
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic generation of ortho-quinone dimethides via donor/donor rhodium carbenes†
Mingchun
Gao
 ,
Jose M.
Ruiz
,
Emily
Jimenez
,
Anna
Lo
,
Croix J.
Laconsay
,
James C.
Fettinger
,
Dean J.
Tantillo
and
Jared T.
Shaw
*
,
Jose M.
Ruiz
,
Emily
Jimenez
,
Anna
Lo
,
Croix J.
Laconsay
,
James C.
Fettinger
,
Dean J.
Tantillo
and
Jared T.
Shaw
*
Department of Chemistry, University of California, One Shields Avenue, Davis, California 95616, USA. E-mail: jtshaw@ucdavis.edu
First published on 11th May 2023
Abstract
Substrates engineered to undergo a 1,4-C–H insertion to yield benzocyclobutenes resulted in a novel elimination reaction to yield ortho-quinone dimethide (o-QDM) intermediates that undergo Diels–Alder or hetero-Diels–Alder cycloadditions. The analogous benzylic acetals or ethers avoid the C–H insertion pathway completely and, after hydride transfer, undergo a de-aromatizing elimination reaction to o-QDM at ambient temperature. The resulting dienes undergo a variety of cycloaddition reactions with high diastereo- and regio-selectivity. This is one of the few examples of catalytic generation of o-QDM without the intermediacy of a benzocyclobutene and represents one of the mildest, ambient temperature processes to access these useful intermediates. This proposed mechanism is supported by DFT calculations. Moreover, the methodology was applied to the synthesis of (±)-isolariciresinol in 41% overall yield.
Introduction
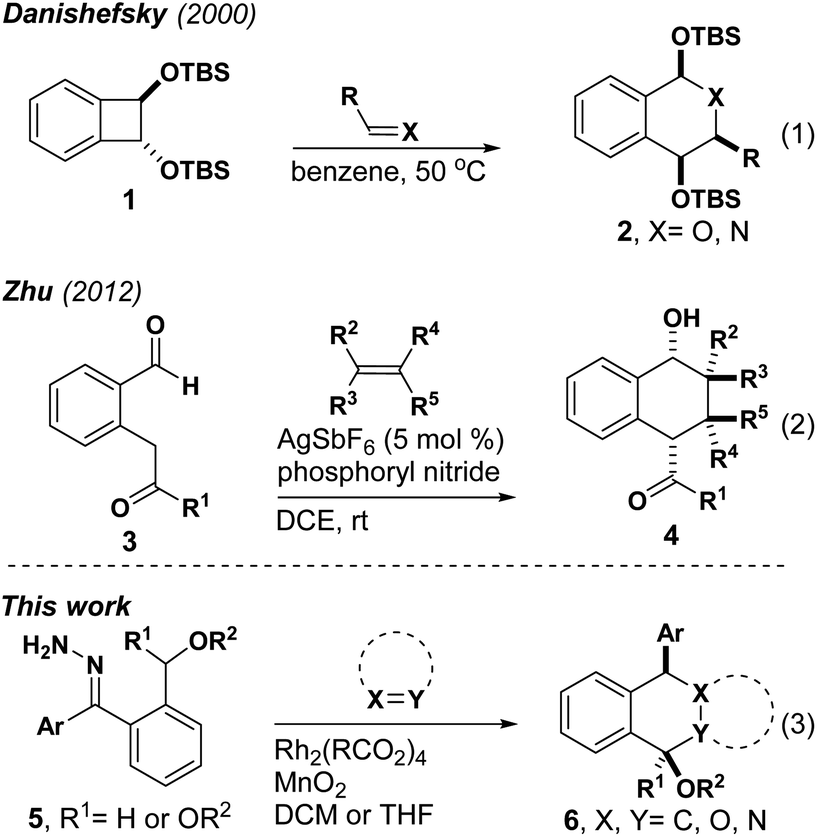
Metal carbenes are useful intermediates for a variety of reactions that assemble complex molecules with high efficiency and atom economy.1–6 While early studies involved carbenes appended with electron-withdrawing groups, donor/acceptor carbenes developed by Davies and co-workers opened up new avenues for reactivity and selectivity.3,5 Recent advances with donor/donor carbenes have revealed complementary reactivity, increased functional group tolerance and an evolving level of reaction diversity.1 Importantly, donor/donor carbenes (DDCs) resist attack by heteroatom nucleophiles, which enables the design of reactions that would normally suffer from side reactions that limit the range of reactivity observed for more electrophilic carbenes.ortho-Quinone dimethide (o-QDM) was first discovered in 1957.7 These reactive dienes are typically formed by the thermal ring opening of benzocyclobutenes. While this process can involve thermolysis at high temperatures, Danishefsky reported an extremely useful substrate that forms o-QDM at 50 °C (Scheme 1).8 Given that multistep syntheses are often required to access o-QDM precursors, recent work has focused on accessing these useful intermediates using catalysis. Zhu has reported that a silver catalyst forms o-QDM from dicarbonyl substrate 3.9 During our investigations of C–H insertion reactions to form benzocyclobutenes, we observed direct formation of o-QDMs from donor/donor carbenes. These intermediates could be generated and trapped in situ with electron deficient alkenes, carbonyls and imines in stereoselective Diels–Alder and hetero-Diels–Alder reactions, respectively. This is the first example of generating o-QDMs from carbene intermediates and one of small number of examples of accessing these intermediates catalytically9–15 at ambient temperature (Scheme 2).
