
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic insight on impact of environment on natural photoprotectants†
Abigail L.
Whittock
 ab,
Xuefei
Ding
c,
Xavier E.
Ramirez Barker
c,
Nazia
Auckloo
ad,
Rebecca A.
Sellers
a,
Jack M.
Woolley
c,
Krishnan
Tamareselvy
e,
Marine
Vincendet
f,
Christophe
Corre
ad,
Emma
Pickwell-MacPherson
c and
Vasilios G.
Stavros
*ag
ab,
Xuefei
Ding
c,
Xavier E.
Ramirez Barker
c,
Nazia
Auckloo
ad,
Rebecca A.
Sellers
a,
Jack M.
Woolley
c,
Krishnan
Tamareselvy
e,
Marine
Vincendet
f,
Christophe
Corre
ad,
Emma
Pickwell-MacPherson
c and
Vasilios G.
Stavros
*ag
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk
bAnalytical Science Centre for Doctoral Training, Senate House, University of Warwick, Coventry, CV4 7AL, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
dWarwick Intergrative Synthetic Biology Centre, School of Life Sciences, University of Warwick, Coventry, CV4 7AL, UK
eLubrizol Advanced Materials Inc., 377 Hoes Lane, Suite 210, Piscataway, New Jersey 08854, USA
fLubrizol Life Science Beauty, Calle Isaac Peral, 17 Pol. Ind. Camí Ral, 08850 Barcelona, Spain
gSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: v.stavros@bham.ac.uk
First published on 26th May 2023
Abstract
Biomimicry has become a key player in researching new materials for a whole range of applications. In this study, we have taken a crude extract from the red algae Palmaria palmata containing mycosporine-like amino acids – a photoprotective family of molecules. We have applied the crude extract onto a surface to assess if photoprotection, and more broadly, light-to-heat conversion, is retained; we found it is. Considering sunscreens as a specific application, we have performed transmission and reflection terahertz spectroscopy of the extract and glycerol to demonstrate how one can monitor stability in real-world applications.
Introduction
Over the ages, humans have turned to natural sources for their basic needs and survival.1 It is no wonder therefore that research in academia and industry has gravitated towards biomimicry. In particular, natural products have gained traction as staple ingredients in various industries including food, agriculture, pharmaceutical and cosmetics.2–6 This work is no exception as we explore the photoprotective capabilities of a crude extract from the red algae Palmaria palmata (P. palmata) in different environments.There are many naturally occurring photoprotective compounds; examples span melanin, flavonoids, polyphenols and mycosporine-like amino acids (MAAs).7 The latter are a family of molecules synthesized by many micro- and macroorganisms that have the basic core structure of a cyclohexenone or cyclohexenimine.8 MAAs are differentiated by the various substituents at the imino position, with a recent database reporting over 70 different structures.9 The zwitterionic nature and the various substituents result in a peak absorption for MAAs spanning 310–362 nm; a broad range within both the ultraviolet (UV) A (400–315 nm) and UVB (315–280 nm) regions.9–11 This is significant given that UVA and UVB is the highest energy radiation that reaches the Earth's surface and has damaging implications to nature and humans.12,13Fig. 1 demonstrates this broad band spectral coverage of a number of common MAAs found in P. palmata.
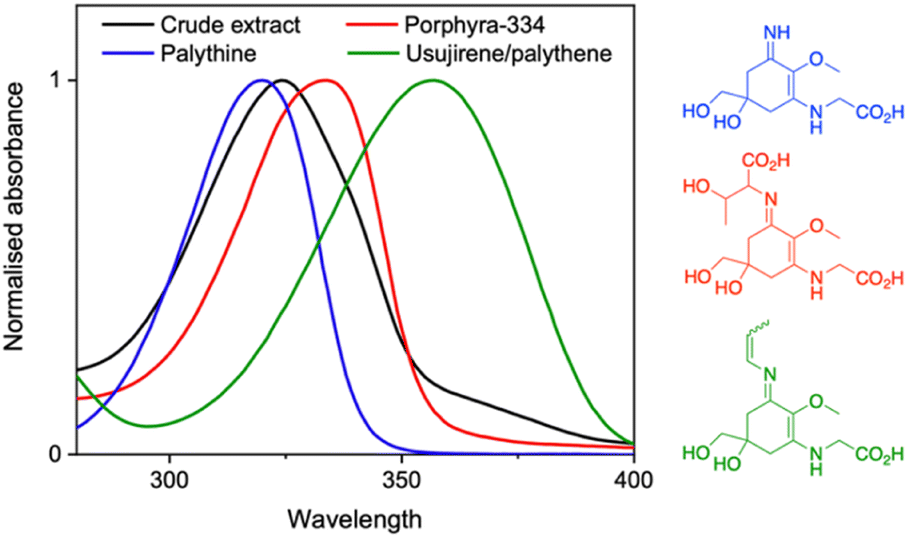 | ||
| Fig. 1 Normalised UV-visible spectra, over the UVA and UVB regions, of the crude extract (black) and common MAAs found in P. palmata; palythine (blue), porphyra-334 (red), usujirene and palythene (green) along with their structures. | ||
The impressive photoprotective properties of several MAAs in aqueous solutions has been linked to their very short lifetime in the excited state with high relaxation efficiency.14,15 This, alongside computational studies revealing the planar to non-planar ring flexing MAAs undergo to traverse the S1/S0 conical intersection (CI) with a barrierless non-radiative decay,15–19 deepens our understanding of MAA photoprotection in nature. The aforementioned studies have been done in solution where there is a lot of unrestricted molecular motion. However, photoprotection is often required in other more restrictive environments including more viscous solvents and on a surface. Therefore, understanding dynamics in these different environments is crucial.
