
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocycle of a cyanobacteriochrome: a charge defect on ring C impairs conjugation in chromophore†
Lisa
Köhler
 a,
Wolfgang
Gärtner
a,
Georgeta
Salvan
b,
Jörg
Matysik
*a,
Christian
Wiebeler
ac and
Chen
Song
*a
a,
Wolfgang
Gärtner
a,
Georgeta
Salvan
b,
Jörg
Matysik
*a,
Christian
Wiebeler
ac and
Chen
Song
*a
aInstitut für Analytische Chemie, Universität Leipzig, 04103 Leipzig, Germany. E-mail: joerg.matysik@uni-leipzig.de; chen.song@uni-leipzig.de
bInstitut für Physik, Technische Universität Chemnitz, 09126 Chemnitz, Germany
cWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, 04103 Leipzig, Germany
First published on 11th May 2023
Abstract
A large number of novel phytochromes named cyanobacteriochromes (CBCRs) have been recently identified. CBCRs appear to be attractive for further in-depth studies as paradigms for phytochromes because of their related photochemistry, but simpler domain architecture. Elucidating the mechanisms of spectral tuning for the bilin chromophore down to the molecular/atomic level is a prerequisite to design fine-tuned photoswitches for optogenetics. Several explanations for the blue shift during photoproduct formation associated with the red/green CBCRs represented by Slr1393g3 have been developed. There are, however, only sparse mechanistic data concerning the factors controlling stepwise absorbance changes along the reaction pathways from the dark state to the photoproduct and vice versa in this subfamily. Conventional cryotrapping of photocycle intermediates of phytochromes has proven experimentally intractable for solid-state NMR spectroscopy within the probe. Here, we have developed a simple method to circumvent this hindrance by incorporating proteins into trehalose glasses which allows four photocycle intermediates of Slr1393g3 to be isolated for NMR use. In addition to identifying the chemical shifts and chemical shift anisotropy principal values of selective chromophore carbons in various photocycle states, we generated QM/MM models of the dark state and photoproduct as well as of the primary intermediate of the backward-reaction. We find the motion of all three methine bridges in both reaction directions but in different orders. These molecular events channel light excitation to drive distinguishable transformation processes. Our work also suggests that polaronic self-trapping of a conjugation defect by displacement of the counterion during the photocycle would play a role in tuning the spectral properties of both the dark state and photoproduct.
Introduction
CBCRs comprise a subgroup in the phytochrome superfamily which photoregulates many aspects of cyanobacterial lifestyle such as phototaxis or the transition between planktonic and sessile lifestyles.1 Their photochemically active linear tetrapyrrole chromophores, such as phycocyanobilin (PCB), phytochromobilin (PΦB), phycoviolobilin (PVB) and biliverdin (BV), are covalently linked to a conserved Cys residue in a c![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) phosphodiesterase/
phosphodiesterase/![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) denylate cyclase/
denylate cyclase/![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) hlA (GAF) domain via a thioether linkage.1 Common to all phytochromes, the C15,C16-double bond (see Fig. 1A for numbering) isomerization is the primary photochemical event upon switching between the dark state and photoproduct.2 Unlike canonical phytochromes, CBCRs combine all phytochrome-specific properties in a single bilin-dependent GAF domain instead of the
hlA (GAF) domain via a thioether linkage.1 Common to all phytochromes, the C15,C16-double bond (see Fig. 1A for numbering) isomerization is the primary photochemical event upon switching between the dark state and photoproduct.2 Unlike canonical phytochromes, CBCRs combine all phytochrome-specific properties in a single bilin-dependent GAF domain instead of the ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) er/
er/![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) RNT/
RNT/![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) im (PAS)–GAF–ph
im (PAS)–GAF–ph![[y with combining low line]](https://www.rsc.org/images/entities/char_0079_0332.gif) tochrome (PHY) tridomain construct. Moreover, CBCRs exhibit an astonishing spectral variety covering the entire spectral range from near UV to near infrared,3 whereas canonical phytochromes predominantly respond to the red/far-red spectral region.2,4 The compact size as well as the spectral diversity makes CBCRs suited experimental paradigms to study structure–function relations in canonical phytochromes in general, and also attractive templates for developing optogenetic tools;3 however, the spectral tuning accompanying the phototransformations is not sufficiently well understood.3
tochrome (PHY) tridomain construct. Moreover, CBCRs exhibit an astonishing spectral variety covering the entire spectral range from near UV to near infrared,3 whereas canonical phytochromes predominantly respond to the red/far-red spectral region.2,4 The compact size as well as the spectral diversity makes CBCRs suited experimental paradigms to study structure–function relations in canonical phytochromes in general, and also attractive templates for developing optogenetic tools;3 however, the spectral tuning accompanying the phototransformations is not sufficiently well understood.3
 | ||
| Fig. 1 Structure characteristics of Slr1393g3. (A) Schematic of an Slr1393g3-bound PCB chromophore adopting a ZZZssa geometry in the Pr dark state. The possible π-conjugated system is highlighted in red. (B) QM/MM-optimized models of Slr1393g3 in the Pr dark state (magenta) and the Pg photoproduct (green). The chromophore-binding pocket was subjected to QM/MM geometry optimization using 310 and 331 atoms in the QM region for Pr and Pg, respectively (see Fig. S1 and S2†). (C) Zoomed-in views of the PCB chromophore and its interaction with the binding pocket in both photostates (color coding as in (B)). Hydrogen bonds are indicated by black dashed lines. The Pg model suggests an influx of water molecules (shown as green spheres) into the pocket, whereas the Pr chromophore is more shielded from the solvent (note the limited water molecules shown as magenta spheres). | ||
Notable progress in this aspect has recently been made using the third GAF domain of one representative red/green CBCR, Slr1393 from Synechocystis PCC6803, composed of three GAF domains and carrying a histidine kinase signaling module at its C-terminal end.5 Only the third GAF domain (g3) binds a PCB chromophore, and the spectral properties of the full-length protein are preserved, even if this domain (Slr1393g3) is selectively expressed (Fig. 1B and C). Slr1393g3 exhibits a strong photochromicity, i.e., a shift of 116 nm between the absorption maxima (λmax) of the red-absorbing Pr dark state and the green-absorbing Pg photoproduct.6 Recently resolved crystal structures of Slr1393g3 in both photostates7 served as a basis for high-quality quantum mechanics/molecular mechanics (QM/MM) calculations to simulate λmax values which are in exact agreement with the experimental data.8,9 The theoretical work on this protein also demonstrated that the effective length of the conjugated system and the planarity of the chromophore are decisive for the spectral tuning of the photoproduct, consistent with the original ‘D-ring control’ model proposed by Rockwell, Lagarias and colleagues for both teal-DXCF and red/green CBCR photoproducts.10,11
Despite tantalizing clues about the spectral tuning mechanism for this CBCR subfamily, the basis to provide a universally valid explanation of the electronic origin of the absorption spectra and drastic spectral changes is still insufficient due to the lack of a fundamental understanding of the factors that modulate the absorption properties of the photocycle intermediates. In both Pr → Pg and Pg → Pr phototransformations of red/green CBCRs, a total of at least four intermediates have been characterized by transient absorption and vibrational spectroscopy under cryoconditions.12–16 In principle, magic-angle spinning (MAS) NMR spectroscopy provides details with outstanding precision for conformational heterogeneity and the electronic structure of the chromophore as well as its interactions with the binding pocket in phytochromes and CBCRs.17–20 Moreover, a number of open questions such as charge distribution and protonation dynamics associated with the chromophore are experimentally accessible through the analysis of its chemical shifts and chemical shift anisotropy (CSA) principal values. However, to date it remains a daunting challenge to trap the photocycle intermediates within the pre-cooled MAS probe with in situ illumination.21 This holds in particular for the forward-reaction of Slr1393g3, in which cooling the sample to −20 °C causes the onset of a thermal conversion of its red-absorbing Pr dark state to an orange-absorbing form.22 Moreover, severe photochemical heterogeneity was reported for the Pr state of this protein,12,23 making NMR experiments under cryotrapping conditions more complicated than considered so far.
The present study exploits the benefits of our recently developed trehalose glass (TG) sample preparation method24 to explore both transformation processes of Slr1393g3 aiming at a deeper and more generalizing understanding of the factors that modulate the absorption properties of red/green CBCRs. The advantages of TG matrices allowed isolation of individual transient reaction intermediates of Slr1393g3 and their measurement separately under noncryogenic conditions. On the plateau of this success, we performed UV-vis and MAS NMR measurements on six TG-trapped Slr1393g3 photocycle states including four intermediates (with distinct absorption ‘colors’) for absorption properties, chemical shifts and CSA principal values of the chromophore during the entire photocycle. In parallel, further refined QM/MM-optimized structures of the Pr dark state and Pg photoproduct (Fig. 1B) as well as an analog model for the primary intermediate of the backward-reaction, Lumi-G, were generated. Besides the prevailing ‘D-ring control’ hypothesis,8 our work underscores the significance of the positive charge distribution inside the chromophore to the conjugation length during the photocycle, particularly for the absorbance changes associated with the final transition from Meta-R to Pg as well as that from Meta-G to Pr.
