
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric imino-acylation of alkenes enabled by HAT-photo/nickel cocatalysis†
Rui
Wang
and
Chuan
Wang
 *
*
Hefei National Laboratory for Physical Science at the Microscale, Department of Chemistry, Center for Excellence in Molecular Synthesis, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, P. R. China. E-mail: chuanw@ustc.edu.cn
First published on 22nd May 2023
Abstract
By merging nickel-mediated facially selective aza-Heck cyclization and radical acyl C–H activation promoted by tetrabutylammonium decatungstate (TBADT) as a hydrogen atom transfer (HAT) photocatalyst, we accomplish an asymmetric imino-acylation of oxime ester-tethered alkenes with readily available aldehydes as the acyl source, enabling the synthesis of highly enantioenriched pyrrolines bearing an acyl-substituted stereogenic center under mild conditions. Preliminary mechanistic studies support a Ni(I)/Ni(II)/Ni(III) catalytic sequence involving the intramolecular migratory insertion of a tethered olefinic unit into the Ni(III)–N bond as the enantiodiscriminating step.
Introduction
Cyclization of oxime derivative-tethered alkenes1 has emerged as a powerful tool for the synthesis of pyrrolines, which are a characteristic structural motif found in numerous natural products and biologically active compounds.2 Mechanistically, these ring closure reactions either proceed via the transition metal-promoted aza-Heck reaction3 or are mediated by iminyl radicals under photoredox catalysis.4 Among various imino-cyclization reactions, imino-acylation of oxime ester-tethered alkenes provides pyrrolines containing a synthetically highly useful carbonyl moiety as the products and thus has attracted considerable attention from synthetic chemists in recent years (Scheme 1A). In 2015, Bower reported a palladium-catalyzed redox-neutral three-component imino-acylation of alkenes incorporating a pendant oxime ester with carbon monoxide and organoborons as coupling partners.5 In 2019, our group developed a nickel-catalyzed olefin imino-acylation employing acid chlorides or anhydrides as the acylating agent under reductive conditions.6 Very recently, Zhao7 and Ye8 successfully applied redox N-heterocyclic carbene catalysis in diastereoselective olefin imino-acylation using aromatic aldehydes or acyl imidazoles as the acyl source. Despite impressive advances, none of these aforementioned imino-acylation reactions could yield highly enantioenriched products. Therefore, establishing a new mode for asymmetric olefin imino-acylation is still highly desired.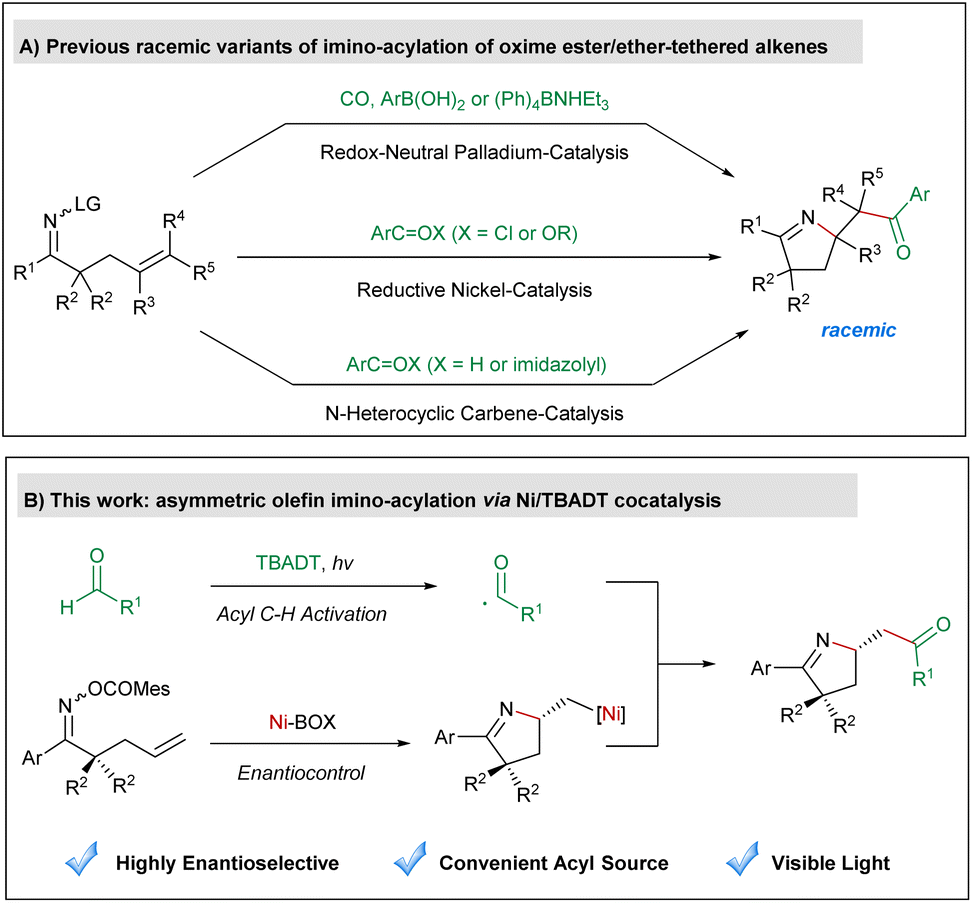 | ||
| Scheme 1 (A) Previous racemic variants of imino-acylation of oxime ester/ether-tethered alkenes; (B) asymmetric olefin imino-acylation via Ni/TBADT cocatalysis. | ||
As a hydrogen-atom-transfer (HAT) photocatalyst, tetrabutylammonium decatungstate (TBADT) is able to promote the homolytic cleavage of the acyl C–H bond of aldehydes as well as the aliphatic C–H bond of hydrocarbons, to produce nucleophilic carbon-centered radicals, which can be used as coupling partners with various electrophiles.9–11 Recently, the scope of C–H functionalization reactions involving TBADT has been significantly expanded through the combination of transition-metal catalysis.12 We focus our research on the asymmetric variants of these reactions13 and accomplished enantioselective olefin acyl-carbamoylation14 and acyl C–H allylation,15 which are the only two precedents in asymmetric cooperative catalysis of transition metals and TBADT to the best of our knowledge. As a continuation of our ongoing interest in this research area, we herein envision an asymmetric imino-acylation reaction of oxime ester-tethered alkenes with aldehydes through merging nickel-mediated enantioselective aza-Heck cyclization and TBADT-promoted acyl C–H activation, to allow for the synthesis of diverse pyrrolines bearing an acyl-substituted stereogenic center in a highly enantioselective fashion (Scheme 1B). The major challenge of the anticipated transformation lies in the avoidance of iminyl radical-mediated uncontrolled ring closure.