Previous photodynamical studies on photoprotective molecules have revealed a variable environment-dependency. Firstly, increased solvent viscosity impacted the photoisomerisation lifetime of p-hydroxycinnamates to a small extent and sinapoyl malate to a large extent. The authors attributed the results to the alternative isomerisation mechanisms in operation: for small changes in lifetime, an in-plane isomerisation was proposed, and for large increases in lifetime, an out-of-plane isomerisation was proposed.20,21 The latter was said to experience increased friction due to the larger amplitude in nuclear motion ultimately resulting in a viscosity-dependent lifetime.20 In addition to viscosity effects, other intermolecular interactions can impact dynamics. Three exemplar cases are briefly presented: (i) the intramolecular proton transfer mechanism in oxybenzone, which facilitates photoprotection, is broken when sodium cations are present;22,23 (ii) relaxation to the electronic ground state of cinnamates is impeded by internal conversion to an optically dark state when in vacuum, but is facilitated when in water due to reverse ordering of the electronic states;23,24 and (iii) recent work examining the impact of depositing such photoprotective molecules on surfaces mimicking skin have shown some variability to their photodynamics depending on surface.25–27
The variability in the findings presented supra certainly warrants further investigations to be conducted. With specific focus on MAAs, experiments probing the photodynamics in different environments have been sparse. Orallo et al.28 have performed studies of MAAs in micellar solutions and observed increased fluorescence quantum yields and lower decomposition upon UVR exposure. The authors attributed their findings to the electrostatic attractions between the carboxylate groups and the cationic heads of the micelles hindering the reorientation molecular movements of the MAAs. Such hindrance also blocks other reactive channels. As such, garnering how the photodynamics of MAAs are impacted by viscous and restricted environments is both intriguing from a molecular-level understanding of photoprotection, as well as of vital importance from an applications-perspective of MAAs. As such, this is the focus of the present work.
Here, due to the time consuming and low yielding process of MAA purification, we have taken a crude extract from P. palmata containing several MAAs. We have assessed its photostability firstly in water to compare to purified MAAs and then in glycerol and on a surface. We add that if the promising photostability is retained in the crude extract, then this may be of interest to several industries owing to the ease of sample preparation. The chosen surface environment is a skin mimic with a rough surface that enables transmission of light for our experimental techniques. Note that we believe our finding can be extended to a number of surface environments. Using terahertz (THz) transmission and reflection spectroscopy, we can glean further insight into the stability of the solution and, with the application of sunscreen in mind, we demonstrate how this technique could be used to evaluate the stability of the solution once applied to skin.
