
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotophysical studies for Cu(I)-based halides: broad excitation bands and highly efficient single-component warm white-light-emitting diodes†
Shuigen
Zhou
,
Yihao
Chen
,
Kailei
Li
,
Xiaowei
Liu
,
Ting
Zhang
,
Wei
Shen
 ,
Ming
Li
,
Lei
Zhou
* and
Rongxing
He
*
,
Ming
Li
,
Lei
Zhou
* and
Rongxing
He
*
Key Laboratory of Luminescence Analysis and Molecular Sensing (Southwest University), Ministry of Education, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China. E-mail: zhoulei25@swu.edu.cn; herx@swu.edu.cn
First published on 2nd May 2023
Abstract
Designing and synthesizing cuprous halide phosphors unifying efficient low-energy emission and a broad excitation band is still a great challenge. Herein, by rational component design, three novel Cu(I)-based metal halides, DPCu4X6 [DP = (C6H10N2)4(H2PO2)6; X = Cl, Br, I], were synthesized by reacting p-phenylenediamine with cuprous halide (CuX), and they show similar structures, consisting of isolated [Cu4X6]2− units separated by organic layers. Photophysical studies uncover that the highly localized excitons and rigid environment give rise to highly efficient yellow-orange photoluminescence in all compounds with the excitation band spanning from 240 to 450 nm. The bright PL in DPCu4X6 (X = Cl, Br) originates from self-trapped excitons due to the strong electron–phonon coupling. Intriguingly, DPCu4I6 features a dual-band emissive characteristic, attributed to the synergistic effect of halide/metal-to-ligand charge-transfer (X/MLCT) and triplet cluster-centered (3CC) excited states. Benefiting from the broadband excitation, a high-performance white-light emitting diode (WLED) with a high color rendering index of 85.1 was achieved using single-component DPCu4I6 phosphor. This work not only unveils the role of halogens in the photophysical processes of cuprous halides, but also provides new design principles for high-performance single-component WLEDs.
Introduction
Organic–inorganic hybrid metal halides have attracted great interest because of their special electronic structures and optical properties.1–4 Different combinations of organic cations and metal–halide clusters enable rich structures with various physical and optical properties.5–10 In addition, hybrid metal halides have many advantages such as high defect tolerance, adjustable energy bands and convenient synthesis methods, making them a great commercial prospect in solid-state lighting (SSL), scintillators, anti-counterfeit encryption, etc.1,11–15 Due to the unique 6s2 lone-pair electronic configuration of Pb2+, lead-based metal halides tend to show excellent optoelectronic performance.16 In fact, a large number of lead-halide perovskites with outstanding luminescence properties have been developed in recent years.17–19 However, the inherent toxicity of lead is a great threat to the environment and people's health, and thus lead-free metal halides have attracted enormous interest.20Among the alternatives, cuprous-based halides have developed rapidly due to their low price, low toxicity and outstanding optical performance, and are one of the promising candidates for the new generation of light-emitting materials.21–23 Although multiple cuprous-based materials with admirable photoluminescence quantum yield (PLQY) including (Gua)3Cu2I5,24 [N(C2H5)4]2Cu2Br4,25 (MA)4Cu2Br6,26 and (AEP)2Cu2I6·2I·2H2O27 have been reported, their further development faces at least the following two problems to be solved: (1) most reported Cu(I)-based halides with high PLQY are blue-green and white emitters; (2) they typically require deep ultraviolet (UV) excitation, in other words, their PL excitation (PLE) spectrum is not broad enough to meet the excitation requirement. Therefore, it is of great significance to develop low-energy light emitting cuprous-based materials with a broad excitation band.
Compared with all-inorganic metal halides, organic–inorganic hybrid metal halides are more flexible and convenient in selecting cations to be paired with metal–halogen clusters, thus inspiring researchers to design and synthesize crystalline materials with different structural dimensionality and photophysical properties. In fact, some principles for selecting appropriate organic cations have been proposed to achieve excellent optoelectronic properties. For example, large distorted polyhedral clusters and low-dimensional structures are conducive to the formation of self-trapped excitons (STEs), while emissions from STEs accompanied by large Stokes shifts tend to generate low-energy emission bands, so that large-sized organic cations are preferred in order to achieve yellow-red light emission.28–34 Further, moderate structural rigidity is beneficial to reduce lattice vibration and inhibit nonradiative recombination, which is confirmed in many compounds such as (EnrofloH2)SnCl6·H2O,6 (C9H8N)2SnCl6,35etc. Therefore, to enable materials with low-energy emission as well as high PL efficiency, aromatic polyamine compounds with rigid structures are often considered as organic cations to support the low dimensionality. In addition, the emission band can be efficiently tuned by changing the halogen anions.36 For lighting application, broadband excitation as well as emission from a single emitter are highly expected, because it can simplify the device structure and avoids the self-absorption and color instability often observed in mixed and multiple emitters.
Herein, the rigid bis-ammonium aromatic compound p-phenylenediamine was adopted to synthesize a range of novel cuprous-based halides DPCu4X6 [DP = (C6H10N2)4(H2PO2)6; X = Cl, Br, I] by reacting with CuX. As a result, three compounds crystallized in zero-dimensional (0D) structures composed of hydrogen-bonded organic layers and spatially isolated [Cu4X6]2– clusters. The resultant materials achieve a wide PLE spectrum and highly efficient orange/yellow light emission, making these materials attractive for practical application. Comprehensive experimental and theoretical investigations show that the bright luminescence of the chloride and bromide originates from STEs, while the efficient emission of the iodide is attributed to the synergistic effect of halide/metal-to-ligand charge-transfer (X/MLCT) and cluster-centered (3CC) excited states. In addition, considering the excellent optical properties (broad excitation and emission bands) and the remarkable stability of DPCu4I6, a white-light emitting diode (WLED) with a high CRI was successfully manufactured using single-component DPCu4I6 powder excited with a 445 nm blue light chip, demonstrating the practical application potential of these new materials.
