
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen Trifluoride Complexes of Group 10 Transition Metals M(NF3) (M = Pd, Pt)†
Guohai
Deng
*,
Yan
Lu
,
Tony
Stüker
and
Sebastian
Riedel
 *
*
Department of Chemistry and Biochemistry, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany. E-mail: ghdeng@zedat.fu-berlin.de; s.riedel@fu-berlin.de
First published on 18th July 2023
Abstract
The group 10 transition metal atoms Pd and Pt react with nitrogen trifluoride (NF3) forming N-coordination M(NF3) complexes in solid neon and argon matrices. The M(NF3) complexes isomerize to more stable fluoronitrenoid FNMF2 isomers via fluorine migration upon blue LED (λ = 470 nm) light irradiation. These products are characterized on the basis of infrared absorption spectroscopy with isotopic substitutions and theoretical frequency calculations. The analysis of the electronic structure of nitrogen trifluoride complexes indicates that the bonding between metal and nitrogen trifluoride can be described as σ donation from the HOMO of nitrogen trifluoride to the empty metal dz2 orbital and π back-donation from the metal dxz/yz orbitals to the LUMO of nitrogen trifluoride, the latter of which stabilized the metal ligand bond and destabilized the ligand N–F bond. In FNMF2, the FN ligand doubly bonded to the metal and bear imido character.
Introduction
Metal–ligand bonding is crucial to transition metal chemistry. The nature and strength of bonding between the metal and ligand determine the stability, geometry and reactivity of intermediates in catalytic reactions.1 Thus, understanding the nature of the metal–ligand bond is very important.2,3 Nitrogen trifluoride (NF3) is a stable and long known compound and was first synthesized by Otto Ruff and co-workers via the electrolysis of a HF/NH4F mixture in 1928.4 Although it has been widely used as a reactant in gas-phase reactions5 and synthetic chemistry,6 the coordination chemistry of this molecule as a Lewis base has received less attention, which is most likely due to its weak Lewis base character. Nevertheless, the proton affinity of NF3 has been experimentally measured many times since 1980.7 Experimental and theoretical investigation on the complexes of NF3 with Lewis acids, such as BH3–nFn (n = 0–3)8 and AlH3 (ref. 9), indicated that nitrogen trifluoride can act as a nitrogen donor to form H3B–NF3, F3B–NF3 and H3Al–NF3 adducts. Nitrogen trifluoride complexes of coinage metals Cu, Ag and Au supported by ligands such as cyanogen and halides were studied by density functional theory.10 In addition, theoretical studies on the metal ligand back-bonding in Pd(NF3) and LPd(NF3) (L = NH3, PH3, CO) complexes using NBO analysis demonstrated that the NF3 ligand also has a strong π acceptor character.11 Besides the neutral complexes, some nitrogen trifluoride ion complexes have also been studied computationally.12 It was found that the H+, Li+, Na+ and K+ nitrogen trifluoride complexes appear to form ion–dipole interactions and the Li+, Mg+ and Be+–NF3 cations could be detected as stable products in the gas phase.Recently, reactions of group 4, 6, 8, 9, and 11 metals, Th, and U with nitrogen trifluoride in excess neon and argon have been studied, and difluoroamino, fluoronitrenoid and nitride metal complexes (F2NMF, FNMF2, and NMF3) have been prepared and characterized by matrix-infrared spectroscopy and theoretical calculations.13–16 In contrast to the nitrogen trifluoride reactions reported above, the analogue ammonia molecule reacted with metal atoms to form M(NH3) complexes first, which rearranged to HMNH2via N–H bond insertion upon light excitation.17 The weak N–F bond, which is only about half as strong as the N–H bond in the NH3 molecule,18 makes the insertion reaction easier than that for the N–H bond. This may result in the spontaneous formation of N–F bond insertion complexes. Moreover, fluorine is the most electronegative element, the lone pair electronic density in the nitrogen atom of NF3 is decreased, and the dipole moment direction is also reversed as compared with those of NH3,19 both of which make it less favourable to give the M(NF3) complex with a M–N interaction. To the best of our knowledge, there are no experimental reports on the nitrogen trifluoride metal coordination in which the NF3 ligand does not immediately react further. Here we report the preparation of the nitrogen trifluoride complexes of group 10 metals M(NF3) (M = Pd, Pt) from laser ablated metal atoms with NF3 molecules and their spectroscopic identification in low-temperature neon and argon matrices. The nitrogen-coordination complexes will further isomerize to the more stable insertion products FNMF2 upon blue LED (λ = 470 nm) light irradiation.