Results and discussion
For optimization of the reaction conditions, we utilized butyraldehyde (1a) and the mesitoyl oxime ester 2a incorporating a terminal olefinic unit as the standard substrates (Table 1). After the systematic screening of various reaction parameters, we identified the optimized reaction conditions as follows: Ni(ClO4)2·6H2O (15 mol%), the BOX ligand L1 (18 mol%), TBADT (5 mol%), and NaClO4 (0.5 equiv.) under irradiation at 390 nm for 24 h in MeCN at room temperature. In this case, the desired imino-acylation product 3aa was obtained in 66% yield and 99% ee (entry 1), wherein the major by-products are the ketone 2a′ (9%) and the hydroimination product 3aa′ (11%, 15% ee). Subsequently, we varied the reaction parameters to demonstrate their influence on the outcome of the target transformation. Only 7% ee could be achieved in the case of the PHOX L2 as the ligand, which has been proven to be the best ligand in our previously reported Ni/TBADT-cocatalyzed asymmetric reactions14,15 (entry 2). In the case of the PyBox L3, the BiOX L4, the BOX L5, or the PyrOx L6, the imino-acylation reaction proceeded with very poor or no asymmetric induction (entries 3–6). The substitution pattern of the bridging methylene of the BOX ligands turned out to be vital for the enantioselectivity of the studied reaction. Compared to the chiral BOX L1, ligands L7–L9 resulted in substantially lower enantioselectivities (entries 7–9). The use of other nickel precatalysts including NiI2, Ni(BF4)2·6H2O, NiBr2·glyme, Ni(acac)2, and Ni(cod)2 gave rise to inferior results (entries 11–15). Replacing MeCN with acetone as the solvent led to a decrease in both yield and enantiomeric excess of 3aa (entry 16). Without the addition of NaClO4, both the efficiency and the enantiocontrol of the desired reaction were slightly attenuated (entry 17). In the absence of nickel, TBADT, or irradiation, no desired imino-acylation occurred, confirming their crucial roles in this cooperative catalysis (entry 18).|
|
|||
|---|---|---|---|
| Entry | Deviation from optimized conditions | Yield 3aab (%) | eec (%) |
| a Unless otherwise specified, the reactions were performed on a 0.2 mmol scale of the oxime ester 2a using 3 equiv. of butyraldehyde (1a), 15 mol% Ni(ClO4)2·6H2O, 18 mol% ligand L1, 5 mol% TBADT, and 0.5 equiv. of NaClO4 in 1 mL MeCN under irradiation at 390 nm for 24 h (two lamps 5 cm away, with adequate fans and a water bath to keep the reaction at room temperature). b Yields of isolated products through column chromatography. c Determined by HPLC-analysis on a chiral stationary phase. | |||
| 1 | None | 66 | 99 |
| 2 | L2 instead of L1 | 78 | 7 |
| 3 | L3 instead of L1 | 40 | 0 |
| 4 | L4 instead of L1 | 65 | 0 |
| 5 | L5 instead of L1 | 26 | 9 |
| 6 | L6 instead of L1 | 70 | 0 |
| 7 | L7 instead of L1 | 62 | 10 |
| 8 | L8 instead of L1 | 47 | 63 |
| 9 | L9 instead of L1 | 73 | 13 |
| 11 | NiI2 instead of Ni(ClO4)2·6H2O | 40 | 96 |
| 12 | Ni(BF4)2·6H2O instead of Ni(ClO4)2·6H2O | 62 | 98 |
| 13 | NiBr2·glyme instead of Ni(ClO4)2·6H2O | 33 | 96 |
| 14 | Ni(acac)2 instead of Ni(ClO4)2·6H2O | 10 | 94 |
| 15 | Ni(cod)2 instead of Ni(ClO4)2·6H2O | 43 | 99 |
| 16 | Acetone instead of MeCN | 25 | 91 |
| 17 | w/o NaClO4 | 61 | 98 |
| 18 | w/o Ni(ClO4)2·6H2O or TBADT or light | 0 | — |