Results and discussion
DPCu4X6 (X = Cl, Br, I) single crystals (SCs) were prepared using a facile cooling crystallization method. Briefly, p-phenylenediamine and cuprous halide CuX (X = Cl, Br, I) were dissolved in a mixture of acetonitrile and hypophosphorous acid (H3PO2) solution at 120 °C to form a colorless transparent solution. By slowly decreasing the temperature of the mixture to room temperature (RT), target compounds were obtained (see details in the ESI†). Although the CuX precursors used for the synthesis of the three compounds are different, the resultant products (DPCu4Cl6, DPCu4Br6, and DPCu4I6) show similar morphologies, being square and transparent shapes with a side length of about 5 mm (Fig. S1†). Under natural light, DPCu4Cl6 and DPCu4Br6 SCs are colorless, while DPCu4I6 SCs are yellow. Under 365 nm ultraviolet (UV) lamp irradiation, DPCu4Br6 and DPCu4I6 SCs emit bright yellow light, while DPCu4Cl6 SCs emit orange light. The structures of the three compounds were analyzed by a single-crystal X-ray diffraction (SCXRD) technique. DPCu4Cl6 crystallizes in the orthorhombic structure system with the space group of Ccca, and DPCu4Br6 and DPCu4I6 belong to the tetragonal system with space groups I41/acd and P4/m, respectively. Because of the almost similar synthetic methods and conditions for them, the structures of DPCu4X6 have some identical features. As shown in Fig. 1a and S2,† through the alternate stacking of organic and inorganic layers, all these crystals form a sandwich structure. The organic layers are composed of p-phenylenediamine cations ([C6H10N2]2+) and hypophosphite anions ([H2PO2]−), in which the oxygen atoms on the [H2PO2]− are closely linked to the (C6H10N2)2+ through hydrogen bonds whose length is less than 3 Å (Fig. 1c and Table S2†). It is worth mentioning that since we did not use hydrohalic acid as a solvent during the synthesis, the amount of halogen ions in the solution was not sufficient to balance the positive charge, so the added [H2PO2]− anions not only play a role in the Cu2+ reduction, but also participate in the structural construction. The introduction of [H2PO2]− anions may bring some beneficial effects for efficient luminescence. First, it can form abundant hydrogen bonds between the components of the crystal, thereby enhancing the rigidity of the crystal structure;37 second, it can lengthen the distance between clusters, leading to stronger exciton localization. Although these crystals resemble planar two-dimensional structures, metal–halide polyhedrons in the inorganic layers are indeed isolated, and the [Cu4X6]2− clusters are periodically embedded in the organic matrix to form a unique 0D structure (Fig. 1b). In such structures, each Cu+ is coordinated with three X− to form a plane triangular configuration of CuX3, and then four congruent CuX3 units are interconnected and shared through triangular edge nodes to form a hollow Cu-halogen octahedral cluster (Fig. 1c, S2c, and S2f†). Similarly, it can also be understood that six halogen atoms form a slightly distorted octahedron, and four copper atoms are embedded in the four faces of the octahedron. The four Cu atoms of each [Cu4X6]2− (X = Cl or Br) cluster in DPCu4Cl6 and DPCu4Br6 form a Cu4 tetrahedron, while the Cu site in a [Cu4I6]2− anion has 50% occupancy, i.e., the two groups of non-edge-sharing octahedron faces form two Cu4 tetrahedra, one of which is inverted with respect to the other, resulting in the half-weighted Cu. The Cu–Cu bond distance in DPCu4I6 is 2.51–2.96 Å, which shows that the Cu–Cu metal bond exists in the iodide and suggests strong Cu–Cu bonding interactions in this cluster.38 The average Cu–Cu bond distances for DPCu4Cl6 and DPCu4Br6 are 2.82 Å and 2.89 Å, respectively, indicating that the Cu–Cu interaction in these two compounds is weaker than the iodide. Furthermore, the distance between two adjacent [Cu4X6]2− clusters is over 6 Å, excluding electronic interactions between them. The powder X-ray diffraction (PXRD) patterns of DPCu4X6 agree well with those from SCXRD, suggesting the high purity and uniformity of the as-synthesized crystals (Fig. 1d). In addition, the hydrogen nuclear magnetic resonance (1H-NMR) spectra of C6H10N2Cl2 and DPCu4Cl6 shown in Fig. S3 and S4† show that the peaks in the spectra are almost identical without peaks of impurity appearing, further confirming the high purity of the synthesized compounds. X-ray photoelectron spectroscopy (XPS) measurements in Fig. S5† confirm that DPCu4X6 (X = Cl, Br, I) contains the elements of C, H, N, O, P, Cu and X, which agrees with the SCXRD analysis. The high-resolution Cu 2p spectra of DPCu4X6 show two peaks at 931.8 eV and 951.8 eV, verifying the +1 oxidation state of Cu.39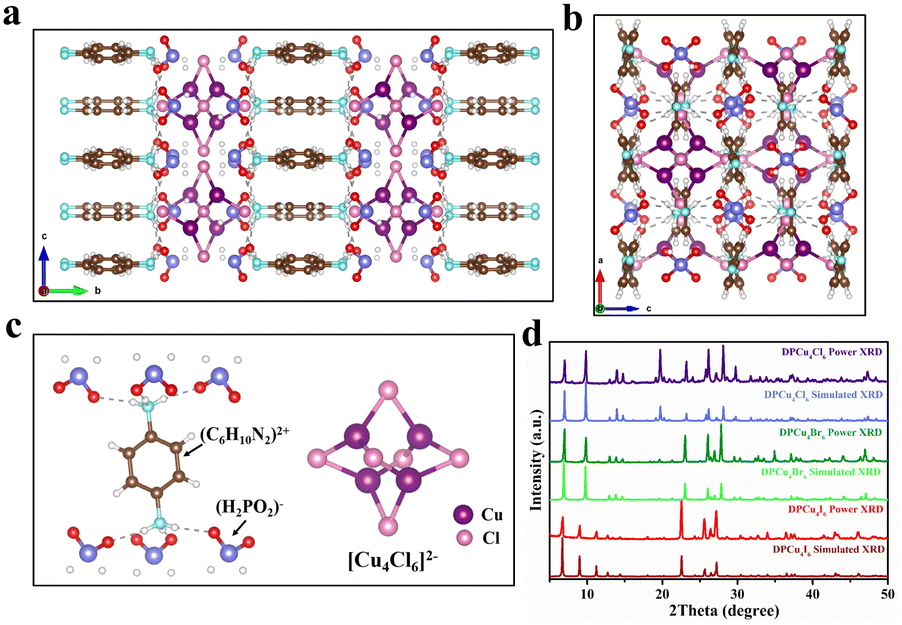 | ||
| Fig. 1 Crystal structure of DPCu4Cl6 viewed along the (a) a axis and (b) b axis (Cu, purple; Cl, pink; P, blue; O, red; C, brown; N, cyan; H, white). (c) Individual [Cu4Cl6]2− cluster and [C6H10N2]2+ organic cation. Note that each [C6H10N2]2+ links with six [H2PO2]− anions through hydrogen bonds. (d) PXRD and SCXRD patterns of DPCu4X6 (X = Cl, Br, I) SCs. | ||
In order to explore the optical properties of DPCu4X6 SCs, steady-state PL/PLE spectra of the as-synthesized SCs were characterized at RT (Fig. 2a). Surprisingly, all three compounds exhibit broad-band PLE spectra covering 240–450 nm, which is beneficial for the SSL applications. It can be clearly observed that the PLE spectra of DPCu4Cl6 and DPCu4Br6 have similar shapes and characters, while that of DPCu4I6 displays an opposite shape with a wider bandwidth. The DPCu4Cl6 SCs exhibit a single broad-band emission peaked at 612 nm with a full width at half-maximum (FWHM) of 148 nm and a large Stokes shift of 245 nm. While both the DPCu4Br6 and DPCu4I6 SCs emit bright yellow light with the same emission peak at 575 nm. The Stokes shift of DPCu4Cl6 and DPCu4I6 is 198 and 282 nm, respectively. The FWHM of DPCu4I6 is 185 nm, which is broader than that of DPCu4Br6 (123 nm). Accordingly, the Commission Internationale de L'Eclairage (CIE) chromaticity coordinate is calculated to be (0.52, 0.45) for DPCu4Cl6, (0.45, 0.51) for DPCu4Br6 and (0.43, 0.49) for DPCu4I6, respectively (Fig. 2b). When the temperature is decreased to 80 K, the PL and PLE spectra of the three halides become narrower (Fig. 2c). Both DPCu4Cl6 and DPCu4Br6 maintain a Gaussian-shaped single peak. The maximum peak position of the bromide remains unchanged, while that of the chloride red shifts to 650 nm. The shape and characteristics of the PLE spectra of DPCu4Cl6 and DPCu4Br6 have hardly changed. However, the spectral characteristics of DPCu4I6 SCs have changed at 80 K. Herein, its PLE spectrum splits to multiple peaks and its emission spectrum contains dual-band emissions with a high-energy (HE) one peaked at 600 nm and a low-energy (LE) peaked at 703 nm. The transient PL (TRPL) decay dynamics was also measured at RT to probe the optical properties of the compounds. As revealed in Fig. 2d, both DPCu4Cl6 and DPCu4Br6 have a long lifetime more than 40 μs monitored at 600 nm, similar to some low-dimensional Cu(I)-based metal halides, such as CuCl-am40 and (18-crown-6)2Na2(H2O)3Cu4Br6.41 The PL decay time of DPCu4I6 is significantly faster, with a lifetime of 4.30 μs monitored at 600 nm and 4.62 μs monitored at 700 nm (Fig. S6†). It is worth noting that the PLQY of the three halides is very high, 89.76% for DPCu4Cl6, 93.52% for DPCu4I6, and even 95.04% for DPCu4Br6 SCs (Fig. S7 and Table S3†). Most reported Cu(I)-based metal halides usually exhibit blue or green emission. Even if there are a few reported materials emitting yellow/orange light, their excitation bands are either narrow or located in the high-energy UV region (<350 nm), which hinders the further lighting applications (Table S3†). Therefore, low-energy emissive (yellow to red light) Cu(I)-based materials with a broad PLE spectrum are highly attractive. Fortunately, DPCu4X6 SCs can meet the above requirements well.
