
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoting water formation in sulphate-functionalized Ru for efficient hydrogen oxidation reaction under alkaline electrolytes†
Chaoyi
Yang
,
Yunbo
Li
,
Jianchao
Yue
,
Hengjiang
Cong
 and
Wei
Luo
*
and
Wei
Luo
*
College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: wluo@whu.edu.cn
First published on 23rd May 2023
Abstract
Improving the sluggish kinetics of the hydrogen oxidation reaction (HOR) under alkaline electrolytes plays a significant role in the practical application of alkaline polymer electrolyte fuel cells (APEFCs). Here we report a sulphate functionalized Ru catalyst (Ru-SO4) that exhibits remarkable electrocatalytic performance and stability toward alkaline HOR, with a mass activity of 1182.2 mA mgPGM−1, which is four-times higher than that of the pristine Ru catalyst. Theoretical calculations and experimental studies including in situ electrochemical impedance spectroscopy and in situ Raman spectroscopy demonstrate that the charge redistribution on the interface of Ru through sulphate functionalization could lead to optimized adsorption energies of hydrogen and hydroxide, together with facilitated H2 transfer through the inter Helmholtz plane and precisely tailored interfacial water molecules, contributing to a decreased energy barrier of the water formation step and enhanced HOR performance under alkaline electrolytes.
Introduction
Hydrogen has been regarded as a clean and sustainable energy carrier to alleviate the current energy crisis and global warming.1,2 Recently, hydrogen-based fuel cell technology is recognized as a promising approach for highly efficient energy conversion.3 Compared with the state-of-the-art proton exchange membrane fuel cells (PEMFCs), alkaline polymer electrolyte fuel cells (APEFCs) have attracted growing attention due to the development of anion exchange membranes and the potential utilization of nonprecious metal catalysts in the cathodic oxygen reduction reaction (ORR).4,5 However, the kinetics of the anodic hydrogen oxidation reaction (HOR) is much complex in alkaline electrolytes, resulting in 2 to 3 orders of magnitude slower kinetics than that in acid electrolytes, which severely limits the practical application of APEFCs.6–8 Despite considerable efforts, the origin of the kinetic pH effect of the HOR is still intractable to reach a consensus. Therefore, underlining the mechanism of the HOR under alkaline electrolytes and developing highly efficient cost-effective electrocatalysts are extremely desirable and remain challenging.The elementary steps including the Tafel–Volmer or Heyrovsky–Volmer process for the HOR in alkaline electrolytes are interpreted as follows:9,10
| Tafel: H2 + 2* ↔ 2H* |
| Heyrovsky: H2 + OH− + * ↔ H* + H2O + e− |
| Volmer: H* + OH− ↔ * + H2O + e− |
The hydrogen binding energy (HBE) theory confirms the key role of adsorbed hydrogen, suggesting an ideal zero Gibbs free energy contributing to the highest HOR activity.11–13 On the other hand, the bifunctional mechanism stresses that both the optimized adsorption energy of hydrogen and hydroxyl can facilitate the sluggish kinetics of alkaline HOR.14–16 Recently, considering the Volmer step as the rate determining step (RDS) for alkaline HOR,17–19 the role of interfacial water molecules has been recognized for the pH dependent kinetics of the HOR.17,20–24 Koper and co-workers found that the rigid interfacial water in alkaline media is difficult to reorganize, thereby leading to the unfavorable transfer of reactants (e.g. H2, OH−) to the surface of the catalyst during hydrogen electrocatalysis.22 Chen et al. demonstrated that the pH-dependent kinetics of the HOR originates from water connectivity in the electrical double layer through ab initio molecular dynamics simulations and in situ surface-enhanced infrared absorption spectroscopy.23 Xu et al. found that the interfacial water molecules on the surface of Pt with weak hydrogen bonding ability are essential for promoting solvent reorganization during the alkaline HOR process.24 Although the function of water in affecting reactants and intermediates has been widely recognized, the role of water as the product of the Volmer step is still worthy of further study. It is expected that simultaneously accelerating the hydrogen molecule transfer through the inter Helmholtz plane and modifying interfacial water molecules on the surface of catalysts could promote the formation of water and boost the kinetics of alkaline HOR, but have been rarely reported so far.
Herein, we demonstrate that sulphate functionalized Ru nanosheets (Ru-SO4) can significantly boost the alkaline HOR performance, with a current density of 0.548 mA cmPGM−2 and mass activity of 1182.2 mA mgPGM−1, which is three- and four-fold enhancement compared with that of pristine Ru, respectively. Combining experimental results and density functional theory (DFT) calculations, we understand that the introduction of sulphate could modulate the charge distribution on the surface of Ru, leading to optimized adsorption energies of hydrogen and hydroxide. More importantly, the concentration of hydrogen molecules and the structure of interfacial water molecules on the surface of Ru could be further effectively modified through sulphate functionalization, resulting in a much lowered energy barrier of water formation and enhanced alkaline HOR performance.