 | ||
| Scheme 1 o-QDM generation methods. | ||
 | ||
| Scheme 2 Discovery of Rh-catalyzed o-ODM generation. | ||
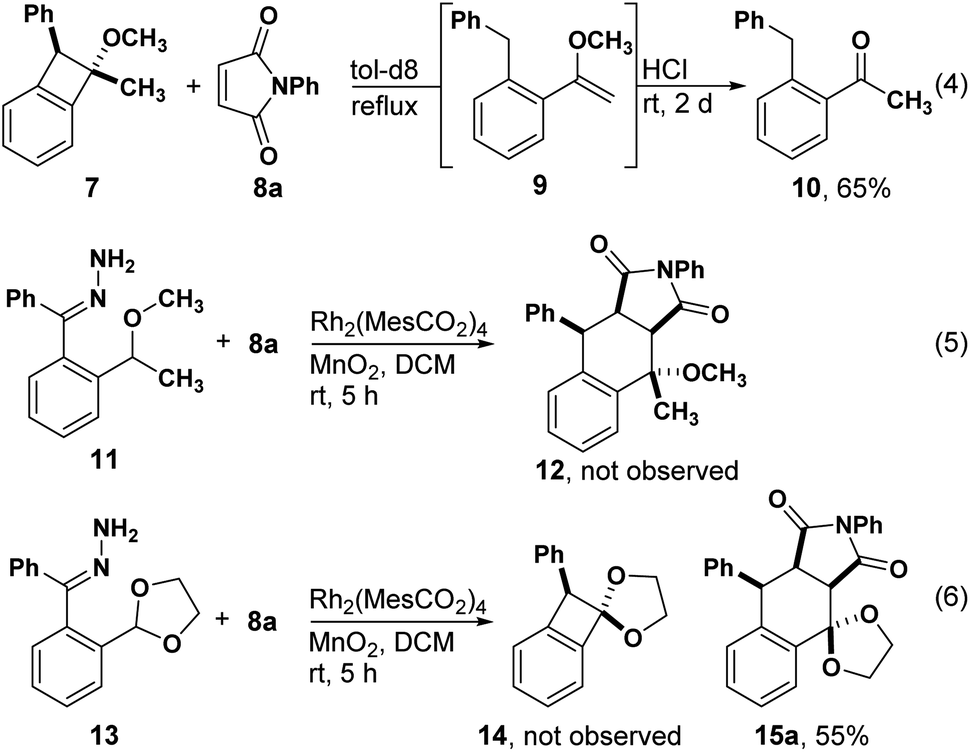
During our studies of enantioselective synthesis of isochromans by C–H insertion, we observed benzocyclobutene 7 as a side product. Recognizing the potential for this product to serve as a precursor to o-QDM, we heated it in the presence of N-phenyl maleimide 8 with the hope of observing a stereoselective Diels–Alder reaction. Acetophenone 10 was the only observed product, which we presume emanates from enol ether 9 through a known process.16,17 We next attempted to intercept the presumed o-QDM intermediate through a one-pot process and did not observe any Diels–Alder products. Finally, we prepared acetal 13, which would avoid enol ether formation. Although we were unable to isolate any products of C–H insertion, e.g. benzocyclobutene 14, Diels–Alder product 15 was isolated in good yield, suggesting that o-QDM was forming directly without the intermediacy of 14. Herein we report the development of this o-QDM-forming method for acetals and ethers and propose a mechanism for its formation based on DFT calculations.
Results and discussion
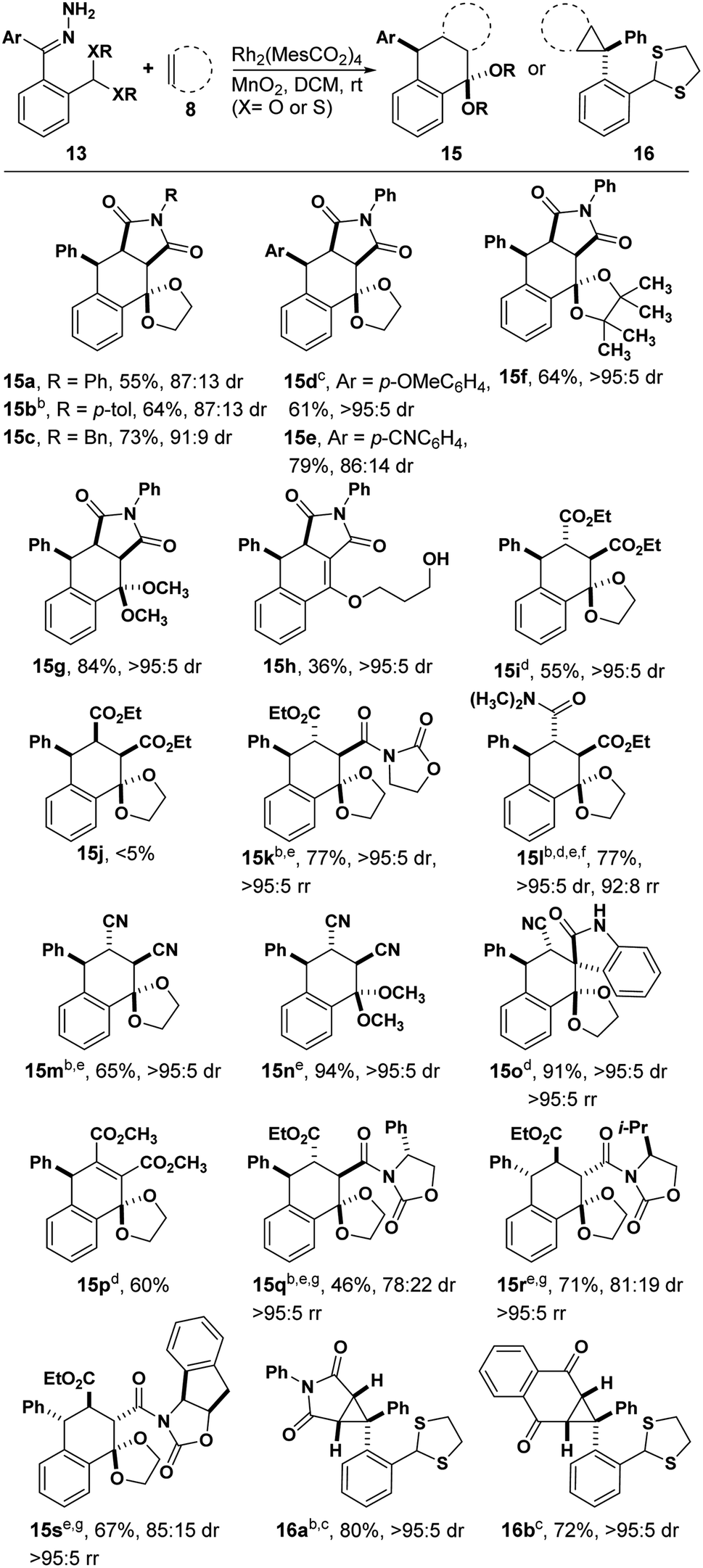
We began our studies by treating hydrazone 13a with manganese dioxide, Rh2(MesCO2)4 and N-phenyl maleimide in DCM obtaining the desired tetralin 15a in 55% yield with 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 dr (Table 1). Encouraged by this result, we next examined the substrate scope of dienophiles in this reaction (Table 1). We were pleased to isolate the desired Diels–Alder products from various N-substituted maleimides (15a–c) and hydrazones with different electronic properties (15d and 15e), respectively. The X-ray structure of 15b demonstrated the endo-product as the major configuration. In addition to the glycol derived hydrazone, substrates with pinacol-derived or dimethoxy acetal groups could also afford the corresponding products 15f and 15g. When a hydrazone with propanediol-derived acetal was used, the product was unstable to column chromatography, affording vinyl ether product 15h. Diethyl fumarate led the cyclization product in 55% yield, while the analogous maleate substrate was far less reactive (15i and 15j). Notably, unsymmetric alkenes containing oxazolidinone groups showed good regioselectivity and diastereoselectivity (15k), and the reaction was feasible for amide substituted alkenes in THF (15l). Fumaronitrile proved to be another good dienophile (15m and 15n). An isatin-derived alkene offered spiro compound 15o in 91% yield, and