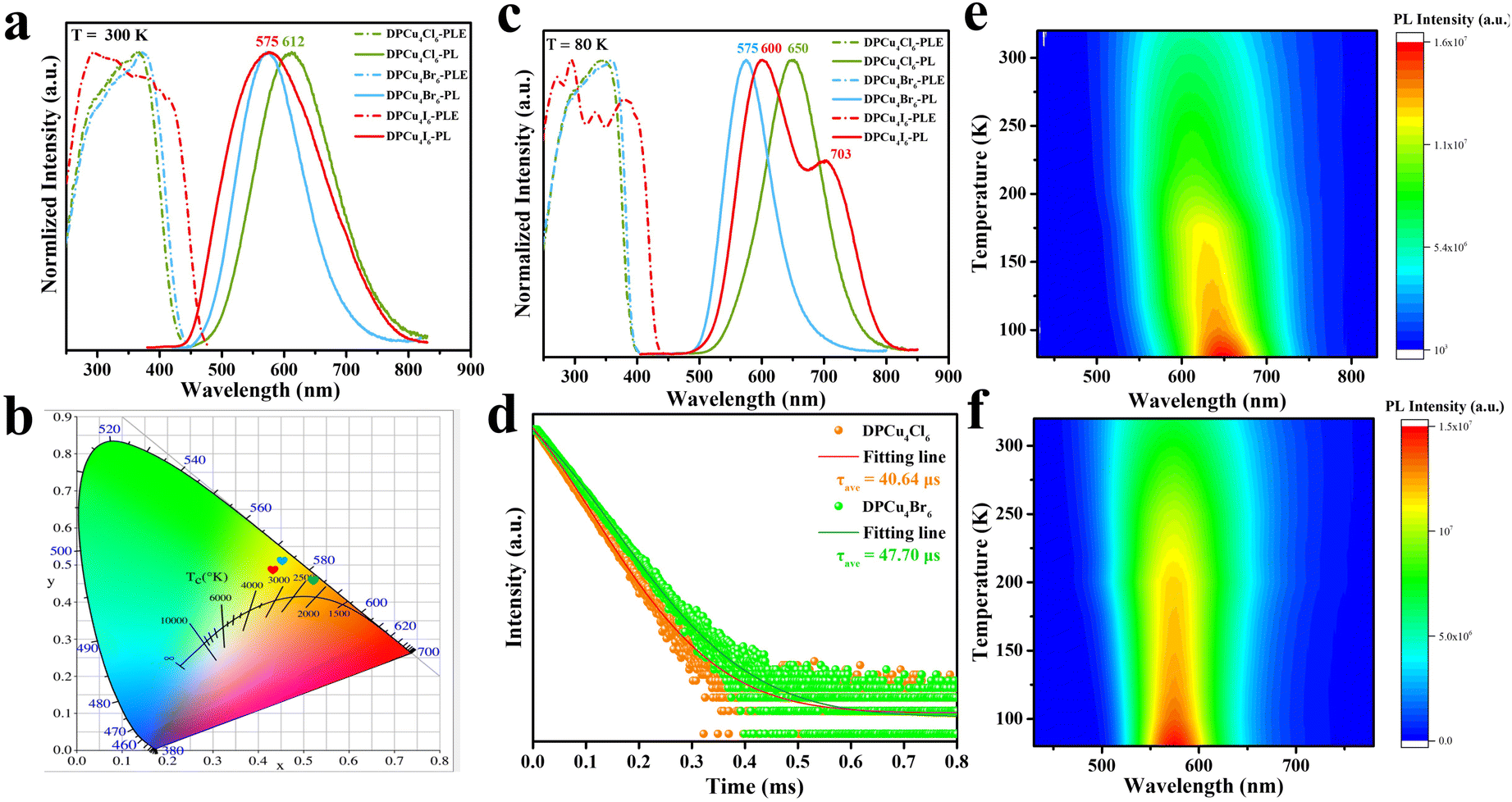 | ||
| Fig. 2 PL and PLE spectra of DPCu4X6 (X = Cl, Br, I) SCs measured at (a) 300 K and (c) 80 K (excited at 320 nm, monitored at 600 nm). (b) CIE chromaticity diagram of DPCu4X6 (DPCu4Cl6, green; DPCu4Br6, red; DPCu4I6, blue). (d) The RT-TRPL spectra of DPCu4X6 (X = Cl, Br) SCs probed at 600 nm. Temperature-dependent PL spectra of (e) DPCu4Cl6 and (f) DPCu4Br6. | ||
Spectroscopic characterization at RT and 80 K indicates that DPCu4Cl6 and DPCu4Br6 have similar PL emission characteristics, while a complex luminescence mechanism may be hidden within the DPCu4I6 SCs. To further explore the changes of the radiative mechanism caused by the halogen substitution, the potential photophysical mechanism of DPCu4Cl6 and DPCu4Br6 was first studied. As shown in Fig. S8,† wavelength-dependent PL/PLE spectra of DPCu4X6 (X = Cl or Br) were recorded at RT, where the identical features of the spectrum suggest that the broad-band emission of the halide should stem from the same excited state. The PL spectra of DPCu4X6 (X = Cl or Br) measured at 80 K with different excitation wavelengths have the same profile, further confirming such an inference (Fig. S9†). The PL intensity of the chloride and bromide increases monotonically and linearly with the increase of excitation power, which excludes the effect of permanent defects on the emission (Fig. S10†). In a word, the typical optical features with a large Stokes shift, broad emission band, high PLQY and long PL lifetime strongly support that the bright orange/yellow emission from DPCu4Cl6 and DPCu4Br6 is related to the STEs.2,42–45 The strong coupling between the exciton and lattice could lead to the elastic lattice to deform transiently, and then form photoinduced trap states below the conduction band. Once the photogenerated excitons are produced, they will be rapidly self-trapped by the STE energy level, and then the excitons return to the ground state through emitting photons with a large Stokes shift and microsecond lifetime.3
Subsequently, the temperature-dependent PL measurement of DPCu4Cl6 and DPCu4Br6 was carried out at 80–300 K to further understand the emission mechanism (Fig. 2e and f). It is found that the PL spectra of the two materials have only one emission peak in the whole temperature range, indicating the single radiative mechanism. Note that the maximum emission wavelength of DPCu4Cl6 gradually increases with the decrease of test temperature, while that of DPCu4Br6 is almost independent of temperature. In fact, this phenomenon that the emission peak position of chloride is more temperature-dependent than that of bromide has been observed in many all-inorganic and organic–inorganic hybrid Cu(I)-based materials.40,46 The red-shift of the emission peak at low temperature is generally attributed to the influence of lattice thermal expansion.47 When the temperature decreases, the energy level of the valence band will increase with the shrinkage of the lattice constant, resulting in a smaller band gap and thus the red-shift emission. Therefore, compared with DPCu4Br6, DPCu4Cl6 has a “softer” lattice.