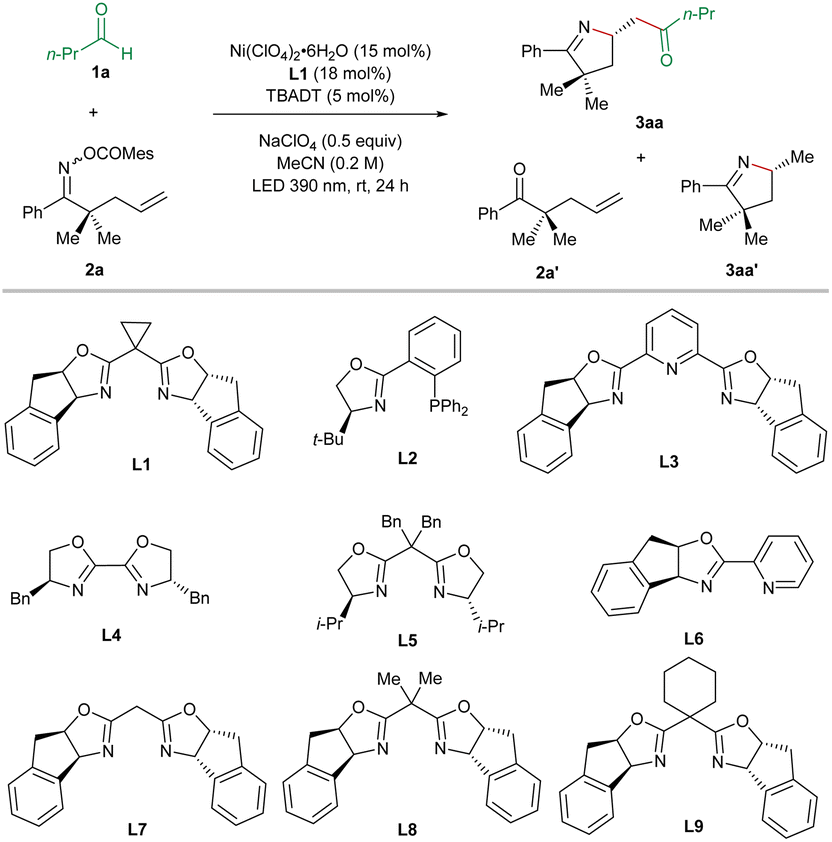
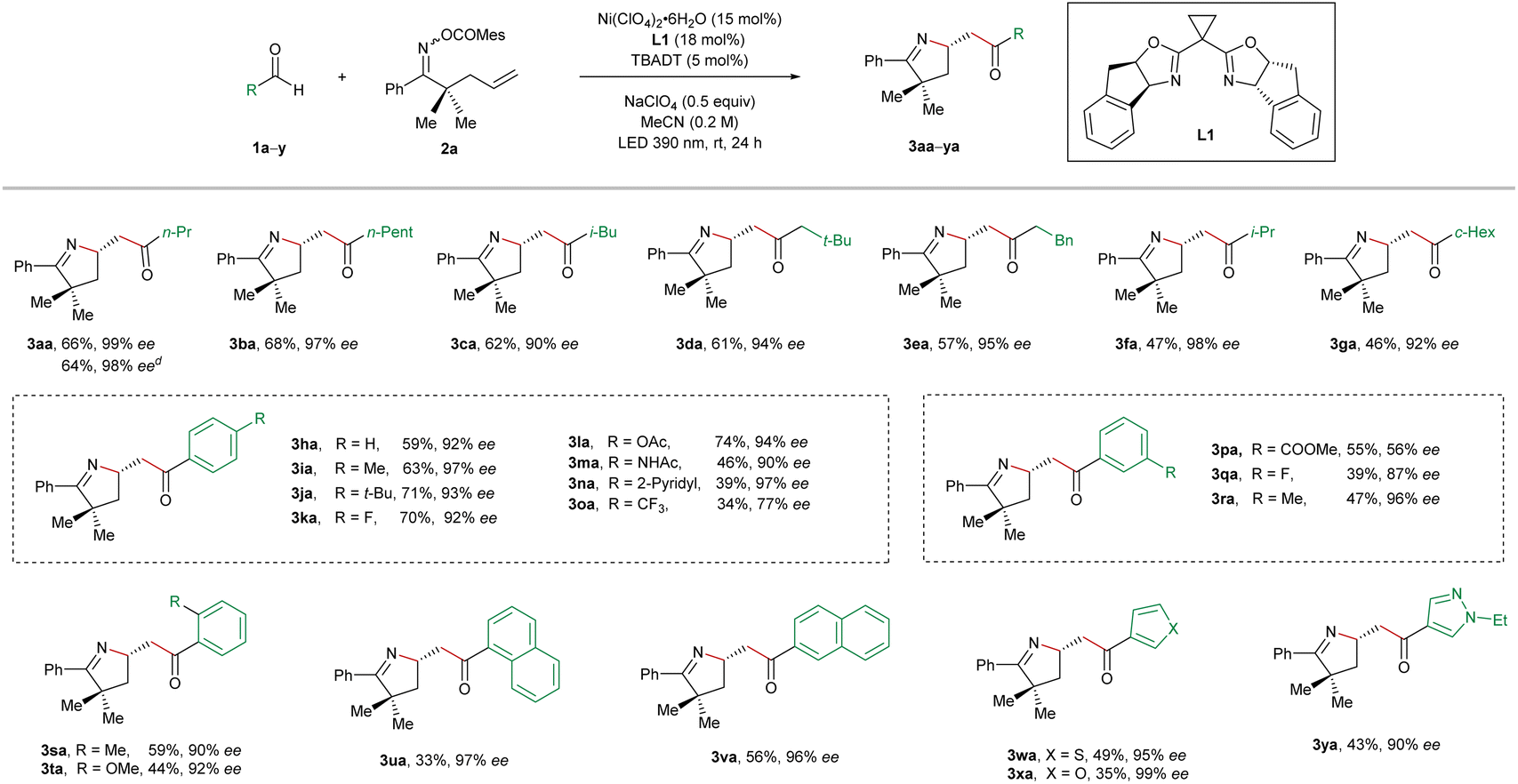
With the optimized reaction conditions in hand, we started to evaluate the substrate scope of this nickel/photo-cocatalyzed olefin imino-acylation. First, various aldehydes were reacted with the oxime ester-tethered alkene 2a under standard conditions (Table 2). The aliphatic aldehydes without α-substitution (1a–1e) provided the products 3aa–3ea in moderate to good yields and excellent enantioselectivities.
| a Unless otherwise specified, the reactions were performed on a 0.2 mmol scale of the oxime ester 2a using 3 equiv. of the aldehydes 1a–1y, 15 mol% Ni(ClO4)2·6H2O, 18 mol% ligand L1, 5 mol% TBADT, and 0.5 equiv. of NaClO4 in 1 mL MeCN under irradiation at 390 nm for 24 h (two lamps 5 cm away, with adequate fans and a water bath to keep the reaction at room temperature). b Yields of the isolated products after column chromatography. c Enantiomeric excesses were determined by HPLC-analysis on a chiral stationary phase. d The reaction was performed on a 2 mmol scale of the oxime ester 2a. |
|---|
|
In the case of bulkier α-branched aldehydes (1f and 1g), the products 3af and 3ag were obtained in slightly lower yields while the asymmetric induction remained high. In contrast, the reactions employing α-trisubstituted aldehydes such as pivalaldehyde and 2,2-diphenylpropanal failed to yield the desired products. Subsequently, the generality of aromatic aldehydes for the studied reaction was interrogated. Benzaldehyde (1h) and its derivatives bearing either electron-donating or weak electron-withdrawing substitution on the para, meta, or ortho positions of the phenyl ring (1i–1n and 1q–1t) were all found to be pertinent precursors, furnishing the products 3ia–3na and 3qa–3ta in a highly enantioselective manner. In contrast, strong electron-withdrawing substituents on the phenyl ring turned out to have a detrimental effect on the enantiocontrol (3oa and 3pa). Furthermore, the naphthaldehydes 1u and 1v, as well as the heteroaryl aldehydes bearing a furan (1w), thiophene (1x), or pyrazole unit (1y) were also suitable precursors, providing the corresponding products 3ua–3ya in high enantiocontrol. In addition, the reaction on a 2 mmol scale of 2a toward the synthesis of compound 3aa provided a similar result in terms of both efficiency and asymmetric induction (64% yield, 98% ee).
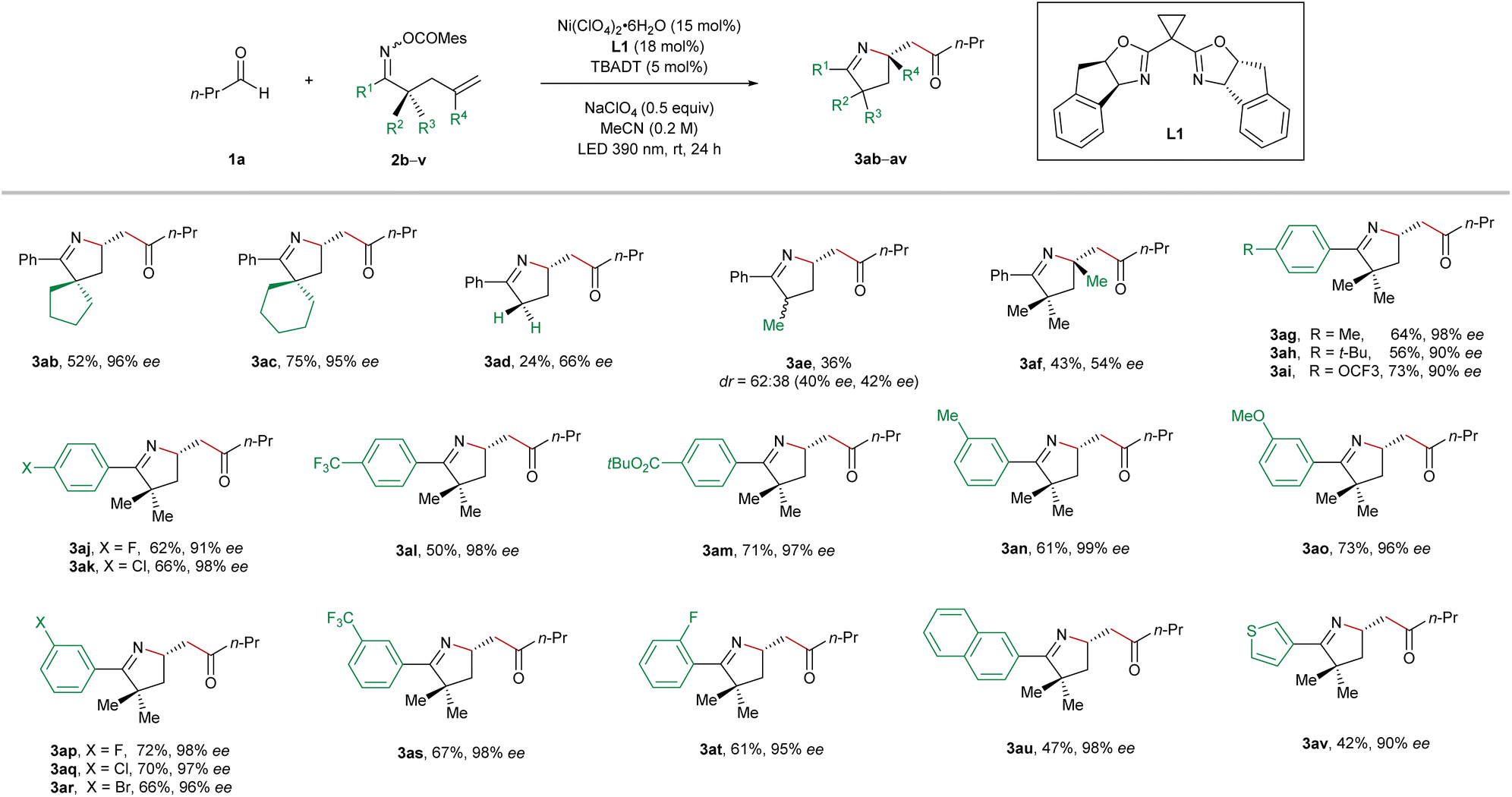
Next, we continued to explore the substrate scope by varying the structure of the oxime ester-tethered alkenes in the reactions with butyraldehyde (1a), and the results are summarized in Table 3. Starting from the cyclic substrates 2b and 2c, the highly enantioenriched pyrrolines with a spirocyclic scaffold (3ab and 3ac) were synthesized in excellent enantioselectivity and in 52% and 75% yield, respectively. Unfortunately, the absence of geminal substitution of the linker between oxime ester and alkene led to a dramatic decrease in both efficiency and enantiocontrol (3ad). Furthermore, the reaction using the α-monomethyl substituted oxime ester 2e delivered the imino-acylation product 3ae in a low diastereomeric ratio and low enantiomeric excesses. In the case of 1,1-disubstituted alkene, a quaternary stereogenic center could be constructed albeit