Results and discussion
A dried P. palmata sample was commercially sourced (The Cornish Seaweed Company) and a simple extraction process was followed to obtain a crude extract; experimental details can be found in the ESI.†Fig. 2 is the two-dimensional spectrum of UV chromatogram vs. time obtained from ultra high performance liquid chromatography-high resolution mass spectrometry (UHPLC-HRMS) analysis of the crude extract. From this, it is clear that several compounds absorb above 300 nm and, accordingly, eight MAAs were identified in the sample based on the accurate m/z values being consistent with their molecular formula; mycosporine-glycine, palythine, asterina-330, palythinol, shinorine, porphyra-334, usujirene and palythene (Fig. S1–S9, ESI†). Our identification was guided by previous literature that has investigated MAA content in P. palmata in addition to a recently published MAA database.9,29,30 Furthermore, our UHPLC-HRMS analysis found that palythine and porphyra-334 were present in the greatest yields (Fig. S1, ESI†), again in line with what has been found previously,29,30 and consequently palythine and porphyra-334 were purified and studied to support this work. Using the known molar extinction coefficients for palythine and porphyra-334, we extracted an approximate dried weight yield for MAAs within the crude extract which was ∼0.7%. As the MAAs make up a very small percentage of the crude extract, our studies aim at elucidating whether upon photoexcitation, the MAAs interact with other components in the crude extract which subsequently effect the ultrafast dynamics and overall photostability.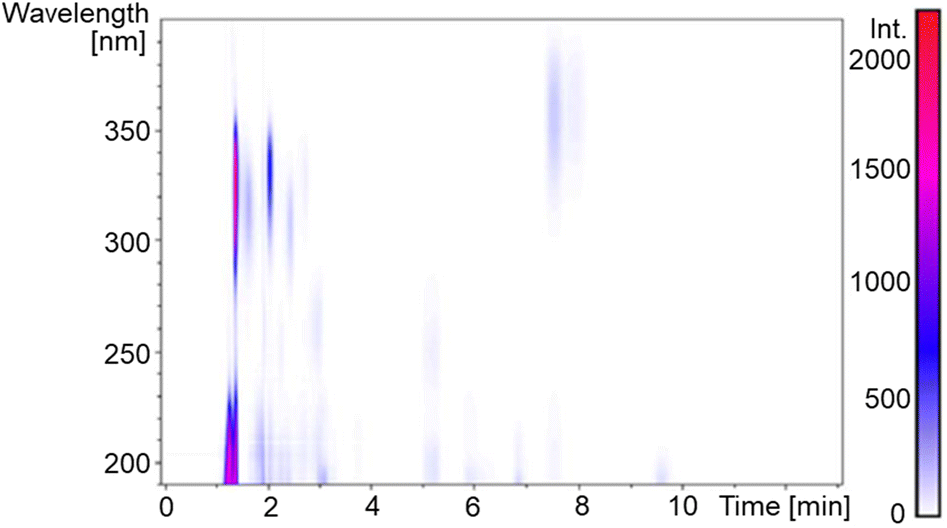 | ||
| Fig. 2 Two-dimensional (UV × UHPLC) plot of P. palmata methanolic extract. | ||
Returning to Fig. 1, we can see that the crude extract peak absorption (λmax) centres somewhere between palythine and porphyra-334's λmax and because of the large array of MAAs present, the overall width of the absorption is broader. An extended UV-visible spectrum out to 700 nm of the crude extract in water can be found in the ESI (Fig. S10).† This absorption is assigned to an S1 ← S0 transition with ππ* character for all MAAs based on previous literature.14,15,17,18Table 1 presents the λmax for each environment presented in this work.
| Sample | λ max (nm) | AUCI |
|---|---|---|
| Palythine (water) | 320 | 0.976 |
| Porphyra-334 (water) | 334 | 0.973 |
| Usujirene/palythene (water) | 357 | 0.961 |
| Crude extract (water) | 324 | 0.897 |
| Crude extract (glycerol) | 327 | 0.940 |
| Crude extract (skin I) | 327 | 0.877 |
| Crude extract (skin II) | 327 | 0.969 |
Table 1 reports the long-term photostability results obtained in this work and our previous work (see ESI† for details).15 All purified MAAs from P. palmata were dissolved in water and subsequently exposed to solar simulated light (equivalent to the power and spectrum of the sun at the Earth's surface) for 2 hours (Fig. S11, ESI†). The same was conducted for the crude extract in water for direct comparison and then we also used glycerol as a solvent for the crude extract due to its increased viscosity. The final two environments focus on the progression of our experiments to more real-life surface environments and for this we used a skin mimic, Vitro Corneum. The first is the crude extract in glycerol applied onto Vitro Corneum (referred to as skin I) and the second is crude extract in water applied onto Vitro Corneum before being left to dry (referred to as skin II). Our choice to conduct studies on two skin environments was because skin I would be in a thin film with a measured thickness of ∼40 μm (Fig. S12, ESI†), however, we could not be certain that we would be investigating the surface effect of the skin and not bulk solvent. Therefore, skin II was studied whereby the crude extract was dried onto the surface. The different described environments are depicted in Fig. 3a. The area under the curve index (AUCI) between 280 and 400 nm after 2 hours of irradiation was calculated and is presented in Fig. 3b, see details in the ESI.† All studied environments had an AUCI > 0.8 making them all classed as photostable following a criterion for photostability that has been used by others (with regards to sunscreens).31 A slightly lower AUCI was found for the crude extract in water compared to the purified MAAs which may be related to the increased complexity of the sample with many other molecules that the excited MAAs can interact with. Comparing this to skin II, we see that the AUCI matches that of purified MAAs possibly suggesting that the more restrictive environment hinders the molecular motions of any alternative decay pathways and favours the photoprotective mechanism.