In addition, as the temperature decreases from 300 to 80 K, the PL intensity of DPCu4Cl6 and DPCu4Br6 increases monotonically, which is attributed to the suppression of nonradiative-decay.48,49 The exciton activation energy (Ea) can be fit using the Arrhenius equation:42
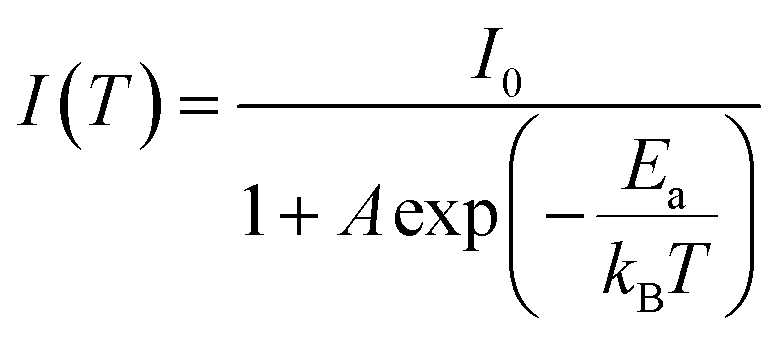 | (1) |
 | (2) |
Although DPCu4I6 has a similar structure to DPCu4Cl6 and DPCu4Br6, the great differences in spectra indicate that their PL mechanisms are different, which prompts us to conduct more detailed photophysical characterization for DPCu4I6 SCs. As shown is Fig. 3a, the emission bands become asymmetric and red-shifted as the excitation wavelength increases from 275 nm to 425 nm. In addition, the PLE spectra also show different shapes when the detection wavelength is from 500 nm to 750 nm (Fig. 3b). The different characteristics of excitation/emission wavelength-dependent PL/PLE spectra at RT imply that the yellow luminescence of DPCu4I6 originates from multiple excited states. However, the changes of emission/excitation-dependent PLE/PL spectra are small and the lifetimes monitored at different emission wavelengths are similar, indicating that the height of the energy barriers between the excited states is low, and thus, excitons can easily cross these barriers, leading to uniform emission due to thermal equilibrium. In other words, the multiple excited states in DPCu4I6 are easily coupled at RT to result in the bright yellow emission, and a similar behavior is also observed in (1,3-dppH2)2Cu4I8·H2O57 and Na4(18-crown-6)5In2Cu4Br14·8H2O.58
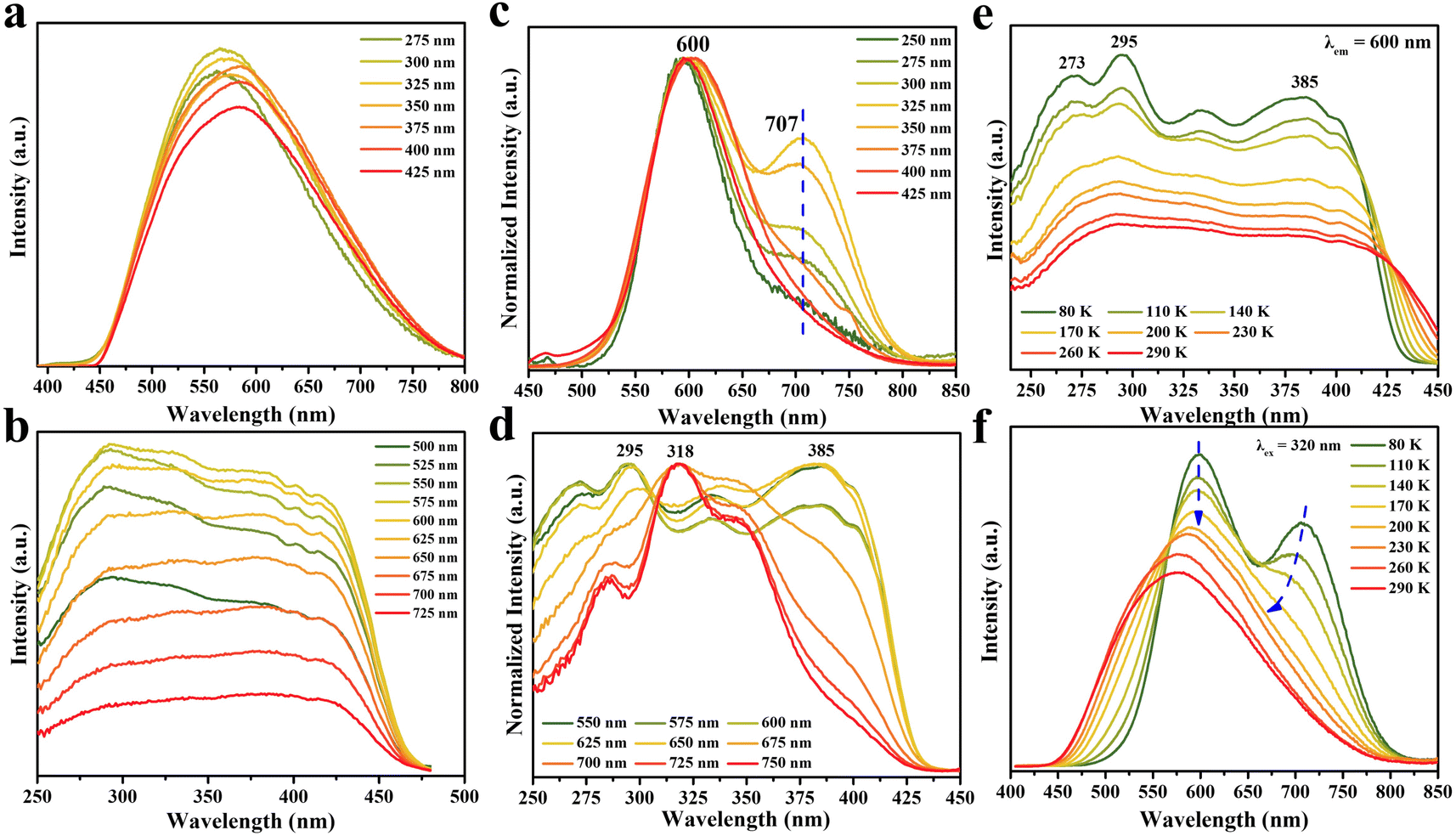 | ||
| Fig. 3 Excitation and emission-wavelength dependent (a) PL and (b) PLE spectra of DPCu4I6 SCs at RT. Normalized excitation and emission-wavelength dependent (c) PL and (d) PLE spectra of DPCu4I6 SCs at 80 K. Temperature-dependent (e) PLE and (f) PL spectra of DPCu4I6 SCs. | ||
Moreover, the excitation wavelength-dependent PL spectra at 80 K clearly reveal the attractive dual-band emission of DPCu4I6 (Fig. 3c). Under 250 nm excitation, the PL spectrum shows a main peak on the high energy side (HE peak, ∼600 nm) and a shoulder peak on the low energy side (LE peak, ∼707 nm). We note that with the decrease of excitation energy (i.e., increasing excitation wavelength), the relative intensity ratio of the two emission peaks (i.e., LE peak intensity/HE peak intensity) gradually increases and reaches a maximum at 325 nm excitation. The ratio decreases with increasing the excitation wavelength over 325 nm and the low energy shoulder almost disappears at excitation wavelengths >375 nm. Moreover, the emission wavelength-dependent PLE spectra at 80 K further elucidate this dual-band emission property (Fig. 3d). By monitoring at 550–650 nm (HE emission band) and 675–750 nm (LE emission band), the resultant PLE spectra are distinctly different, although they exhibit similar PLE spectral ranges. Thus, these spectral features obtained at 80 K, on the one hand, illustrate that there are two emission centers in the DPCu4I6 SCs, and on the other hand, the same PLE spectral range indicates that the excited states corresponding to the HE and LE emissions are interrelated. The TRPL of DPCu4I6 was studied at 80 K, and the decay lifetimes monitored at 600 nm or 700 nm increase with the temperature cooling, which is due to the suppression of nonradiative recombination (Fig. S12†).59 Notably, the decay lifetimes of DPCu4I6 monitored at the two wavelengths (600 nm and 700 nm) show distinct differences at 80 K, whereas the lifetimes at RT are almost identical, demonstrating that low temperature can unveil the dual emission center and implies that temperature can change the emission states of DPCu4I6.
To explore the transition of the electronic structure from low temperature to RT more intuitively and uncover the photophysical mechanism of the double emission, temperature-dependent PLE/PL spectra of DPCu4I6 were measured in the range of 80–290 K. As shown in Fig. 3e and S13,† the PLE spectrum of DPCu4I6 shows multiple peaks whether monitored at 600 or 700 nm at low temperature, however with the increase of test temperature, the PLE profiles gradually become flat, which is attributed to the coupling of excited states in DPCu4I6.60 The temperature-dependent PL spectrum of DPCu4I6 excited at 320 nm is shown in Fig. 3f. As noted above, DPCuI6 shows a distinct dual-emission band at 80 K, and as the temperature increases, the position of the HE peak remains virtually unchanged, whereas the LE peak gradually blueshifts. When the temperature is greater than 170 K, the dual-emission profile of DPCu4I6 turns into a broadband single peak, which is consistent with the change of temperature-dependent PLE spectra (i.e., the shapes of the PLE spectra monitored at 600 nm and 700 nm are different at low temperatures and tend to be consistent at high temperatures) and confirms that the dual-excited state gradually transitions from nonequilibrium at low temperature to near equilibrium at RT. Remarkably, the CIE chromaticity coordinate of DPCu4I6 shifts from yellow (0.43, 0.49) to orange (0.55, 0.44) when the temperature is decreased from RT to 80 K (Fig. S14†). Furthermore, as illustrated in Fig. S15,† under 400 nm excitation, the PL spectra of DPCu4I6 consistently show only one emission peak over the whole measured temperature range (80–290 K). The single peak is Gaussian symmetric and matches well the HE peak of the dual-band emission (upon 320 nm excitation), indicating that a low excitation energy can only promote electrons to the level corresponding to the HE band at low temperatures. At temperatures above 170 K, the characteristics of the PL spectra recorded at 400 and 320 nm excitation are identical, indicating that thermal energy can change the excited state (i.e., lowering the energy barrier), and thus the electrons, although excited by a low energy, can still reach the excitation energy level corresponding to the LE band at higher temperatures (Fig. S16†).