in a relatively low enantiomeric excess (3af). Subsequently, the permutation of different substitutions on the phenyl ring of the oxime esters 2g–2t was carried out. In general, in the case of both electron-donating and electron-withdrawing substituents, all the reactions proceeded smoothly, furnishing the products 3ag–3at in moderate to good yields with excellent enantiocontrol. Moreover, the naphthyl and thienyl oxime esters 2u and 2v also turned out to be suitable precursors for this cocatalyzed reaction, providing the coupling products 3au and 3av in high enantiomeric excesses. Unsuccessful substrates for this reaction include internal olefins and oxime esters derived from dialkyl ketones.
| a Unless otherwise specified, the reactions were performed on a 0.2 mmol scale of the oxime esters 2b–2v using 3 equiv. of butyraldehyde (1a), 15 mol% Ni(ClO4)2·6H2O, 18 mol% ligand L1, 5 mol% TBADT, and 0.5 equiv. of NaClO4 in 1 mL MeCN under irradiation at 390 nm for 24 h (two lamps 5 cm away, with adequate fans and a water bath to keep the reaction at room temperature). b Yields for the isolated products after column chromatography. c Enantiomeric excesses were determined by HPLC-analysis on a chiral stationary phase. |
|---|
|
To demonstrate the utility of this method, various derivatizations starting from imino-acylation product 3aa or 3ha were carried out (Scheme 2). First, Wittig olefination successfully converted compound 3aa into a chiral alkene (4) in 61% yield and 98% ee. Next, compound 3aa was reacted with tosylhydrazine to afford a Z-configured hydrazone (5) in 53% yield and 96% ee, the absolute configuration of which was unambiguously determined to be S through X-ray crystallography (CCDC number: 2246373). By means of mCPBA-mediated oxidation of compound 3aa, a chiral oxaziridine (6) was obtained as a single diastereomer in 68% yield and 98% ee. Notably, such structural motifs are often used as O- or N-atom transferring agents and the precursors for [3 + 2] cycloadditions.16 Furthermore, DIBAL-H turned out to be able to promote the chemo- and diastereoselective reduction of the carbonyl group of compound 3aa in DCM at −78 °C, leading to the formation of a chiral secondary alcohol (7) in 54% yield, 98% ee, and >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 dr. Moreover, treatment of compounds 3aa and 3ha with the Grignard reagents provided the corresponding chiral tertiary alcohols 8 (88% yield, 96% ee) and 9 (60% yield, 90% ee), respectively. In addition, the latter was subjected to NaBH(OAc)3-mediated reduction, furnishing a chiral diphenyl homoprolinol (10) in 75% yield, 10:1 dr, and 92% ee, which might find applications as a catalyst or a catalyst precursor since it is structurally analogous to CBS-17 and the Jørgensen–Hayashi18 catalyst.
:2 dr. Moreover, treatment of compounds 3aa and 3ha with the Grignard reagents provided the corresponding chiral tertiary alcohols 8 (88% yield, 96% ee) and 9 (60% yield, 90% ee), respectively. In addition, the latter was subjected to NaBH(OAc)3-mediated reduction, furnishing a chiral diphenyl homoprolinol (10) in 75% yield, 10:1 dr, and 92% ee, which might find applications as a catalyst or a catalyst precursor since it is structurally analogous to CBS-17 and the Jørgensen–Hayashi18 catalyst.