Results and discussion
The title complexes were produced by the reactions of laser-ablated palladium, platinum atoms and nitrogen trifluoride in solid argon, and were recorded by FTIR absorption spectroscopy with isotopic substitutions. The IR spectra in the 1150–550 cm−1 region using a 0.5% NF3/Ar mixture are represented in Fig. 1 and 2. After 60 min of sample deposition, common absorption bands due to NF and NF2 radicals are observed,20 which decrease on annealing but slightly increase on blue LED (λ = 470 nm) light irradiation. No obvious absorption bands of molecular metal fluorides are observed. Besides these common product absorption bands, new metal dependent absorption bands were also detected, which can be classified into two groups based on their identical annealing and photochemical behaviours (labeled A and B in Fig. 1 and C and D in Fig. 2). Since some absorption bands are partially overlapped by the nitrogen trifluoride absorption bands, a difference IR spectrum taken from the spectrum after blue LED (λ = 470 nm) light irradiation minus the spectrum after 20 K annealing is obtained and represented in Fig. S1 and S2.† Similar experiments were also performed using an isotopically labelled compound 15NF3 and a 14NF3 + 15NF3 mixture. The IR spectra in the selected region are shown in Fig. S1 and S2.† All observed band positions are listed in Table 1.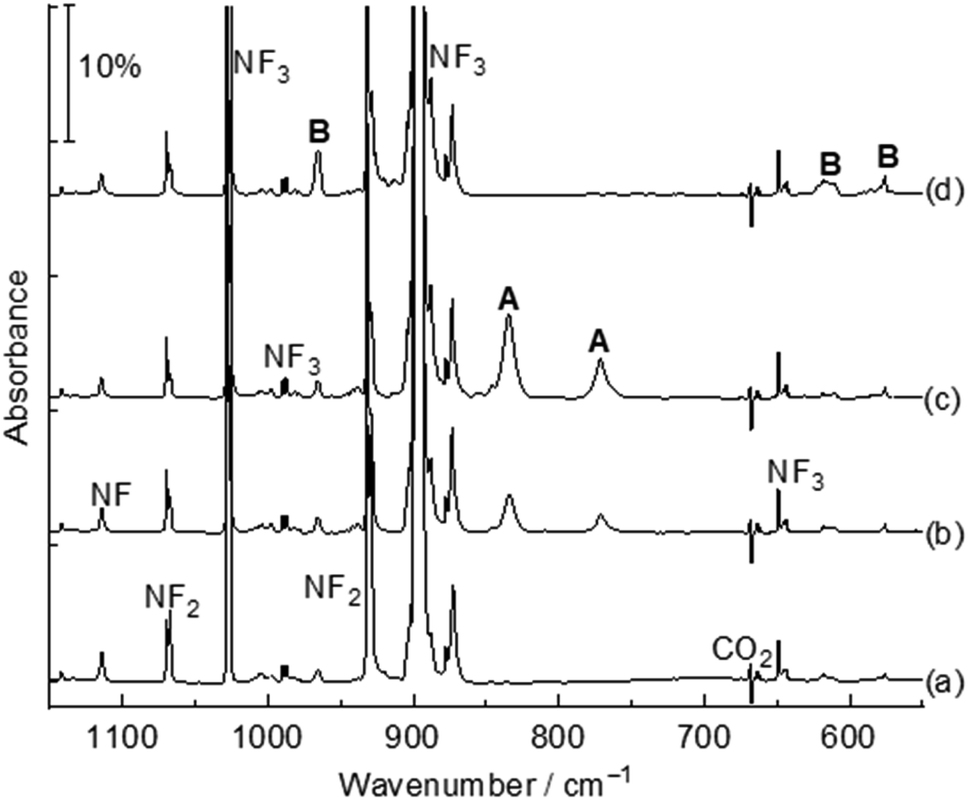 | ||
| Fig. 1 Infrared spectra in the 1150–550 cm−1 region from co-deposition of laser-ablated Pd atoms with 0.5% NF3 in argon. (a) After 60 min of sample deposition, (b) after annealing to 15 K, (c) after annealing to 20 K, and (d) after 10 min of blue LED (λ = 470 nm) light irradiation. A: Pd(NF3); B: FNPdF2. | ||
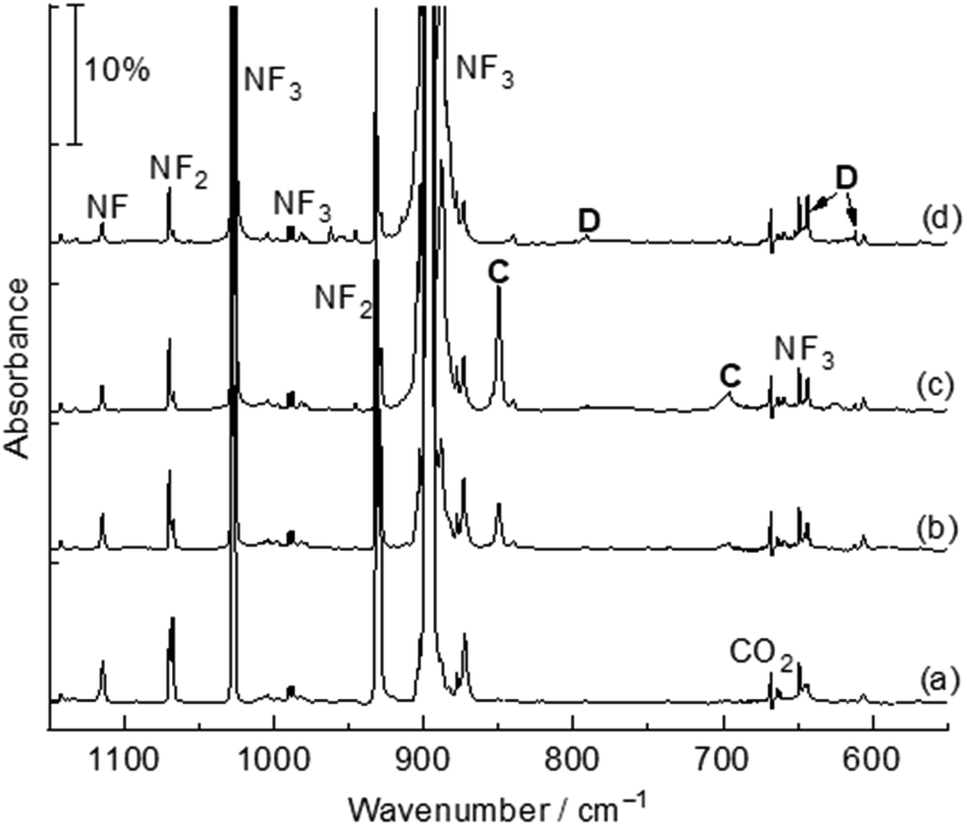 | ||
| Fig. 2 Infrared spectra in the 1150–550 cm−1 region from co-deposition of laser-ablated Pt atoms with 0.5% NF3 in argon. (a) After 60 min of sample deposition, (b) after annealing to 15 K, (c) after annealing to 20 K, and (d) after 10 min of blue LED (λ = 470 nm) light irradiation. C: Pt(NF3); D: FNPtF2. | ||
| Complexes | Obs. | Calc. | Assignmentsc | ||
