
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular metallotecton for engineering permanently porous frameworks: supernumerary role of ancillary ion†
Deepak
Gupta
 ,
Géraldine
Chanteux
,
Gulshan
Kumar‡
,
Koen
Robeyns
and
Alexandru
Vlad
*
,
Géraldine
Chanteux
,
Gulshan
Kumar‡
,
Koen
Robeyns
and
Alexandru
Vlad
*
Institute of Condensed Matter and Nanosciences, Molecular Chemistry, Materials and Catalysis, Université catholique de Louvain, Louvain-la-Neuve B-1348, Belgium. E-mail: alexandru.vlad@uclouvain.be
First published on 24th August 2023
Abstract
The formation of robust supramolecular frameworks built from hetero-polytopic metal complexes and interacting with different ancillary ions remains a long-standing and underexplored desire. Herein, the secondary sphere interaction chemistry of [Ru(5-oxido-6-hydroxy-1,10-phenanthroline)a(5,6-dihydroxy-1,10-phenanthroline)(3−a)]−(a−2) (1) (a = 1, 3) coordination ion is reported, where the π-conjugated phenanthroline ligands are functionalized with catecholate groups used as H-bond donors and ligands. The deprotonation of the catechols is found to control the overall charge stoichiometry in 1, acting as a metallotecton to interact with anions of different basicity (Cl− in 1.Cl and Br− in 1.Br) as well as with Li+ cations (in 1.Li+). These interactions lead to the formation of 2D porous honeycomb networks without any significant alteration in the molecular packing. This implies that the self-assembly process is controlled by complementary intermolecular non-covalent interactions making the choice of the ancillary ion insignificant. The robust porous structure of the frameworks is established by uptake of D2O and I2 molecules within the microporous channels. This work demonstrates that supramolecular frameworks appear as flexible candidates for applications in gas sorption, separation and chemical sensing.
Introduction
Combining chemical entities in a controlled and modular fashion is the fundamental principle to achieve porous extended solids.1–3 The domain of reticular chemistry is dominated by strong directional bonding between linkers and Secondary Building Units (SBUs); however structure predictions remain highly challenging while dealing with non-covalent interactions.4–6 Although the formation of open frameworks by combination of metal-ions and polytopic ligands (i.e. MOFs) is a relatively well-developed and controlled strategy, the rules governing the assembly of frameworks based on supramolecular interactions remain poorly understood, yet highly desirable.7–9 Indeed, supramolecular frameworks can find appealing applications in gas separation, chemical sensing, catalysis and proton/electron conduction. These materials, stabilized by weak interactions, may benefit from superior flexibility and reversibility upon external stimuli as compared to stronger covalent bonded materials. From the synthesis point of view, the ultimate challenge is to modulate the strength and nature of the weaker interactions (for instance H-bonding, π–π stacking) in order to precisely control the framework composition and topology.In this context, several examples of crystalline molecular solids in which hydrogen bonds are exploited to create supramolecular assemblies of organic molecules can be found in the literature. However, low thermal and mechanical stability of the framework are typically acknowledged as a potential drawback.8 Moreover, organic molecules have the tendency to exhibit a variety of hydrogen-bonding patterns through functionalization of the primary skeleton which leads to the problem of polymorphism having different physical properties.10–12 In contrast, coordination compounds offer various advantages as metal may exhibit multiple coordination geometries as well as a wide range of physical properties. For example, in addition to structural rigidity and fast nucleation kinetics, metal complexes may impart magnetic, electronic, and catalytic properties leading to functional supramolecular materials.13 The assembly of such supramolecular frameworks is mainly directed by H-bond donor/acceptor ability of the exocyclic functional groups on the metal complex, making these suitable to interact with either hard anions (oxo, halide etc.) or iminium cations.14 The strategy of using a combination of metal–ligand bond and ionic hydrogen bonding as supramolecular glue to form extended layers has served as a suitable platform to gain control over molecular packing in engineering crystalline solids.15–18 For example, dianionic bis(2,6-pyridinedicarboxylate)M(II) complexes have been reported as supramolecular building blocks to form well-defined structures dominated by strong ionic hydrogen bonding between imidazolium ions and the carboxylate ends.19 It was found that the metal ion does not impact the geometry of the complex, with the packing arrangement of the frameworks being identical despite metal ion modification. The fixed octahedral geometry of the metal complex was governed by the tris-chelating nature of 2,6-pyridinedicarboxylate ligand which imposed a coordination mode irrespective of the type of the central metal ion.
Another approach is to design building blocks with a fixed geometry that can resist any chemical modification occurring at the secondary sphere interaction to form robust networks. From this perspective, the tris-chelated metal complexes with octahedral geometry have emerged as versatile building blocks.20 In these complexes, the ligand chelation not only imparts stability and geometrical rigidity to the complex, but the presence of the external conjugated binding sites allows secondary sphere interactions such as hydrogen bonding or coordination acting as a metallotecton for supramolecular assemblies. One representative example is the tris-biimidazolate Ni(II) complex where the bidentate biimidazolate ligands bind with Ni(II) ion as well as possess three complementary sites for outer sphere interactions.21 Since the external binding sites are placed at a fixed angle of nearly 120 °C, this tripodal building block assembles in a two-dimensional honeycomb network (6,3 net, with or without ancillary ions) preferably, thereby providing a handy tool to control the molecular arrangement. However, controlled assembly by rational design still remains ambiguous as this building block undergoes supramolecular polymorphism and isomerism.22,23 This is due to the conformational variation of the C–C single bond between the planar imidazolate rings, weakening the rigidity of the ligand skeleton and losing planarity. Therefore, the choice of the templating counterion as well the crystallization conditions primarily regulate the formation of 2D or 3D network.