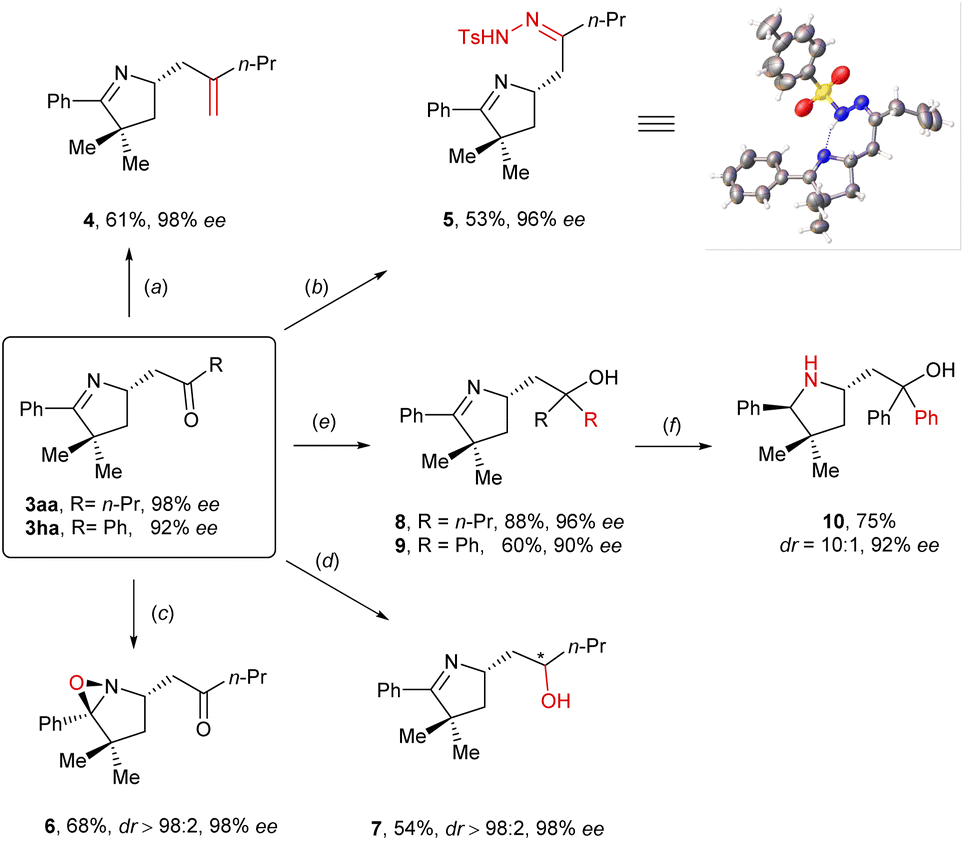 | ||
| Scheme 2 Derivatizations of imino-acylation products. | ||
In order to shed light on the mechanism of this reaction, we carried out a set of control experiments and cyclic voltammetry studies (Scheme 3). Under the standard reaction conditions, the oxime esters bearing benzoate (2a-1) or acetate (2a-2) as the leaving group were utilized as the precursors instead of mesitoate (2a), yielding the product 3aa in significantly lower enantiomeric excess (Scheme 3A). Subsequently, 1,4-cyclohexadiene was employed instead of butanal in this reaction, and the hydroimination product 3aa′ was afforded in 16% ee (Scheme 3B), which is consistent with the ee of the isolated by-product in the imino-acylation reaction demonstrated in Table 1. The aforementioned results confirm that the enantiocontrol in the cyclization process depends on both the properties of the leaving group and the presence of an acyl source, indicating that acyl and carboxylate are both probably attached to the nickel center when it performs the enantiodiscriminating intramolecular migratory insertion with the pendant olefin. Moreover, we conducted the reaction between the oxime ester 2a and butanal (1a) under the sole photocatalysis of TBADT (Scheme 3C). No hydroacylation occurred in this case, suggesting that the addition of an acyl radical to the terminal olefinic unit could not serve as an elementary step for the initial C–C bond formation in this cocatalyzed imino-acylation reaction. Cyclic voltammetry studies reveal that the half-wave potential of [W]5−/[W]6− (E1/2 = −1.41 V vs. Ag/AgCl)19 is more negative than the one of Ni(II)/Ni(I) (E1/2 = −0.81 V vs. Ag/AgCl) but less negative than the one of Ni(I)/Ni(0) (E1/2 = −1.99 V vs. Ag/AgCl) in acetonitrile, indicating that Ni(I) species can be generated in situ starting from the Ni(II) precatalyst through reduction by [W]6− (Scheme 3D). Besides, the half-wave potential of the oxime ester 2a (E1/2 = −0.86 V vs. Ag/AgCl) turned out to be slightly more negative than the one of Ni(II)/Ni(I), accounting for the preferred reduction of Ni(II) by [W]6− in the presence of oxime esters in the reaction mixture. In addition, the stoichiometric reaction of the oxime ester 2a with the Ni(0) complex generated from Ni(COD)2 and L1 did not yield any ring closure product, which also argues against the reaction sequence of Ni(0)-mediated oxidative addition followed by Ni(II)-mediated cyclization (Scheme 3E).
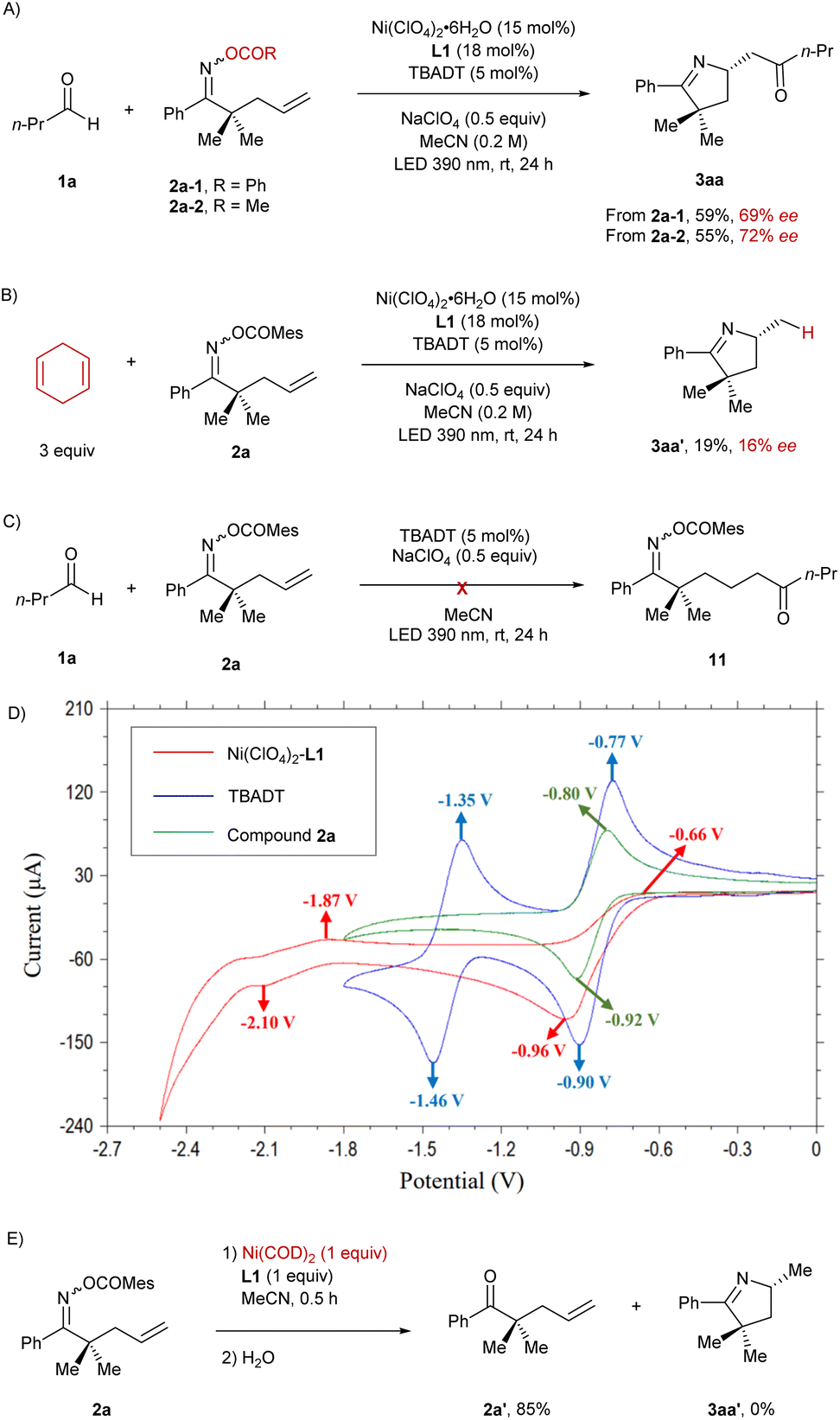 | ||
| Scheme 3 Control experiments and cyclic voltammetry studies. | ||
On the basis of the results of the preliminary mechanistic investigations, a plausible reaction mechanism exemplified by butyraldehyde (1a) and the oxime ester-tethered alkene 2a is proposed in Scheme 4. Initially, decatungstate [W]4− is activated to [W]4−* under irradiation at 390 nm, which subsequently abstracts a hydrogen atom from butyraldehyde (1a), leading to the formation of corresponding acyl radical A and [W]5−H+. Disproportionation of the latter affords original [W]4− for the next TBADT catalytic cycle and [W]6−2H+, which is able to reduce Ni(II) to Ni(I) via single-electron transfer (SET). In the nickel-catalytic cycle, the in situ generated Ni(I) is oxidized by the acyl radical A, to provide the acyl-Ni(II) species B, which is reduced to the acyl-Ni(I) species C by [W]6−2H+via SET. Next, the oxime ester 2a performs oxidative addition to the Ni(I) complex C, to afford the Ni(III) intermediate D, the pendant olefinic unit of which undergoes the enantiodetermining migratory insertion into the Ni–N bond. After facile reductive elimination from the resultant Ni(III) complex E, the corresponding imino-acylation product 3aa is furnished and Ni(I) is regenerated for the next catalytic cycle.