|---|---|---|---|---|---|
| Ne | Ar | B3LYPb | CCSD (T)b | ||
| a Absorption bands not observed or too weak. b Harmonic frequencies calculated at the B3LYP/aug-cc-pVTZ(-PP) and CCSD(T)/aug-cc-pVTZ(-PP) levels are listed for the 106Pd and 195Pt isotopologues. The complete set of vibrational frequencies is provided in ESI Tables S1 and S2. For the CCSD(T) calculations no intensities are available. c Tentative assignments based on calculated vibrational displacement vectors. | |||||
| Pd(NF3) (A) | 838.6 (20.5) | 834.4 (20.6) | 849 (22.1) [731] | 878 (21.3) | sym. N–F str. |
| 778.1 (17.5) | 771.3 (17.6) | 703 (17.8) [159] × 2 | 813 (18.7) | asym. N–F str. | |
| —a | —a | 610 (0.9) [11] | 629 (1.5) | ||
| —a | —a | 445 (0.1) [10] × 2 | 475 (0.5) | ||
| Pt(NF3) (C) | 869.3 (24.7) | 849.5 (24.1) | 901 (26.3) [811] | 854 (23.4) | sym. N–F str. |
| 703.9 (17.5) | 696.3 (17.7) | 667 (17.9) [121] × 2 | 750 (18.5) | asym. N–F str. | |
| —a | —a | 624 (0.3) [42] | 634 (0.1) | ||
| —a | —a | 452 (0.1) [23] × 2 | 476 (0.5) | ||
| FNPdF2 (B) | 968.0 (19.2) | 965.6 (19.9) | 994 (22.9) [232] | — | N–F str. |
| —a | —a | 820 (20.7) [60] | — | N–Pd str. | |
| 625.5 (0) | 612.3 (0) | 610 (<0.1) [104] | — | asym. F–Pd–F str. | |
| 588.0 (0.3) | 575.7 (0.4) | 579 (0.2) (94) | — | sym. F–Pd–F str. | |
| FNPtF2 (D) | 890.4 (20.7) | 883.6 (21.2) | 925 (26.6) [160] | 933 (20.2) | N–F str. |
| 798.7 (20.2) | 790.8 (20.2) | 833 (18.0) [119] | 804 (23.0) | N–Pt str. | |
| 653.6 (0.3) | 643.4 (0) | 640 (<0.1) [109] | 608 (<0.1) | asym. F–Pt–F str. | |
| 622.6 (0) | 611.5 (0.2) | 608 (<0.1) [79] | 578 (<0.1) | sym. F–Pt–F str. | |
Experiments were also carried out with laser-ablated palladium, platinum atoms and NF3 in excess neon. The spectra with 0.1% NF3 and isotopic substitutions in neon are shown in Fig. S3–S6.† The same products as detected in argon are presented, but the Pd(NF3) product absorption bands are weaker than those detected in solid argon. The product absorption bands in solid neon are given in Table 1. The band positions are blue-shifted to higher wave numbers compared to those of the argon matrix.
