal., 2022, 416, 39–46 CrossRef CAS.
- C. Bianchini, M. Peruzzini and F. Zanobini, J. Organomet. Chem., 1988, 354, C19–C22 CrossRef CAS.
- C. Bianchini, A. Meli, M. Peruzzini, P. Frediani, C. Bohanna, M. A. Esteruelas and L. A. Oro, Organometallics, 1992, 11, 138–145 CrossRef CAS.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed.
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed.
- G. Wienhöfer, F. A. Westerhaus, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, Chem. Commun., 2012, 48, 4827–4829 RSC.
- G. Wienhöfer, F. A. Westerhaus, K. Junge, R. Ludwig and M. Beller, Chem.–Eur. J., 2013, 19, 7701–7707 CrossRef PubMed.
- G. Wienhöfer, F. A. Westerhaus, K. Junge and M. Beller, J. Organomet. Chem., 2013, 744, 156–159 CrossRef.
- M.-C. Fu, R. Shang, Z. Huang and Y. Fu, Synlett, 2014, 25, 2748–2752 CrossRef CAS.
- G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- G. Wienhöfer, M. Baseda-Krüger, C. Ziebart, F. A. Westerhaus, W. Baumann, R. Jackstell, K. Junge and M. Beller, Chem. Commun., 2013, 49, 9089–9091 RSC.
- W. Liu, W. Li, A. Spannenberg, K. Junge and M. Beller, Nat. Catal., 2019, 2, 523–528 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760–773 CrossRef CAS.
- C. J. Burrows, J. B. Harper, W. Sander and D. J. Tantillo, J. Org. Chem., 2022, 87, 1599–1601 CrossRef CAS PubMed.
- D. Jaiswal, A. P. De Souza, S. Larsen, D. S. LeBauer, F. E. Miguez, G. Sparovek, G. Bollero, M. S. Buckeridge and S. P. Long, Nat. Clim. Change, 2017, 7, 788–792 CrossRef.
- L. M. Rossi, J. M. R. Gallo, L. H. C. Mattoso, M. S. Buckeridge, P. Licence and D. T. Allen, ACS Sustainable Chem. Eng., 2021, 9, 4293–4295 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS PubMed.
- T. Kondo and T.-a. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed.
- J. K. Dunleavy, Platinum Met. Rev., 2006, 50, 110 CrossRef CAS.
- A. Kolpin, G. Jones, S. Jones, W. Zheng, J. Cookson, A. P. E. York, P. J. Collier and S. C. E. Tsang, ACS Catal., 2017, 7, 592–605 CrossRef CAS.
- R. Xiong, W. Ren, Z. Wang and M. Zhang, ChemCatChem, 2021, 13, 548–552 CrossRef CAS.
- G. Kortüm and W. Vogel, Dissociation Constants of Organic Acids in Aqueous Solution, Butterworths, London, 1961 Search PubMed.
- H. G. Kuivila and K. V. Nahabedian, J. Am. Chem. Soc., 1961, 83, 2159–2163 CrossRef CAS.
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed.
- P. R. B. Fallavena and E. E. S. Schapoval, Int. J. Pharm., 1997, 158, 109–112 CrossRef CAS.
- G. S. Vanier, Synlett, 2007, 2007, 0131–0135 CrossRef.
- P. K. Mandal and J. S. McMurray, J. Org. Chem., 2007, 72, 6599–6601 CrossRef CAS PubMed.
- F.-X. Felpin and E. Fouquet, Chem.–Eur. J., 2010, 16, 12440–12445 CrossRef CAS PubMed.
- P. R. Sultane, T. B. Mete and R. G. Bhat, Tetrahedron Lett., 2015, 56, 2067–2070 CrossRef CAS.
- S. Lange, D. Formenti, H. Lund, C. Kreyenschulte, G. Agostini, S. Bartling, S. Bachmann, M. Scalone, K. Junge and M. Beller, ACS Sustainable Chem. Eng., 2019, 7, 17107–17113 CrossRef CAS.
- G. Poce, R. H. Bates, S. Alfonso, M. Cocozza, G. C. Porretta, L. Ballell, J. Rullas, F. Ortega, A. De Logu, E. Agus, V. La Rosa, M. R. Pasca, E. De Rossi, B. Wae, S. G. Franzblau, F. Manetti, M. Botta and M. Biava, PLoS One, 2013, 8, e56980 CrossRef CAS PubMed.
- G. Poce, S. Consalvi, M. Cocozza, R. Fernandez-Menendez, R. H. Bates, F. O. Muro, D. B. Aguirre, L. Ballell and M. Biava, Eur. J. Pharm. Sci., 2017, 99, 17–23 CrossRef CAS PubMed.
- M. Biava, G. C. Porretta, G. Poce, A. De Logu, M. Saddi, R. Meleddu, F. Manetti, E. De Rossi and M. Botta, J. Med. Chem., 2008, 51, 3644–3648 CrossRef CAS PubMed.
- C. Lu, Q. Zhu, X. Zhang, H. Ji, Y. Zhou, H. Wang, Q. Liu, J. Nie, W. Han and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 8542–8553 CrossRef CAS.
- X. Deng, B. Qin, R. Liu, X. Qin, W. Dai, G. Wu, N. Guan, D. Ma and L. Li, J. Am. Chem. Soc., 2021, 143, 20898–20906 CrossRef CAS PubMed.
- L. Huang, F. Tang, F. Hao, H. Zhao, W. Liu, Y. Lv, P. Liu, W. Xiong and H. Luo, ACS Sustainable Chem. Eng., 2022, 10, 2947–2959 CrossRef CAS.
- E. M. Gordon, M. A. Gallop and D. V. Patel, Acc. Chem. Res., 1996, 29, 144–154 CrossRef CAS.
- Á. Cores, J. Clerigué, E. Orocio-Rodríguez and J. C. Menéndez, Pharmaceuticals, 2022, 15, 1009 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of isolated compounds. See DOI: https://doi.org/10.1039/d3sc02879h |
| This journal is © The Royal Society of Chemistry 2023 |