Results
Photocycle intermediates of Slr1393g3 in TGs
We previously reported the long-term preservation of diverse short-lived photoproducts of phytochromes and CBCRs by embedding the proteins into TG matrices at room temperature (RT).24 The initial motivation for generating binary trehalose–phytochrome/CBCR glasses was the extraordinary bioprotective capabilities of the trehalose towards labile proteins with respect to desiccation and denaturation as well as freezing and osmotic stresses.25–27 Here, taking advantages of trehalose matrices in protein immobilization and extraordinary bioprotection against denaturation at RT,24,25 we isolated four individual photocycle intermediates of Slr1393g3 (Fig. 2) and moreover, the trapped intermediate samples could be stored at room and even higher temperatures for periods as long as weeks. TG-embedded samples of all six photocycle states including four intermediates present different colors (Fig. 2) which are associated with the large absorbance changes during the photocycle with maxima lying in between 652 and 536 nm corresponding to Pr and Pg, respectively. | ||
| Fig. 2 Photocycle of trehalose-embedded Slr1393g3. Conditions for generating and trapping all photocycle states in both forward- and backward-reactions and their corresponding UV-vis spectra measured in TGs at RT are shown. Photographs of all six TG samples under ambient light and temperature conditions to present their absorption ‘colors’ are shown individually with the UV-vis spectra and assembled at the center of the cycle. The ‘Lumi-R-only’ spectrum (Lumi-R, thin line) was obtained by subtracting the Pr spectrum from that of the photoequilibrium mixture of Pr and Lumi-R (thick line). The line fitting of the Meta-R spectrum revealed three components (dashed lines). Detailed experimental procedures are presented in the ESI.† | ||
For the TG sample in the Pr dark state at 100% occupancy (Fig. 2, Pr), irradiation with 650 nm light at RT yields a steady-state mixture of Pr and the primary intermediate, Lumi-R of the Pr → Pg forward-reaction (Fig. 2, Lumi-R), as indicated by its broad absorption band centered at ∼643 nm. Complete removal of the Pr spectral contribution by line-fitting revealed the spectrum of ‘pure’ Lumi-R intermediate with a characteristic blue-shifted absorption band relative to Pr, in accordance with the transient absorption data of the same protein.12 By exploiting the formerly observed inverse temperature effect of the TG preparation,24 the transition to the late Meta-R intermediate was initialized upon cooling down the Lumi-R sample to −20 °C, however, the maximal Meta-R occupancy attainable at the equilibrium mixture was rather low (∼10%). It appears that the Lumi-R intermediate at low temperatures favors a thermal reaction back to Pr while not proceeding to Meta-R. This situation is not analogous to those in the solutions of phytochromes and CBCRs, in which the Meta-R intermediates can be enriched thermally.21,28,29 For the TG-embedded sample, the maximal Meta-R occupancy was achieved by illumination of the Pr-state TG with 650 nm light at −20 °C which causes the Pr transition to proceed beyond the Lumi-R intermediate. Line fitting of the resulting spectrum (Fig. 2, Meta-R) revealed the major component with λmax of 578 nm for Meta-R, together with two minor components at 652 and 536 nm originating from the residual Pr form and the subsequent Pg photoproduct, respectively. The appearance of the Pr component in the Meta-R spectrum provides further evidence for a rapid thermal decay from Lumi-R to Pr. This observation might explain why the low photochemical quantum yield of the forward-reaction amounted to only 0.08.30 Again, in contrast to solution samples, the final transition to form the Pg photoproduct of the TG sample can only proceed well upon further cooling to −80 °C. The resulting Pg absorption spectrum recorded at RT (Fig. 2, Pg) highly resembles that of the solution state (Fig. S3†).
Analogous to the forward-reaction, the Pg → Pr backward-reaction occurs via two intermediates as revealed by absorption spectroscopy. The backward-reaction, however, is not a mirror-symmetric process of the forward-reaction with the two intermediates Lumi-G and Meta-G absorbing at 564 and 579 nm, respectively (Fig. 2). Moreover, the Pg → Lumi-G primary photoreaction is initiated during the irradiation step of the Pg-state TG glass with 530 nm light at RT, and the subsequent transitions to form Pr proceed well with gradual cooling of the sample. Specifically, once the Lumi-G intermediate is formed (Fig. 2, Lumi-G), a decrease in sample temperature to −20 °C enables the progression to the late Meta-G intermediate with λmax of 579 nm (Fig. 2, Meta-G). Besides that, a broad shoulder centered at ∼650 nm is visible in the spectrum which can be ascribed to partial formation of the Pr state. Upon further cooling of the TG sample to −80 °C, the Meta-G intermediate is fully converted back to Pr (Fig. 2). Compared with the two intermediates in the Pr → Pg phototransformation, the absorption spectra of Lumi-G and Meta-G show higher occupancies with less ‘contamination’ of either residual Pg or subsequent Pr forms. Moreover, the unidirectional ratchet-like reaction pathway from Pg to Pr may account for the three-fold higher photochemical quantum yield relative to that of the Pr → Pg transformation.30 All TG intermediates described herein show almost identical spectral characteristics (e.g., λmax and absorbance ratios) as previous transient absorption data reported for this protein in the solution state.6,12,23
Conformational changes of the chromophore during the photocycle
Utilization of [5-13C]-δ-aminolevulinic acid (ALA) during biosynthesis of the PCB chromophore allows incorporation of 13C at all three methine bridge carbons (C5, C10, and C15) as well as at C4, C9, C11 and C19 of rings A–D (Fig. 3A). Complete 13C assignments of these PCB carbons in all six photocycle states trapped in TGs were achieved mainly using 13C–13C dipolar-assisted rotational resonance (DARR) experiments and the associated chemical shift changes (ΔδC) in both Pr → Pg and Pg → Pr reaction pathways are shown in Fig. 3B and Table S1.† The most prominent ΔδC changes during the forward-reaction was detected for the C10-methine carbon bridging the two inner rings B and C that are of −1.8, +2.4, and +3.7 ppm in the three transitions to form Pg. The overall large upfield shift of C10 upon Pg photoproduct formation (+4.3 ppm) is also evident in other red/green CBCR representatives such as AnPixJg2 from Anabaena31 and NpR6012g4 from Nostoc.32 However, detailed fractionized ΔδC changes during the formation of the intermediates could so far not been detected. Surprisingly, during the first two transitions, the neighboring carbons of rings B (C9) and C (C11) showed only minor ΔδC changes. It is thus clear that a deprotonation and reprotonation cycle associated with the chromophore (protonation dynamics) in the Meta-R intermediate and subsequent transition, as proposed for both canonical phytochromes6,13–15 and the red/green CBCR AnPixJg231 can be excluded for Slr1393g3, otherwise a drastic ΔδC change of C9 would be observed.33,34 Moreover, although it is widely accepted that the isomerization of the C15,C16-double bond is solely linked to the formation of the two primary intermediates, Lumi-R in the forward-reaction and Lumi-G in the backward-reaction,21 the ΔδC changes associated with the C15-methine carbon and D-ring carbonyl C19 in the Pr → Lumi-R photoreaction (−0.2 and +0.1 ppm, respectively) are much smaller than those seen in canonical phytochromes,21 implying that ring D is twisted away from the B–C plane in both states. Intriguingly, a larger ΔδC change of −0.6 ppm is seen for C19 in the subsequent Lumi-R → Meta-R transition which could be interpreted as arising due to a single-bond rotation around C14–C15.21 Pronounced ΔδC changes associated with this transition also occur further away at C4 (−1.4 ppm) and at the C5-methine bridge (+0.5 ppm), indicating an A-ring structural modification which may result from the twist of both C4–C5 and C5–C6 bonds. The overall change of the A-ring tilt angle relative to ring B is more than 40° during the Pr → Pg phototransformation, as found for the QM/MM structures of the Pr and Pg states (Fig. 1C and see ref. 7 for the calculation of the absolute tilt angle between two pyrrole rings). Amongst the three transitions of the forward-reaction, the formation of Pg from Meta-R causes the most significant ΔδC changes for all selected carbon atoms (Fig. 3B and Table S1†). Unlike C10 that is downshifted by +3.7 ppm, the C9 and C11 atoms associated with this methine bridge are oppositely shifted by −2.7 and −1.2 ppm, respectively. Moreover, there is a gradual decrease during the forward-reaction in peak intensities of DARR correlations from C9 and C10 to C4, particularly for the C4/C9 correlation that is essentially not resolved in the Pg spectrum (Fig. S4 and Table S2†). Considering that the interatomic distances between C4 and C9/C10 are nearly identical in the Pr and Pg photostates, a gradual increase in the chromophore mobility within its binding pocket would thus account for the observed intensity losses. Apart from the ΔδC changes around the C10-methine bridge, C4 and C5 are further shifted in the last transition by +1.0 and −1.6 ppm, respectively, suggesting that ring A is not yet fully relaxed in Meta-R. | ||
| Fig. 3 Changes in the electronic structure and conformational distributions of the chromophore during the photocycle. (A) PCB atoms labeled with [5-13C]-ALA (highlighted in yellow). (B) 13C chemical shifts for selected carbon atoms during both forward- and backward-reactions. Observed signal splittings of 13C resonances are indicated by small lines and the mean chemical shift values of multiple resonances from a given carbon are depicted as solid bars (summarized in Table S1†). For the sake of clarity, the resonances of C9 and C11 are shown as inverted and regular triangles, respectively. No correlations involving C15 and C19 are resolved in the DARR spectra (see Fig. S4† for the full spectra with complete assignment of all trapped states), only their chemical shifts are shown in the dashed boxes. (C) Changes in conformational flexibility of the C10-methine bridge. Enlarged DARR contour plots displaying the correlations of C9/C10 and C10/C11 in the Pr and Pg photostates (upper panel) as well as in the forward- (middle panel) and backward- (lower panel) reaction intermediates. Possible conformational states attained by the C9–C11 segment are superscripted with a, b, c, and so on (from high- to low-field). | ||
During the backward-reaction, however, the major ΔδC changes of most carbons are associated with the primary photoreaction from Pg to Lumi-G (Fig. 3B and Table S1†). The largest shifts of +2.9, −4.8, and +1.8 ppm were found for C9–C11 (Table S1†). Such an alternating shift pattern in this region was also observed in the last transition of the forward-reaction to form Pg, however, the sign of the pattern is inverted. Besides C9–C11, C5 is also affected during this transition, upshifted by 1.3 ppm. Most of the selected carbons in the subsequent transitions are shifted with |ΔδC| ≤ 0.5 ppm, the notable exceptions being C4 and C10. Following up the drastic change during the primary photoreaction (−4.8 ppm), C10 is again shifted by +1.2 and −0.7 ppm in the last two transitions back to Pr, as is also C4 in the last transition by +0.7 ppm.