In this study, we present the secondary sphere interaction of the coordination complex ion [Ru(5-oxido-6-hydroxy-1,10-phenanthroline)a(5,6-dihydroxy-1,10-phenanthroline)(3−a)]−(a−2)·X (1) (when a = 1; X = Cl− (1.Cl)/Br− (1.Br) and when a = 3; X = Li+ (1.Li)) with counter ions. The derived 5,6-hydroxy-1,10-phenanthroline ligand is rigid, fully conjugated and aromatic, and prevents any chance of polymorphism. The counter ion varies upon deprotonation of the catechol groups where the complex ion (1) interacts with halide ions via H-bonding, or coordinates with alkali cations to form a series of hexagonal porous supramolecular frameworks (PSFs). When one of the catechol ligands is deprotonated (a = 1 in the formula of the complex ion), the complex (1) attains a formal positive charge and act as cationic metallotecton to undergo H-bonding interactions with anions of different basicity (Cl− and Br− in the present study). The robust porous structure of these frameworks was established by exchange with D2O and I2 molecules within the microporous channels. Furthermore, upon complete deprotonation of the catechol groups (a = 3), the negatively charged complex forms a bimetallic porous framework with Li+ cations. This modular ligand offers tunable binding sites on the metal complex and allows the formation of ordered 2D networks with different compositions and similar topologies via weak supramolecular interactions as well as coordinating bonds. This is the first report on crystal structures of metal complexes based on [Ru(5-oxido-6-hydroxy-1,10-phenanthroline)a(5,6-dihydroxy-1,10-phenanthroline)(3−a)]−(a−2) complex ion and its potential as building block to assemble supramolecular frameworks with different ancillary ions.
Results and discussion
Synthesis of 1.Cl, 1.Br and 1.Li
The 5,6-dihydroxy-1,10-phenanthroline ligand (H2L) possesses a conjugated aromatic core that imparts structural rigidity to the building block as well as offers possibility for non-covalent interactions through H-bond donor catechol groups. Interestingly, the catechol groups have the ability to act as anion binders (to form supramolecular structures via H-bonding interactions) and also as metal ion chelators.23 Thus, complex cation (1) was selected as a potential building block for hierarchical assembly into diverse superstructures. Metal complexes 1.Cl and 1.Br were synthesized by chemical reduction of [Ru(1,10-phenanthroline-5,6-dione)3]Cl2 followed by anion exchange; whereas 1.Li was prepared by reacting 1.PF6 (prepared by exchanging Cl− ions in 1.Cl with PF6− anions) with lithium methoxide at RT (Fig. 1 and Schemes S1–S3†).24 In FTIR spectrum of all the three complexes (1.Cl, 1.Br and 1.Li), the disappearance of the carbonyl band in the starting material at 1694 cm−1 confirms the reduction of the carbonyl groups, whereas the presence of broad νO–H stretches in the range of 3230–3260 cm−1 attributed to the hydroxyl groups confirms the formation of 1.Cl and 1.Br (Fig. S1†). The reduction of the complexes was also confirmed by 1H NMR where the downfield shift of the signals for H1 protons and upfield shift of the H2–H3 protons indicate alterations in the electronic conjugation in the aromatic structure of the phenanthroline ligands (Fig. S2 and S3†). Slow diffusion of diethylether into a methanolic solutions of 1.Cl and 1.Br at RT in dark for several days resulted in orange-colored needle-shaped single crystals suitable for X-ray diffraction (SC-XRD) analysis (Table S1†). Single crystals of 1.Li were grown in a sealed vial by layering a saturated methanolic solution of 1.Li over DMF for several weeks at RT, and under inert atmosphere. The crystals were kept in the mother liquor under inert atmosphere and picked under a stream of N2 for X-ray diffraction analysis. The complete description of the crystallization methods is detailed in the “Crystallization Procedures” part of the ESI.†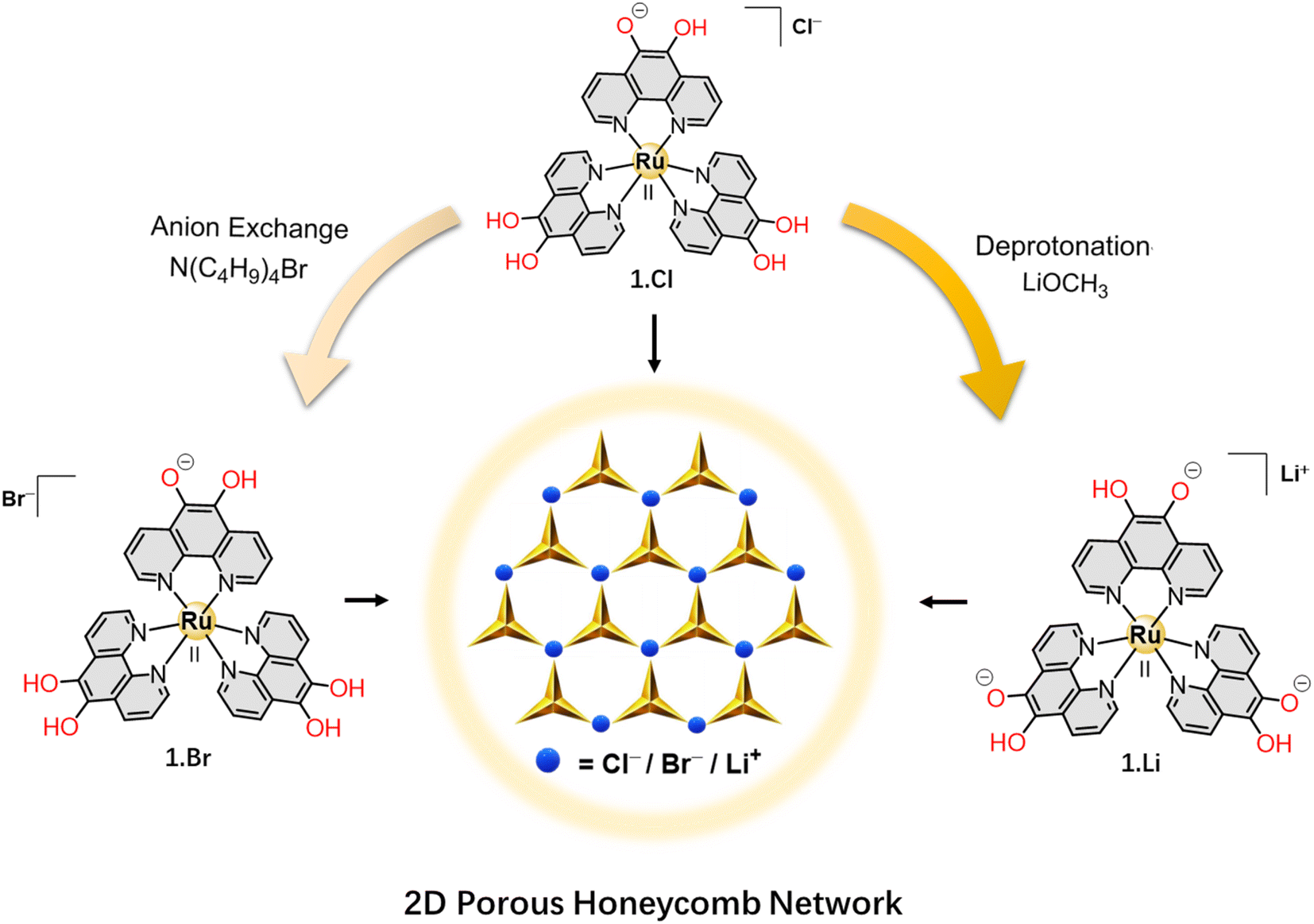 | ||