The absorption bands at 834.4 and 771.3 cm−1 labeled A (Fig. 1, trace c) are assigned to the Nitrogen-coordination Pd(NF3) complex. These absorption bands appeared after sample annealing to 15 K and increase remarkably when the matrix samples are annealed to 20 K, but disappeared totally under 10 min blue LED (λ = 470 nm) light irradiation. Both bands are red-shifted compared to the N–F stretching vibration (1028.1 and 898.1 cm−1) modes of free NF3 in argon.15 In the experiment using the isotopic-labeled 15NF3 sample, the absorption bands shift to 813.8 and 753.7 cm−1 with a 14/15N isotopic shift of 20.6 and 17.6 cm−1 respectively. The band position and isotopic splitting in the mixed 14NF3 + 15NF3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) experiment (Fig. S1†) confirm that only one NF3 ligand is involved. Accordingly, the two absorption bands 834.4 and 771.3 cm−1 are assigned to the symmetric and antisymmetric N–F stretching modes. The Pd(NF3) complex in solid neon is observed at 838.6 and 778.1 cm−1, which are only 4.2 and 6.8 cm−1 blue-shifted from those in argon, indicating a weak matrix effect. This type of 1:1 metal–ligand complex with a metal coordinated by a nitrogen atom of the ligand is common for molecular complexes.21
:1) experiment (Fig. S1†) confirm that only one NF3 ligand is involved. Accordingly, the two absorption bands 834.4 and 771.3 cm−1 are assigned to the symmetric and antisymmetric N–F stretching modes. The Pd(NF3) complex in solid neon is observed at 838.6 and 778.1 cm−1, which are only 4.2 and 6.8 cm−1 blue-shifted from those in argon, indicating a weak matrix effect. This type of 1:1 metal–ligand complex with a metal coordinated by a nitrogen atom of the ligand is common for molecular complexes.21
In addition to the Pd(NF3) absorption bands, another group of absorption bands at 965.6, 612.3 and 575.5 cm−1 labeled B (Fig. 1, trace d) are produced after sample co-deposition. These absorption bands increased under blue LED (λ = 470 nm) light irradiation at the expense of group A absorption bands, indicating that product B is a structural isomer of product A, and is assigned to the FNPdF2 complex. The 965.6 cm−1 absorption band is mainly a F–N stretching vibration with a large 14/15N shift (19.9 cm−1). The 14/15N shift is close to those of 21.4 cm−1 in FNCoF2 and 18.9 cm−1 in FNRhF2.16 This band position is red-shift by 153.8 cm−1 compared to the free NF radical (1119.4 cm−1),12 indicating a strong interaction between nitrogen and palladium. Two low bands are attributed to the PdF2 antisymmetric and symmetric stretching modes, which are lower than 652.1 and 586.1 cm−1 in OPdF2.22 The palladium isotope pattern of these bands is not resolved, and only the band at 575.5 cm−1 displays a small 0.4 cm−1 shift with the 15NF3 sample. The Pd–N stretch is too weak to be observed. The FNPdF2 complex in solid neon is observed at 968.0, 625.5 and 588.0 cm−1. The band positions are 2.4, 13.2 and 2.3 cm−1 blue-shifted from argon to neon. The observation of B directly after sample deposition indicates that it can be generated during the sample deposition process initiated by the excitation from the ablation plume.
Similar absorption bands are also observed in the platinum experiments. The product absorption bands at 849.5 and 696.3 cm−1 labeled C (Fig. 2, trace c) are assigned to the Pt(NF3) complex. These bands shifted to 825.4 and 678.6 cm−1 with 15NF3, and only the pure isotopic counterparts were observed in the 1:1 mixed 14NF3 + 15NF3 experiment. The band positions and isotopic frequency shifts (24.1 and 17.7 cm−1) are close to those of the Pd(NF3) complex, indicating similar bonding mode between palladium and platinum. The bands at 883.6, 790.8, 643.4 and 611.5 cm−1 labeled D (Fig. 2, trace d) that appeared upon co-deposition and increased after blue LED (λ = 470 nm) light irradiation are assigned to the FNPtF2 molecule. The 15NF3 counterparts are detected at 862.4, 770.6, 643.4 and 611.3 cm−1 with isotopic shifts of 21.2, 20.2, 0 and 0.2 cm−1. These absorption bands belong to the F–N and Pt–N stretching vibration as well as PtF2 antisymmetric and symmetric stretching modes. The Pt(NF3) and FNPtF2 complexes are detected at 849.5, 696.3 and 890.4, 798.7, 653.6, and 622.6 cm−1 respectively in solid neon. The photoisomerization reaction of metal nitrogen trifluoride complexes (A and C) to FNMF2 (B and D) is reproducible in the neon matrix. No similar product absorption bands are observed in the Ni + NF3 experiments.