Changes in chromophore conformational states during the photocycle
In addition to the state-related ΔδC changes of the PCB carbon atoms in both phototransformations, variations in distributions of their conformational states (subpopulations) are also seen, as represented by signal splittings of 13C resonances (Fig. 3B and Table S1†). During the forward-reaction, both the number of 13C split components and their resonance separations (defined as the largest ΔδC difference for all split components of a given carbon) are increased for almost all selected carbon atoms in a stepwise manner towards the Pg photoproduct. This observation implies a greater structural heterogeneity of the chromophore as a whole generated in the reaction course which strongly correlates with the modification of its interactions with the protein surroundings.7,11,35 In Pr, the C10 resonances are evidently tripled with a separation of 2.7 ppm, however, no structural variations of C9 and C11 were detected (Fig. 2C, upper panel). The resonance splittings of C9 and C11 were revealed in the first two transitions via their correlations with C10 (Fig. 3C, middle panel). Moreover, during the two transitions, the C10 resonances show a gradually increased number of split components (Fig. 3B and S4†). Intriguingly, although the 13C resonance splitting pattern at C10 in the last transition to form Pg is retained, their separation is 3.2 ppm larger than that in the second transition (Table S1†). For C4 and C5, the last two transitions are associated with the appearance of additional split components and larger resonance separations (Fig. 3B and Table S1†). The Pg photoproduct is the most structurally heterogeneous form in the photocycle which manifests itself as pronounced 13C resonance splittings. Specifically, a total of 21 split components in the Pg state were detected (Fig. 3B and Table S1†).In general, the backward-reaction leads to a reversed trend with an overall reduction in the number of 13C split components and their separations (Fig. 3B and C, bottom panels). The Pg → Lumi-G photoreaction, however, has little effect on decreasing the number of 13C split components but their separations are substantially narrowed. Taking C10 as an example, although two additional split components are found in Lumi-G, the overall separation for a total of seven split components is 2.8 ppm more narrower than that in Pg (Table S1†). The second transition of Lumi-G → Meta-G shows a subset of four split components for C10 with a 0.7 ppm reduction of resonance separation which is further reduced by 1.1 ppm during the last transition back to Pr, implying a stepwise release of the mechanical tension stored on this methine bridge.
QM/MM models for Pr, Pg and a Lumi-G intermediate and spectroscopic properties of the calculated structures
In addition to the NMR analysis, we performed QM/MM simulations on the Pr and Pg photostates as well as on a ‘Pg-like’ intermediate of Slr1393g3 (Fig. 4A). Briefly, the three structural models were constructed as follows: (i) generation of starting models based on the crystal structures (PDB codes 5DFX, 5M82, and 5M85 for Pr, Pg, and the ‘Pg-like’ intermediate, respectively), (ii) simulations along the lines of our recently established computational protocol,8 and (iii) QM/MM geometry optimizations of the chromophore-binding pocket using a large QM region including at least 310 atoms (see ESI, Experimental†). Based on the QM/MM-optimized solvated structural models, we calculated excited states and absolute shielding constants that provide further insights into the chromophore geometry, the extent of the conjugated system, and its interaction with the surrounding protein in the respective states. The QM/MM-optimized model of the ‘Pg-like’ intermediate retains nearly all structural features of the protein moiety in the Pg state (rmsd 0.45 Å over 157 Cα atoms). However, the chromophore is already isomerized to adopt a Z,anti-geometry at C15 (Fig. 4A). As in canonical phytochromes21 and red/green CBCRs,12,14 the C15-E/Z isomerization of the chromophore is known to occur during the first transition of backward-reaction, this hybrid structure would therefore be tentatively assigned to Lumi-G. As seen in the three models, ring D undergoes a facial disposition change in the reaction course from Pg (α-face) to Lumi-G (β-face) and thence to Pr (α-face). | ||
| Fig. 4 Structural and spectroscopic properties of Lumi-G. (A) QM/MM-optimized model of the Lumi-G intermediate in the Pg → Pr backward-reaction (dark brown) based on the Slr1393g3 5M85 structure using a QM region of 340 atoms (Fig. S5†). The Lumi-G intermediate model is superimposed on those of Pg (green) and Pr (magenta) and changes in geometry and D-ring facial disposition of the chromophore during the Pg → Pr backward-reaction are highlighted. (B) Calculated absorption spectra for the three QM/MM models (solid lines) as displayed in (A) superimposed on their experimental counterparts (dashed lines). (C) Calculated 13C chemical shift changes of all selected PCB carbon atoms between Pr, Lumi-G, and Pg photocycle states (i.e. Pg–Pr, Lumi-G–Pr, and Lumi-G–Pg) were plotted versus the experimental δiso changes upon transitions (summarized in Table S3†). Data were fitted by linear regression and the corresponding R2 coefficients, together with the P-values, are shown in the inset. | ||
These QM/MM-optimized structures were then taken as input geometries to calculate excited states.8,9 The convoluted spectra for both Pr and Pg fit the experimental data reasonably well (Fig. 4B), as we employed one single optimized geometry for each state which does not allow the line shape of absorption bands to be simulated. Despite that, the deviation of the Q-band λmax between the calculated and experimental values in the Pr state is 15 nm (0.05 eV) and is even negligible in the Pg state. The Q-band λmax obtained for the hybrid structure (most likely corresponding to Lumi-G) is at 594 nm, with a deviation of 30 nm (0.11 eV) from the experimental value, slightly larger than that of Pr, but still in reasonable agreement given the accuracy of excited state calculations. The extent of conjugation in the optimized structures thus seems to be a reasonable approximation for the conjugation of the chromophore in all three states. To further evaluate these models, the 13C absolute shielding constants were calculated with density functional theory (DFT) employing the gauge-including atomic orbital (GIAO) method which represents a more sensitive indicator of the chromophore geometry.36,37 The corresponding absolute shielding constants were validated with linear regression by plotting against the experimental isotropic chemical shifts, δiso (Fig. S6†). As we modeled only one optimized structure per photocycle state, we took the dominant subpopulation in case of structural heterogeneity for comparison, by extracting the experimental δiso values of the selected PCB carbons from the most intense 13C correlations in the DARR spectra (Fig. S4†). In both Pr and Pg photostates, the calculated absolute shielding constants of the selected PCB carbons correlate very well with the experimental values with squared correlation coefficients R2 ≥ 0.9972 and a deviation of ≤±0.09 from the ideal slope of −1. Taking the absolute shielding constants from one of the two models, the highest linear correlation with the experimental δiso values is only obtained when correlating it with the δiso data from the same form, i.e., either Pr or Pg. As expected, the shielding constants from the hybrid structure indeed show the strongest linear correlation with the experimental shifts from the Lumi-G intermediate (Fig. S6,† brown line) with the R2 value of 0.9970. It is now clear that this QM/MM-optimized model serves as a close structural approximation of Lumi-G (Fig. 4A). Although we demonstrated good agreement with respect to optical absorption and correlation of chemical shielding constants for all three photocycle states of Pr, Pg, and Lumi-G, the changes in chemical shifts associated with transitions are more intricate to describe. Performing linear regression analyses between experimental and theoretical changes in chemical shifts between two states (Fig. 4C), we obtained P-values of 0.003 (Pg–Pr), 0.030 (Lumi-G–Pr), and 0.067 (Lumi-G–Pg). Assuming that a P-value ≤0.050 is statistically significant, this is only fulfilled for the former two transitions involving the Pr dark state. Despite these discrepancies, the QM/MM-derived structural models provide at least some guidance in the discussion of the experimental results.