| Fig. 1 Schematic diagram showing the formation of 1.Br, 1.Li and 1.Cl. | ||
Structural features of 1.Cl, 1.Br and the effect of counter anion
The single crystal X-ray diffraction data revealed that a discrete 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex was formed for both compositions. The compound 1.Cl {(RuL2L−)·Cl} crystallizes in trigonal space group P31c. The asymmetric unit consists of one Ru(II) cation coordinated with one ligand unit (L−) through pyridyl nitrogens, and one Cl− as counter-anion (Table S1†). Unlike 1.Cl, the asymmetric unit in 1.Br {(RuL2L−)·Br} (space group P21/n) represents one [Ru(5-oxido-6-hydroxy-1,10-phenanthroline)(5,6-dihydroxy-1,10-phenanthroline)2]Br complex unit where the Br− anion is disordered over two positions with 75:25% occupancy.
:1 complex was formed for both compositions. The compound 1.Cl {(RuL2L−)·Cl} crystallizes in trigonal space group P31c. The asymmetric unit consists of one Ru(II) cation coordinated with one ligand unit (L−) through pyridyl nitrogens, and one Cl− as counter-anion (Table S1†). Unlike 1.Cl, the asymmetric unit in 1.Br {(RuL2L−)·Br} (space group P21/n) represents one [Ru(5-oxido-6-hydroxy-1,10-phenanthroline)(5,6-dihydroxy-1,10-phenanthroline)2]Br complex unit where the Br− anion is disordered over two positions with 75:25% occupancy.
In the crystal structure of 1.Cl and 1.Br, the bond parameters corresponding to the octahedral metallo-ligand are in agreement with the literature reports on similar complexes where the average Ru–N bond distances are 2.066 Å and 2.047 Å, with phenanthroline bite angles of 80.5(3)° and 80.8–79.8°, respectively (Fig. S4†).24 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths (1.329 Å for 1.Cl, 1.358–1.342 Å for 1.Br) in the catechol subunits indicate that the extent of charge delocalization is different in the two complexes. This can be understood by the fact that chloride ions being stronger H-bond acceptors due to higher electronegativity than the bromide anions. The existence of only one counter anion in the asymmetric units of 1.Cl and 1.Br confirms that only one out of all the six hydroxyl groups is deprotonated, generating the [Ru(II)L2L−]+ mono-cation (Fig. 2A). As a result, the negative charge on the ligands surrounding the Ru(II) cation is distributed over all three dihydroxy-phenanthroline ligands (detailed discussion on charge distribution can be found in Fig. S5†).The partial deprotonation of the hydroxyl groups in Ru polypyridyl complex is feasible since the coordination with a first anion might deactivate the complex and reduce its affinity to interact with the second anion. Despite being a weaker H-bond acceptor, Br− ions displayed similar anion-template effect in 1.Brvia weaker O–H⋯anion interactions with four nearby hydroxyl groups from three building block units (avg. O⋯Br ∼3.127 Å, avg. O–H⋯Br ∼2.373 Å, avg. ∠BrHO ∼154.46°) (Fig. 2). In this H-bonded quadruple, one of the building blocks forms convergent hydrogen bonds to the Br− ion through both of the hydroxyl groups (Fig. S6†).