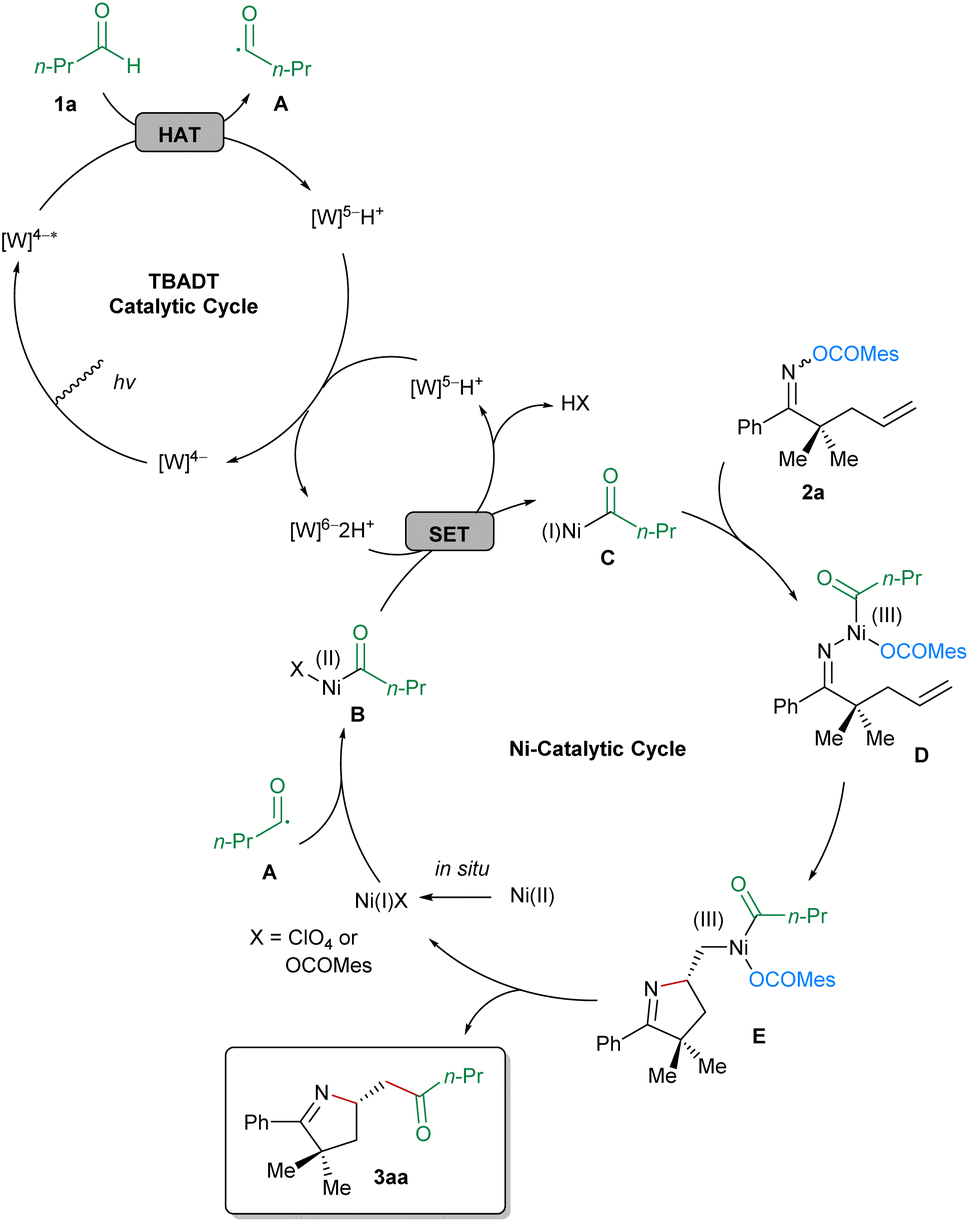 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed an asymmetric imino-acylation reaction of oxime ester-tethered alkenes with aldehydes, which proceeds under the cooperative catalysis of a nickel-BOX complex and TBADT as a HAT photocatalyst, providing a new method to construct a chiral pyrroline scaffold. The synthetic diversification potential of the imino-acylation products has been demonstrated through various derivatizations based on the chemoselective transformations of the incorporated carbonyl and imino moieties. On the basis of the results of control experiments and cyclic voltammetry, a catalytic cycle with Ni(I) as the initial operating species has been proposed, in which Ni(III)-mediated aza–Heck cyclization serves as the enantiodetermining step.Data availability
General information, detailed experimental procedures, characterization data for all new compounds, NMR spectra, and HPLC chromatograms are in the ESI.† Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC 2246373.Author contributions
C. W. designed the project and wrote the paper. R. W. carried out the experimental work. Both authors analyzed the data, discussed the results, and commented on the manuscript.Conflicts of interest
There is no conflict to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22071230 and 22271267).Notes and references
- For reviews on cyclization of oxime ester-tethered alkenes, see: (a) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268–277 CrossRef CAS PubMed; (b) N. J. Race, I. R. Hazelden, A. Faulkner and J. F. Bower, Chem. Sci., 2017, 8, 5248–5260 RSC; (c) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem. Eur J., 2018, 24, 12154–12163 CrossRef CAS PubMed; (d) C. Chen, J. Zhao, X. Shi, L. Liu, Y.-P. Zhu, W. Sun and B. Zhu, Org. Chem. Front., 2020, 7, 1948–1969 RSC; (e) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066–1083 CrossRef CAS PubMed; (f) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed; (g) C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- (a) V. C. Clark, C. J. Raxworthy, V. Rakotomalala, P. Sierwald and B. L. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11617–11622 CrossRef CAS PubMed; (b) A. Adams and N. De Kimpe, Chem. Rev., 2006, 106, 2299–2319 CrossRef CAS PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njadarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (d) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213–7256 CrossRef CAS.