The structures and vibrational frequencies of M(NF3) and FNMF2 (M = Pd, Pt) in the closed shell singlet ground state were calculated at the B3LYP/aug-cc-pVTZ(-PP) and CCSD(T)/aug-cc-pVTZ(-PP) levels (for details see the ESI†). Among the four isomers, the FNMF2 isomer has the lowest energy (Fig. S7†). As shown in Fig. 3, the Pd(NF3) and Pt(NF3) complexes possess an 1A1 electronic ground state and nitrogen trifluoride is coordinated to the metal center through nitrogen forming a C3v structure. The triplet states of [Pd(NF3)] and [Pt(NF3)] are less stable by 33.0 and 25.1 kcal mol−1 at the CCSD(T)/aug-cc-pVTZ(-PP)//B3LYP/aug-cc-pVTZ(-PP) level. The N–F bond distance in the nitrogen trifluoride ligand is significantly longer (1.399 Å for Pd and 1.409 Å for Pt) than in free nitrogen trifluoride (1.371 Å).23 The calculated Pd–N distance in the complex is slightly longer (2.073 Å) than that of PdNCCN (1.948 Å)24 and PdNNO (2.003 Å).25 The predicted Pt–N bond distance is 1.885 Å and close to that of 1.893 Å in the PtNNO complex.25 The FNPdF2 and FNPtF2 molecules are predicted to have an 1A′ ground state with a Cs symmetric structure, and are more stable than the Pd(NF3) and Pt(NF3) isomers by 41.6 and 80.9 kcal mol−1, respectively. The triplet FNPdF2 and FNPtF2 species are 9.8 and 16.6 kcal mol−1 higher in energy than the corresponding molecules in the singlet state. The computed Pd–N distance of 1.705 Å in FNPdF2 is significantly shorter with respect to the Pd–NDip bond length of 1.872(3) Å in the (CAAC)Pd(Dip) compound [CAAC = cyclic (alkyl)(amino)carbene, Dip = 2,6-diisopropylphenyl]26 and in the range of those for calculated palladium nitrenes (1.8–2.0 Å).27 The predicted Pt–N bond length of 1.743 Å and shorter than that of the linear two-coordinated platinum complex Pt(NCtBu2)2 [1.814(4) Å, 1.818(4) Å],28 but longer than that of the binary molecule PtN (1.703 Å).29 The short bond length and large 14/15N isotopic shift indicate that the M–N bond in FNMF2 is a double bond with a nitrene structure in which the metal shows a formal oxidation state +II, which are similar to the Rh–N bond in the FNRhF2 molecule.16 The calculated structures of the F2NMF and NMF3 (M = Pd, Pt) isomers are shown in Fig. S8.†
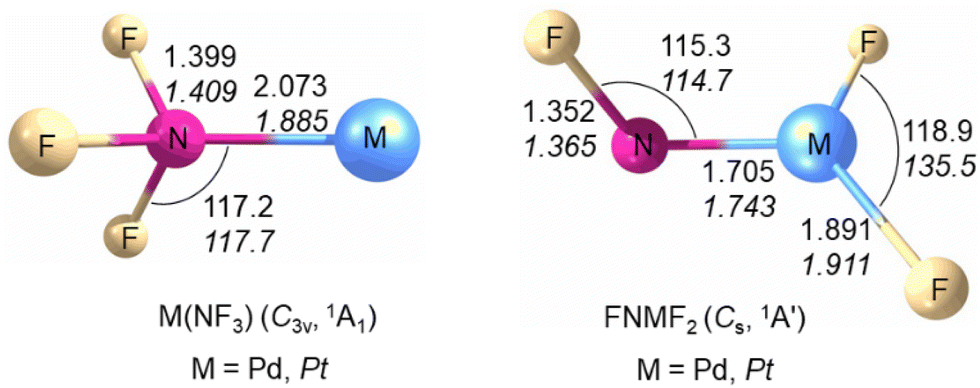 | ||
| Fig. 3 Calculated structures (bond lengths in Ångstroms and bond angles in degrees) of M(NF3) (M = Pd, Pt) and FNPtF2 at the CCSD(T)/aug-cc-pVTZ(-PP) level and of FNPdF2 at the B3LYP/aug-cc-pVTZ(-PP) level of theory. | ||
The assignment of the M(NF3) and FNMF2 (M = Pd, Pt) complexes is supported by theoretical calculations at the B3LYP and CCSD(T) levels of theory. As listed in Table 1, similar to the FNRhF2 complex,16 the calculated absorption band positions and observed experimental values of the F–N and N–M stretching frequencies in the FNMF2 complexes do not match perfectly due to strong vibrational coupling between these two vibrational modes. Additionally, the predicted wavenumbers as well as isotopic shifts in other vibrational stretching modes in the complexes M(NF3) and FNMF2 (M = Pd, Pt) show good agreement with the experimental values. All in all, the comparison between the computed and observed IR spectra strongly supports that the experimentally observed products are the nitrogen trifluoride complex M(NF3) and FNMF2 (M = Pd, Pt) molecules. The calculated vibrational frequencies and intensities of the possible further isomers F2NMF and NMF3 are shown in Tables S1 and S2.† However, both hypothetical isomers show different spectral signatures than our observed spectra, which further supports our assignment of the experimental bands. The relative energies (kcal mol−1) of different isomers of F3NM (M = Pd, Pt) are given in Table S3.†