To gain further insights into the electron transfer mechanism of DPCu4X6, the band structures of the halides were calculated based on the density functional theory (DFT). Because the crystal structures and optical properties of the chloride and bromide are similar, we selected DPCu4Cl6 as a representative for comparison with DPCu4I6. As shown in Fig. 4a and b, the calculations show that the DPCu4Cl6 and DPCu4I6 SCs have direct band characteristics, and the bandgaps are 1.41 eV for DPCu4Cl6 and 1.55 eV for DPCu4I6. The projected density of states (PDOS) of the above two halides, shown in Fig. 4c and d, indicates that the valence band maxima (VBM) of the two materials are mainly derived from Cu-3d and X-p orbitals and their conduction band minima (CBM) are entirely derived from the orbitals of the organic cation (C6H10N2)2+. Furthermore, the differential charge densities corresponding to the VBM and CBM states of DPCu4X6 (X = Cl, I) also confirm that electrons are located in the organic part (C6H10N2)2+ and holes are sited in the [Cu4X6]2− (X = Cl, I) clusters (Fig. S17†). Moreover, it can be noted from the bands that the VBM of the above two crystals are almost flat, whereas the CBM is more dispersed. This difference can be understood based on the following reasons: (1) the neighboring [Cu4X6]2− (X = Cl, I) anions are completely isolated in space, and their interactions are very weak; (2) the distances between the organic cation (C6H10N2)2+ are relatively short, which can enable orbital coupling, resulting in electron delocalization.25,61 In addition, the ultra-long PL lifetime (>40 μs) of DPCu4Cl6 may be related to the carrier distribution, i.e., the photo-excited electrons and holes are spatially separated, thus prolonging the exciton lifetime. However, the RT lifetime of DPCu4I6 is only ∼4 μs, which is much shorter than that of DPCu4Cl6, although they have similar band structures. The difference in emission lifetime suggests that the radiative mechanism of the iodide is unlike that of DPCu4Cl6 (from STEs).
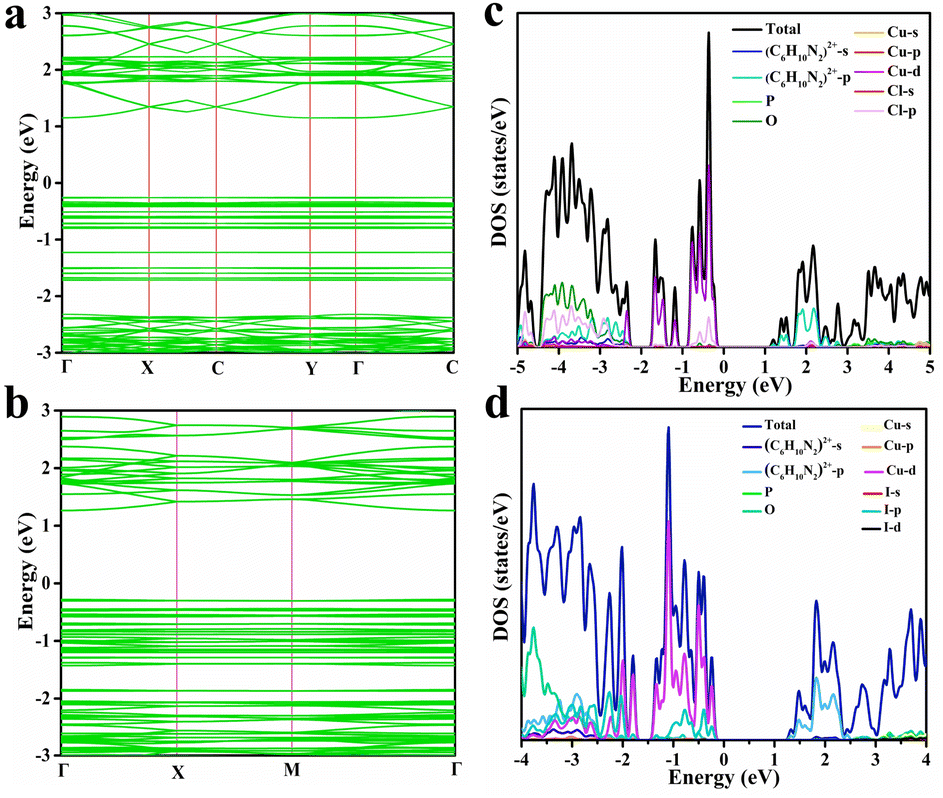 | ||
| Fig. 4 Band structures of (a) DPCu4Cl6 SCs and (b) DPCu4I6 SCs. Total and orbital-projected DOSs for (c) DPCu4Cl6 SCs and (d) DPCu4I6 SCs. | ||
As mentioned above, the PL color of DPCu4I6 changes with temperature. In fact, a similar phenomenon of thermochromic luminescence exists in tetranuclear cuprous iodide clusters Cu4I4L4 and their derivatives, which is usually attributed to dual-channel electron transfer, including 3CC emission and X/MLCT.38,60,62–65 The 3CC emission is a combination of halide-to-metal charge transfer (3XMCT) and copper-centered d → s, p transitions and is essentially independent of the ligand nature. According to the previous reports, 3CC emission is usually observed as LE emission ranging from 600 to 800 nm and with lifetimes less than 20 μs.38,60 In most cases, the Cu–Cu bonds of Cu(I)-based luminophores in the excited state have a bonding character. During the cooling process, their distances become shorter, thus stabilizing the 3CC excited state and shifting the emission to the red region.63 The HE band is usually associated with X/MLCT, which is considered to involve transfer of electrons from π* orbitals in the organic component to inorganic metal polyhedrons. A prominent feature of this band is the stable peak position over a wide range of temperatures.38,58Fig. 3f shows the variation of the double emission band of DPCu4I6 with temperature, and the spectral features allow us to attribute the observed emission at 707 nm to the 3CC excited state and the one at 600 nm to the X/MLCT state. The shorter Cu–Cu distance of DPCu4I6 compared with that of DPCu4X6 (X = Cl or Br) suggests that 3CC emission is more likely to be realized in DPCu4I6. Besides, the PL intensity of DPCu4I6 is directly proportional to the power density, indicating the negligible contribution of permanent defects to emission (Fig. S18†). Fig. S19† illustrates the normalized PL spectra at different temperatures of the three single crystals, and it can be found that there is a large thermal broadening of the emission for chloride and bromide, while the FWHM of iodide remains unchanged essentially, implying that the emission of DPCu4I6 is not triggered by the intrinsic STE transition.42 In fact, it is possible for the three halides to form similar self-trapped states due to their similar structures, however, compared with chlorides and bromides, iodides usually have lower self-trapping energy, which may be attributed to their different lattice deformation potentials induced by different M–X bonding.66 The lower self-trapping energy of DPCu4I6 implies that the STEs can be thermally de-trapped even at low temperatures, and thus the bright emission of DPCu4I6 is dominated by the 3CC and X/MLCT synergistic excited states. Because of the large de-trapping barrier of the chloride and bromide, STEs are more readily formed in these two halides, so they exhibit similar STE emission properties including ultra-long decay lifetimes and broadband single-peaked emission.