C bond lengths (1.329 Å for 1.Cl, 1.358–1.342 Å for 1.Br) in the catechol subunits indicate that the extent of charge delocalization is different in the two complexes. This can be understood by the fact that chloride ions being stronger H-bond acceptors due to higher electronegativity than the bromide anions. The existence of only one counter anion in the asymmetric units of 1.Cl and 1.Br confirms that only one out of all the six hydroxyl groups is deprotonated, generating the [Ru(II)L2L−]+ mono-cation (Fig. 2A). As a result, the negative charge on the ligands surrounding the Ru(II) cation is distributed over all three dihydroxy-phenanthroline ligands (detailed discussion on charge distribution can be found in Fig. S5†).The partial deprotonation of the hydroxyl groups in Ru polypyridyl complex is feasible since the coordination with a first anion might deactivate the complex and reduce its affinity to interact with the second anion. Despite being a weaker H-bond acceptor, Br− ions displayed similar anion-template effect in 1.Brvia weaker O–H⋯anion interactions with four nearby hydroxyl groups from three building block units (avg. O⋯Br ∼3.127 Å, avg. O–H⋯Br ∼2.373 Å, avg. ∠BrHO ∼154.46°) (Fig. 2). In this H-bonded quadruple, one of the building blocks forms convergent hydrogen bonds to the Br− ion through both of the hydroxyl groups (Fig. S6†).
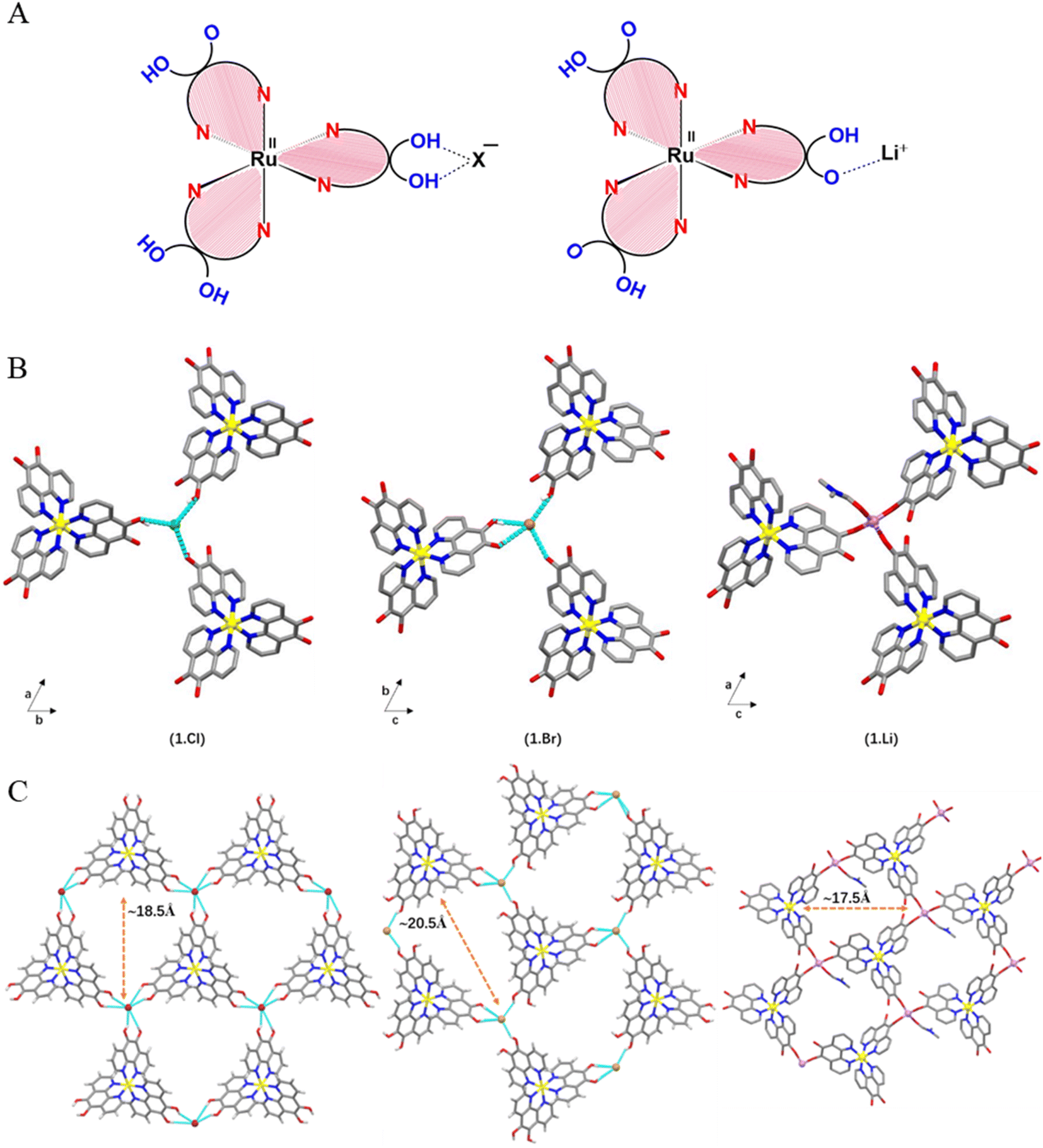 | ||
| Fig. 2 (A) Schematic diagram of the metal complexes (1) highlighting the different types of outer sphere interaction between the peripheral catecholate groups and the counter ions. (B) One ancillary ion is interacting with three metallotecton (building blocks) units via hydrogen bonding in 1.Cl and 1.Br and ionic interaction in 1.Li. (C) Views of one 2D layer highlighting H-bonded framework consisting of ancillary ion in 1.Cl, 1.Br and 1.Li. Color codes: Ru (yellow), Cl (green), Br (brown), N (blue), O (red), H-bonds (cyan). Hydrogen atoms other than those involved in H-bonding interactions have been omitted for clarity. | ||
The intermolecular organization in the crystal structure of both the complexes can be best understood in terms of supramolecular assembly of the building blocks via non-covalent interactions between three 5,6-dihydroxy-1,10-phenanthroline units and the ancillary ions (Fig. 2B). In the crystal lattice of 1.Cl, one of the hydroxyl groups from each catecholate of three neighboring metallo-ligands points toward a single Cl− ion and forms short O–H⋯Cl hydrogen bonds (O⋯Cl: 3.043 Å, O–H⋯Cl: 2.281 Å, ∠ClHO: 150.69°) in a trifurcated manner, which extends in the ab plane (Fig. 2C). The average O–H⋯Cl hydrogen bond distance is ca. 76% vdW which is in accordance with the previously reported catechol-chloride H-bonded organic frameworks.24 This supramolecular association extends in the ab-plane to form honeycomb (hcb) network with hexagonal pores consisting of Ru(II) cations and halide anions as corners and aromatic phenanthroline ligands as edges (Fig. 2). The large perfect hexagonal pores have edge length of ca. 9.266 Å (Ru⋯Cl distance) leading to high pore window of ∼223 Å2 where the phenanthroline rings are out of the plane connecting Ru and Cl ions by ca. 30° (Fig. S6†). Therefore, each Cl− ion receives three O–H hydrogen bonds from three different metallo-ligands. This H-bonded triad represents a perfect tetrahedron where the Cl− ions lie ca. 0.63 Å above the basal plane formed by three hydroxyl oxygens (∠OClO ∼109.6°; O⋯O ∼4.974 Å) (Fig. S7†). The powder XRD diffraction patterns of 1.Cl and 1.Br are matching with the simulated single crystal data which proves the purity of the samples (Fig. S8 and S9†).