The bonding of Pd(NF3) and Pt(NF3) complexes is of particular interest. By taking Pt(NF3) as an example. Fig. 4 shows the highest-lying occupied molecular orbitals. The HOMO and HOMO−1 are metal based 6s and 5dxy/x2−y2 nonbonding orbitals. The HOMO−2 is doubly degenerate metal dπ orbital and composed of metal 5dxz/yz and the LUMO of nitrogen trifluoride. The HOMO−3 and HOMO−4 are only composed of ligand orbitals. The HOMO−5 is a ligand-based orbital that comprises some weak ligand to metal σ donation. Previous reports have proved that the bonding of lone-pair ligands to transition metals is generally consisted of σ donation from the ligand to the metal and π back-donation from the metal to the ligand.2 Based on this assumption, the Pt 5dz2 orbital should be considered as an empty σ orbital to accept electron donation of the NF3 HOMO and the back-donation of the Pt π electrons to the NF3 LUMO upon the formation of the Pt(NF3) complex. The donation interaction between the HOMO of NF3 and a vacant Pt 5dz2 orbital with σ symmetry stabilizes the Pt–N and N–F bonds, while the back-donation from the Pt dπ orbital to the NF3 LUMO causes the destabilization of the N–F bond. Consistent with this notion, the N–F stretching frequencies of the Pt(NF3) complex are red-shifted compared to the neutral nitrogen trifluoride. The Pd(NF3) complex has a quite similar bonding situation to Pt(NF3). The palladium complex shows a lower N–F stretching frequency and the comparison with the platinum species suggests that the palladium complex has an even stronger back-donation. This shift can be explained by the different energy levels of the valence (n – 1)d and ns orbitals of the 4d and 5d transition metal atoms, which has been discussed for group 10 transition metal nitrous oxide complexes25 as well as group 11 transition metal carbonyl complexes.30
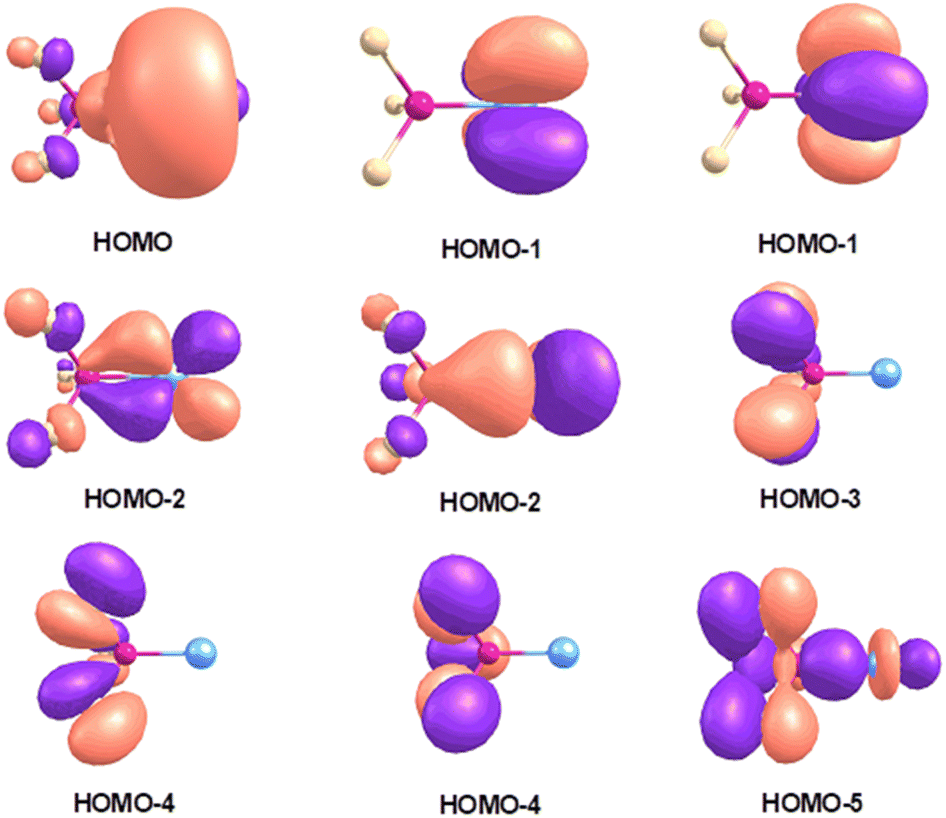 | ||
| Fig. 4 Shape of the selected highest-lying occupied molecular orbitals of Pt(NF3) at the B3LYP/aug-cc-pVTZ level. | ||
The spectra illustrated in Fig. 1 and 2 clearly suggest that the N-coordination M(NF3) complexes are generated by the reactions of ground state palladium (1S) and platinum (3D) atoms with nitrogen trifluoride in solid neon and argon matrices. These association reactions are calculated to be exothermic by 11.0 (Pd), and 28.5 kcal mol−1 (Pt) [CCSD(T)/aug-cc-pVTZ level], respectively, and require negligible activation energy, as the M(NF3) absorption bands increase on annealing, while a similar reaction product for nickel is predicted to be endothermic and was not observed. The bonding energies of nitrogen trifluoride are very close to those of the PdNNO and PtNNO complexes,25 which are produced between Pd/Pt metals and nitrous oxide, but lower than that of PdNCCN and PtNCCN cyanogen complexes.24 The M(NF3) complexes rearrange to the FNMF2 isomers under blue LED (λ = 470 nm) light irradiation. These isomerization reactions are computed to be exothermic by 41.6 (Pd) and 80.9 (Pt) kcal mol−1 at the CCSD(T)/aug-cc-pVTZ(-PP)//B3LYP/aug-cc-pVTZ(-PP) level of theory. The isomeric pathway is predicted to proceed via two transition states involving the sequence M(NF3) → F2NMF → FNMF2. Fig. S9† shows the pathway where the first step M(NF3) → F2NMF involves an activation energy barrier of 24.6 (Pd) and 19.1 (Pt) kcal mol−1. The second step F2NMF → FNMF2 requires to cross a lower barrier of 10.0 (Pd) and 17.6 (Pt) kcal mol−1. Both two energy barriers 34.6 (Pd) and 36.7 (Pt) kcal mol−1 are lower than the energy of LED (λ = 470 nm) light (60.8 kcal mol−1). Consistent with the low bonding energy, the M(NF3) complexes underwent decomposition to form metal atoms and NF3 molecules when the matrix samples were subjected to blue LED (λ = 470 nm) light photolysis and result in low efficiency to yield FNMF2 complexes. Further work is currently underway to study the reactions of group 10 metal atoms with phosphorus trifluoride (PF3).