On the basis of the results of spectral characterization and theoretical calculations, the energy level diagrams and electron transfer paths are proposed to describe the photophysical processes of DPCu4X6 SCs. As illustrated in Fig. 5a and b, upon photoexcitation, the electrons of [Cu4X6]2− (X = Cl or Br) units transition from the ground state to the excited state and then become free excitons (FEs). The FEs are subsequently captured by the self-trapped states resulting from lattice distortion caused by strong electron–phonon coupling. Then, the electrons located at self-trapped states return to the ground state and recombine with the holes, thereby emitting bright orange/yellow light. Owing to the “softer” lattice of DPCu4Cl6, it is prone to thermal expansion, which leads to the downward shift of its valence band and thus the blue-shifted emission with the increase of temperature. Because of the Cu–Cu distance of the two halides close to the sum of the van der Waals radii of copper(I) (2.8 Å) and the ultra-wide PLE profiles, the 3CC and X/MLCT states probably exist in DPCu4Cl6 and DPCu4Br6. However, due to the formation of the self-trapping states (below the conduction band), excited electrons are easily trapped, and therefore, the 3CC and X/MLCT states have negligible contribution to the luminescence of DPCu4Cl6 and DPCu4Br6. For DPCu4I6, the lower self-trapping energy makes STEs difficult to form, and the shorter Cu–Cu distance makes 3CC and X/MLCT states more significant, so a synergistic effect by the 3CC and X/MLCT states is proposed to understand the dual-band emission of the iodide (Fig. 5c). When the iodide is excited by high-energy UV light with wavelengths less than 320 nm, electrons in [Cu4I6]2− are pumped to the X/MLCT state, producing an HE emission band, and then a few electrons can relax into the relatively low excited state (3CC state), resulting in the LE emission band. When the excitation wavelength is larger than 320 nm, the excited electrons must undergo an internal conversion process to reach the 3CC state from the X/MLCT state, which decreases the population of the 3CC state and weakens the LE emission, such that even LE emission disappears upon 400 nm excitation. In addition, temperature can change the excited states of DPCu4I6, and at high temperatures the two excited states of the iodide will couple leading to a lowering of the energy barrier, finally resulting in near-unify broadband yellow-light emission. In addition, long times are needed for the chloride and bromide to undergo the processes of self-trapping and de-trapping, while the iodide has almost no such processes, so it is understandable that the emission lifetime of DPCu4I6 is much shorter than that of DPCu4X6 (X = Cl or Br).
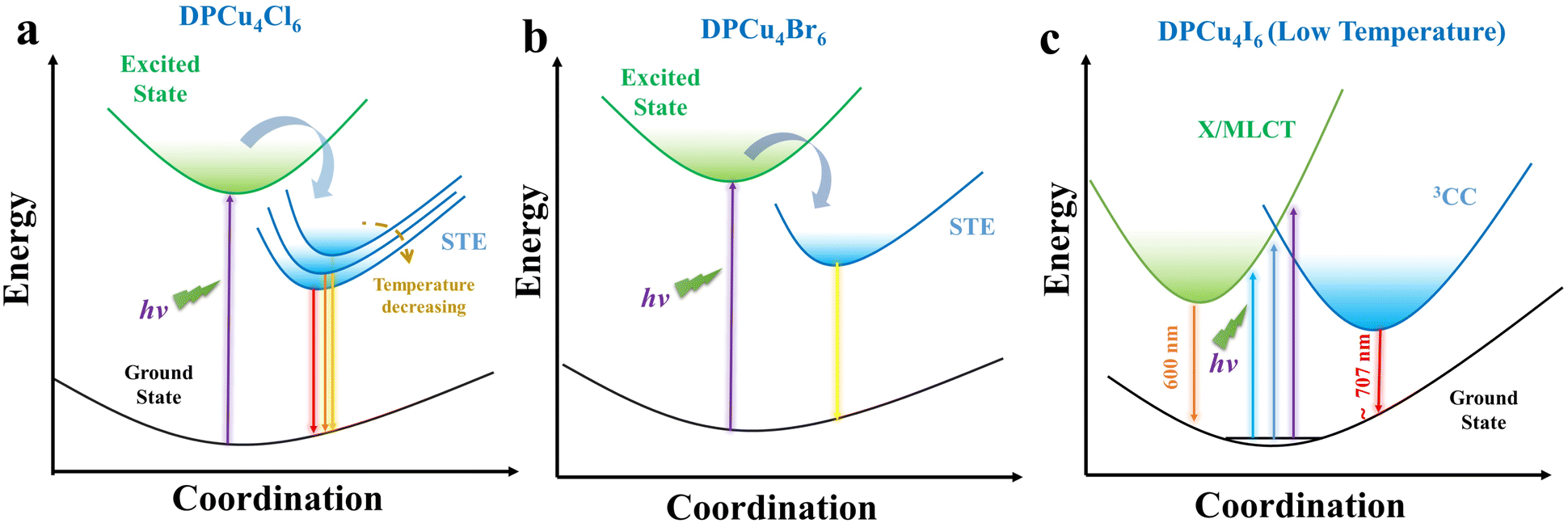 | ||
| Fig. 5 Schematic of the photophysical processes in (a) DPCu4Cl6, (b) DPCu4Br6, and (c) DPCu4I6 SCs. | ||
Normally, stability is an important index for SSL applications but is generally poor for cuprous-based metal halides. We first conducted thermal stability tests on the three as-synthetic crystals. The results show that the decomposition temperatures of all three halides are above 175 °C, indicating their good thermal stability (Fig. S20a†). When exposing the three crystals to air with 75% humidity, DPCu4Cl6 SCs can easily decompose and deteriorate, while DPCu4Br6 and DPCu4I6 SCs maintain their structural stability and more than 80% emission efficiency within two months (Fig. S20b and S20c†), suggesting that they have excellent air stability.
Benefiting from the high efficiency broadband yellow emission under the blue-light excitation, and the excellent thermal/environmental stability of DPCu4I6 SCs, a high-performance single-component WLED device was fabricated using a commercial blue light chip (445 nm) as the excitation source, as shown in Fig. 6a. Notably, high-performance warm-white-light without adding additional commercial phosphors was achieved, with a CIE color coordinate of (0.36, 0.35), a correlated color temperature (CCT) of 4415 K, a luminous efficiency of 18.41 lm W−1 and a color rendering index (CRI) of 85.1 (Fig. 6b), outperforming many single-component free-lead halide-based WLEDs (Table S4†). In addition, the fabricated WLED has a similar electroluminescence spectral profile at different driving currents (20–320 mA), indicating its good electroluminescence stability (Fig. 6c). More importantly, after the WLED operates at 20 mA for 9 h, the emission intensity of the device can be maintained at 85%, revealing excellent operational stability (Fig. 6d). Hence, the above results illustrate that the as-synthesized halide DPCu4I6 is a potential candidate for a single-component white light emitter.
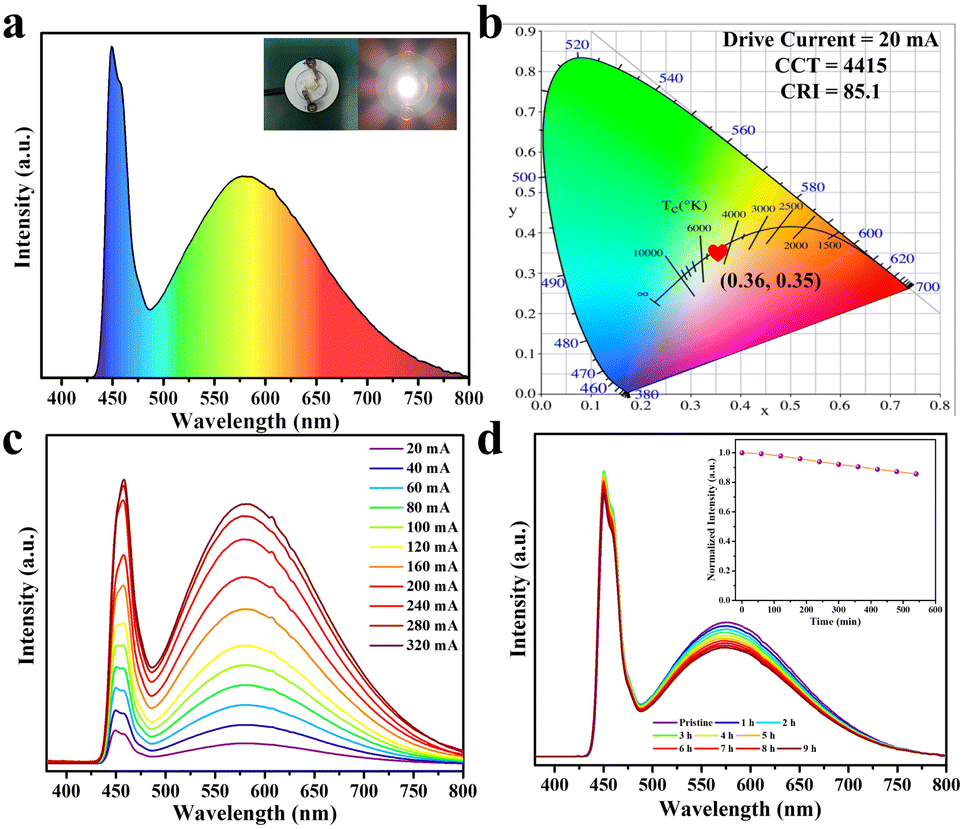 | ||
| Fig. 6 (a) Luminescence spectrum of the WLED device fabricated with DPCu4I6 powder excited with a 450 nm blue light chip. The insets show photographs of a WLED with power off (left) and power on (right). (b) CIE coordinate of the WLED. (c) Drive current-dependent electroluminescence spectra. (d) Emission stability of the WLED at 20 mA driving current for 9 h. The inset shows the integrated luminous intensity of the WLED device working over different periods. | ||
Conclusions
In summary, three new cuprous metal halides, DPCu4X6 (X = Cl, Br, I), were successfully synthesized by a simple solution method. In the DPCu4Cl6 and DPCu4Br6 SCs, the unique crystal structures and strong electron–phonon coupling promote the formation of self-trapped excited states, leading to near-unity bright yellow/orange emission. Owing to the structural changes caused by halogen substitution (shorter Cu–Cu distance and lower self-trapping energy), the DPCu4I6 SCs exhibit dual-band emission at low temperatures, which is attributed to the synergistic effects of the 3CC and X/MLCT excited states. Moreover, a WLED with a high CRI of 85.1 was fabricated by combining the DPCu4I6 powders and a blue light chip (445 nm), displaying the great potential of the obtained new materials for application in SSL. This work provides new insights into the rational design of emissive metal halides with broad excitation/emission bands, which is crucial to the development of high-performance lighting applications.Data availability
All data are available in the manuscript and in the ESI.†Author contributions
S. G. Zhou designed and carried out the experiments. S. G. Zhou, Y. H. Chen, X. W. Liu, and K. L. Li collected and analyzed characterization data. S. G. Zhou and T. Zhang wrote the manuscript. L. Zhou and R. X. He supervised the work and gave guidance. All authors participated in a discussion of the data. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (22109130), the Fundamental Research Funds for the Central Universities (SWU-KT22031), the Natural Science Foundation of Chongqing (cstc2020jcyj-msxmX0332 and cstc2021jcyj-msxm2853), the Innovation Research 2035 Pilot Plan of Southwest University (SWU-XDZD22011) and the Program for Innovation Team Building at Institutions of Higher Education in Chongqing (CXTDX201601011).Notes and references
- T. T. Xuan and R. J. Xie, Chem. Eng. J., 2020, 393, 124757 CrossRef CAS.