Despite different hydrogen bonding arrangements in 1.Cl and 1.Br, 2D sheets are formed in each case. Unlike 1.Cl, the pores in 1.Br are not perfectly hexagonal and rather skewed which is probably due to higher ionic radii of Br− ions where the distance between the Ru(II) and the Br− corners lies in the range of 9.09–9.45 Å (Fig. S10†). Despite the fact that crystallization of 1.Cl and 1.Br took place in methanol, which is a highly competitive hydrogen-bonding solvent, the multitude of weaker non-covalent intermolecular interactions e.g., O–H⋯X made the 2-dimensional assembly thermodynamically favorable by sufficiently increasing the entropy component.25 Thus, the halide ions (Cl− in 1.Cl and Br− in 1.Br) act as template to interact with three stereochemically similar metal complex units (Δ/Λ) and vice versa (Fig. S11†). The details on the distribution and occupancies of atoms in 1.Br is provided in Fig. S12.† While the framework formation with bromide anions is known to be ambiguous due to weaker H-bonding interactions, 1.Br represents one of the rare examples of bromide-templated metallotecton based porous supramolecular framework.26
Structural features of 1.Li and the effect of counter cation
In order to establish the multifaceted binding ability of the metallo-ligand (1), we investigated the deprotonation of the catecholate sites and coordination of 1 with Li+ ions. When the three catecholate functionalities are mono-deprotonated, the metallotecton (1) attains an overall one negative charge and acts as efficient cation receptor (Fig. 1). The asymmetric unit in 1.Li consists of one Li+ cation coordinated with catecholate fragment of one complex unit. The presence of one Li+ ion per molecule (instead of 6Li+ ions in case of Li6[Ru(L6−)](Cl)2 or 4Li+ ions for Li4[Ru(L6−)]) confirms that Li+[Ru(II)(HL−)3]− is the formula unit where each catecholate ligand is mono-protonated (Fig. 2A and Table S1†). The Li+ ion resides in a tetragonal configuration formed by three ligands through C–O− and one oxygen from a coordinating DMF solvent molecule (additional details on the geometry around the Li+ and charge balance can be found on Fig. S13†).In the crystal structure of 1.Li, the formation of a 2D porous layered network (similar to 1.Cl and 1.Br) was observed consisting of Ru coordination complex [Ru(II)(5-oxido-6-hydroxy-1,10-phenanthroline)3]− and Li+ cations. The honeycomb layered network is planar where the layers stack in AB manner along the b-axis (Fig. 2C and S14†). The hexagonal pores are slightly distorted where the three adjacent sides are of different length while the pairs of sides facing each other are of similar dimensions. Worth to be noted, the powder sample of 1.Li was found to be amorphous whereas little amount of single crystals were obtained by crystallization which precluded an elaborate PXRD investigation. Additionally, contrary to 1.Cl and 1.Br, 1.Li is not air stable and single crystals where found to react rapidly when exposed to air, which did not facilitate single crystal XRD measurements and detailed structure resolution. This instability justifies the essential presence of DMF molecules and no further powder XRD analysis could be performed. Notably, to the best of our knowledge, this is the only report on bimetallic porous framework consisting of Li-ions.