13C CSA tensor elements of C9 and C11 in various photocycle states
Besides the analysis of δiso changes of the chromophore in both phototransformations (Fig. 3 and 4), the CSA tensor elements for C9 and C11 were determined (Fig. 5). For Pr and the Lumi-R and Meta-R intermediates of the forward-reaction, the tensors were extracted from CSA line shapes in the indirect dimension of 2D separation of undistorted powder patterns by effortless recoupling (SUPER)38 spectra of the corresponding TGs. For the backward-reaction, we directly employed the calculated absolute shielding constants of Pr, Pg, and the Lumi-G intermediate for further analysis. The orientations of the experimental principal components (δ11, δ22, and δ33) for C9 and C11 could readily be assigned based on the results of the GIAO calculations (Fig. 5A). As depicted, δ11 and δ22 are in the plane of the pyrrole rings, with δ33 aligned normal to the plane. Specifically, the δ11 component lies approximately along the C–N σ-bond axis, while δ22 is oriented tangential to the inner rings. Unlike the two in-plane components, δ33 is dominated by the C–C σ-electronic structure. It should be noted that the three principal values are not affected by different line broadening applied to the best-fitted spectra of Pr, Lumi-G, and Meta-G (Fig. 5B). A plausible explanation for this difference is that both the two intermediates cannot be only trapped at 100% occupancy like Pr (Fig. 2), the reduced sample amount of the ‘pure’ Lumi-R and Meta-R would degrade the spectral resolution, and thus larger line broadening parameters were applied for suppressing noise.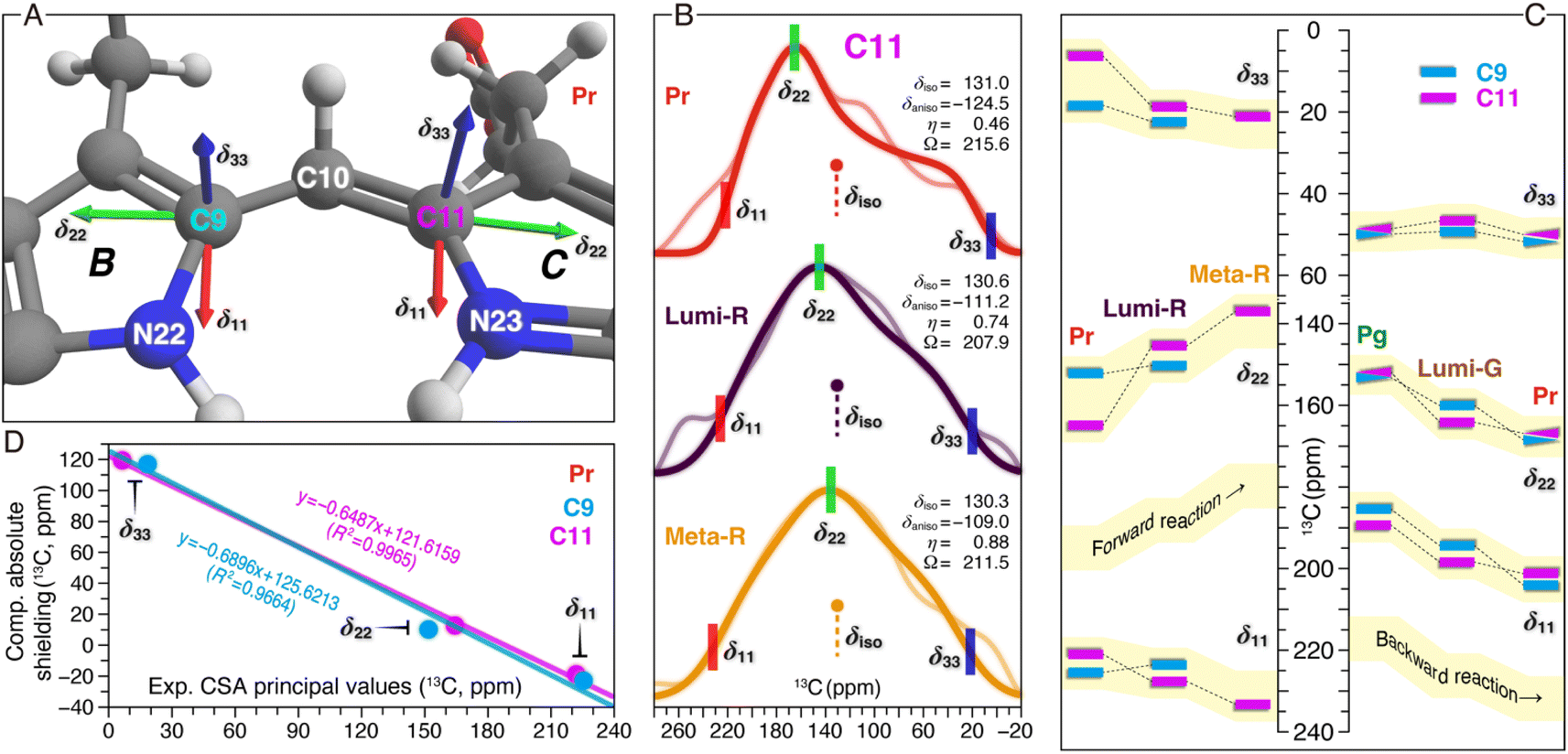 | ||
| Fig. 5 13C CSA tensor values of C9 and C11 in both reaction pathways. (A) Calculated orientations of the principal axes on the molecular frame of C9 (connecting N22 of ring B and C10) and C11 (connecting N23 of ring C and C10). For calculation details of the CSA tensors, see ESI, Experimental.† (B) SUPER pseudostatic powder patterns of C11 in the Pr state and the two intermediates of the forward-reaction, Lumi-R and Meta-R (thin lines) with best-fit simulations (thick lines). The applied line broadening for the three simulated spectra of Pr, Lumi-R, and Meta-R was 200, 300, and 350 Hz, respectively. For each photocycle state, the isotropic value (δiso in ppm, defined as (δ11 + δ22 + δ33)/3), anisotropy parameter (δaniso in ppm, defined as |δ11 − δiso| for describing the largest separation from the center of gravity of the line shape), asymmetry parameter (η, defined as (δ22 − δ33)/δaniso indicating how much the line shape deviated from that of an axially symmetric tensor), and span (Ω in ppm, defined as |δ11 − δ33| for describing the maximum width of the powder pattern) are indicated. The best-fit uncertainties are ±1.2 ppm for δaniso and ±0.05 for η. (C) Changes in the three experimental principal values of C9 and C11 during the Pr → Lumi-R and Lumi-R → Meta-R transitions of the forward-reaction are shown (left panel). For C9 only the changes associated with the Pr → Lumi-R transition are determined (for line shape analysis, see Fig. S7†). Calculated principal values are shown for Pg, Lumi-G, and Pr in the backward-reaction (right panel). All CSA principal values are summarized in Tables S4–S6.† (D) Correlations between the calculated and experimental principal values of the chemical shift tensors for C9 (cyan) and C11 (magenta) in the Pr state. The equations from linear regression and corresponding R2 coefficients are shown in the inset. | ||
Despite minor changes in the δiso values of the C-ring C11 in the first two transitions of the forward reaction (Fig. 3B and Table S1†), both anisotropy (δaniso) and asymmetry (η) parameters of the CSA line shapes for this carbon are greatly affected during the reaction course (Fig. 5B and Tables S4–S7†). The δaniso in Meta-R is attenuated by 15.5 ppm in total as compared to that of Pr which could arise from increased conformational flexibility of C11, consistent with the larger number of conformational states attained by this carbon in Meta-R (Fig. 3B and Table S1†). The asymmetry parameter η differs more significantly during the two transitions: η is 0.46 for Pr but 0.74 for Lumi-R, and the line shape further collapses to a nearly axial symmetric pattern with η of 0.88 for Meta-R, implying modifications in the electronic and molecular environment of this site due to the influence of the adjacent NH group in the same ring, for example. Besides these, small changes are observed for the tensor span Ω for the first two transitions (−7.7 and +3.4 ppm, respectively). To further evaluate the local structural and electronic changes of C11 and adjacent PCB atoms, the in-plane anisotropy δin-plane (defined as |δ11 − δ22|) is used as a qualitative measure for the π-electron distribution and the bond order between C11 and adjacent atoms within the conjugated system.39 It is also known that δin-plane is strongly affected by electrostatic interactions such as effects of charge in the vicinity. Here, the Pr → Lumi-R transition causes a significant increase in δin-plane (24.5 ppm), implying an increase in bond length and associated bond order adjustment around C11 as well as redistribution of the positive charge in the chromophore. The size of the δin-plane is further enlarged by 13.7 ppm during the second transition. The overall Δδin-plane increase (38.2 ppm) is caused by the opposite trends of the individual components δ11 and δ22 during the two transitions (Fig. 5C). Specifically, δ11 gets +10.8 ppm larger, while δ22 moves in an opposite direction with a greater variation of −27.4 ppm. The perpendicular component δ33 becomes larger (+14.8 ppm) which is predominantly caused by the first transition (+12.9 ppm), unlike the stepwise increases in δ11 and δ22 (Fig. 5C). The Δδ33 of C11 indicates an overall increase in the single-bond character of the C–C bonds around this carbon. By contrast, C9 is less affected, e.g., in the transition from Pr to Lumi-R, none of the principal values are shifted by more than 3.8 ppm.