Results and discussion
Through a low temperature sulfidation, amorphous sulphate-functionalized Ru nanosheets can be obtained (denoted as Ru-SO4, detailed experimental procedures are presented in the ESI†). As shown in Fig. 1a, no apparent peaks in powder X-ray diffraction (XRD) for both Ru and Ru-SO4 can be observed, indicating that the amorphous structure is maintained well during the sulfidation process.25 Meanwhile, as shown in the Raman spectra (Fig. 1b), a distinct characteristic peak located at ∼1025 cm−1 can be observed, implying that the sulphate is successfully anchored to the surface of Ru.26,27 X-ray absorption spectroscopy (XAS) is employed to verify the structure of Ru-SO4(Fig. 1c and S1†). The Ru K-edge X-ray adsorption near-edge structure (XANES) curves of Ru foil, RuO2 and Ru-SO4 are depicted in Fig. 1c. The near-edge adsorption energy of Ru in Ru-SO4 is located between those of Ru foil and RuO2, implying that the valence state of Ru for Ru-SO4 is lower than +4. This oxidation state in Ru-SO4 may originate from the electron transformation between Ru and surface sulfate species. The Fourier transformed (FT) k2-weighted-extended X-ray absorption fine structure (EXAFS) of Ru-SO4 shows peaks around 1.6 Å and 2.4 Å, which can be assigned to Ru–O and Ru–Ru coordination, respectively (Fig. S1†). Fig. S2† presents the X-ray photoelectron spectroscopy (XPS) spectra of Ru-SO4 in the Ru 3p regions with the binding energies of 462.6 eV and 484.8 eV, contributing to the 3p3/2 and 3p1/2 of Ru, respectively.28 In addition, the metal–sulphide bond usually located at around 161.5 eV cannot be observed in Fig. 1d.29 The binding energies at 168.2 eV (2p3/2) and 169.3 eV (2p1/2) are assigned to sulphate due to the oxidized S species in the S 2p XPS spectrum (Fig. 1d).30 The Ru–O bond located at 529.7 eV can also be observed in O 1s spectra (Fig. S3†). The catalyst maintains the nanosheet morphology before and after sulfidation as depicted in transmission electron microscopy (TEM) of Ru (Fig. S4†) and Ru-SO4 (Fig. 1e). As shown in Fig. S5 and 6,† the high-resolution TEM image and corresponding selected area electron diffraction (SAED) patterns of Ru and Ru-SO4 further indicate the amorphous structure.25,31 The energy-dispersive X-ray (EDX) mapping in Fig. 1f reveals the uniform distribution of sulphate over the whole nanosheet.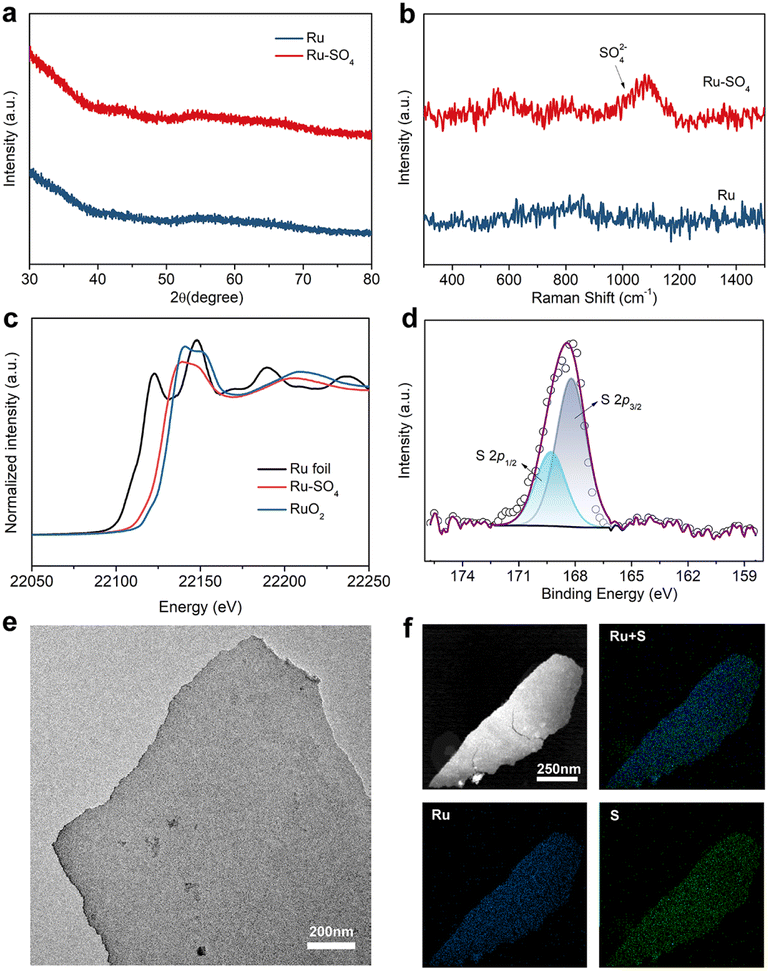 | ||
| Fig. 1 (a) X-ray diffraction patterns of Ru and Ru-SO4. (b) Raman spectra of Ru and Ru-SO4. (c) Ru K-edge XANES of Ru foil, Ru-SO4 and RuO2. (d) XPS spectra of S 2p for Ru-SO4. (e) Typical TEM image and (f) EDX mapping images for Ru-SO4. | ||