Thus, each of the anions (Cl−, Br−) and cation (Li+) are found to interact in a similar manner with the Ru complex units and establish the versatile secondary sphere interaction ability of this building block (1). These interactions prevail uniformly in 2-dimensions to form porous honeycomb networks. The ability of the ligand to undergo selective deprotonation of the catechol units in coordination complex (1) dictates the formal charge on the complex and accordingly the choice of the ancillary anion or cation. While the deprotonation of the first hydroxyl proton in 1 was performed by weakly basic solvent (ethanol) resulted in 1.Cl and 1.Br, the deprotonation of the remaining groups to achieve 1.Li could only be performed with strong bases such as lithium methoxide (LiOMe). This implies that these hydroxyl protons are poorly acidic which provides a handy tool to control deprotonation of 1 as well as framework composition.
To understand the simplified underlying nets in the three porous architectures, topological calculations were performed revealing two-dimensional, binodal three-connected net with hcb topology having {63} point symbol for the networks for all three porous networks (Fig. 2B and S15†).20 Important to note, often replacement of one of the components in a self-assembled multi-component network leads to changes in inter-component interactions and hence the topology.27 Such topological invariance between 1.Cl, 1.Br and 1.Li is unique and implies that matrix aspects prevail over second-sphere interactions in the self-assembly process. Each 2D layer in 1.Cl and 1.Br represents a perfectly planar and robust hydrogen bonded network where Ru and halide ions lie in the same plane. However, the presence of three free oxygen sites from the catechol and one extra DMF solvent molecule bonded to the Li+ cation confers a 2D quasi-planar network in 1.Li. The layers are homochiral, where all the Ru complex units have the same configuration in one layer (either Δ or Λ) which is opposite in the adjacent ones. Thus the ABAB stacking mode is based on the stereochemical configuration of 1. The layers are stacked in an eclipsed manner to form hexagonal channels where the complex subunits of one layer lie exactly above the trimeric non-covalent halide-phenanthroline nodes of the next layer (the Ru cations and halide anions are super imposed in alternate layers). This stacking is facilitated by weak non-covalent interactions, such as aromatic stacking between the close-lying phenanthroline ligands and H-bonding (Fig. S16†). The pore dimensions are different in both the frameworks because the Ru and Cl ions lie on the C3 axis of symmetry in 1.Cl which allows the formation of highly symmetrical hexagonal channels which is not the case for 1.Br and 1.Li (Fig. 2C and S11†).
Guest exchange study in 1.Cl and 1.Br
The vertical channels in the three porous frameworks are occupied by solvent molecules which can be replaced by external guest molecules without disrupting the supramolecular assembly. To prove the existence of solvents molecules within the framework, thermogravimetric analyses were performed on 1.Cl and 1.Br. Thermograms displayed mass loss in the low temperature range (<100 °C) typical to residual low boiling solvents used during the synthesis such as ethanol, diethylether or traces of water (Fig. S17 and S18†). These observations point towards the existence of weaker interactions between the building units and the solvent molecules. The Ru complex and the Cl− ions exist on the C3 axis of symmetry in 1.Cl whereas in 1.Br, the Br− ions also exist on the similar location. Therefore, the interlayer spacing (4.7–5 Å) being too small to accommodate solvent molecules, these must be located within the hexagonal channels which can be responsible for the existence of unidentified electron density peaks observed in single crystal XRD measurements. At this stage, it remains hard to predict the contribution of such interactions towards the stability of the frameworks, yet we attempted to replace the solvent molecules and to utilize the channels for molecular sorption.Crystallites of 1.Cl were exposed to D2O or I2 environment where the solvent molecules were exchanged (details are provided in ESI†).28 A clear appearance of broad signal at 2200–2600 cm−1 in the FTIR spectra for the O–D stretch and disappearance of signals corresponding to hydroxyl groups of the trapped solvents e.g. CH3OH or H2O confirmed the replacement of solvent molecules by D2O (Fig. 3A and B). The resemblance of the peak positions in the powder X-ray diffraction patterns of 1.Cl and 1.Br after D2O and iodine exchange further confirmed the framework stability (Fig. 3E and S19†). However, despite that the peak position remained generally unaltered, the relative intensities as well as peak widths significantly changed upon guest inclusion (Fig. 3C–E). Additional peaks in the diffractogram (above 15°) appear for both 1.Cl and 1.Br, likely suggesting lattice distortion or inclusion of ordered guest molecules. While the increase in peak width might be the result of higher lattice strain due to the absence of opportune non-covalent interactions and reduced crystallites size (powder samples were used for these experiments) upon solvent exclusion, the change in relative intensities of the peaks can be attributed to guest inclusion. Upon removal of supportive interactions, the crystallites might attain statistically random distribution of crystal plane orientations accounting for the reduction in peak intensity belonging to the 100 planes (6.28°). This guest exchange ability was further supported by incorporation of I2 molecules. When crystallites of 1.Cl and 1.Br were suspended in a 1000 ppm solution of I2 in diethylether (orange color), the solution was found to be completely colorless after 48 h. The equilibrium absorption amount was found to be 135.56 mg g−1 and 123.52 mg g−1 for 1.Cl and 1.Br respectively. The sharp color change and increase in weight of the crystallites with no change in FTIR spectrum and PXRD pattern of the two frameworks further substantiated the absorption of I2 (Fig. 3D and E). The crystallinity of 1.Cl was found to be significantly reduced after I2 incorporation and new peaks appeared at higher angles (>15°), which can be attributed to ordered trapping of I2 molecules in the porous framework (Fig. 3E). The same phenomena was observed for 1.Br, with a moderate loss of crystallinity, indicating that 1.Br is comparatively more stable upon I2 incorporation than 1.Cl (Fig. S19†). Thus, these versatile and robust hydrogen bonded frameworks (1.Cl and 1.Br) act as efficient hosts for a variety of guests and display robust and porous structures.