The calculated principal values for the absolute shielding tensors of C9 and C11 associated with the backward-reaction were converted into chemical shifts by the use of linear regression functions inserted in Fig. 5D. Although a significant correlation was found between the experimental and calculated principal values, the spans of the calculated tensor components are more compressed compared to the experimental values (Fig. 5C). The deviation might be caused by a lack of flexibility in the basis set used in our quantum chemical calculations for describing this property and/or due to the use of a single structure per form instead of taking ensemble averages. With respect to the former and given the size of the QM region, however, it was not feasible to employ a larger pcsSeg basis set40 which is optimized for isotropic absolute shielding calculations. The backward-reaction causes major changes in δ11 and δ22 of C9 and C11, whereas the δ33 changes are rather limited (Fig. 5C). For both carbons, δ22 becomes larger during the reaction course: C9 increases stepwise in the Pg → Lumi-G → Meta-R transitions (+6.9 and +8.6 ppm, respectively). For C11, however, the δ22 change during the primary photoreaction is much larger than the subsequent transitions to form Pr (+11.8 and +2.9 ppm, respectively). The δ22 changing trend of C11 is remarkably similar to that observed in the forward-reaction, but shows an opposite sign (Fig. 5C). Despite the δin-plane of both carbons being marginally altered during the backward-reaction (Table S7†), their overall trend and signs are opposite to those in the forward-reaction. The opposite pattern of the Δδin-plane could arise from the difference in the charge and the electronic structure of the chromophore π-conjugated system. Moreover, the backward-reaction leads to an overall reduction of δaniso of C9 and C11 by −9.2 and −7.9 ppm, respectively, again opposite to that determined for the forward-reaction (Table S6†).
Discussion
TG-trapping of Slr1393g3 intermediates
Besides the Pr dark state and Pg photoproduct, four intermediates in both reaction pathways were trapped by incorporating protein into TG under non-cryogenic conditions (see Fig. 3 for photographs of all six states), demonstrating its extraordinary capability to immobilize the embedded protein. As compared to the conventional cryotrapping with in situ illumination of phytochrome solutions, the use of trehalose–phytochrome glasses is particularly advantageous as it ensures a homogeneous penetration of actinic light throughout the diluted sample (see ESI, Experimental†), thus achieving the largest proportion of intermediate forms. Moreover, the extremely low overall water content of the glass allows the MAS rotor volume to be filled completely. This offers large gains in sensitivity as high as tenfold over frozen solutions,24 an efficient factor to collect more detailed structural insights into the photocycle and information on molecular dynamics within the NMR time scales. We also demonstrated that TGs exhibit a superior efficacy in blocking thermal averaging among conformational substates and dark relaxation of Slr1393g3 as well as in preventing its thermal denaturation at RT (Fig. S8†). In this study, all NMR measurements on intermediate samples were carried out at RT (see ESI, Experimental†).Motion of the three methine bridges
Our QM/MM-optimized Pr model for Slr1393g3 shows that the inner rings B and C of the chromophore are nearly coplanar, whereas the two outer rings (A and D) are twisted away from the B–C plane in the α-facial disposition with the tilt angles of 22.1° and 40.1° for rings A and D, respectively. Although the Za → Ea isomerization of the C15,C16-double bond is the initial structural event in the Pr → Lumi-R photoreaction, the D-ring rotation associated with this step is hindered due to steric repulsion induced by the bulky sidechain of W496 on the chromophore carbons in the D-ring region (Fig. 6, Lumi-R). This might reconcile the small ΔδC at C15 and C19 occurring during the transition (Fig. 3B and Table S1†). W496 connects in the Pr state both outer rings by forming π-stacking interactions with ring D via its indole ring and by hydrogen-bonding to the A-ring carbonyl group (Fig. 6, Pr). In Lumi-R, the indole ring might still be positioned in close proximity to, but not directly over, ring D and thereby maintains the Pr-like interactions with ring A (Fig. 6, Lumi-R). Indeed, this hypothesis is consistent with the minor δC changes observed at C4 and C5 in the first transition, most likely associated with an undistorted geometry of the C5-methine bridge. In the subsequent Lumi-R → Meta-R transition, however, both carbons are strongly affected (Fig. 3B and Table S1†), implying that structural changes now involve this methine bridge (Fig. 6, Meta-R). It is also reasonable to expect the occurrence of an A-ring movement observed in the reaction course towards Pg that is accompanied by a substantial twist in the C5-methine bridge towards the β-face of the chromophore (Fig. 6, Pr → Pg). Such a geometric rearrangement could be facilitated by an outward movement of W496 which disrupts the constraining interaction of C1(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)⋯W496Nε1. Specifically, this Trp residue moves towards the hydrophobic side of ring A, and eventually locates in proximity to C528 in the Pg photoproduct, consequently expanding the chromophore cavity to allow further rotation of ring D. As implied by the drastic blue shift associated with the Lumi-R → Meta-R transition (643 nm vs. 578 nm), ring D seems to be already highly twisted in Meta-R (Fig. 6) which shortens the effective conjugation length, thus resulting in a blue shift in the absorption.8 However, other factors might also contribute to this surprisingly large absorbance change (see below). The rotation around the C14,C15-single bond would extend through the final formation of Pg from Meta-R, as demonstrated by the ΔδC changes of C15 and C19 associated with this transition (Fig. 3B and Table S1†). However, such a geometric adjustment around C15 alone might not be sufficient to explain the pronounced blue shift upon photoproduct formation (578 nm vs. 536 nm).
O)⋯W496Nε1. Specifically, this Trp residue moves towards the hydrophobic side of ring A, and eventually locates in proximity to C528 in the Pg photoproduct, consequently expanding the chromophore cavity to allow further rotation of ring D. As implied by the drastic blue shift associated with the Lumi-R → Meta-R transition (643 nm vs. 578 nm), ring D seems to be already highly twisted in Meta-R (Fig. 6) which shortens the effective conjugation length, thus resulting in a blue shift in the absorption.8 However, other factors might also contribute to this surprisingly large absorbance change (see below). The rotation around the C14,C15-single bond would extend through the final formation of Pg from Meta-R, as demonstrated by the ΔδC changes of C15 and C19 associated with this transition (Fig. 3B and Table S1†). However, such a geometric adjustment around C15 alone might not be sufficient to explain the pronounced blue shift upon photoproduct formation (578 nm vs. 536 nm).
 | ||
| Fig. 6 Changes of the PCB chromophore and its binding pocket during the photocycle of Slr1393g3. The conjugated π system of the chromophore is colored red as Pr; the color bleaching during the forward-reaction represents a reduction of the effective conjugation length. The sidechains of relevant residues in the binding pocket are depicted in blue. Water molecules are shaded in cyan. Hydrogen bonds are indicated by the dashed green lines and the corresponding distances derived from the QM/MM models for Pr, Pg, and Lumi-G are labeled. | ||
Contrary to the large overall rotations of rings A and D in the reaction course to form Pg, the QM/MM models predict that the inner rings B and C are only slightly more twisted in the photoproduct with the tilt angle enlarged by 6.0° as in the Pr state (6.1° vs. 12.1° in Pr and Pg, respectively), consistent with resonance Raman data on the cryotrapped intermediates of AnPixJg2, indicating no torsional and deformation modes of the central C10-methine bridge.14 During the first two transitions, C9 and C11 show only minor δC changes (≤0.4 ppm), whereas the Meta-R → Pg transition has a major effect on the entire C9–C11 region (Fig. 3B and Table S1†). Hence, we expect that the twist of the C10-methine bridge around its two dihedral angles does not take place in a stepwise manner but is specifically associated with the formation of the final photoproduct. This could be viewed as being caused by the gradually increasing motion of the two inner rings on the way to form Pg. Indeed, we first detected the conformational flexibility of C9 in Lumi-R and of C11 in the subsequent Meta-R state (Fig. 3B and Table S1†). These observations suggest that the release of the inner rings from their tensed states occurs in a specific order that may arise indirectly due to restructuring of the binding pocket deriving from the D-ring isomerization. In Pr, the ring B is held tightly in place from the α-face of the chromophore by the imidazole of H529. Upon photoconversion, however, this residue is freed from the water-mediated hydrogen-bonding interaction with ring D (Fig. 1C) which in turn would partially release the steric constraints prohibiting conformational dynamics of ring B. For ring C, however, its local conformational flexibility is most likely initialized by the further D-ring rotation in the transition to form Meta-R which is accompanied by a stretched-to-bent transformation of the C-ring propionate (Fig. 6, Meta-R). This change is not in fact essential for phytochromes,41,42 but contributes to the formation of a steric blockade that finalizes the twisted D-ring geometry of the Pg photoproduct in red/green CBCRs.7,11,31,43 As a further consequence of the C-ring propionate bending, a concomitant twist in the C10-methine bridge would occur. Although changes of the tilt angle between rings B and C are predicted to profoundly impact on the peak absorption of the chromophore, in this case, the change appears to be too small (6.0°) for being the decisive factor for the 42 nm blue shift associated with photoproduct formation from Meta-R.