- For selected examples of transition metal-catalyzed aza-Heck cyclization using oxime-tethered alkenes, see: (a) H. Tsutsui and K. Narasaka, Chem. Lett., 1999, 28, 45–46 CrossRef; (b) Y. Koganemaru, M. Kitamura and K. Narasaki, Chem. Lett., 2002, 31, 784–785 CrossRef; (c) A. Faulkner and J. F. Bower, Angew. Chem., Int. Ed., 2012, 51, 1675–1679 CrossRef CAS PubMed; (d) A. Faulkner, N. J. Race, J. S. Scott and J. F. Bower, Chem. Sci., 2014, 5, 2416–2421 RSC; (e) B. C. Lemercier and J. G. Pierce, Org. Lett., 2014, 16, 2074–2076 CrossRef CAS PubMed; (f) C. Chen, L. Hou, M. Cheng, J. Su and X. Tong, Angew. Chem., Int. Ed., 2015, 54, 3092–3096 CrossRef CAS PubMed; (g) H. Su, W. Li, Z. Xuan and W. Yu, Adv. Synth. Catal., 2015, 357, 64–70 CrossRef CAS; (h) N. J. Race, A. Faulkner, M. H. Shaw and J. F. Bower, Chem. Sci., 2016, 7, 1508–1513 RSC; (i) N. J. Race, A. Faulkner, G. Fumagalli, T. Yamauchi, J. S. Scott, M. Rydén-Landergren, S. A. Sparkes and J. F. Bower, Chem. Sci., 2017, 8, 1981–1985 RSC; (j) X. Bao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 9577–9581 CrossRef CAS PubMed; (k) H.-B. Yang, S. R. Pathipati and N. Selander, ACS Catal., 2017, 7, 8441–8445 CrossRef CAS; (l) T. Shimbayashi, K. Okamoto and K. Ohe, Chem.–Asian J., 2018, 13, 395–399 CrossRef CAS PubMed; (m) L. Wang and C. Wang, J. Org. Chem., 2019, 84, 6547–6556 CrossRef CAS PubMed; (n) Y. Zhang and X.-F. Wu, Chem. Commun., 2020, 56, 14605–14608 RSC; (o) L. Feng, L. Guo, C. Yang, J. Zhou and W. Xia, Org. Lett., 2020, 22, 3964–3968 CrossRef CAS PubMed; (p) Y. Zhang, H.-J. Ai and X.-F. Wu, Org. Chem. Front., 2020, 7, 2986–2990 RSC; (q) W.-X. Wei, Y. Li, Y.-T. Wen, M. Li, X.-S. Li, C.-T. Wang, H.-C. Liu, Y. Xia, B.-S. Zhang, R.-Q. Jiao and Y.-M. Liang, J. Am. Chem. Soc., 2021, 143, 7868–7875 CrossRef CAS PubMed; (r) X.-G. Jia, Q.-W. Yao and X.-Z. Shu, J. Am. Chem. Soc., 2022, 144, 13461–13467 CrossRef CAS PubMed; (s) D. Qi, X. Zhang, X. Wang, X. Liu, Z. Zhang, L. Shi and G. Zhang, Org. Lett., 2023, 25, 1126–1130 CrossRef CAS PubMed.
- For selected examples of photo-catalyzed cyclization using oxime-tethered alkenes, see: (a) T. Mikami and K. Narasaka, Chem. Lett., 2000, 29, 338–339 CrossRef; (b) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 4055–4059 CrossRef CAS PubMed; (c) J. Davies, S. J. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS PubMed; (d) S.-H. Cai, J.-H. Xie, S. Song, L. Ye, C. Feng and T.-P. Loh, ACS Catal., 2016, 6, 5571–5574 CrossRef CAS; (e) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 12273–12276 CrossRef CAS PubMed; (f) J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS PubMed; (g) K. M. Nakafuku, S. C. Fosu and D. A. Nagib, J. Am. Chem. Soc., 2018, 140, 11202–11205 CrossRef CAS PubMed; (h) X. Shen, C. Huang, X.-A. Yuan and S. Yu, Angew. Chem., Int. Ed., 2021, 60, 9672–9679 CrossRef CAS PubMed; (i) A. F. Prusinowski, H. C. Sise, T. N. Bednar and D. A. Nagib, ACS Catal., 2022, 12, 4327–4332 CrossRef CAS PubMed.
- A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224–7230 CrossRef CAS PubMed.
- L. Wang and C. Wang, Org. Chem. Front., 2018, 5, 3476–3482 RSC.
- W.-D. Liu, W. Lee, H. Shu, C. Xiao, H. Xu, X. Chen, K. N. Houk and J. Zhao, J. Am. Chem. Soc., 2022, 144, 22767–22777 CrossRef CAS PubMed.
- Y.-X. Dong, C.-L. Zhang, Z.-H. Gao and S. Ye, Org. Lett., 2023, 25, 855–860 CrossRef CAS PubMed.
- For reviews of TBADT-catalyzed reactions, see: (a) C. L. Hill, Synlett, 1995, 1995, 127–132 CrossRef; (b) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC; (c) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed; (d) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS; (e) X. Yuan, G. Yang and B. Yu, Chin. J. Org. Chem., 2020, 40, 3620–3632 CrossRef CAS; (f) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (g) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS.
- For selected examples of TBADT-catalyzed acyl C–H activation reactions of aldehydes, see: (a) S. Esposti, D. Dondi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 2531–2534 CrossRef CAS PubMed; (b) M. D. Tziraki and M. Orfanopoulos, J. Am. Chem. Soc., 2009, 131, 4063–4069 CrossRef PubMed; (c) D. Ravelli, S. Montanaro, M. Zema, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2011, 353, 3295–3300 CrossRef CAS; (d) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS PubMed; (e) F. Bonassi, D. Ravelli, S. Protti and M. Fagnoni, Adv. Synth. Catal., 2015, 357, 3687–3695 CrossRef CAS; (f) L. Capaldo, M. Fagnoni and D. Ravelli, Chem. Eur J., 2017, 23, 6527–6530 CrossRef CAS PubMed; (g) P. Fan, C. Zhang, Y. Lan, Z. Lin, L. Zhang and C. Wang, Chem. Commun., 2019, 55, 12691–12694 RSC; (h) J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2020, 11, 1026–1031 RSC; (i) Y. Kuang, H. Cao, H. Tang, J. Chew, W. Chen, X. Shi and J. Wu, Chem. Sci., 2020, 11, 8912–8918 RSC; (j) J. Dong, F. Yue, X. Wang, H. Song, Y. Liu and Q. Wang, Org. Lett., 2020, 22, 8272–8277 CrossRef CAS PubMed; (k) A. Prieto and M. Taillefer, Org. Lett., 2021, 23, 1484–1488 CrossRef CAS PubMed; (l) X. Wang, Y. Chen, H. Song, Y. Liu and Q. Wang, Org. Lett., 2021, 23, 2199–2204 CrossRef CAS PubMed; (m) S. Le, J. Li, J. Feng, Z. Zhang, Y. Bai, Z. Yuan and G. Zhu, Nat. Commun., 2022, 13, 4734 CrossRef CAS PubMed; (n) Y. Mao, P. Fan and C. Wang, Org. Lett., 2022, 24, 9413–9418 CrossRef CAS PubMed; (o) T. Varlet, D. Bouchet, E. van Eslande and G. Masson, Chem. Eur J., 2022, 28, e202201707 CrossRef CAS PubMed.