computational NMR suggested a trans configuration of phenyl and cyano groups (see ESI†). The observation of product with the indicated regioselectivity can be attributed to steric hindrance between the phenyl and the isatin moiety. Furthermore, an alkyne dienophile is also compatible with this reaction, affording product 15p in 60% yield. Chiral dienophiles afforded their respective tetralins 15q, 15r and 15s in good yields with excellent regioselectivity, albeit with modest levels of chiral induction from the oxazolidinone. The major isomers were isolated and the stereo- and regiochemistry for 15q were confirmed by X-ray crystallography. When a thioacetal-containing hydrazone was used, [4 + 2] cyclization was replaced by cyclopropanation, affording 16a and 16b, respectively. The observation of single stereoisomer suggests that the bulky thioacetal groups blocked one face of the carbene. This result also reveals that changes to the electronic structure of donor/donor carbenes can lead to completely different reaction pathways.18
:13 dr (Table 1). Encouraged by this result, we next examined the substrate scope of dienophiles in this reaction (Table 1). We were pleased to isolate the desired Diels–Alder products from various N-substituted maleimides (15a–c) and hydrazones with different electronic properties (15d and 15e), respectively. The X-ray structure of 15b demonstrated the endo-product as the major configuration. In addition to the glycol derived hydrazone, substrates with pinacol-derived or dimethoxy acetal groups could also afford the corresponding products 15f and 15g. When a hydrazone with propanediol-derived acetal was used, the product was unstable to column chromatography, affording vinyl ether product 15h. Diethyl fumarate led the cyclization product in 55% yield, while the analogous maleate substrate was far less reactive (15i and 15j). Notably, unsymmetric alkenes containing oxazolidinone groups showed good regioselectivity and diastereoselectivity (15k), and the reaction was feasible for amide substituted alkenes in THF (15l). Fumaronitrile proved to be another good dienophile (15m and 15n). An isatin-derived alkene offered spiro compound 15o in 91% yield, and computational NMR suggested a trans configuration of phenyl and cyano groups (see ESI†). The observation of product with the indicated regioselectivity can be attributed to steric hindrance between the phenyl and the isatin moiety. Furthermore, an alkyne dienophile is also compatible with this reaction, affording product 15p in 60% yield. Chiral dienophiles afforded their respective tetralins 15q, 15r and 15s in