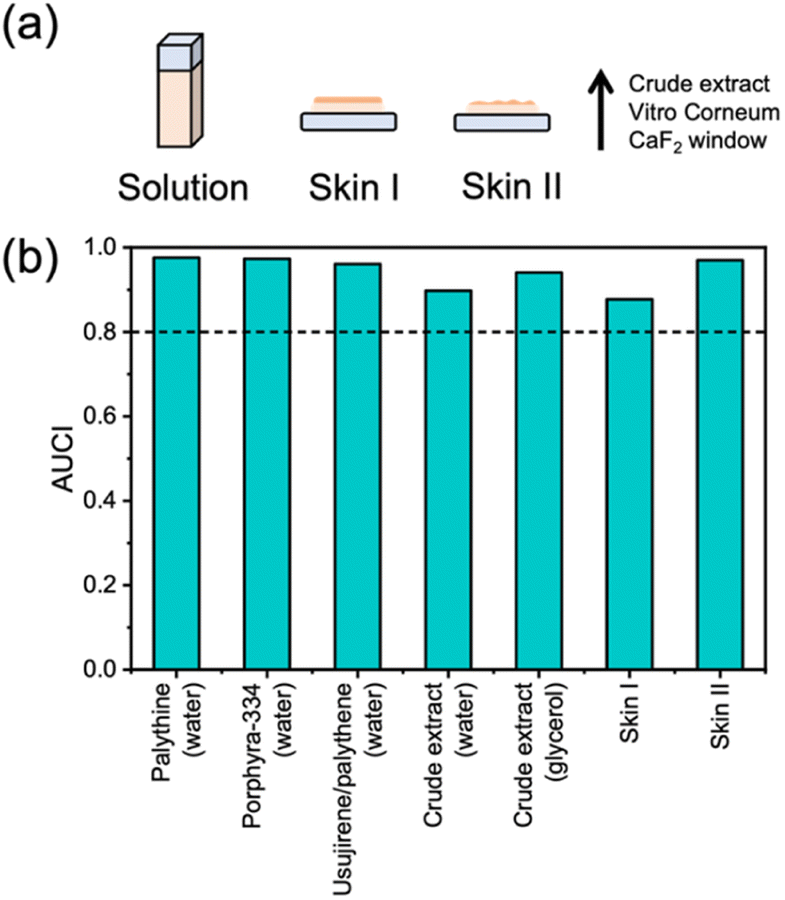 | ||
| Fig. 3 (a) Schematic representation of the studied environments. (b) Summary of AUCI over 2 hours of irradiation with a solar simulator.15 Dotted line at 0.8 is given as the criteria for photostability that has been used by others.31 | ||
We now try to link the long-term photostability with what happens at the very early stages of photon-to-molecule interaction using transient electronic absorption spectroscopy (TEAS), see ESI† for experimental details. Using TEAS, we can glean insight into whether the photoprotective mechanism (planar to non-planar ring flexing, Fig. 4a) that has been proposed computationally in gas-phase or implicit solvent environments is also followed when MAAs are on a surface.15–19 In Fig. 4b–g, the transient absorption spectra (TAS) are presented for (1) the crude extract in glycerol, (2) skin I and (3) skin II (Fig. S13, ESI;† results in water). Note that for skin II we observed an increased amount of scatter because of the extract being dried onto the skin mimic (see inset of Fig. 4f).