A conjugation defect localized around C12
13C chemical shifts for chromophore pyrrolic carbons have been found to be largely determined by the conjugation system and charge pattern in the chromophore itself.44 Recently, Jähnigen and Sebastiani specified that there is a characteristic correlation between the 13C chemical shifts and C–C bond lengths of the chromophore in both Cph1 and AnPixJg2.45 For Slr1393g3, we observed the |δC| changes of ≥1.8 ppm at C10 during all three transitions of the forward-reaction (Fig. 3B and Table S1†). It is therefore tempting to deduce the changes in bond length for the C10-methine bridge in the reaction course. Although no such stepwise δC changes were detected at C9 and C11, the changing patterns of the tangential component (δ22) for C11 fully support this interpretation (Fig. 5C). δ22 lies roughly along the C10,C11-bond (Fig. 5A), thus showing apparently higher geometric sensitivity relative to the changes of δiso values. During the first transition, a δC change of −1.8 ppm was observed for C10, with a strikingly large δ22 change of −19.3 ppm at C11. Such consistent upfield shifts are likely due to the C10,C11-bond elongation which in turn could arise from a charge distribution shift (increased probability) towards the formation of a stabilized mesomeric (resonance) form with a tertiary carbocation centered at C12 in Lumi-R (Fig. S9†). In the Pr state, the dynamic nuclear polarization (DNP)-enhanced MAS NMR study of Cph1 has shown that the positive charge is mainly localized at ring B, not spreading over both inner rings as generally assumed.46 Moreover, the 13C chemical shifts of the pyrrolic carbons in Cph1 show great similarity to those of AnPixJg2 and the closely related NpR6012g4 in their respective Pr dark states,18,31,44 suggesting that the electronic structure of the conjugated systems and charge distribution patterns of the Pr chromophores are comparable for all species. These observations imply the migration of partial positive charge into ring C during the Pr → Lumi-R transition. For the subsequent reaction to Meta-R (Fig. 6), two independent lines of evidence support an appreciable positive charge accumulated at C12. First, the C10,C11-bond would be further elongated, but to a smaller extent than that of the initial photoreaction, as demonstrated by an additional upshift of δ22 (−8.1 ppm). As discussed above, the elongated C10,C11-bond tends to render a decrease in bond length alteration which would promote the charge delocalization to include ring C.45 Perhaps most importantly, downward bending of the C-ring propionate in Meta-R places its carboxylate moiety (C123) close to C12 and the formal negative charge of the carboxylate oxygens may act as an ideal counterion for the carbocation at C12, thus stabilizing the formation of this mesomeric form (Fig. S9†). As the reaction proceeds to its end photoproduct, the stabilization would be enhanced due to the single-bond rotation of ring D (see above) which may reshape the propionate for a further reduction of the interatomic distance between C123 and C12 (Fig. 6). The Pg model shows a C123⋯C12 separation of ∼2.8 Å which is, however, at least 3.9 Å apart in Pr. By contrast, the situation is reversed in the B-ring propionate, with a C83⋯C8 separation of 2.9 and 3.8 Å in Pr and Pg, respectively. The latter observation is consistent with the notion that, in the Pr state, the positive charge is mainly localized at ring B, in which the mesomeric forms with the positive charge at N22 as well as a tertiary carbocation at C8 would be prevailing (Fig. S9†). This carbocation intermediate, however, would become less favorable upon Pg formation because of the upward stretching of B-ring propionate seen in the first transition (Fig. 6, Lumi-R). It is thus reasonable to suspect that the two propionates participate in determining the contribution of the major mesomeric forms with carbocations at either C8 or C12 during the photocycle. This interpretation is consistent with charge distribution extracted from orbital-based Löwdin population analysis for the non-hydrogen PCB atoms involved in mesomeric forms shown in Fig. S9.† The changes in Löwdin charges among the Pr, Pg, and Lumi-G forms are relatively small (Table S8†). Nonetheless, C8 is assigned the highest charge in Pr (−0.008e) relative to Pg (−0.011e) and Lumi-G (−0.026e), whereas C12 shows the lowest charge with −0.005e in Pr, but the highest one with 0.012e in Pg.Multiple lines of evidence consistently indicate charge movement towards ring C occurring in a stepwise manner after the initial photoisomerization. The resulting partial positive charge of ring C would locate preferentially at N23 and its high electronegativity could further shift the distribution towards the formation of a carbocation intermediate at C12 (Fig. S9 and Table S8†). This intra-ring distribution is consistent with the gradually downshifted δ11 of C11 (Fig. 5D) which assumes roughly an aligned orientation to the C11,N23-bond. Moreover, it has been known that a positively charged pyrrole nitrogen like N23 is conjugated with the π-system and that the formation of a carbocation at C12 would interrupt the bond length alternation of the conjugated system.45,47 The latter statement is also borne out by the development of a greater degree of σ-bond character of the C–C bonds around C11 on the pathway towards Pg, as implied by the associated Δδ33 pattern of this carbon (Pr → Pg, Fig. 5D). Such a conjugation defect localized around C12 would weaken the conjugation throughout the two inner rings, therefore leading to a blue shift of the absorption maximum.21,47 Although the Lumi-R intermediate even displays the onset of a positive charge delocalization into ring C and also generation of a resonance-stabilized carbocation intermediate at C12, the ionic interaction offered by the C-ring propionate carboxylate seems to be fully functional until the final transition to form Pg (Fig. 6). As a result, this transition would be associated with the appearance of a conjugation defect localized around C12, thus accounting, at least partially, for the pronounced spectral blue shift from Meta-R (578 nm) to Pg (536 nm).
The backward-reaction is not simply a mirror-symmetric process of the forward-reaction
The Pg photoproduct is characterized by a solvated binding pocket and a highly twisted chromophore (Fig. 1C and 4A), leading to a broad distribution of chromophore conformational states (Fig. 3B and Table S1†). The increased flexibility of the chromophore is consistent with the net loss of chromophore–protein interactions particularly at the two outer rings (Fig. 6, Pg). As predicted by the QM/MM models for Pg and Lumi-G, the initial structural event in the Pg → Pr transformation is an E → Z isomerization of the C15,C16-double bond that flips ring D in the counterclockwise direction (Fig. 4A). Subsequently, ring D moves towards the β-face of the chromophore with the binding pocket still optimized to accommodate the 15Ea geometry of the photoproduct. The decoupling of the chromophore and its binding pocket demonstrates that ring D has to first slip out of the tongs formed by F474 and F536 upon photoexcitation in order to flip (Fig. 6, Pg). The Lumi-G formation is accompanied by a decrease of the tilt angle between rings C and D (from 61.2° to 31.9°). The less twisted D-ring geometry trapped in Lumi-G lengthens the conjugation, thus inducing a red-shift as detected for the first transition (536 nm vs. 564 nm). As Pg evolves into Lumi-G, the tilt angle between rings B and C decreases from 12.1° to 7.6°, indicating a more coplanar arrangement of the two inner rings. However, there is no such concomitant impact on ring C associated with the Pr → Lumi-R photoreaction of the forward-reaction. Instead, the structural changes originating from the D-ring motion extend directly to the surrounding residues, and further to rings A and B in the subsequent transition. The motion of ring C is induced afterwards during the final transition to form Pg (Fig. 6, Pr → Pg).The most significant δC changes of the entire backward-reaction occur at C9–C11 atoms during the Pg → Lumi-G transition (Fig. 3B and Table S1†). Such changes cannot be interpreted as arising solely due to the small twist of the C10-methine bridge which partially relieves the tension of rings B and C, but are most likely associated with the onset to restore the charge distribution of the Pr chromophore, i.e., increase in C–C bond length alternation. This idea is further supported by an exactly opposite 13C shift pattern for the three carbons in the Meta-R → Pg and Pg → Lumi-G transitions which points to conservation of the same electronic factors, but driving in the opposite direction (Fig. 3B and Table S1†). Another line of evidence is the opposite changing patterns of both δ22 and δin-plane anisotropy of C11 in these two transitions (Fig. 5D), indicating an inverted effect on the variation of associated C–C bond lengths, e.g., to restore the elongated C10,C11-bond as observed in the forward-reaction. This circumstance may also explain the increased flexibility of the C10-methine bridge during the Pg → Lumi-G transition (Fig. 3B and Table S1†). Moreover, as discussed above, the C12-carbocation represents the major mesomeric form in the Pg photoproduct which is stabilized by its interaction with the C-ring carboxylate. However, the Lumi-G model reveals little modification of this ionic interaction; we therefore conclude that the C12-carbocation is likely to be still prevalent in this intermediate. The associated conjugation pattern of the two inner rings (with a defect at C12) would remain essentially unchanged, making little to no contribution to the red-shift of the Lumi-G absorbance relative to Pg.
The Lumi-G model exhibits a β-facial disposition of both the two outer rings relative to the B–C plane (Fig. 4A), and the formation of the Pr state requires single-bond rotations around both C5 and C15, during which both outer rings move concertedly towards the α-face. For the analogous backward-reaction of canonical phytochromes, the rotation of the C14,C15-single bond has been proposed to be nearly completed upon formation of the late Meta-F intermediate from Lumi-F, while not extending to the last transition to form Pr.21,42 A similar explanation likely accounts for the minor δC changes along with the formation of Pr from Meta-G in Slr1393g3 for C15 and C19 (≤0.2 ppm). It is thus conceivable that ring D in the Meta-G intermediate adopts a less-twisted Pr-like geometry (Fig. 6). Indeed, this intermediate is further red-shifted relative to Lumi-G with substantial deconjugation of ring D (579 nm vs. 564 nm). However, it should be noted that, in the forward-reaction, the rotation of the C14,C15-single bond is not specifically associated with the formation of Meta-R but also continues in the subsequent transition to form Pg (Fig. 6). On the other side of the chromophore, Lumi-G exhibits a Pg-like twist in the C5-methine bridge, and its structural relaxation would mainly occur in forming Meta-G, during which the A-ring carbonyl group swaps its hydrogen-bonding partners (Fig. 6, Lumi-G → Meta-G). The hydrogen-bonding interactions are presumably stronger in the Meta-G intermediate, consistent with the decreased separation of C4 resonances in this intermediate than that in Lumi-G (Table S1†). The number of 13C split components of C4 and C5 is further reduced during the last transition back to Pr (Fig. 3B) which provides good evidence of a stronger fixation of ring A in the pocket arising from modifications of specific interactions between ring A and its immediate protein surroundings. Moreover, the δC changes in forming Pr are largely localized to the vicinity of the C5-methine bridge, e.g., ΔδC of C4 being 0.7 ppm. Such a large change is incompatible with effects arising solely from structural relaxation in the protein cavity, as occurring in the analogous transition from Meta-F to Pr in Cph1,21 in which only minor ΔδC changes along the C4–C19 conjugated system (≤0.4 ppm) were seen. It is thus a reasonable assumption that the single-bond rotation around C5 would be a stepwise process that can still be incomplete upon the formation of Meta-G, unlike that around C15.