Conclusions
In summary, we described the formation of N-coordination nitrogen trifluoride complexes M(NF3) (M = Pd, Pt) by the reaction of laser-ablated group 10 transition metal atoms with NF3 in solid neon and argon matrices. These molecular complexes were characterized by matrix-isolation infrared absorption spectroscopy and quantum-chemical calculations. The complexes rearrange to the FNMF2 isomer under blue LED (λ = 470 nm) light irradiation, which is identified to be a fluoronitrenoid complex with a N–M double bond. The bonding in these nitrogen trifluoride complexes involves the electron donation of the HOMO of the nitrogen trifluoride to the empty metal dz2 orbital and back-donation of the metal π electrons to the LUMO of nitrogen trifluoride. Although the detected metal nitrogen trifluoride products have low thermodynamic stability and can only be isolated in cryogenic matrices, they enrich and extend the arsenal of transition metal coordination chemistry and provide a deeper understanding of the fluorine specific interactions.Data availability
Data supporting this manuscript is available within the ESI† and available on request.Author contributions
GD planned and performed the experiments and carried out the quantum chemical calculations. He wrote the first draft of the manuscript. YL performed some of the experiments. TS helped with the design of the experiments and proof read the manuscript. SR guided and advised the project and proof read the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Zentraleinrichtung für Datenverarbeitung (ZEDAT) of the Freie Universität Berlin for the allocation of computing resources. We thank the ERC Project HighPotOx as well as the CRC 1349 (SFB 1349) Fluorine Specific Interactions – Project-ID 387284271 – for continuous support. Open access funding was enabled and organized by Projekt DEAL. G. D thanks the Alexander von Humboldt Foundation (AvH) for a research scholarship.Notes and references
- (a) L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364 CrossRef CAS; (b) J. Mas-Roselló, C. J. Cope, E. Tan, B. Pinson, A. Robinson, T. Smejkal and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 15524 CrossRef; (c) C. V. Wilson, D. Kim, A. Sharma, R. X. Hooper, R. Poli, B. M. Hoffman and P. L. Holland, J. Am. Chem. Soc., 2022, 144, 10361 CrossRef CAS.
- G. Frenking and N. Fröhlich, Chem. Rev., 2000, 100, 717 CrossRef CAS PubMed.
- M. F. Zhou and G. Frenking, Acc. Chem. Res., 2021, 54, 3071 CrossRef CAS PubMed.
- O. Ruff, J. Fischer and F. Luft, Z. Anorg. Allg. Chem., 1928, 172, 417 CrossRef CAS.
- (a) P. Antoniotti, L. Operti, R. Rabezzana, F. Turco and G. A. Vaglio, Int. J. Mass Spectrom., 2006, 255–256, 225 CrossRef CAS; (b) P. Antoniotti, P. Benzi, S. Borocci, C. Demaria, M. Giordani, F. Grandinetti, L. Operti and R. Rabezzana, Chem. - Eur. J., 2015, 21, 15826 CrossRef CAS PubMed; (c) P. Antoniotti, P. Benzi, D. Marabello and D. Rosso, ACS Omega, 2020, 5, 4907 CrossRef CAS PubMed.
- R. K. Belter, J. Fluorine Chem., 2015, 175, 110 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413 CrossRef CAS.
- (a) O. de La Luz Rojas, J. Am. Chem. Soc., 1977, 99, 2902 CrossRef CAS; (b) K. R. S. Chandrakumar and S. Pal, J. Phys. Chem. A, 2002, 106, 11775 CrossRef CAS; (c) P. Antoniotti, S. Borocci and F. Grandinetti, Eur. J. Inorg. Chem., 2004, 2004, 1125 CrossRef.
- A. Boutalib, J. Phys. Chem. A, 2003, 107, 2106 CrossRef CAS.