 | ||
| Fig. 4 (a) Computed optimised S0 (opaque) and S1/S0 CI (transparent) geometries of MAAs taken from previous literature.15,17,19 TAS of crude extract in glycerol presented as (b) false colour heat map and (c) lineouts, crude extract (skin I) presented as (d) false colour heat map and (e) lineouts, and crude extract (skin II) presented as (f) false colour heat map and (g) lineouts. Time delays in (b), (d) and (f) are plotted linearly until 5 ps and then as a logarithmic scale from 5 to 1800 ps. In (c), (e) and (g) the inset is of a high average transient at 1800 ps. The inset of (d) and (f) is a picture of the sample setup. | ||
In general, the TAS in each environment displays the same spectral signatures and are consistent with previous work;14,15,32 a ground state bleach at the blue edge of the probe window (<350 nm), a broad stimulated emission centred at ∼450 nm and an excited state absorption (ESA) which grows with a delay from time-zero centred at ∼370 nm. The described features mostly decay within tens of ps leaving a very weak ESA (almost within the noise) below ∼450 nm that persists until beyond the time window of the experiment. It should also be briefly mentioned that a second ESA (that almost merges with the one described above) is evident when solvated in water at the red edge of the probe. This is assigned to solvated electron and has been previously identified as a multiphoton process in MAAs.14,15 Further to this, evidence of solvated electron in glycerol (Fig. 4c) is given by the weak ESA from ∼425 to 650 nm. In glycerol, solvated electron absorption is shifted to shorter wavelengths supporting this assignment.33 We add that we elect not to discuss the solvated electron absorption further given it is a multiphoton process and unlikely to be initiated by the sun. However, we provide further details of its presence in the ESI (Fig. S14).† Other important details about the TAS are that (1) the stimulated emission red-shifts with time (more pronounced in water) which is indicative of rapid evolution along the S1 state, and (2) the ESA blue-shifts with time and is located to longer wavelengths than the GSB which is suggestive of electronic ground state vibrational cooling. Such details are important when assigning lifetimes below.
Returning to the weak ESA below ∼450 nm present at the final time delay (1800 ps and 3000 ps), we tentatively assign this to a very minor portion of either trapped population in the excited state (S1 or T1) or as a photoproduct (such as an isomer). We now present evidence for each of these. Firstly, fluorescence is observed for palythine, porphyra-334 and the crude extract in water suggesting that there is a population that is trapped in the S1 excited state (Fig. S15, ESI†). The fluorescence quantum yield for the crude extract in water is <1% (data not shown). The same applies for palythine and porphyra-334,34,35 meaning any population in the S1 state mostly finds its way to the CI after the final time delay or contributes very little to the persistent ESA. Secondly, triplet–triplet absorption spectra have been reported for palythine and porphyra-334 and appears in a similar spectral region to the ESA and, further to this, a computational study demonstrated that the S1 and T1 energies approach one another along the reaction coordinate.18,34,35 It is noted, however, that the short lifetime the ESA occurs on, particularly in water, does render intersystem crossing less likely. Finally, a computational investigation on protonated palythine found that upon photoexcitation and relaxation via the S1/S0 CI, the formation of a higher in energy conformer was identified in addition to the original electronic ground state geometry.17 As such, isomer formation is also plausible for the persistent ESA. We add that it is this that we feel is the most likely candidate although all three are possibly present to varying degrees. Given that the solar simulator irradiation does not result in an absorption in this region, we suggest that the isomer may be metastable and convert back to the starting isomer beyond the final time delay. Furthermore, this longer-lived species or isomer may go on to degrade causing the AUCI in the solar simulator irradiations to be <1.
Using the software package Glotaran, lifetimes associated with dynamical contributions were extracted using a global 3- or 4-component (depending on system) sequential kinetic model.36 These lifetimes are presented in Table 2. Our lifetime assignment was guided by the evolution associated difference spectra (EADS) which are presented in the ESI (Fig. S16);† quality of the fit can be assessed through the residuals found in Fig. S17, ESI.†
| Sample | t 1 (fs) | t 2 (fs) | t 3 (ps) | t 4 (ns) |
|---|---|---|---|---|
| Palythine (aq.) | 300 ± 60 | — | 0.60 ± 0.06 | >1.8 |
| Porphyra-334 (aq.) | 130 ± 60 | 940 ± 60 | 1.33 ± 0.07 | >3 |
| Crude extract (aq.) | 170 ± 60 | 630 ± 60 | 2.15 ± 0.08 | >1.8 |
| Crude extract (glycerol) | 160 ± 60 | — | 2.52 ± 0.06 | >1.8 |
| Crude extract (skin I) | 110 ± 60 | 290 ± 60 | 3.19 ± 0.14 | >1.8 |
| Crude extract (skin II) | — | — | 4.30 ± 0.24 | >1.8 |
We begin by discussing the extracted lifetimes for palythine, porphyra-334 and the aqueous crude extract. The first lifetime, t1 which is ∼200 fs, is assigned to geometry rearrangement (both solute and solvent) as the population evolves out of the Franck–Condon region towards the S1/S0 CI. The second lifetime t2, for porphyra-334 and the crude extract (aq.), is attributed to population funnelling through the S1/S0 CI and populating the vibrationally hot electronic ground state. We note that in palythine, a 3-component fit is sufficient (and thus t2 is absent) implying that t1 captures both the geometry rearrangement and (to a certain extent) population moving towards and traversing the S1/S0 CI. The crude extract (aq.) lifetimes lie close to those of palythine and porphyra-334. Evidently, upon photoexcitation of the crude extract (aq.) at 324 nm, palythine and porphyra-334 are excited and contribute to the observed dynamics. The third lifetime, t3 is assigned to vibrational cooling along the electronic ground state coordinate via vibrational energy transfer both intra- and intermolecular to the solvent. t3 for palythine is significantly shorter than that of porphyra-334 and the crude extract; we tentatively attribute this to the hydrogen bonding network of water (previously assigned as crucial to the fast and efficient energy dissipation of MAAs)37 having a greater impact in palythine when compared to porphyra-334 and the crude extract. The final lifetime is attributed to the long-lived excited state and/or isomer.