The electrocatalytic performances of Ru and Ru-SO4 catalysts for HOR activities are tested using a rotating disk electrode (RDE) in a H2-saturated 0.1 M KOH electrolyte. As depicted in Fig. 2a, typical peaks near 0.1 V are observed in the cyclic voltammetry (CV) curves of Ru and Ru-SO4, presenting the iconic CV curves of Ru-based catalysts.32 The iR-corrected linear sweep voltammetry (LSV) curves of Ru-SO4, Ru and Pt are acquired at a rotating speed of 1600 rpm in a H2-saturated 0.1 M KOH electrolyte. The electrochemically active surface areas (ECSAs) are conducted by Cu underpotential deposition (Cu-UPD) stripping for calculating the exchange current density (j0,s) (Fig. S7†).33 After normalized by ECSA, Ru-SO4 presents the highest current density of 0.543 mA cmPGM−2 among all samples (Fig. 2b and d), showing great improvement compared with Ru and Pt. Fig. 2c demonstrates the kinetic current density (jk) versus the potential in logarithm on Ru-SO4, Ru and Pt, according to the Butler–Volmer equation.34,35 The corresponding HOR polarization curves of Ru-SO4, Pt and Ru at various rotating rates are collected in Fig. S8, 9 and 10.† With normalizing the kinetic current density (denoted as jk,m) by the actual loading mass of catalysts (which can be collected through the inductively coupled plasma atomic emission spectroscopy (ICP-AES) results shown in Table S1†), Ru-SO4 presents an outstanding mass activity of 1182.2 mA mgPGM−1 at 50 mV, which is four- and two-fold enhancement compared with that of pristine Ru and Pt, respectively (Fig. 2d). It is worth noting that the exchange current density and mass activity of Ru-SO4 outperform most of the reported noble metal-based HOR electrocatalysts (Fig. 2e and Table S2†). Besides, the exchange current density normalized by corresponding metal loadings (j0,m) is also depicted in Fig. S11 and Table S3,† with the fact that the j0,m of Ru-SO4 is higher than that of Ru and Pt. Furthermore, the HOR curve of Ru in 0.1 M KOH with 0.1 mM K2SO4 shows no difference with Ru in 0.1 M KOH (Fig. S12†), highlighting that the HOR performance enhancement originates from the adsorbed sulfate on the surface. The durability of Ru-SO4 is also investigated by the accelerated degradation test (ADT).36 As shown in Fig. 2f, no obvious deviation can be observed in the HOR curves before and after 1000 cycles. Moreover, the 1000th CV curve is also maintained well compared with the 1st CV curve. However, the HOR activity of Ru demonstrates a relatively pronounced attenuation under the same test as depicted in Fig. S13,† implying enhanced stability after introducing sulfate. The XRD and XPS of Ru-SO4 after the ADT show that the amorphous structure and valence of Ru and S are both maintained well (Fig. S14 and 15†). The Raman spectra and EDX mapping also prove that the sulfate still evenly distributes on the surface of Ru. (Fig. S16 and 17†).