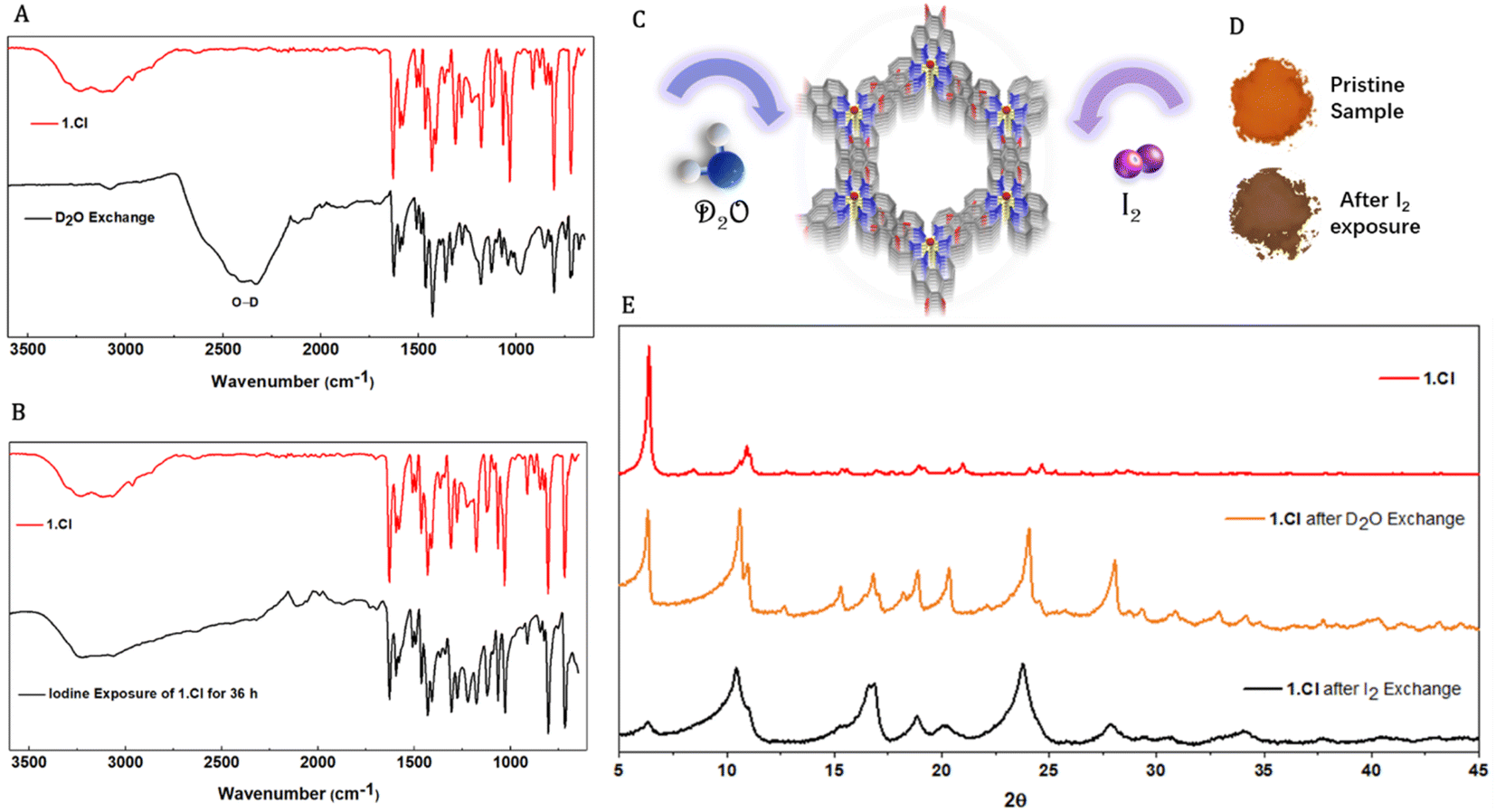 | ||
| Fig. 3 Comparative FTIR spectra of 1.Cl (A) before (red) and after solvent – D2O exchange (black); and (B) before (red) and after I2 exchange (black). (C) Schematic diagram showing one channel sorption of 1.Cl. (D) Change of color of powder of 1.Cl before and after I2 exchange (no change in color was observed after D2O exchange). (E) Comparative PXRD patterns of 1.Cl before (red) and after I2 (black) and D2O (orange) exchange. | ||
Perspective and conclusion
The nearly similar arrangement of building blocks in the crystal packing of 1.Cl and 1.Br demonstrates that the [Ru(5-oxido-6-hydroxy-1,10-phenanthroline)(5,6-dihydroxy-1,10-phenanthroline)2]+ cation is a suitable building block to fabricate permanently porous materials through assembly of anions of different size and basicity; but also of the cations, as demonstrated by 1.Li. By tuning the bonding modes of this coordination complex through acid–base charge stoichiometry modulation, we formed a series of ordered porous frameworks with anions and cations as ancillary ions via non-covalent interactions. This implies that the self-assembly process is controlled by complementary intermolecular non-covalent interactions making the choice of the ancillary ion insignificant. Furthermore, the robustness of the supramolecular frameworks was validated by guest inclusion within the porous structures. This work shows that achieving isostructural supramolecular frameworks, even upon replacement of the constituent moieties is possible, upon judicious selection of the building blocks, and controlling the engaged interactions. Broadly, this work has the potential to achieve novel structural properties in framework materials by transforming the way building blocks interact.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
AV was involved in scheme design, funding procurement, periodic monitoring of the project, manuscript preparation and submission. DG helped in scheme design, performed synthesis of the compounds and manuscript preparation. GK carried out crystallization and absorption experiments. GC collected and analyzed the other characterization data. KR performed Single crystal X-ray diffraction experiments and structure modelling. DG, GK, GC and AV wrote the final manuscript. GC, DG and AV revised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge funding from the European Research Council (ERC) grant-project 770870-MOOiRE. G. C. acknowledges the Belgian Fonds de la recherche scientifique (F.R.S.-FNRS) for FRIA fellowship.References
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH, 1995 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974–987 CrossRef CAS PubMed.