We now move to discuss the lifetime for the crude extract in glycerol, a more viscous solvent than water. The extracted lifetime for t1 is on a similar timescale to that of crude extract (aq.); a 3-component fit is once again sufficient (and thus the absence of t2) to capture the majority of the excited state relaxation prior to vibrational cooling. Within the EADS of t3, a stimulated emission (>425 nm) is present indicating that some population still remains in the excited state, decaying within t3. Broadly speaking, the dynamics of photoprotection are comparable in glycerol and water; very fast and sub-picosecond for the majority of excited state population decay and less than a few ps for vibrational cooling in the electronic ground state. The absence of viscosity-dependent lifetimes suggests that nuclear motion towards the CI does not experience significant friction. Within the ring flexing mechanism, the side chains that come out-of-plane are relatively small, i.e. they are not long hydrocarbon chains, supporting this argument.
Finally, the extracted lifetimes for skin I and skin II should be taken tentatively given the decreased signal-to-noise and increased scatter in their respective TAS. Four lifetimes were extracted for skin I and the results are similar to the crude extract (aq.), therefore we propose the same arguments as supra for the lifetime assignment. For skin II, the fit was only able to extract two lifetimes due to the increased scatter and noise. As such, we believe that the first extracted lifetime in this case, ∼4 ps, is a convolution of the described processes for t1–3 and is hence reported under t3. We note that the vibrational cooling still being on the ps timescale implies that whilst bulk solvation enables fast and efficient dissipation of energy through the solvent network, it is not vital as efficient energy dissipation is maintained in skin I and skin II. In summary, the dynamics when placed on a surface do not qualitatively change compared to when in solution, suggesting that (1) any intermolecular interaction with the synthetic skin (if there are any) and (2) the more restricted environment, such as little to no solvation shells, does not noticeably impact the dynamics at the molecular level.
Further to this, THz reflection imaging has recently demonstrated the potential to evaluate skin products in vivo including moisturizers.38 Although we cannot apply the crude extract solution to the skin in this stage of development, here we show the future potential for monitoring the stability of the crude extract once applied onto skin. Fig. 5a shows the THz transmission spectroscopy results for the crude extract in glycerol before and after irradiation with a solar simulator. There is only a very small amplitude change of 0.82% between the measurements indicating that the extract is stable, consistent with our UV-visible spectroscopy findings. Fig. 5b shows the THz reflection spectroscopy results of the volar forearm before and after applying glycerol, a well-known component of moisturizers, to the skin. We see a clear difference in the THz spectrum of the skin due to the presence of glycerol. Therefore, THz reflection spectroscopy could be used to determine the stability of the extract when applied to the skin as part of a skin product and potentially evaluate other products within surface applications in the future.
 | ||
| Fig. 5 (a) Mean normalised THz spectrum of crude extract in glycerol before and after 2 hours of irradiation with a solar simulator. (b) Mean normalised THz spectrum of the volar forearm before and after applying glycerol to the skin. Insets of (a) and (b) are magnifications of the THz spectra at the peak. The error bars represent the standard deviation. | ||
Conclusions
In summary, we have demonstrated from a photoprotection standpoint that a crude extract from P. palmata is very efficient and behaves like purified MAAs. We have also demonstrated that this photoprotection ability is retained in more viscous and close-to-real-life environments using TEAS; the first study of its kind on MAAs to the best of our knowledge. We attribute this to the lack of friction encountered from the Franck–Condon geometry to the S1/S0 CI and believe this is due to the small amplitude change in nuclear motion of the side chains undergoing the out-of-plane movement, which is particularly small for palythine (NH2). As a result, whilst there are increases in the extracted lifetimes, specifically for vibrational cooling and (we predict) porphyra-334's excited state, the main photoprotective dynamics are over within 10 ps and thus remain ultrafast. Additionally, the minor reaction channel resulting in a long-lived species in the studies here does not adversely impact the long-term photostability of MAAs, however, it may have consequences in some environments warranting further investigations in the future.In conclusion, this work offers new evidence to support MAAs within a crude extract's use as photoprotective materials via efficient light-to-heat conversion in solution and in surface-based applications. Such ultrafast studies can inform on the future generations of light-to-heat converters. Finally, we have demonstrated that THz spectroscopy can be used to assess stability of a sample and is a technique where direct measurements on a surface can be made; a useful prospect for future experiments.