- Q. Luo, Q. S. Li, Y. M. Xie, R. B. King and H. F. Schaefer, J. Chem. Theory Comput., 2011, 7, 131 CrossRef CAS PubMed.
- T. Leyssens, D. Peeters, A. G. Orpen and J. N. Harvey, Organometallics, 2007, 26, 2637 CrossRef CAS.
- K. M. Pei, J. Liang and H. Y. Li, J. Mol. Struct., 2004, 690, 159 CrossRef CAS.
- X. F. Wang, L. Andrews, R. Lindh, V. Veryazov and B. O. Roos, J. Phys. Chem. A, 2008, 112, 8030 CrossRef CAS PubMed.
- (a) L. Andrews, X. F. Wang, R. Lindh, B. O. Roos and C. J. Marsden, Angew. Chem., Int. Ed., 2008, 47, 5366 CrossRef CAS PubMed; (b) L. Andrews, X. F. Wang and B. O. Roos, Inorg. Chem., 2009, 48, 6594 CrossRef CAS PubMed; (c) X. F. Wang and L. Andrews, Dalton Trans., 2009, 9260 RSC; (d) X. F. Wang, J. T. Lyon and L. Andrews, Inorg. Chem., 2009, 48, 6297 CrossRef CAS PubMed; (e) T. Stüker, X. X. Xia, H. Beckers and S. Riedel, Chem. - Eur. J., 2021, 27, 11693 CrossRef.
- Y. Gong and L. Andrews, Inorg. Chem., 2012, 51, 667 CrossRef CAS PubMed.
- T. Stüker, T. Hohmann, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2020, 59, 23174 CrossRef PubMed.
- (a) M. H. Chen, H. Lu, J. Dong, L. Miao and M. Zhou, J. Phys. Chem. A, 2002, 106, 11456 CrossRef CAS; (b) M. F. Zhou, M. H. Chen, L. N. Zhang and H. Lu, J. Phys. Chem. A, 2002, 106, 9017 CrossRef CAS; (c) X. F. Wang, L. Andrews and C. J. Marsden, Chem. - Eur. J., 2008, 14, 9192 CrossRef CAS PubMed; (d) X. F. Wang and L. Andrews, Organometallics, 2008, 27, 4885 CrossRef CAS; (e) X. Liu, X. F. Wang, B. Xu and L. Andrews, Chem. Phys. Lett., 2012, 523, 6 CrossRef CAS; (f) Z. Pu, F. Li, J. W. Qin, B. Y. Ao, P. Shi and M. B. Shuai, J. Phys. Chem. A, 2018, 122, 3541 CrossRef CAS PubMed.
- (a) J. Berkowitz, J. P. Greene, J. Foropoulos and O. M. Nesković, J. Chem. Phys., 1984, 81, 6166 CrossRef; (b) S. T. Gibson, J. P. Greene and J. Berkowitz, J. Chem. Phys., 1985, 83, 4319 CrossRef CAS.
- F. Blanco, I. Alkorta, I. Rozas, M. Solimannejad and J. Elguero, Phys. Chem. Chem. Phys., 2011, 13, 674 RSC.
- M. E. Jacox and D. E. Milligan, J. Chem. Phys., 1967, 46, 184 CrossRef CAS.
- (a) W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018 CrossRef CAS PubMed; (b) H.-J. Himmel and M. Reiher, Angew. Chem., Int. Ed., 2006, 45, 6264 CrossRef CAS PubMed; (c) L. Andrews and A. Citra, Chem. Rev., 2002, 102, 885 CrossRef CAS PubMed.
- L. Li, H. Beckers, T. Stüker, T. Lindič, T. Schlöder, D. Andrae and S. Riedel, Inorg. Chem. Front., 2021, 8, 1215 RSC.
- J. Sheridan and W. Gordy, Phys. Rev., 1950, 79, 513 CrossRef CAS.
- X. T. Chen, Z. X. Xiong, L. Andrews and Y. Gong, Inorg. Chem., 2020, 59, 6489 CrossRef CAS PubMed.
- X. Jin, G. Wang and M. F. Zhou, J. Phys. Chem. A, 2006, 110, 8017 CrossRef CAS PubMed.
- A. Grünwald, B. Goswami, K. Breitwieser, B. Morgenstern, M. Gimferrer, F. W. Heinemann, D. M. Momper, C. W. M. Kay and D. Munz, J. Am. Chem. Soc., 2022, 144, 8897 CrossRef.
- A. Grünwald and D. Munz, J. Organomet. Chem., 2018, 864, 26 CrossRef.
- A. W. Cook, P. Hrobárik, P. L. Damon, D. Najera, B. Horváth, G. Wu and T. W. Hayton, Inorg. Chem., 2019, 58, 15927 CrossRef CAS PubMed.
- A. Citra, X. F. Wang, W. D. Bare and L. Andrews, J. Phys. Chem. A, 2001, 105, 7799 CrossRef CAS.
- P. H. Kasai and P. M. Jones, J. Phys. Chem., 1985, 89, 1147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02313c |
| This journal is © The Royal Society of Chemistry 2023 |