good yields with excellent regioselectivity, albeit with modest levels of chiral induction from the oxazolidinone. The major isomers were isolated and the stereo- and regiochemistry for 15q were confirmed by X-ray crystallography. When a thioacetal-containing hydrazone was used, [4 + 2] cyclization was replaced by cyclopropanation, affording 16a and 16b, respectively. The observation of single stereoisomer suggests that the bulky thioacetal groups blocked one face of the carbene. This result also reveals that changes to the electronic structure of donor/donor carbenes can lead to completely different reaction pathways.18
| a Reaction conditions: 13 (0.1 mmol), 8 (0.12 mmol), Rh2(MesCO2)4 (1 mol%), MnO2 (0.8 mmol) in DCM (6 mL); isolated yields; dr was determined by 1H NMR of unpurified reaction mixture. b The structure and configuration were further determined by X-ray structure. c Rh2(R-PTAD)4 was used as catalyst. d Rh2(MesCO2)4 and dienophile were added after hydrazone and MnO2 had been stirred in solvent for 2 h. e 1.0 equiv. of dienophile was used. f THF was used instead of DCM. g Major diastereoisomer yield. |
|---|
|
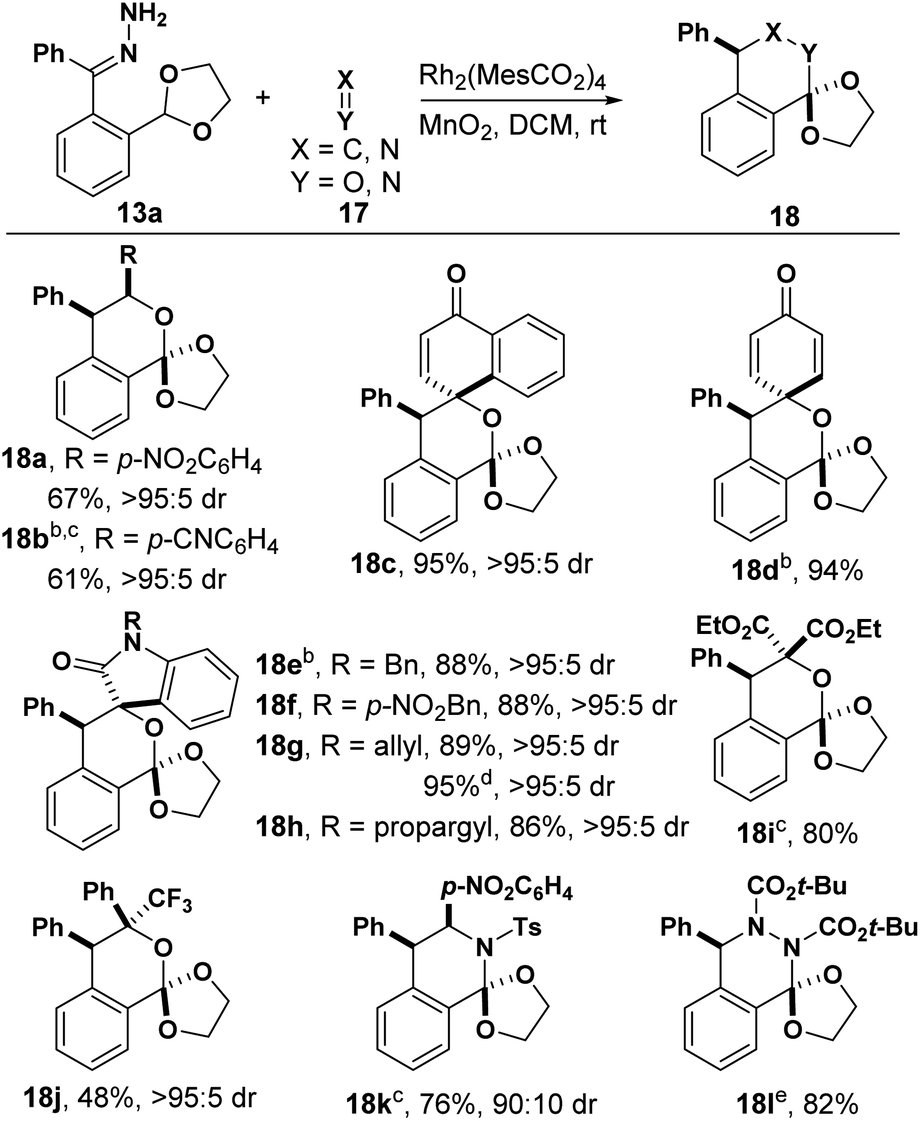
We next investigated hetero-Diels–Alder reactions between o-QDMs formed from hydrazones and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X/XX double bonds (Table 2). Electron-deficient aldehydes lead to isochroman products 18a and 18b, while benzaldehyde failed to react under the same conditions. An X-ray structure of 18a indicated that the phenyl group and p-nitrophenyl (PNP) group were assembled in a cis configuration. Generally, quinones are a class of reactive dipolarophiles because of the activated CC double bond.19,20 Surprisingly, this [4 + 2] cycloaddition occurred on the carbonyl group instead of the activated CC double bond (18c and 18d), which has been observed in a related reaction.19 The structure of 18d was further confirmed by X-ray diffraction (see ESI†). A variety of isatins were examined next (18e–h). The products were isolated in excellent yields and diastereoselectivity in all cases. It should be noted that both alkene and alkyne substitution are tolerated in this transformation (18g and 18h). Furthermore, diethyl ketomalonate afforded the corresponding product 18i in 80% yield. A trifluoromethyl group was successfully introduced into product 18j with trifluoro acetophenone as the dienophile. We were pleased to see N-containing heterocycle 18k was obtained when an imine was used as dienophile, as well as dihydrophthalazine 18l from an azo compound, illustrating the broad substrate scope of this method.