The calculated 13C CSA data show a prominent δ22 shift of +11.8 ppm at C11 during the Pg → Lumi-G photoreaction, but the further changes in the transitions of Lumi-G → Pr only amounted to +2.8 ppm (Fig. 5D). These data imply that restoration of the elongated C10,C11-bond is predominantly associated with the formation of Lumi-G, in contrast to the stepwise elongation of this bond during the forward-reaction. Surprisingly, there is also evidence that the length of the adjacent C9,C10-bond is reduced during the backward-reaction because of the progressive δ22 downshifts of C9, e.g., +6.9 and +8.6 ppm in the transitions of Pg → Lumi-G and Lumi-G → Pr, respectively (Fig. 5D). We thus expect that the formation of Lumi-G only serves to partially relieve the strain of the elongated C9,C10-bond, and its subsequent relaxations in the last two transitions are likely to be coupled to conformational rearrangements of the two propionate sidechains (Fig. 6). On the other hand, for the forward-reaction, elongation of the C9,C10-bond would not occur during the primary Pr → Lumi-R transition due to the minor δ22 upshift of C9 (−2.0 ppm), but is most likely associated with the last two transitions to form Pg (Fig. 6).
Our data provide good evidence of a Pr-like D-ring geometry in the Meta-G intermediate, indicating re-formation of the π-stacking interaction between ring D and the indole ring of W496 during the Lumi-G → Meta-G transition. The associated inward movement of this bulky ‘lid Trp’ (being proximal to C528 in Lumi-G) requires dramatic structural changes in the cavity. Prominent examples here include a two-turn helix-to-loop transition involving residues 486–492 and an overall positional shift of the chromophore slightly out of the protein cleft. The latter process must necessarily be accompanied by breakage of the Lumi-G-stabilizing hydrogen bonds around the two propionate sidechains which may induce the bent-to-stretched transformation of the C-ring propionate and reversed conformational switching of its B-ring counterpart (Fig. 6, Meta-G). We therefore propose that the C-ring carboxylate group in Meta-G is no longer in position to stabilize the C12-carbocation, thus lowering the importance (decreased probability) of this mesomeric form compared to the Lumi-G and Pg photostates. As a further consequence, the positive charge density of the chromophore is at least partially withdrawn from ring C to be shifted further to ring B in the Meta-G formation. Subsequent charge redistribution within ring B is consistent with much more deshielded in-plane components (δ11 and δ22) of C9 during the two last transitions to form Pr (+10.4 and +8.6 ppm for δ11 and δ22, respectively, Fig. 5D). For ring B in Pr, as discussed above, N22 and C8 are the two favorable sites for carrying the positive charge (Fig. S9† and ref. 45). The mesomeric form with the positive charge at N22 would be more stable due to its interaction with the acidic sidechain of D498 at a distance of 2.8 Å, thus prevalent in Pr (Fig. 6). A stabilization of the positive charge to some extent at C8 seems also a reasonable assumption as the back-folding of the B-ring propionate places its carboxylate group closer to C8 in the Meta-G intermediate and eventually at a distance of ∼2.9 Å in Pr. The two B-ring mesomeric forms consequently weaken the effect of the conjugation defect located at C12 and the resulting ‘full conjugation’ of the two inner rings may serve as a reasonable explanation for the formation of a drastically red-shifted Pr state (652 nm) from Meta-G (579 nm). Such a large absorbance change (73 nm) was not detected in the formation of Pg from Meta-R in the forward-reaction (578 nm vs. 536 nm). Moreover, the 65 nm blue shift associated with the Lumi-R → Meta-R transition (643 nm vs. 578 nm) cannot be solely derived from a further twist of ring Dvia the C14,C15-single bond rotation, as judging from the corresponding Lumi-G → Meta-G transition in the backward-reaction in which this D-ring rotation is restored, but accompanied by only a 35 nm red shift (564 nm vs. 579 nm). Taken together, we reason that the prevalence of the C12-carbocation is not only a dominant factor for the blue shift associated with the last transition in forming the Pg photoproduct, but it also contributes significantly to blue-shifting the Meta-R absorption (Fig. 6).
The mobile defect becomes self-trapped
The charge dynamics shown in Fig. S9† can also be expressed in terms of polymer physics. Structures I–III show the (de)localization of positive charge on N22 and N23, either on one of them or spread on both. In structures IV and V, the ‘hole’ has moved into the conjugation chain. One might assume that the ‘hole’ could move rather freely in the chain leading to local distortions of the matrix, implying a polaronic character. The ‘hole’ causes a conjugation defect leading to a significant blue shift of the first absorption band. Passing positions C8 or C12 the polaron causes, considering structures I–III as references, a structural change in the environment is induced by attracting the negatively charged carboxylate group. Here, the polaronic distortion causes structural relaxation and therefore self-trapping of the polaron and the localization of the defect. The absorption ‘color’ of the sample is changed by the conjugation defect as well as by the displacement of the counterion which could control the stability of the LUMO and therefore the energy of the HOMO–LUMO transition. These two effects might provide the key to the strong photochromicity in red/green CBCRs.Conclusions
In contrast to all previous cryotrapping measurements of photocycle intermediates in phytochromes and CBCRs,21,42 the preparation of TG-embedded protein allows isolation and long-term preservation of individual intermediates at RT. This innovative sample preparation is particularly beneficial for the MAS NMR technique by which multiple attempts to trap the entire photocycle within the NMR magnet have failed so far.9 In this work, all four intermediates of Slr1393g3 in both reaction pathways as well as the Pr and Pg photostates have been investigated. The experimental and calculated 13C δiso and CSA principal values of the selected PCB carbons reflect changes in local electronic structure, geometry, and dynamics as well as charge redistribution of the chromophore during the photocycle. The QM/MM-optimized structures of Pr, Pg, and Lumi-G provide further atomic details concerning structural rearrangements in the chromophore-binding pocket. For the Pr → Pg forward-reaction, we present evidence for the twisting of all three methine bridges in the order C15 → C5 → C10 and a gradual redistribution of the positive charge from a stronger localization at ring B to a more delocalized distribution between rings B and C. In addition to the earlier assumption of ‘D-ring control’, the prevalence of the tertiary C12-carbocation associated with the Pg formation is found to be required for blue-shifting of both the late Meta-R intermediate and the final photoproduct by inducing a polaronic defect of the ‘core conjugation’ formed by the two inner rings. Stabilization of this carbocation in the Pg photoproduct is achieved through its interaction with the C-ring carboxylate. This finding rationalizes the role of the C-ring propionate which converts radically from a stretched to a bent conformation in the reaction course from Pr to Pg. Our data also demonstrate that the Pg → Pr backward-reaction is not a mirror-symmetric process of the forward-reaction, although the C15,C16-double bond isomerization is the first step of photoconversion in both cases. Subsequent to this photochemical event, we find first that the twist of the C10-methine bridge is reversed, and second that the structural changes extend to the C5-methine bridge, in the order C15 → C10 → C5. We interpret this difference as arising from radical inward/outward movements of the ‘lid Trp’. Again, unlike the forward-reaction, the positive charge relocalization occurring in the backward-reaction (towards ring B) would be solely responsible for absorbance change of the last transition to form Pr rather than for the red shift of the second transition to form Meta-G. These data offer valuable insight into the molecular events that channel light excitation to drive distinguishable Pr → Pg and Pg → Pr phototransformations with different molecular mechanisms, thus leading to unidirectionality of the photocycle (Fig. 6). We anticipate that the established methods will prove to be equally well suited to locate the positive charge of the chromophore during the photocycles of canonical phytochromes, thus helping to substantiate the role of charge dynamics in regulating the conjugation length in the chromophore as an inherent spectral-tuning mechanism. Indeed, a comprehensive understanding of the factors controlling the photochromic capabilities of bilin chromophores in different protein environments (e.g., CBCRs vs. phytochromes) allows for developing novel tailor-made photoswitches for modern biological and biomedical applications.48Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
J. M. and C. S. conceived the project. L. K., W. G., C. W. and C. S. designed the research. L. K. and C. W. performed the experiments. L. K., W. G., G. S., C. W. and C. S. analyzed the data. All authors wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (Project ID 417685888 to C. S.). The QM/MM optimizations were realized with resources of the Paderborn Center for Parallel Computing (PC2); the excited state and GIAO calculations were performed on resources provided by the Leipzig University Computing Center. The authors thank Prof. F. Ortmann (TU München) for helpful discussions and also the two reviewers for their thoughtful critique of this manuscript.References
- K. Fushimi, M. Hasegawa, T. Ito, N. C. Rockwell, G. Enomoto, N.-N. Win, J. C. Lagarias, M. Ikeuchi and R. Narikawa, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15573–15580 CrossRef CAS PubMed
.