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490 CrossRef CAS.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154–1196 CrossRef.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–907 CrossRef CAS PubMed.
- M. K. Corpinot and D.-K. Bucar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS.
- D. J. Cram, Angew. Chem., Int. Ed., 1988, 27, 1009–1020 CrossRef.
- J.-L. Atwood and J.-M. Lehn, Comprehensive Supramolecular Chemistry, Pergamon, Oxford, 1996, vol. 9 Search PubMed.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed.
- A. M. Beatty, CrystEngComm, 2001, 51, 1–13 Search PubMed.
- M. D. Ward, Chem. Commun., 2005, 5838–5842 RSC.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS PubMed.
- M. Atzori, A. Serpe, P. Deplano, J. A. Schlueter and M. L. Mercuri, Inorg. Chem. Front., 2015, 2, 108–115 RSC.
- G. Mouchaham, N. Roques, C. Duhayon, I. Imaz and J.-P. Sutter, New J. Chem., 2013, 37, 3476 RSC.
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, New York, 1989 Search PubMed.
- P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1(1), 77–87 CrossRef CAS.
- A. Dieter Schlüter, Functional Molecular Nanostructures, Springer, New York, 2005 Search PubMed.
- J. C. McDonald, P. C. Dorrestein, M. M. Pilley, M. M. Foote, J. L. Lundberg, R. W. Henning, A. J. Schultz and J. L. Manson, J. Am. Chem. Soc., 2000, 122, 11692–11702 CrossRef.
- L. Öhrström and K. Larsson, Dalton Trans., 2004, 347–353 RSC.
- M. Tadokoro and K. Nakasuji, Coord. Chem. Rev., 2000, 198, 205–218 CrossRef CAS.
- Y.-H. Tan, L.-F. Yang, M.-L. Cao, J.-J. Wu and B.-H. Ye, CrystEngComm, 2011, 13, 4512–4518 RSC.
- M. W. Hosseini, Coord. Chem. Rev., 2003, 240, 157–166 CrossRef CAS.
- D. Gupta, A. E. Lakraychi, B. D. Boruah, S. D. Kreijger, L. Troian-Gautier, B. Elias, M. D. Volder and A. Vlad, Chem.–Eur. J., 2022, e202201220 CrossRef CAS PubMed.
- M. Morshedi, A. C. Willis and N. G. White, CrystEngComm, 2016, 18, 4281–4284 RSC.
- N. G. White and M. J. MacLachlan, Chem. Sci., 2015, 6, 6245–6249 RSC.
- L.-F. Yang, M.-L. Cao, H.-J. Mo, H.-G. Hao, J.-J. Wo, J.-P. Zhang and B.-H. Ye, CrystEngComm, 2009, 11, 1114–1121 RSC.
- G. Kumar, S. Pandey and R. Gupta, Cryst. Growth Des., 2018, 18, 5501–5511 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2143383–2143385. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02068a |
| ‡ Current address: Fraunhofer Institute of Interfacial Engineering and Biotechnology IGB, Bio-, Electro-, and Chemocatalysis BioCat, Straubing Branch, Schulgasse 11a, 94315, Straubing, Germany |
| This journal is © The Royal Society of Chemistry 2023 |