X/XX double bonds (Table 2). Electron-deficient aldehydes lead to isochroman products 18a and 18b, while benzaldehyde failed to react under the same conditions. An X-ray structure of 18a indicated that the phenyl group and p-nitrophenyl (PNP) group were assembled in a cis configuration. Generally, quinones are a class of reactive dipolarophiles because of the activated CC double bond.19,20 Surprisingly, this [4 + 2] cycloaddition occurred on the carbonyl group instead of the activated CC double bond (18c and 18d), which has been observed in a related reaction.19 The structure of 18d was further confirmed by X-ray diffraction (see ESI†). A variety of isatins were examined next (18e–h). The products were isolated in excellent yields and diastereoselectivity in all cases. It should be noted that both alkene and alkyne substitution are tolerated in this transformation (18g and 18h). Furthermore, diethyl ketomalonate afforded the corresponding product 18i in 80% yield. A trifluoromethyl group was successfully introduced into product 18j with trifluoro acetophenone as the dienophile. We were pleased to see N-containing heterocycle 18k was obtained when an imine was used as dienophile, as well as dihydrophthalazine 18l from an azo compound, illustrating the broad substrate scope of this method.
| a Reaction conditions: 13a (0.1 mmol), 17 (0.12 mmol), Rh2(MesCO2)4 (1 mol%), MnO2 (0.8 mmol) in DCM (6 mL); isolated yields; dr was determined by 1H NMR of unpurified reaction mixture. b The structure and configuration were determined by X-ray structure. c Rh2(MesCO2)4 and dienophile were added after hydrazone and MnO2 had been stirred in DCM for 2 h. d 0.5 mmol scale. e 0.2 mmol scale. |
|---|
|
Motivated by the positive results obtained above, we decided to test the effect on reactivity induced by replacing acetal with methyl ether (Table 3). Treating 13f with N-phenyl maleimide at 55 °C using THF as the solvent and Rh2(MesCO2)4 as the catalyst afforded the desired tetralin 19a in 69% with >95:5 dr. The relative stereochemistry of 19a was confirmed by X-ray crystallography.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | [Rh] Cat. | Solvent | T (°C) | Yieldb | drc |
| a Reaction conditions: 13f (0.1 mmol), 8a (0.1 mmol), MnO2 (0.8 mmol), solvent (6 mL), [Rh] (1 mol%), reaction mixture stirred for 1 h before adding [Rh]. b Isolated yield. c dr was determined by 1H NMR of unpurified reaction mixture. | |||||
| 1 | Rh2(MesCO2)4 | DCM | rt | 25% | >95:5 |
| 2 | Rh2(PTCC)4 | DCM | rt | 40% | >95:5 |
| 3 | Rh2(PTCC)4 | THF | rt | 60% | >95:5 |
| 4 | Rh2(PTCC)4 | THF | 55 | 65% | >95:5 |
| 5 | Rh2(MesCO2)4 | THF | 55 | 69% | >95:5 |
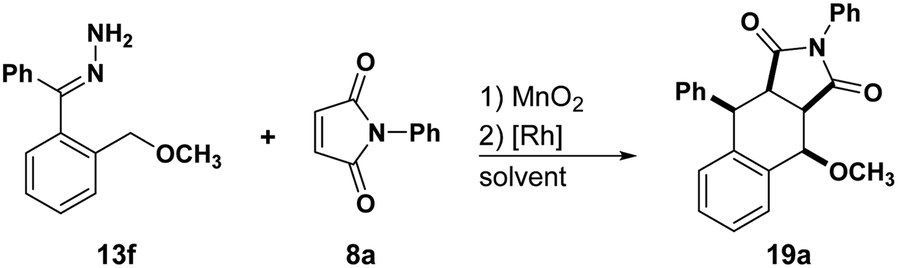
With these optimized conditions, we began developing the substrate scope by testing hydrazones with different electronic properties (Table 4). Electron-poor hydrazone afforded tetralin 19b in 77% yield with perfect diastereoselectivity. On the other hand, electron-rich hydrazones did not yield the desired tetralin, instead a complex mixture of products was observed. Reacting 13f with different fumarates afforded tetralins (19c–e) in good yields with high diastereoselectivity. Surprisingly, the reaction with trimethyl aconitate and t-butyl acrylate, which did not show any reactivity with 13a, afforded tetralins 19f and 19g respectively in moderate yields with great regio- and diasteroselectivity. Fumaronitrile was less reactive and the diastereoselectivity of its corresponding tetralin product 19i decreased to 82:18 with respect to 15m and 15n. Reactions with asymmetric dienophiles afforded the desired tetralins (19i–k) in moderate to excellent yields with >95:5 dr and 72:28 to >95:5 rr. We also investigated the reactivity of 13f towards carbonyls to afford their respective heterocycles. Moderate reactivity and diastereoselectivity were observed in the reaction with p-cyanobenzaldehyde as the desired isochroman 20a obtained in 67% yield with 77:23 dr. Reaction with ketones and isatins afforded cycloadducts (20b–d) in moderate to high yields with high diastereoselectivity.