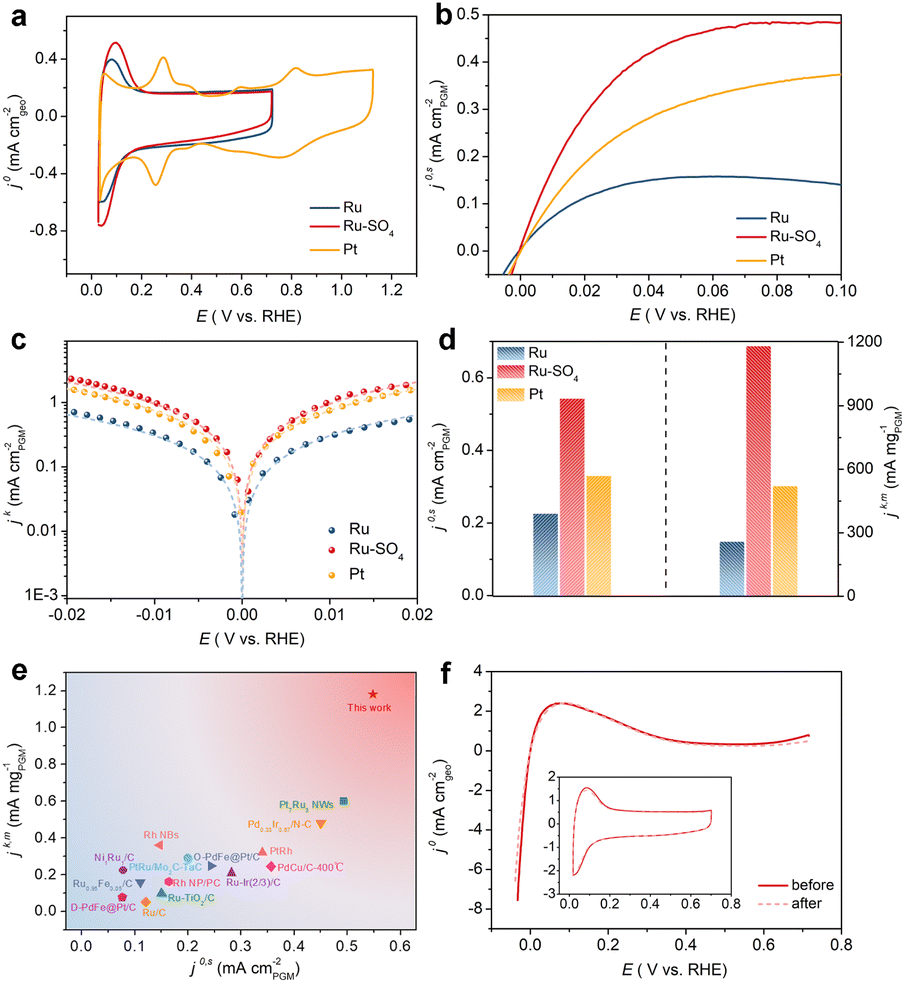 | ||
| Fig. 2 (a) CV curves of Ru, Ru-SO4 and Pt catalysts at a scanning rate of 50 mV s−1 in Ar-saturated 0.1 M KOH. (b) The HOR polarization curves of Ru, Ru-SO4 and Pt catalysts with a scan rate of 10 mV s−1 at the rotating speed of 1600 rpm in H2-saturated 0.1 M KOH. (c) Tafel plots derived from HOR polarization curves of Ru, Ru-SO4 and Pt fitted by the Butler–Volmer equation. (d) Comparation of the exchange current densities (j0,s) and the mass activities (jk,m) (@ 50 mV) for Ru, Ru-SO4 and Pt catalysts. (e) Comparation of jk,m and j0,s of Ru-SO4 with those of noble metal-based electrocatalysts reported in the literature. Details are summarized in Table S2.† (f) The HOR polarization curves at the rotating speed of 1600 rpm in H2-saturated 0.1 M KOH before and after 1000 cycles. The inset figure shows the CV curves in Ar-saturated 0.1 M KOH before and after 1000 cycles. | ||
To identify adsorption behaviors after introducing sulfate, in situ electrochemical impedance spectroscopy (EIS) measurements are conducted. Fig. 3a shows the Nyquist plots of the impedance of the reaction procedure on Ru and Ru-SO4 in the potential range from 0 V to 0.1 V with an interval of 0.01 V. The Nyquist plots are fitted as a double-parallel equivalent circuit model with uncompensated solution resistance (Rs) as shown in Fig. 3b.37,38 The small semicircle represents a high frequency section (CPE1 and Rct), while the large semicircle serves as a low frequency section (CPE2 and Rp). The migration of reactants and intermediates to the active sites on the electrolyte–catalyst interface can be reflected at the high frequency section.39 The values of Rct for Ru-SO4 are much lower than those of pristine Ru at all potentials (Fig. S18†), suggesting that the reactant H2 molecules can move more easily from the bulk electrolyte across the inter Helmholtz plane (IHP) to the surface of the catalyst, thereby leading to an enhanced reaction process.40,41 In addition, as shown in Fig. 3b, the double layer capacitances (Cdl) at all potentials related to the high frequency time constant of Ru-SO4 are higher than those of Ru,42 revealing the enhancement of oxygen-containing species coverage, which is consistent with the calculation results (vide infra).43 The low frequency time constant depicts the hydrogen adsorption behavior on the interface, as reflected by Rp.44–46 As shown in Fig. 3c, Ru-SO4 displays much higher resistance values at relatively low potentials (insert of Fig. 3c) after introducing sulphate species, suggesting a decreased amount of adsorbed H* on the surface of Ru-SO4 compared to pristine Ru.11,18,47 Furthermore, CO stripping