- For selected examples of TBADT-catalyzed aliphatic C–H activation reactions of hydrocarbons, see: (a) D. Dondi, M. Fagnoni, A. Molinari, A. Maldotti and A. Albini, Chem. Eur J., 2004, 10, 142–148 CrossRef CAS PubMed; (b) D. Dondi, M. Fagnoni and A. Albini, Chem. Eur J., 2006, 12, 4153–4163 CrossRef CAS PubMed; (c) M. D. Tzirakis and M. Orfanopoulos, Org. Lett., 2008, 10, 873–876 CrossRef CAS PubMed; (d) D. Ravelli, A. Albini and M. Fagnoni, Chem. Eur J., 2011, 17, 572–579 CrossRef CAS PubMed; (e) S. D. Halparin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690–4693 CrossRef PubMed; (f) M. Okada, T. Fukuyama, K. Yamada, I. Ryu, D. Ravelli and M. Fagnoni, Chem. Sci., 2014, 5, 2893–2898 RSC; (g) D. Ravelli, M. Zoccolillo, M. Mella and M. Fagnoni, Adv. Synth. Catal., 2014, 356, 2781–2786 CrossRef CAS; (h) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed; (i) V. I. Supranovich, V. V. Levin and A. D. Dilman, Org. Lett., 2019, 21, 4271–4274 CrossRef CAS PubMed; (j) K. Yahata, S. Sakurai, S. Hori, S. Yoshioka, K. Hasegawa and S. Akai, Org. Lett., 2020, 22, 1199–1203 CrossRef CAS PubMed; (k) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed; (l) P. J. Sarver, N. B. Bissonnette and D. W. C. MacMillan, J. Am. Chem. Soc., 2021, 143, 9737–9743 CrossRef CAS PubMed; (m) L. Capaldo and D. Ravelli, Org. Lett., 2021, 23, 2243–2247 CrossRef CAS PubMed; (n) T. E. Schirmer, A. B. Rolka, T. A. Karl, F. Holzhausen and B. König, Org. Lett., 2021, 23, 5729–5733 CrossRef CAS PubMed; (o) Q. Liu, Y. Ding, Y. Gao, Y. Yang, L. Gao, Z. Pan and C. Xia, Org. Lett., 2022, 24, 7983–7987 CrossRef CAS PubMed.
- For selected examples of transition metal/TBADT-cocatalyzed reactions, see: (a) I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed; (b) P. J. Sarver, V. Bacauanu, D. M. Shultz, D. A. DiRocco, Y.-h. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed; (c) H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS PubMed; (d) L. Wang, T. Wang, G.-J. Cheng, X. Li, J.-J. Wei, B. Guo, C. Zheng, G. Chen, C. Ran and C. Zheng, ACS Catal., 2020, 10, 7543–7551 CrossRef CAS; (e) P. Fan, C. Zhang, L. Zhang and C. Wang, Org. Lett., 2020, 22, 3875–3878 CrossRef CAS PubMed; (f) S. Xu, H. Chen, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2021, 60, 7405–7411 CrossRef CAS PubMed; (g) P. Fan, Y. Jin, J. Liu, R. Wang and C. Wang, Org. Lett., 2021, 23, 7364–7369 CrossRef CAS PubMed; (h) P. Fan, R. Wang and C. Wang, Org. Lett., 2021, 23, 7672–7677 CrossRef CAS PubMed; (i) V. Murugesan, A. Ganguly, A. Karthika and R. Rasappan, Org. Lett., 2021, 23, 5389–5393 CrossRef CAS PubMed; (j) D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma and T. Noël, Angew. Chem., Int. Ed., 2021, 60, 21277–21282 CrossRef CAS PubMed; (k) B. Maity, C. Zhu, M. Rueping and L. Cavallo, ACS Catal., 2021, 11, 13973–13982 CrossRef CAS; (l) D. Wang and L. Ackermann, Chem. Sci., 2022, 13, 7256–7263 RSC; (m) Y. Jin, E. W. H. Ng, T. Fan, H. Hirao and L.-Z. Gong, ACS Catal., 2022, 12, 10039–10046 CrossRef CAS; (n) W. Liu, C. Liu, M. Wang and W. Kong, ACS Catal., 2022, 12, 10207–10221 CrossRef CAS; (o) Q. Wang, S. Ni, L. Yu, Y. Pan and Y. Wang, ACS Catal., 2022, 12, 11071–11077 CrossRef CAS; (p) P. Fan, Y. Mao and C. Wang, Org. Chem. Front., 2022, 9, 4649–4653 RSC; (q) W. Liu, Y. Ke, C. Liu and W. Kong, Chem. Commun., 2022, 58, 11937–11940 RSC; (r) X. Li, Y. Mao, P. Fan and C. Wang, Eur. J. Org. Chem., 2022, 2022, e202200214 CAS; (s) V. Murugesan, A. Muralidharan, G. V. Anantharaj, T. Chinusamy and R. Rasappan, Org. Lett., 2022, 24, 8435–8440 CrossRef CAS PubMed; (t) E. Mao and D. W. C. MacMillan, J. Am. Chem. Soc., 2023, 145, 2787–2793 CrossRef CAS PubMed.
- For reviews on asymmetric transition metal/photo cocatalysis, see: (a) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed; (b) C. Jiang, W. Chen, W.-H. Zheng and H. Lu, Org. Biomol. Chem., 2019, 17, 8673–8689 RSC; (c) H. H. Zhang, H. Chen, C. Zhu and S. Yu, Sci. China Chem., 2020, 63, 637–647 CrossRef CAS; (d) A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2021, 60, 1714–1726 CrossRef CAS PubMed; (e) Y. Li, Z. Ye, J. Cai and L. Gong, Synthesis, 2021, 53, 1570–1583 CrossRef CAS; (f) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC; (g) Z. Li, C. Li, Y. Ding and H. Huo, Coord. Chem. Rev., 2022, 460, 214479 CrossRef CAS; (h) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- P. Fan, Y. Lan, C. Zhang and C. Wang, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed.
- R. Wang, P. Fan and C. Wang, ACS Catal., 2023, 13, 141–146 CrossRef CAS.
- K. S. Williamson, D. J. Michaelis and T. P. Yoon, Chem. Rev., 2014, 114, 8016–8036 CrossRef CAS PubMed.
- C. J. Helal and M. P. Meyer, The Corey–Bakshi–Shibata reduction: mechanistic and synthetic considerations – bifunctional Lewis base catalysis with dual activation, in Lewis Base Catalysis in Organic Synthesis, Wiley, Hoboken, 2016, ch. 11, pp. 387–389 Search PubMed.
- K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS PubMed.
- T. Yamase, N. Tapakayashi and M. Kaji, J. Chem. Soc., Dalton Trans., 1984, 793–799 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectral data, NMR-data, and HPLC-data. CCDC 2246373. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01945d |
| This journal is © The Royal Society of Chemistry 2023 |