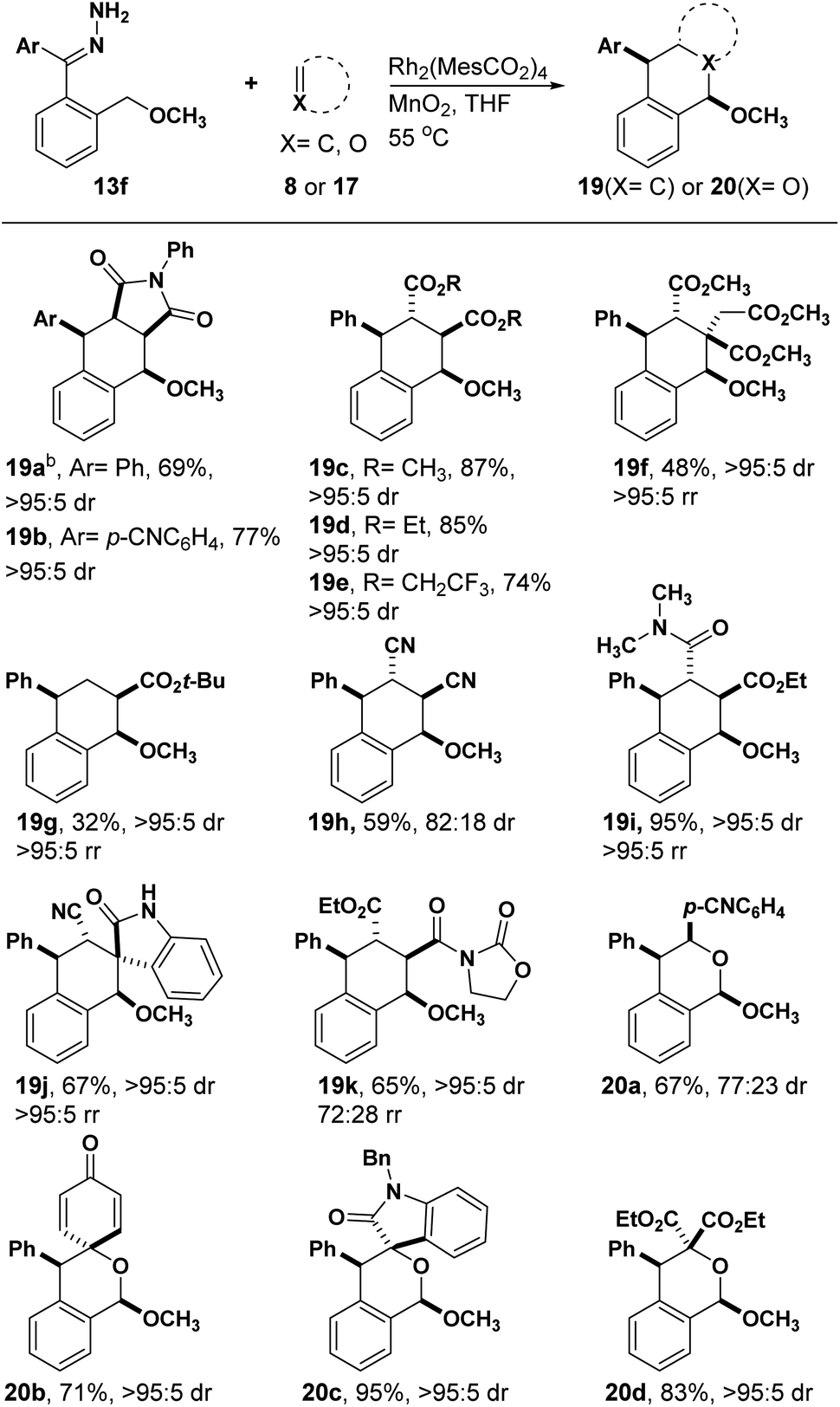
We next tried to make lactones through cycloaddition and successive acidic deprotection (Scheme 3). For example, compound 21a and 21b were separated in moderate yield and excellent diastereoselectivity, respectively (eqn (7) and (8)). Using the presented method followed by deprotection with Hg(OAc)2, aldehyde 21c was provided smoothly with the cyclopropane untouched (eqn (9)).