- M. Z. Li and Z. G. Xia, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- L. Zhou, J. F. Liao and D. B. Kuang, Adv. Opt. Mater., 2021, 9, 2100544 CrossRef CAS.
- T. Bai, X. Wang, Z. Wang, S. Ji, X. Meng, Q. Wang, R. Zhang, P. Han, K. L. Han, J. Chen, F. Liu and B. Yang, Angew. Chem., Int. Ed., 2023, 62, e202213240 CAS.
- C. Zhu, J. B. Jin, M. Y. Gao, M. C. Folgueras, Y. Zhang, C. K. Lin, P. D. Yang and A. M. Oddo, J. Am. Chem. Soc., 2022, 144, 12450–12458 CrossRef CAS PubMed.
- B. B. Su, G. M. Song, M. S. Molokeev, N. N. Golovnev, M. K. Lesnikov, Z. S. Lin and Z. G. Xia, J. Phys. Chem. Lett., 2021, 12, 1918–1925 CrossRef CAS PubMed.
- Y. Han, S. J. Yue and B. B. Cui, Adv. Sci., 2021, 8, 2004805 CrossRef CAS PubMed.
- V. Morad, Y. Shynkarenko, S. Yakunin, A. Brumberg, R. D. Schaller and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 9764–9768 CrossRef CAS PubMed.
- L. Mao, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS PubMed.
- B. Luo, Y. Guo, Y. Xiao, X. Lian, T. Tan, D. Liang, X. Li and X. Huang, J. Phys. Chem. Lett., 2019, 10, 5271–5276 CrossRef CAS PubMed.
- S. Y. Zhao, W. S. Cai, H. X. Wang, Z. G. Zang and J. Z. Chen, Small Methods, 2021, 5, 2001308 CrossRef CAS PubMed.
- Q. Wang, M. Q. Lyu, M. Zhang, J. H. Yun and L. Z. Wang, J. Mater. Chem. A, 2017, 5, 902–909 RSC.
- Z. J. Shi, J. Guo, Y. H. Chen, Q. Li, Y. F. Pan, H. J. Zhang, Y. D. Xia and W. Huang, Adv. Mater., 2017, 29, 1605005 CrossRef PubMed.
- E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718–1721 CrossRef CAS PubMed.
- Z. Luo, Y. Liu, Y. Liu, C. Li, Y. Li, Q. Li, Y. Wei, L. Zhang, B. Xu, X. Chang and Z. Quan, Adv. Mater., 2022, 34, 2200607 CrossRef CAS PubMed.
- X. Huang, X. T. Li, Y. Tao, S. H. Guo, J. Z. Gu, H. L. Hong, Y. G. Yao, Y. Guan, Y. A. Gao, C. Li, X. J. Lu and Y. P. Fu, J. Am. Chem. Soc., 2022, 144, 12247–12260 CrossRef CAS PubMed.
- C. Q. Jing, Q. L. Liu, C. H. Zhao, Y. Y. Zhao, C. Y. Yue and X. W. Lei, J. Mater. Chem. C, 2021, 9, 15047–15055 RSC.
- C. K. Zhou, H. R. Lin, M. Worku, J. Neu, Y. Zhou, Y. Tian, S. Lee, P. Djurovich, T. Siegrist and B. Ma, J. Am. Chem. Soc., 2018, 140, 13181–13184 CrossRef CAS PubMed.
- Y. L. Jiang and H. H. Fei, Adv. Opt. Mater., 2022, 10, 2102148 CrossRef CAS.
- Z. W. Xiao, Z. N. Song and Y. F. Yan, Adv. Mater., 2019, 31, 1803792 CrossRef CAS PubMed.
- Y. Y. Li, Z. C. Zhou, N. Tewari, M. Ng, P. Geng, D. Z. Chen, P. K. Ko, M. Qammar, L. Guo and J. E. Halpert, Mater. Chem. Front., 2021, 5, 4796–4820 RSC.
- H. B. Li, Y. Lv, Z. N. Zhou, H. Tong, W. Liu and G. F. Ouyang, Angew. Chem., Int. Ed., 2022, 61, e202115225 CAS.
- S. Li, J. Xu, Z. G. Li, Z. C. Zeng, W. Li, M. H. Cui, C. C. Qin and Y. P. Du, Chem. Mater., 2020, 32, 6525–6531 CrossRef CAS.
- H. Peng, X. X. Wang, Y. Tian, B. S. Zou, F. Yang, T. Huang, C. Y. Peng, S. F. Yao, Z. M. Yu, Q. R. Yao, G. H. Rao and J. P. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 13443–13451 CrossRef CAS PubMed.
- X. Y. Liu, F. Yuan, C. R. Zhu, J. R. Li, X. Q. Lv, G. C. Xing, Q. Wei, G. Wang, J. F. Dai, H. Dong, J. Xu, B. Jiao and Z. X. Wu, Nano Energy, 2022, 91, 106664 CrossRef CAS.
- H. Peng, S. F. Yao, Y. C. Guo, R. N. Zhi, X. X. Wang, F. J. Ge, Y. Tian, J. P. Wang and B. S. Zou, J. Phys. Chem. Lett., 2020, 11, 4703–4710 CrossRef CAS PubMed.
- J. H. Wei, Y. W. Yu, J. B. Luo, Z. Z. Zhang and D. B. Kuang, Adv. Opt. Mater., 2022, 10, 2200724 CrossRef CAS.
- B. B. Su, S. N. Geng, Z. W. Xiao and Z. G. Xia, Angew. Chem., Int. Ed., 2022, 61, e202208881 CAS.
- D. Y. Li, Y. Cheng, Y. H. Hou, J. H. Song, C. J. Sun, C. Y. Yue, Z. H. Jing and X. W. Lei, J. Mater. Chem. C, 2022, 10, 3746–3755 RSC.
- J. L. Li, Y. F. Sang, L. J. Xu, H. Y. Lu, J. Y. Wang and Z. N. Chen, Angew. Chem., Int. Ed., 2022, 61, e202113450 CrossRef CAS PubMed.
- D. Y. Li, Y. M. Sun, Z. Y. Xu, P. C. Xiao, J. H. Song, X. W. Lei, G. Chen and C. Y. Yue, Chem. Commun., 2022, 58, 9084–9087 RSC.