experiments and zeta potential characterization are carried out to investigate the adsorption ability of OH*. As shown in Fig. 3d, the CO-stripping peak of Ru-SO4 is obviously lower than that of Ru, implying an enhancement of adsorption of OH species through sulphate functionalization.48,49 Zeta potential tests shown in Fig. 3e indicate that Ru-SO4 possesses a much lower potential than the pristine Ru, further indicating promoted OH adsorption.48,50 Moreover, interfacial water on the surface of the catalyst is investigated by in situ Raman spectroscopy in the potential range of 0 mV to 140 mV (Fig. 3f). With the increase of potential, the intensity of two peaks located at ∼3200 cm−1 and ∼3400 cm−1 is gradually enhanced. It can be seen unambiguously that the peak located at ∼3200 cm−1 shows much higher intensity for Ru, suggesting that tetrahedrally coordinated H-bonded water molecules are mainly formed on the surface of Ru.51,52 In contrast, after introducing sulphate, a peak is located around 3400 cm−1 which corresponds to active trihedrally coordinated H-bonded water molecules mainly formed on the surface, suggesting the promoted formation process of water.24,51,52
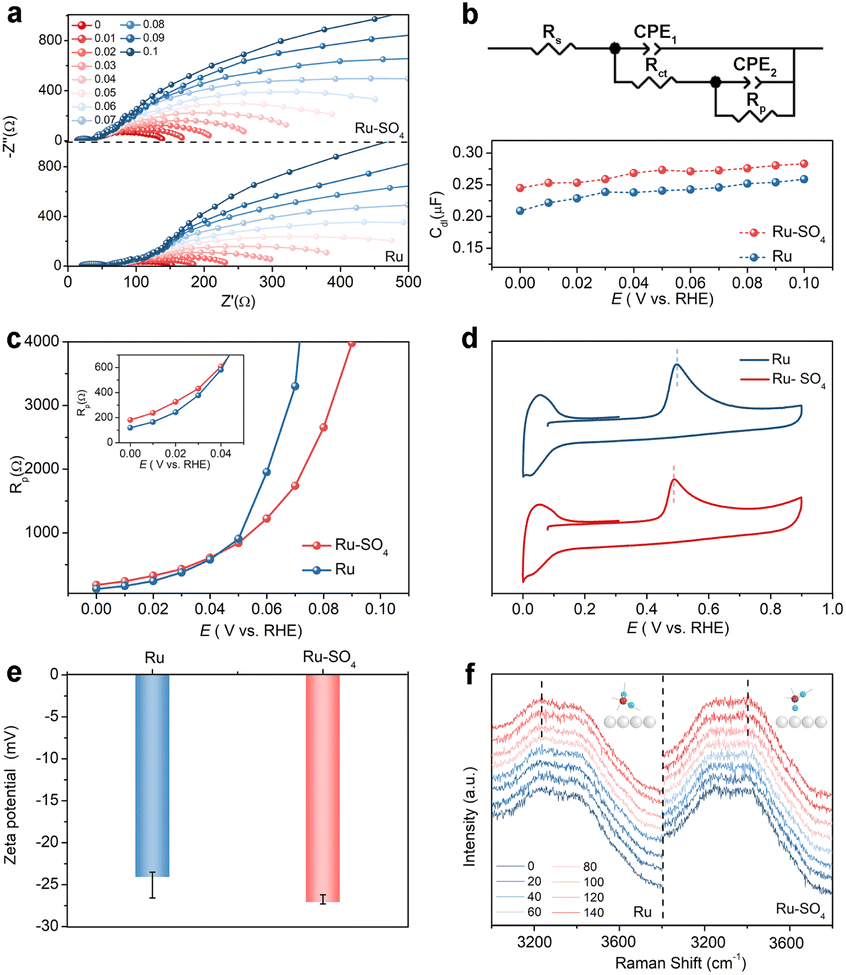 | ||
| Fig. 3 (a) Nyquist plots of Ru and Ru-SO4 in the potential range from 0 V to 0.1 V. (b) The equivalent circuit and the Cdl at different potentials for Ru and Ru-SO4. (c) The Rp at different potentials for Ru and Ru-SO4. (d) CO stripping curves in CO-statured 0.1 M KOH. (e) Zeta potentials of Ru and Ru-SO4. (f) In situ Raman spectra of interfacial water on Ru and Ru-SO4 in 0.1 M KOH at 0 mV to 140 mV. | ||
Based on the improvement of HOR activities of Ru-SO4, the hydrogen evolution reaction (HER) activities under an alkaline electrolyte were also explored. Fig. S19a† shows the HER polarization curves of Ru, Ru-SO4 and Pt in 0.1 M KOH. Ru-SO4 exhibits much faster reaction kinetics than Ru and Pt, with an overpotential of 41.77 mV at a current density of 10 mA cm−2. In Fig. S19b,† the Tafel slope value of Ru-SO4 is also smaller than that of Ru and Pt, implying accelerated reaction kinetics after sulphate functionalization.53 Fig. S19c and d† show that Ru-SO4 possesses the best mass activities at different potentials. The same tendency is shown when the electrolyte is changed to 1 M KOH. Ru-SO4 displays the best activity, with an overpotential of 16.86 mV at 10 mA cm−2 (Fig. S20†). The alkaline HER activity of Ru-SO4 outperforms most of the PGM-based HER catalysts in previous literature (Table S4†). In addition, Ru-SO4 can maintain a constant overpotential at a current density of 10 mA cm−2 for 6 h with unchanged structure in both 0.1 M KOH and 1 M KOH (Fig. S21 and 22†), showing excellent stability.