 | ||
| Scheme 3 Two-pot cycloaddition and acetal deprotection. | ||
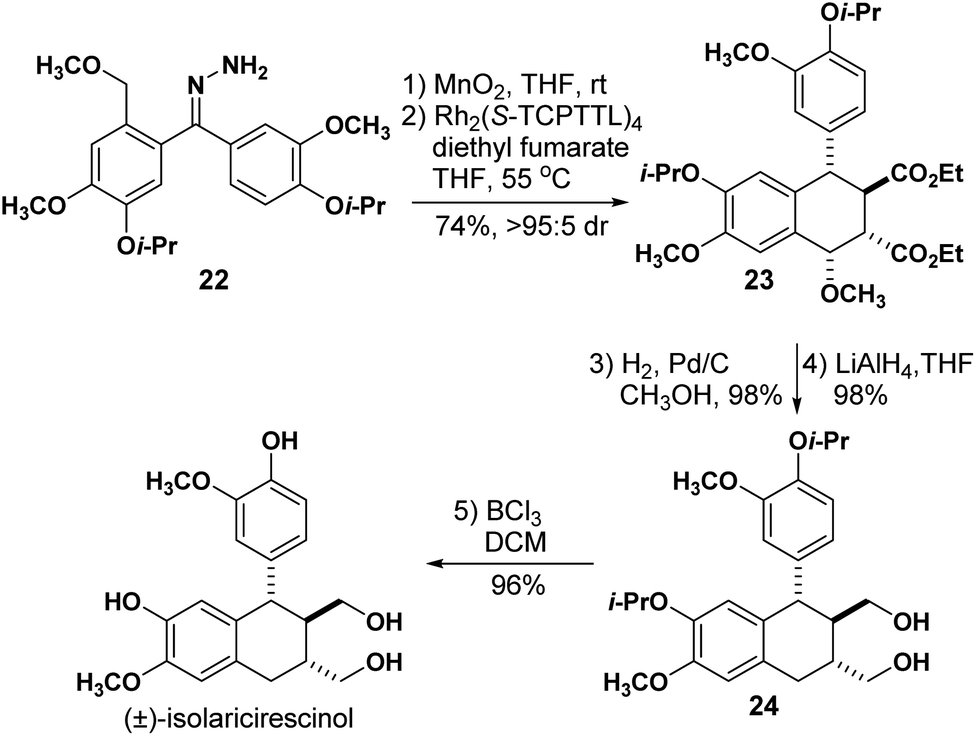
To further illustrate the application of this method, we next completed the stereoselective synthesis of isolariciresinol (Scheme 4), a natural product found in different plant species and is believed to possess anti-inflammatory and antioxidant properties.21–23 From the relative stereochemistry observed in tetralins 15i and (19c–e), it became obvious to us that the natural product tetralin core could be readily accessed using our methodology and it would take only a few transformations to arrive at the final target. Hydrazone 22 was readily obtained from vanillin in 7 steps and subjected to our one-pot sequential oxidation-carbene formation reaction. Using Rh2(S-TCPTTL)4 as the catalyst, the reaction with diethyl fumarate afforded tetralin 23 in 74% yield and >95:5 dr.
 | ||
| Scheme 4 Stereoselective synthesis of isolariciresinol. | ||
While developing the methyl ether substrate scope (Table 4), we did not observe the desired tetralin for the electron-rich hydrazone. Although 22 is an electron-rich hydrazone, we believe that the additional meta alkoxy groups facilitated the o-QDM formation needed for the cycloaddition to take place. Treatment with H2 and Pd/C allowed for the benzylic methoxy cleavage in 98% yield, and ester reduction with LiAlH4 yielded diol 24 in 99% yield. Finally, isolariciresinol was obtained after cleaving the isopropyl protecting groups with BCl3 in 96% yield resulting in a 41% overall yield. This synthesis installs all of the stereogenic centers in a single step and is completed in higher overall yield than the only previous synthesis reported to date.24
The mechanism was further investigated with density functional theory calculations (SMD(DCM)-B3LYP-D3/Def2-TZVPP//SMD(DCM)-B3LYP-D3/Def2-SVP) to examine the formation of o-QDM (Fig. 1, see ESI† for details). Our calculations predict that the hydride shift event is facile to the ylide intermediate 26 and occurs with the metal bound. At this stage, loss of Rh- catalyst reveals a metal-free ortho-quinone dimethide (o-QDM, 27) which acts as the diene in the Diels–Alder step to obtain the final product 15a.
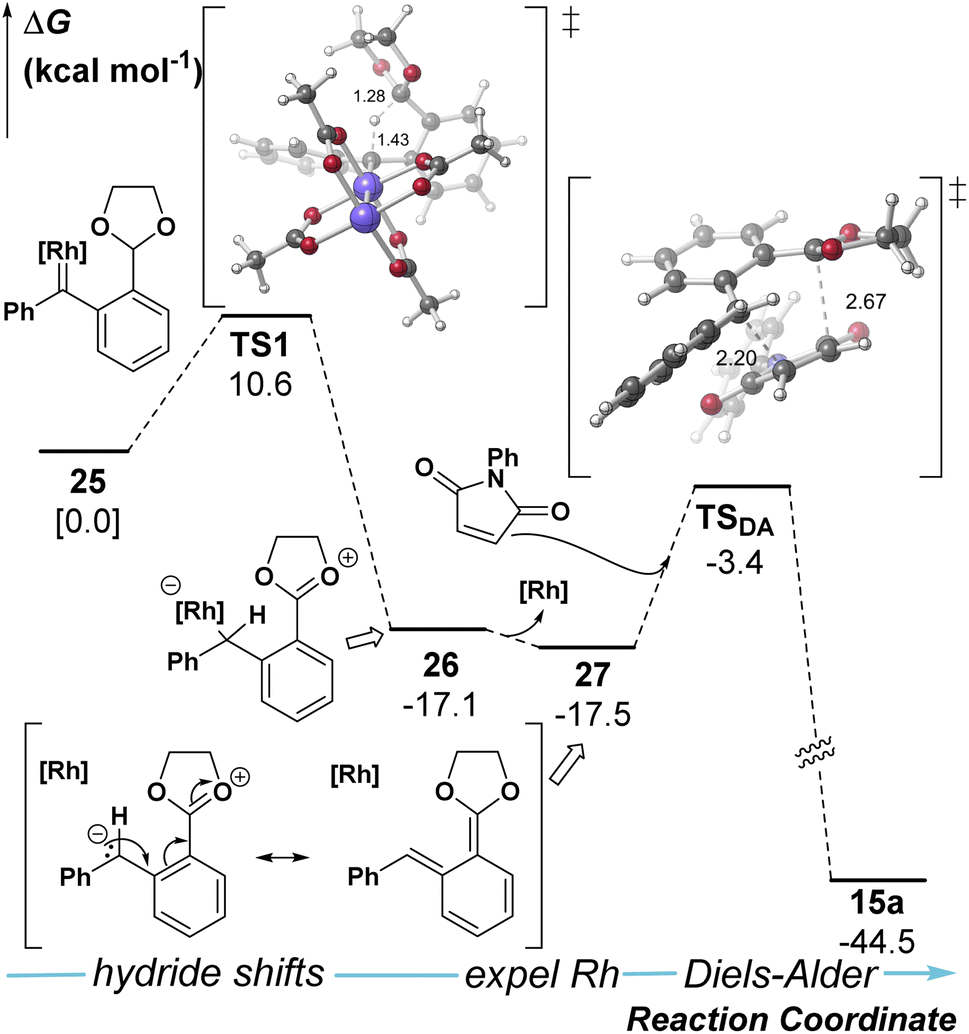 | ||
| Fig. 1 DFT calculations for QDM generation and Diels–Alder reaction. | ||
Conclusion
In summary, we report a novel and easy access to o-QDM dienes that react with various dienophiles, affording tetralins and benzo-fused heterocycles in moderate to good yields. This method features broad substrate scope, excellent regio- and diastereoselectivity, as well as mild reaction conditions. Computational results revealed ortho-quinone dimethide as the key intermediate during the reaction. Application of this methodology afforded the racemic synthesis of isolariciresinol in 41% overall yield.Data availability