Data availability
The datasets underpinning this study are available on request from the corresponding author.Author contributions
A. L. W., X. D., X. E. R. B., N. A., R. A. S. and C. C. conducted the investigations. A. L. W. and X. D. carried out the formal analysis assisted by J. M. W., C. C., E. P.-M. and V. G. S. J. M. W., K. T., M. V., C. C., E. P.-M., and V. G. S. provided supervision. The writing of the original manuscript draft was by A. L. W. A. L. W., X. D., X. E. R. B., N. A., J. M. W., C. C., E. P.-M. and V. G. S. reviewed and edited the writing of the manuscript. All authors approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Warwick Centre for Ultrafast Spectroscopy Research Technology Platform for use of the transient electronic absorption spectroscopy, UV-visible spectrometer, solar simulator and spectrofluorometer. A. L. W. thanks the University of Warwick and Lubrizol for funding a PhD studentship through the Centre for Doctoral Training in Analytical Science. N. A. thanks the University of Warwick for a PhD studentship though the Chancellor Scholarship. J. M. W. thanks the Engineering and Physical Sciences Research Council (EPSRC) (EP/V007688/1) for funding. V. G. S. thanks the Royal Society for a Royal Society Industry Fellowship and HO2020 for the support of FetOpen grant BoostCrop (Grant Agreement 828753). This work was supported by Grant BB/M017982/1 from the UK Biotechnology and Biological Sciences Research Council (BBSRC) and Grant EP/S021442/1 from the EPSRC.Notes and references
- D. A. Dias, S. Urban and U. Roessner, Metabolites, 2012, 2, 303–336 CrossRef CAS PubMed.
- S. K. Mahesh, J. Fathima and V. G. Veena, in Natural Bio-active Compounds, ed. M. K. Swamy and M. S. Akhtar, Springer, Singapore, 2019, vol. 2, pp. 215–250 Search PubMed.
- T. C. Sparks, F. J. Wessels, B. A. Lorsbach, B. M. Nugent and G. B. Watson, Pestic. Biochem. Physiol., 2019, 161, 12–22 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- G. D. Mogoşanu, A. M. Grumezescu, C. Bejenaru and L. E. Bejenaru, in Food Preservation, ed. A. M. Grumezescu, Academic Press, 2017, pp. 365–411 Search PubMed.
- N. Saewan and A. Jimtaisong, J. Cosmet. Dermatol., 2015, 14, 47–63 CrossRef PubMed.
- E. P. Balskus and C. T. Walsh, Science, 2010, 329, 1653–1656 CrossRef CAS PubMed.
- V. Geraldes and E. Pinto, Pharmaceuticals, 2021, 14, 63 CrossRef CAS PubMed.
- R. Lucas, T. McMicheal, W. Smith and B. Armstrong, in Environmental Burden of Disease Series, No. 13, World Health Organization, Geneva, Switzerland, 2006 Search PubMed.
- J. I. Carreto and M. O. Carignan, Mar. Drugs, 2011, 9, 387–446 CrossRef CAS PubMed.
- P. A. Morganroth, H. W. Lim and C. T. Burnett, Am. J. Lifestyle Med., 2013, 7, 168–181 CrossRef.
- P. Weihs, A. W. Schmalwieser and G. Schauberger, in Environmental Toxicology, ed. E. A. Laws, Springer New York, 2013, pp. 609–688 Search PubMed.
- A. L. Whittock, N. Auckloo, A. M. Cowden, M. A. P. Turner, J. M. Woolley, M. Wills, C. Corre and V. G. Stavros, J. Phys. Chem. Lett., 2021, 12, 3641–3646 CrossRef CAS PubMed.
- A. L. Whittock, J. M. Woolley, N. Auckloo, C. Corre and V. G. Stavros, Molecules, 2022, 27, 2272 CrossRef CAS PubMed.