To further investigate the origin of enhanced HOR activity on Ru-SO4, density functional theory (DFT) calculations are employed. We expand the surfaces in the (001) direction for Ru and add sulphate on the surface of Ru as the model of Ru-SO4 (Fig. S23†). As presented in Fig. 4a, O atoms in sulphate tend to draw electrons from Ru atoms at the surface, resulting in charge redistribution on the interface, as well as modulated adsorption of intermediates.54 As shown in Fig. 4b and c, with the functionalization of sulphate, the distance from H2 to the surface of Ru is reduced from 3.38 Å to 1.50 Å, indicating the key role of sulphate species in facilitating the transfer of reactant H2.41,55 In addition, Fig. S24 and 25† show that the calculated hydrogen adsorption free energy (ΔGH) of Ru-SO4 is much close to 0 compared with pristine Ru. Fig. 4d and S26† indicate that the OH binding energy (OHBE) on Ru-SO4 is calculated to be −0.377 eV, much lower than that of pristine Ru. These results indicate that introducing sulphate species could lower the hydrogen binding energy and promote the OH binding energy of Ru, agreeing well with the in situ EIS observations. The projected density of states (PDOS) shown in Fig. 4e indicates that the d-band center (Ed) of Ru-SO4 is much close to the Fermi level (Ef) compared with Ru, suggesting fewer antibonding orbitals occupied on Ru-SO4, which is responsible for the enhanced adsorption of OH.56,57 The calculation of more sulfate on the Ru surface has also been taken into consideration. (Fig. S27–31†) The increased amount of sulfate may intensify the charge distribution on the surface, further optimizing the OHBE and HBE. Furthermore, the reaction pathways of Ru and Ru-SO4 for alkaline HOR are presented in Fig. 4f.58 The first two steps of adsorption of OH* or H* and adsorption of H* + OH* are both spontaneous for Ru and Ru-SO4 (Fig. S32†), while the water desorption steps for Ru (0.05 eV) and Ru-SO4 (0.572 eV) are endothermic (Fig. S33†). The water formation step is endothermic for Ru with a potential barrier of 0.58 eV, while it is exothermic for Ru-SO4. In addition, the activation energies of the transition state (TS) of water formation for Ru and Ru-SO4 are calculated to be 1.27 eV and 0.93 eV (Fig. S34 and 35†), implying that the rate determining steps for both catalysts contribute to the water formation step.59 The apparent lower energy barriers of the RDS for Ru-SO4 after introducing sulphate species lead to the enhancement of HOR performance.
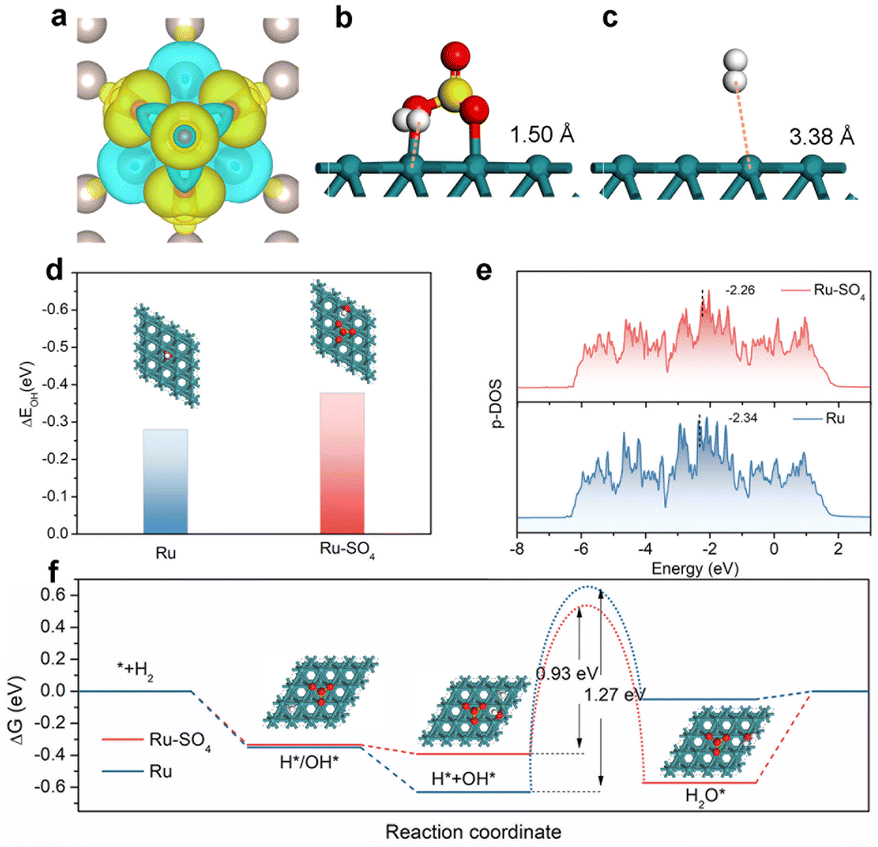 | ||
| Fig. 4 (a) Calculated differential charge density for Ru-SO4. The blue and yellow areas correspond to the depletion and accumulation of electrons, respectively. The optimal theoretical structure of H2 on Ru-SO4 (b) and Ru (c). (d) The adsorption energy of OH* on Ru-SO4 and Ru with the corresponding optimal theoretical structure. The projected density of states (e) and the reaction pathways (f) of Ru and Ru-SO4. | ||
Conclusions
In conclusion, we successfully constructed sulfate functionalized Ru nanosheets (Ru-SO4) through a simple low temperature sulfidation approach. As expected, the obtained Ru-SO4 displays much enhanced stability and electrocatalytic performance toward the HOR under an alkaline electrolyte, with a mass activity of 1182.2 mA mgPGM−1, which is a four-fold enhancement compared with that of pristine Ru. Experimental studies including in situ electrochemical impedance spectroscopy and in situ Raman spectrscopy, and density functional theory (DFT) calculations reveal that the redistribution of electrons on the interface of Ru-SO4 derived from sulphate functionalization could significantly optimize the adsorption energies of H and OH, facilitate H2 molecule transfer from the bulk electrolyte across the inter Helmholtz plane to the surface of the catalyst, and regulate the structure of interfacial water molecules, which lead to a decreased energy barrier of water formation, contributing to an enhanced HOR performance under alkaline electrolytes. This strategy sheds new light for rational design of advanced alkaline HOR electrocatalysts through regulating water formation and can be extended to precise modification of other electrocatalysts for various applications.Data availability