Experimental and computational work can be found in the ESI.†Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R01GM124234). JMR Thanks the National Institutes of Health for support in the form of an administrative supplement to support his PhD training. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank the National Science Foundation (Grant CHE-1531193) for the Dual Source X-ray diffractometer and additional support (XSEDE).Notes and references
- B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864–6878 CrossRef CAS PubMed
.
- Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024–10030 CrossRef CAS PubMed
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed
- E. Nakamura, N. Yoshikai and M. Yamanaka, J. Am. Chem. Soc., 2002, 124, 7181–7192 CrossRef CAS PubMed
- M. P. Cava and D. R. Napier, J. Am. Chem. Soc., 1957, 79, 1701–1705 CrossRef CAS
- M. F. Hentemann, J. G. Allen and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 1937–1940 CrossRef CAS PubMed
- S. Zhu, Z. Guo, Z. Huang and H. Jiang, Chem. – Eur. J., 2014, 20, 2425–2430 CrossRef CAS PubMed
- S. Zhu, Z. Zhang, X. Huang, H. Jiang and Z. Guo, Chem. – Eur. J., 2013, 19, 4695–4700 CrossRef CAS PubMed
- S. Zhu, R. Liang, H. Jiang and W. Wu, Angew. Chem., Int. Ed., 2012, 51, 10861–10865 CrossRef CAS PubMed
- S. Zhu, Y. Xiao, Z. Guo and H. Jiang, Org. Lett., 2013, 15, 898–901 CrossRef CAS PubMed
- J. Zhang, Y. Xiao, K. Chen, W. Wu, H. Jiang and S. Zhu, Adv. Synth. Catal., 2016, 358, 2684–2691 CrossRef CAS
- H. Luo, C. He, H. Jiang and S. Zhu, Chin. J. Chem., 2020, 38, 1052–1056 CrossRef CAS
- S. Zhu, H. Huang, Z. Zhang, T. Ma and H. Jiang, J. Org. Chem., 2014, 79, 6113–6122 CrossRef CAS PubMed
- T. Peez, V. Schmalz, K. Harms and U. Koert, Org. Lett., 2019, 21, 4365–4369 CrossRef CAS PubMed
- W. Oppolzer, Synthesis, 1978, 11, 793–802 CrossRef
- H. M. L. Davies, J. Org. Chem., 2019, 84, 12722–12745 CrossRef CAS PubMed
- K. Benda, W. Regenhardt, E. Schaumann and G. Adiwidjaja, Eur. J. Org. Chem., 2009, 2009, 1016–1021 CrossRef
- M. C. Pirrung and K. P. Kaliappan, Org. Lett., 2000, 2, 353–355 CrossRef CAS PubMed
- B. Baderschneider and P. Winterhalter, J. Agric. Food Chem., 2001, 49, 2788–2798 CrossRef CAS PubMed
- J. Y. Cho, O. Paper, A. R. Kim and M. H. Park, Planta Med., 2000, 67, 312–316 CrossRef PubMed
- L. P. Meagher, G. R. Beecher, V. P. Flanagan and B. W. Li, J. Chem. Sci., 1999, 47, 3173–3180 CAS
- M. Sampei, M. A. Arai and M. Ishibashi, J. Nat. Med., 2018, 72, 651–654 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2087127–2087130, 2087132–2087135, 2087415, 2145030, 2240749. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00734k |
| This journal is © The Royal Society of Chemistry 2023 |