- N. C. Rockwell and J. C. Lagarias, New Phytol., 2020, 225, 2283–2300 CrossRef PubMed
- K. Fushimi and R. Narikawa, Curr. Opin. Struct. Biol., 2019, 57, 39–46 CrossRef CAS PubMed
- K. Fushimi, M. Ikeuchi and R. Narikawa, Photochem. Photobiol., 2017, 93, 903–906 CrossRef CAS PubMed
- Y. Chen, J. Zhang, J. Luo, J.-M. Tu, X.-L. Zeng, J. Xie, M. Zhou, J.-Q. Zhao, H. Scheer and K.-H. Zhao, FEBS J., 2012, 279, 40–54 CrossRef CAS PubMed
- X.-L. Xu, A. Gutt, J. Mechelke, S. Raffelberg, K. Tang, D. Miao, L. Valle, C. D. Borsarelli, K.-H. Zhao and W. Gärtner, ChemBioChem, 2014, 15, 1190–1199 CrossRef CAS PubMed
- X. Xu, A. Höppner, C. Wiebeler, K.-H. Zhao, I. Schapiro and W. Gärtner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2432–2440 CrossRef CAS PubMed
- C. Wiebeler, A. G. Rao, W. Gärtner and I. Schapiro, Angew. Chem., Int. Ed., 2019, 58, 1934–1938 CrossRef CAS PubMed
- C. Wiebeler and I. Schapiro, Molecules, 2019, 24, 1720 CrossRef CAS PubMed
- N. C. Rockwell, S. S. Martin, A. G. Gulevich and J. C. Lagarias, Biochemistry, 2014, 53, 3118–3130 CrossRef CAS PubMed
- S. Lim, Q. Yu, S. M. Gottlieb, C.-W. Chang, N. C. Rockwell, S. S. Martin, D. Madsen, J. C. Lagarias, D. S. Larsen and J. B. Ames, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4387–4392 CrossRef CAS PubMed
- D. Buhrke, K. T. Oppelt, P. J. Heckmeier, R. Fernández-Terán and P. Hamm, J. Chem. Phys., 2020, 153, 245101 CrossRef CAS PubMed
- Y. Fukushima, M. Iwaki, R. Narikawa, M. Ikeuchi, Y. Tomita and S. Itoh, Biochemistry, 2011, 50, 6328–6339 CrossRef CAS PubMed
- F. Velazquez Escobar, T. Utesch, R. Narikawa, M. Ikeuchi, M. A. Mroginski, W. Gärtner and P. Hildebrandt, Biochemistry, 2013, 52, 4871–4880 CrossRef CAS PubMed
- P. W. Kim, L. H. Freer, N. C. Rockwell, S. S. Martin, J. C. Lagarias and D. S. Larsen, J. Am. Chem. Soc., 2012, 134, 130–133 CrossRef CAS PubMed
- A. J. Jenkins, S. M. Gottlieb, C.-W. Chang, R. J. Hayer, S. S. Martin, J. C. Lagarias and D. S. Larsen, Photochem. Photobiol. Sci., 2019, 18, 2539–2552 CrossRef CAS PubMed
- B. Borucki, D. von Stetten, S. Seibeck, T. Lamparter, N. Michael, M. A. Mroginski, H. Otto, D. H. Murgida, M. P. Heyn and P. Hildebrandt, J. Biol. Chem., 2005, 280, 34358–34364 CrossRef CAS PubMed
- C. Song, R. Narikawa, M. Ikeuchi, W. Gärtner and J. Matysik, J. Phys. Chem. B, 2015, 119, 9688–9695 CrossRef CAS PubMed
- H. Saitô, I. Ando and A. Ramamoorthy, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 181–228 CrossRef PubMed
- D. Huster, X. Yao and M. Hong, J. Am. Chem. Soc., 2002, 124, 874–883 CrossRef CAS PubMed
- T. Rohmer, C. Lang, C. Bongards, K. B. S. S. Gupta, J. Neugebauer, J. Hughes, W. Gärtner and J. Matysik, J. Am. Chem. Soc., 2010, 132, 4431–4437 CrossRef CAS PubMed
- D. Buhrke, G. Battocchio, S. Wilkening, M. Blain-Hartung, T. Baumann, F.-J. Schmitt, T. Friedrich, M.-A. Mroginski and P. Hildebrandt, Biochemistry, 2020, 59, 509–519 CrossRef CAS PubMed
- C. Slavov, X. Xu, K.-H. Zhao, W. Gärtner and J. Wachtveitl, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 1335–1344 CrossRef CAS PubMed
- L. Köhler, W. Gärtner, J. Matysik and C. Song, ChemPhotoChem, 2022, 6, e202100220 CrossRef
- M. Malferrari, A. Savitsky, W. Lubitz, K. Möbius and G. Venturoli, J. Phys. Chem. Lett., 2016, 7, 4871–4877 CrossRef CAS PubMed
- K. Möbius, A. Savitsky, M. Malferrari, F. Francia, M. D. Mamedov, A. Y. Semenov, W. Lubitz and G. Venturoli, Appl. Magn. Reson., 2020, 51, 773–850 CrossRef
- G. Camisasca, M. De Marzio and P. Gallo, J. Chem. Phys., 2020, 153, 224503 CrossRef CAS PubMed
- C. Kneip, P. Hildebrandt, W. Schlamann, S. E. Braslavsky, F. Mark and K. Schaffner, Biochemistry, 1999, 38, 15185–15192 CrossRef CAS PubMed
- P. Eilfeld and W. Rüdiger, Z. Naturforsch., C: J. Biosci., 1985, 40c, 109–114 CrossRef CAS
- F. Pennacchietti, A. Losi, X.-L. Xu, K.-H. Zhao, W. Gärtner, C. Viappiani, F. Cella, A. Diaspro and S. Abbruzzetti, Photochem. Photobiol. Sci., 2015, 14, 229–237 CrossRef CAS PubMed
- S. Altmayer, L. Köhler, P. Bielytskyi, W. Gärtner, J. Matysik, C. Wiebeler and C. Song, Photochem. Photobiol. Sci., 2022, 21, 447–469 CrossRef CAS PubMed
- N. C. Rockwell, S. S. Martin, S. Lim, J. C. Lagarias and J. B. Ames, Biochemistry, 2015, 54, 2581–2600 CrossRef CAS PubMed
- T. Rohmer, C. Lang, W. Gärtner, J. Hughes and J. Matysik, Photochem. Photobiol., 2010, 86, 856–861 CrossRef CAS PubMed
- M. Stanek and K. Grubmayr, Chem. Eur. J., 1998, 4, 1660–1666 CrossRef CAS
- L. K. Scarbath-Evers, S. Jähnigen, H. Elgabarty, C. Song, R. Narikawa, J. Matysik and D. Sebastiani, Phys. Chem. Chem. Phys., 2017, 19, 13882–13894 RSC
- T. Watermann, H. Elgabarty and D. Sebastiani, Phys. Chem. Chem. Phys., 2014, 16, 6146–6152 RSC
- D. A. Forsyth and A. B. Sebag, J. Am. Chem. Soc., 1997, 119, 9483–9494 CrossRef CAS
- S.-F. Liu, J.-D. Mao and K. Schmidt-Rohr, J. Magn. Reson., 2002, 155, 15–28 CrossRef CAS PubMed
-
D. M. Grant, Chemical Shift Tensors, in Solid-State NMR Studies of Biopolymers, ed. A. E. McDermott and T. Polenova, Wiley, 2010, pp. 49–82 Search PubMed
- F. Jensen, J. Chem. Theory Comput., 2015, 11, 132–138 CrossRef CAS PubMed
- C. Song, M. A. Mroginski, C. Lang, J. Kopycki, W. Gärtner, J. Matysik and J. Hughes, Front. Plant Sci., 2018, 9, 498 CrossRef PubMed
- X. Yang, Z. Ren, J. Kuk and K. Moffat, Nature, 2011, 479, 428–432 CrossRef CAS PubMed
- R. Narikawa, T. Ishizuka, N. Muraki, T. Shiba, G. Kurisu and M. Ikeuchi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 918–923 CrossRef CAS PubMed
- N. C. Rockwell, S. S. Martin, S. Lim, J. C. Lagarias and J. B. Ames, Biochemistry, 2015, 54, 3772–3783 CrossRef CAS PubMed
- S. Jähnigen and D. Sebastiani, Molecules, 2020, 25, 5505 CrossRef PubMed
- D. Stöppler, C. Song, B.-J. van Rossum, M.-A. Geiger, C. Lang, M.-A. Mroginski, A. P. Jagtap, S. T. Sigurdsson, J. Matysik, J. Hughes and H. Oschkinat, Angew. Chem., Int. Ed., 2016, 128, 16017–16020 CrossRef PubMed
- S. Altmayer, S. Jähnigen, L. Köhler, C. Wiebeler, C. Song, D. Sebastiani and J. Matysik, J. Phys. Chem. B, 2021, 125, 1331–1342 CrossRef CAS PubMed
- K. Lehtinen, M. S. Nokia and H. Takala H, Front. Cell. Neurosci., 2022, 15, 778900 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00636k |
| This journal is © The Royal Society of Chemistry 2023 |