The data underlying this study are available in the ESI.†Author contributions
CY, YL, and JY performed the material synthesis and electrochemical tests. CY performed the DFT calculations. HC performed TEM characterization. WL supervised the work. CY and WL wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support from the National Natural Science Foundation of China (22272121 and 21972107), National Key Research and Development program of China (2021YFB4001200), Fundamental Research Funds for the Central Universities (2042022kf1179), and Natural Science Foundation of Hubei Province (2020CFA095). We thank the Core facility of Wuhan University for the measurements of ICP-AES and XPS. We also thank the Core Research Facilities of College of Chemistry and Molecular Sciences of Wuhan University. The numerical calculations have been done on the supercomputing system in the Supercomputing Center of Wuhan University. The authors also thank the BL14W1 in the Shanghai Synchrotron Radiation Facility (SSRF). We acknowledge the great help from Prof. L. Zhuang at Wuhan University for helpful measurements and discussion.Notes and references
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- N. Govindarajan, G. Kastlunger, H. H. Heenen and K. Chan, Chem. Sci., 2022, 13, 14–26 RSC.
- B. P. Setzler, Z. Zhuang, J. A. Wittkopf and Y. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS PubMed.
- H. E. Kim, J. Kwon and H. Lee, Chem. Sci., 2022, 13, 6782–6795 RSC.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- W. C. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- X. Xiao, L. Tao, M. Li, X. Lv, D. Huang, X. Jiang, H. Pan, M. Wang and Y. Shen, Chem. Sci., 2018, 9, 1970–1975 RSC.
- L. Su, D. Gong, Y. Jin, D. Wu and W. Luo, J. Energy Chem., 2022, 66, 107–122 CrossRef CAS.
- L. Gao, Y. Wang, H. Li, Q. Li, N. Ta, L. Zhuang, Q. Fu and X. Bao, Chem. Sci., 2017, 8, 5728–5734 RSC.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- J. Zheng, W. C. Sheng, Z. B. Zhuang, B. J. Xu and Y. S. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- E. Skulason, V. Tripkovic, M. E. Bjorketun, S. Gudmundsdottir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jonsson and J. K. Norskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- J. Li, S. Ghoshal, M. K. Bates, T. E. Miller, V. Davies, E. Stavitski, K. Attenkofer, S. Mukerjee, Z. F. Ma and Q. Jia, Angew. Chem., Int. Ed., 2017, 56, 15594–15598 CrossRef CAS PubMed.
- F. Yang, P. Han, N. Yao, G. Cheng, S. Chen and W. Luo, Chem. Sci., 2020, 11, 12118–12123 RSC.
- M. Li, L. Li, X. Huang, X. Qi, M. Deng, S. Jiang and Z. Wei, J. Phys. Chem. Lett., 2022, 13, 10550–10557 CrossRef CAS PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- S. Intikhab, J. D. Snyder and M. H. Tang, ACS Catal., 2017, 7, 8314–8319 CrossRef CAS.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900–911 CrossRef CAS.
- K. Zhao, X. Chang, H. S. Su, Y. Nie, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202207197 CrossRef CAS PubMed.
- G. Wu, X. Zheng, P. Cui, H. Jiang, X. Wang, Y. Qu, W. Chen, Y. Lin, H. Li, X. Han, Y. Hu, P. Liu, Q. Zhang, J. Ge, Y. Yao, R. Sun, Y. Wu, L. Gu, X. Hong and Y. Li, Nat. Commun., 2019, 10, 4855 CrossRef PubMed.
- F. H. Saadi, A. I. Carim, W. S. Drisdell, S. Gul, J. H. Baricuatro, J. Yano, M. P. Soriaga and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 12927–12930 CrossRef CAS PubMed.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- S. Anantharaj, M. Jayachandran and S. Kundu, Chem. Sci., 2016, 7, 3188–3205 RSC.
- Y. Zhu, H. A. Tahini, Y. Wang, Q. Lin, Y. Liang, C. M. Doherty, Y. Liu, X. Li, J. Lu, S. C. Smith, C. Selomulya, X. Zhang, Z. Shao and H. Wang, J. Mater. Chem. A, 2019, 7, 14222–14232 RSC.
- Y. Xue, J. Fang, X. Wang, Z. Xu, Y. Zhang, Q. Lv, M. Liu, W. Zhu and Z. Zhuang, Adv. Funct. Mater., 2021, 31, 2101405 CrossRef CAS.
- G. Mirabello, M. GoodSmith, P. H. H. Bomans, L. Stegbauer, D. Joester and G. de With, Chem. Sci., 2021, 12, 9458–9465 RSC.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef CAS PubMed.