- R. Losantos, I. Funes-Ardoiz, J. Aguilera, E. Herrera-Ceballos, C. García-Iriepa, P. J. Campos and D. Sampedro, Angew. Chem., Int. Ed., 2017, 56, 2632–2635 CrossRef CAS PubMed.
- D. Sampedro, Phys. Chem. Chem. Phys., 2011, 13, 5584–5586 RSC.
- M. Hatakeyama, K. Koizumi, M. Boero, K. Nobusada, H. Hori, T. Misonou, T. Kobayashi and S. Nakamura, J. Phys. Chem. B, 2019, 123, 7649–7656 CrossRef CAS PubMed.
- M. Hatakeyama and S. Nakamura, J. Phys. Chem. A, 2022, 126, 7460–7467 CrossRef CAS PubMed.
- M. D. Horbury, W.-D. Quan, A. L. Flourat, F. Allais and V. G. Stavros, Phys. Chem. Chem. Phys., 2017, 19, 21127–21131 RSC.
- A. Espagne, D. H. Paik, P. Changenet-Barret, M. M. Martin and A. H. Zewail, ChemPhysChem, 2006, 7, 1717–1726 CrossRef CAS PubMed.
- J. A. Berenbeim, N. G. K. Wong, M. C. R. Cockett, G. Berden, J. Oomens, A. M. Rijs and C. E. H. Dessent, Phys. Chem. Chem. Phys., 2020, 22, 19522–19531 RSC.
- N. G. K. Wong and C. E. H. Dessent, Front. Chem., 2022, 9, 812098 CrossRef PubMed.
- E. M. M. Tan, M. Hilbers and W. J. Buma, J. Phys. Chem. Lett., 2014, 5, 2464–2468 CrossRef CAS PubMed.
- M. D. Horbury, E. L. Holt, L. M. M. Mouterde, P. Balaguer, J. Cebrián, L. Blasco, F. Allais and V. G. Stavros, Nat. Commun., 2019, 10, 4748 CrossRef PubMed.
- T. T. Abiola, B. Rioux, S. Johal, M. M. Mention, F. Brunissen, J. M. Woolley, F. Allais and V. G. Stavros, J. Phys. Chem. A, 2022, 126, 8388–8397 CrossRef CAS PubMed.
- T. T. Abiola, N. d. N. Rodrigues, C. Ho, D. J. L. Coxon, M. D. Horbury, J. M. Toldo, M. T. do Casal, B. Rioux, C. Peyrot, M. M. Mention, P. Balaguer, M. Barbatti, F. Allais and V. G. Stavros, J. Phys. Chem. Lett., 2021, 12, 337–344 CrossRef CAS PubMed.
- D. E. Orallo, S. G. Bertolotti and M. S. Churio, Photochem. Photobiol. Sci., 2017, 16, 1117–1125 CrossRef CAS PubMed.
- Y. V. Yuan, N. D. Westcott, C. Hu and D. D. Kitts, Food Chem., 2009, 112, 321–328 CrossRef CAS.
- Y. Nishida, Y. Kumagai, S. Michiba, H. Yasui and H. Kishimura, Mar. Drugs, 2020, 18, 502 CrossRef CAS PubMed.
- H. Gonzalez, N. Tarras-Wahlberg, B. Strömdahl, A. Juzeniene, J. Moan, O. Larkö, A. Rosén and A.-M. Wennberg, BMC Dermatol., 2007, 7, 1–9 CrossRef PubMed.
- J. M. Woolley, M. Staniforth, M. D. Horbury, G. W. Richings, M. Wills and V. G. Stavros, J. Phys. Chem. Lett., 2018, 9, 3043–3048 CrossRef CAS PubMed.
- S. Arai and M. C. Sauer Jr, J. Chem. Phys., 1966, 44, 2297–2305 CrossRef CAS.
- F. R. Conde, M. S. Churio and C. M. Previtali, J. Photochem. Photobiol., B, 2000, 56, 139–144 CrossRef CAS PubMed.
- F. R. Conde, M. S. Churio and C. M. Previtali, Photochem. Photobiol. Sci., 2007, 6, 669–674 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- K. Koizumi, M. Hatakeyama, M. Boero, K. Nobusada, H. Hori, T. Misonou and S. Nakamura, Phys. Chem. Chem. Phys., 2017, 19, 15745–15753 RSC.
- H. Lindley-Hatcher, A. I. Hernandez-Serrano, J. Wang, J. Cebrian, J. Hardwicke and E. Pickwell-MacPherson, J. Phys.: Photonics, 2020, 3, 014001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01875j |
| This journal is © The Royal Society of Chemistry 2023 |