- J. W. Lin, K. J. Liu, H. Ruan, N. Sun, X. Chen, J. Zhao, Z. N. Guo, Q. L. Liu and W. X. Yuan, J. Phys. Chem. Lett., 2022, 13, 198–207 CrossRef CAS PubMed.
- L. Zhou, L. Zhang, H. Li, W. Shen, M. Li and R. X. He, Adv. Funct. Mater., 2021, 31, 2108561 CrossRef CAS.
- L. Zhou, M. X. Ren, R. X. He and M. Li, Inorg. Chem., 2022, 61, 5283–5291 CrossRef CAS PubMed.
- S. Zhou, L. Zhou, Y. Chen, W. Shen, M. Li and R. He, J. Phys. Chem. Lett., 2022, 13, 8717–8724 CrossRef CAS PubMed.
- B. M. Benin, D. N. Dirin, V. Morad, M. Worle, S. Yakunin, G. Raino, O. Nazarenko, M. Fischer, I. Infante and M. V. Kovalenko, Angew. Chem., Int. Ed., 2018, 57, 11329–11333 CrossRef CAS PubMed.
- Y. C. Peng, S. H. Zhou, J. C. Jin, Q. Gu, T. H. Zhuang, L. K. Gong, Z. P. Wang, K. Z. Du and X. Y. Huang, Dalton Trans., 2022, 51, 4919–4926 RSC.
- E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS.
- H. Peng, Y. H. Xiao, Y. Tian, X. X. Wang, T. Huang, T. T. Dong, Y. T. Zhao, J. P. Wang and B. S. Zou, J. Mater. Chem. C, 2021, 9, 16014–16021 RSC.
- X. Y. Liu, Y. Y. Li, T. Y. Liang, W. J. Liu and J. Y. Fan, J. Phys. Chem. Lett., 2022, 13, 1373–1381 CrossRef CAS PubMed.
- M. S. Chen, C. Y. Ye, C. Q. Dai, R. J. Qi, H. M. Fu, C. H. Luo, H. Peng and H. C. Lin, Adv. Opt. Mater., 2022, 10, 2200278 CrossRef CAS.
- L. Zhou, J. F. Liao, Z. G. Huang, J. H. Wei, X. D. Wang, H. Y. Chen and D. B. Kuang, Angew. Chem., Int. Ed., 2019, 58, 15435–15440 CrossRef CAS PubMed.
- B. Chen, Y. Guo, Y. Wang, Z. Liu, Q. Wei, S. X. Wang, A. L. Rogach, G. C. Xing, P. Shi and F. Wang, J. Am. Chem. Soc., 2021, 143, 17599–17606 CrossRef CAS PubMed.
- J. C. Jin, Y. H. Peng, Y. T. Xu, K. Han, A. R. Zhang, X. B. Yang and Z. G. Xia, Chem. Mater., 2022, 34, 5717–5725 CrossRef CAS.
- Z. Y. Li, C. Y. Zhang, B. Li, C. Lin, Y. Li, L. Wang and R. J. Xie, Chem. Eng. J., 2021, 420, 129740 CrossRef CAS.
- L. Y. Lian, M. Y. Zheng, P. Zhang, Z. Zheng, K. Du, W. Lei, J. B. Gao, G. D. Niu, D. L. Zhang, T. Y. Zhai, S. Y. Jin, J. Tang, X. W. Zhang and J. B. Zhang, Chem. Mater., 2020, 32, 3462–3468 CrossRef CAS.
- R. S. Zeng, L. L. Zhang, Y. Xue, B. Ke, Z. Zhao, D. Huang, Q. L. Wei, W. C. Zhou and B. S. Zou, J. Phys. Chem. Lett., 2020, 11, 2053–2061 CrossRef CAS PubMed.
- B. B. Su, M. Z. Li, E. H. Song and Z. G. Xia, Adv. Funct. Mater., 2021, 31, 2105316 CrossRef CAS.
- D. Y. Li, J. H. Song, Y. Cheng, X. M. Wu, Y. Y. Wang, C. J. Sun, C. Y. Yue and X. W. Lei, Angew. Chem., Int. Ed., 2022, 61, e202206437 CAS.
- J. H. Wei, J. F. Liao, L. Zhou, J. B. Luo, X. D. Wang and D. B. Kuang, Sci. Adv., 2021, 7, eabg3989 CrossRef CAS PubMed.
- Y. Y. Jing, Y. Liu, X. X. Jiang, M. S. Molokeev, Z. S. Lin and Z. G. Xia, Chem. Mater., 2020, 32, 5327–5334 CrossRef CAS.
- Q. D. Ran, Y. Zhang, J. Yang, R. X. He, L. Zhou and S. S. Hu, J. Mater. Chem. C, 2022, 10, 1999–2007 RSC.
- G. Zhang, P. Dang, H. Xiao, H. Lian, S. Liang, L. Yang, Z. Cheng, G. Li and J. Lin, Adv. Opt. Mater., 2021, 9, 2101637 CrossRef CAS.
- X. Meng, S. Ji, Q. Wang, X. Wang, T. Bai, R. Zhang, B. Yang, Y. Li, Z. Shao, J. Jiang, K. Han and F. Liu, Adv. Sci., 2022, 9, 2203596 CrossRef CAS PubMed.
- X. Z. Lao, Z. Yang, Z. C. Su, Z. L. Wang, H. G. Ye, M. Q. Wang, X. Yao and S. J. Xu, Nanoscale, 2018, 10, 9949–9956 RSC.
- V. Turck, S. Rodt, O. Stier, R. Heitz, R. Engelhardt, U. W. Pohl, D. Bimberg and R. Steingruber, Phys. Rev. B, 2000, 61, 9944–9947 CrossRef CAS.
- B. B. Su, J. Jin, Y. H. Peng, M. S. Molokeev, X. B. Yang and Z. G. Xia, Adv. Opt. Mater., 2022, 10, 2102619 CrossRef CAS.
- X. Liu, Y. Li, L. Zhou, M. Li, Y. Zhou and R. He, Adv. Opt. Mater., 2022, 10, 2200944 CrossRef CAS.
- L. Zhou, J. F. Liao, Y. C. Qin, X. D. Wang, J. H. Wei, M. Li, D. B. Kuang and R. X. He, Adv. Funct. Mater., 2021, 31, 2102654 CrossRef CAS.
- J. L. Huang, B. B. Su, E. H. Song, M. S. Molokeev and Z. G. Xia, Chem. Mater., 2021, 33, 4382–4389 CrossRef CAS.
- L. Y. Lian, X. Wang, P. Zhang, J. S. Zhu, X. W. Zhang, J. B. Gao, S. Wang, G. J. Liang, D. L. Zhang, L. Gao, H. S. Song, R. Chen, X. Z. Lan, W. X. Liang, G. D. Niu, J. Tang and J. B. Zhang, J. Phys. Chem. Lett., 2021, 12, 6919–6926 CrossRef CAS PubMed.
- S. Perruchas, C. Tard, X. F. Le Goff, A. Fargues, A. Garcia, S. Kahlal, J. Y. Saillard, T. Gacoin and J. P. Boilot, Inorg. Chem., 2011, 50, 10682–10692 CrossRef CAS PubMed.
- Q. Benito, X. F. Le Goff, G. Nocton, A. Fargues, A. Garcia, A. Berhault, S. Kahlal, J. Y. Saillard, C. Martineau, J. Trebosc, T. Gacoin, J. P. Boilot and S. Perruchas, Inorg. Chem., 2015, 54, 4483–4494 CrossRef CAS PubMed.
- Y. Thefioux, M. Cordier, F. Massuyeau, C. Latouche, C. Martineau-Corcos and S. Perruchas, Inorg. Chem., 2020, 59, 5768–5780 CrossRef CAS PubMed.
- B. Li, H. T. Fan, S. Q. Zang, H. Y. Li and L. Y. Wang, Coord. Chem. Rev., 2018, 377, 307–329 CrossRef CAS.
- R. Gautier, M. Paris and F. Massuyeau, J. Am. Chem. Soc., 2019, 141, 12619–12623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental materials and methods for sample preparation; computational methods; detailed crystallographic results; supplementary figures including digital photos of the samples, CIE, PLE, PL, PLQY, TRPL, XPS. CCDC 2236271, 2236273 and 2253788. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01762a |
| This journal is © The Royal Society of Chemistry 2023 |