- C. L. Green and A. Kucernak, J. Phys. Chem. B, 2002, 106, 1036–1047 CrossRef CAS.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, e1808066 CrossRef PubMed.
- Y. Xue, L. Shi, X. Liu, J. Fang, X. Wang, B. P. Setzler, W. Zhu, Y. Yan and Z. Zhuang, Nat. Commun., 2020, 11, 5651 CrossRef CAS PubMed.
- H. Wang and H. D. Abruna, J. Am. Chem. Soc., 2017, 139, 6807–6810 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- A. Damian and S. Omanovic, J. Power Sources, 2006, 158, 464–476 CrossRef CAS.
- C. Xie, W. Chen, S. Du, D. Yan, Y. Zhang, J. Chen, B. Liu and S. Wang, Nano Energy, 2020, 71, 104653 CrossRef CAS.
- L. Tang, M. Xia, S. Cao, X. Bo, S. Zhang, Y. Zhang, X. Liu, L. Zhang, L. Yu and D. Deng, Nano Energy, 2022, 101, 107562 CrossRef CAS.
- Q. Wen, Y. Lin, Y. Yang, R. Gao, N. Ouyang, D. Ding, Y. Liu and T. Zhai, ACS Nano, 2022, 16, 9572–9582 CrossRef CAS PubMed.
- H. Yang, Y. Zhao, Q. Wen, Y. Mi, Y. Liu, H. Li and T. Zhai, Nano Res., 2021, 14, 4814–4821 CrossRef CAS.
- R. L. Doyle and M. E. G. Lyons, J. Electrochem. Soc., 2013, 160, H142–H154 CrossRef CAS.
- R. Simpraga, G. Tremiliosi-Filho, S. Y. Qian and B. E. Conway, J. Electroanal. Chem., 1997, 424, 141–151 CrossRef CAS.
- N. V. Krstajic, B. N. Grgur, N. S. Mladenovic, M. V. Vojnovic and M. M. Jaksic, Electrochim. Acta, 1997, 42, 323–330 CrossRef CAS.
- J. Li, H.-X. Liu, W. Gou, M. Zhang, Z. Xia, S. Zhang, C.-R. Chang, Y. Ma and Y. Qu, Energy Environ. Sci., 2019, 12, 2298–2304 RSC.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- L. F. Shen, B. A. Lu, X. M. Qu, J. Y. Ye, J. M. Zhang, S. H. Yin, Q. H. Wu, R. X. Wang, S. Y. Shen, T. Sheng, Y. X. Jiang and S. G. Sun, Nano Energy, 2019, 62, 601–609 CrossRef CAS.
- S. Lu and Z. Zhuang, J. Am. Chem. Soc., 2017, 139, 5156–5163 CrossRef CAS PubMed.
- Y. Duan, Z. Y. Yu, L. Yang, L. R. Zheng, C. T. Zhang, X. T. Yang, F. Y. Gao, X. L. Zhang, X. Yu, R. Liu, H. H. Ding, C. Gu, X. S. Zheng, L. Shi, J. Jiang, J. F. Zhu, M. R. Gao and S. H. Yu, Nat. Commun., 2020, 11, 4789 CrossRef CAS PubMed.
- L. F. Shen, B. A. Lu, Y. Y. Li, J. Liu, Z. C. Huang-Fu, H. Peng, J. Y. Ye, X. M. Qu, J. M. Zhang, G. Li, W. B. Cai, Y. X. Jiang and S. G. Sun, Angew. Chem., Int. Ed., 2020, 59, 22397–22402 CrossRef CAS PubMed.
- K. Ataka, T. Yotsuyanagi and M. Osawa, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- L. Peng, M. Liao, X. Zheng, Y. Nie, L. Zhang, M. Wang, R. Xiang, J. Wang, L. Li and Z. Wei, Chem. Sci., 2020, 11, 2487–2493 RSC.
- K. Wu, K. Sun, S. Liu, W.-C. Cheong, Z. Chen, C. Zhang, Y. Pan, Y. Cheng, Z. Zhuang, X. Wei, Y. Wang, L. Zheng, Q. Zhang, D. Wang, Q. Peng, C. Chen and Y. Li, Nano Energy, 2021, 80, 105467 CrossRef CAS.
- Z. Luo, H. Zhang, Y. Yang, X. Wang, Y. Li, Z. Jin, Z. Jiang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2020, 11, 1116 CrossRef CAS PubMed.
- B. Hammer, O. H. Nielsen and J. K. Norskov, Catal. Lett., 1997, 46, 31–35 CrossRef CAS.
- B. Hammer and J. K. Norskov, Adv. Catal., 2000, 45, 71–129 CAS.
- X. Yang, Y. Wang, X. Wang, B. Mei, E. Luo, Y. Li, Q. Meng, Z. Jin, Z. Jiang, C. Liu, J. Ge and W. Xing, Angew. Chem., Int. Ed., 2021, 60, 26177–26183 CrossRef CAS PubMed.
- Y. Zhao, D. Wu and W. Luo, ACS Sustainable Chem. Eng., 2022, 10, 1616–1623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02144k |
| This journal is © The Royal Society of Chemistry 2023 |