DOI:
10.1039/D3SC00035D
(Edge Article)
Chem. Sci., 2023,
14, 5425-5430
Regulating supramolecular interactions in dimeric macrocycles†
Received
4th January 2023
, Accepted 25th April 2023
First published on 26th April 2023
Introduction
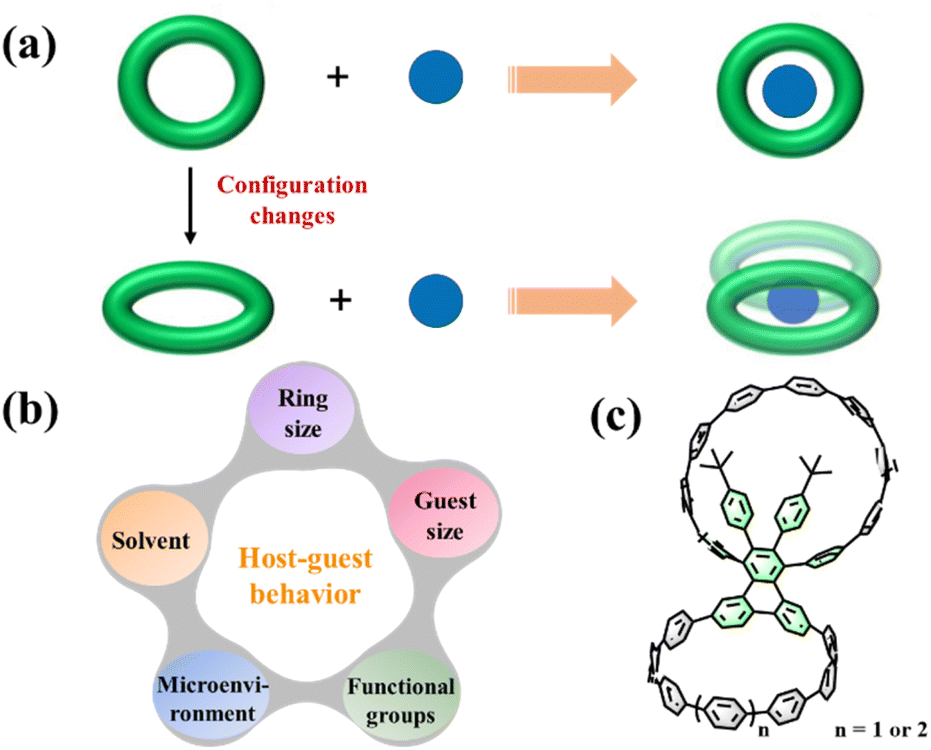
Fullerene is one of the most extensively studied carbon nanomaterials in recent decades due to its unique chemical and electrical properties.1–3 In particular, the encapsulation of fullerenes by some specific host molecules via π–π interaction has attracted much attention because of their potential applications in the separation, solubilization, and chemical modification of fullerenes.4–6 Generally, an ideal host molecule should satisfy both the structural and electronic complementarity with spherical fullerenes.7 Thus electron-rich macrocyclic structures, such as azacrown ethers,8 calixarenes,9 tetrathiafulvalenes-based macrocycles,10 pillararenes,11 and porphyrinylene nanohoops,12 have long been considered as promising hosts in fullerene-containing host–guest systems. Their shape complementarity greatly improves the recognition abilities and selectivities toward fullerenes.10,13 Over the past decade, carbon-rich cycloparaphenylene macrocycles and their derivatives were reported as interesting host molecules in the study of π–π interactions with C60 or other fullerenes.14–18 These macrocycles usually exhibit good π–π interactions with fullerenes or their derivatives in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. It would be intriguing if one could manipulate the supramolecular characteristics of these macrocycles to construct novel supramolecular donor–acceptor–donor (D–A–D) systems,19 for example, two macrocycles cooperate to bind a fullerene molecule rather than the typical one-to-one complexing (Fig. 1a).
:1 ratio. It would be intriguing if one could manipulate the supramolecular characteristics of these macrocycles to construct novel supramolecular donor–acceptor–donor (D–A–D) systems,19 for example, two macrocycles cooperate to bind a fullerene molecule rather than the typical one-to-one complexing (Fig. 1a).
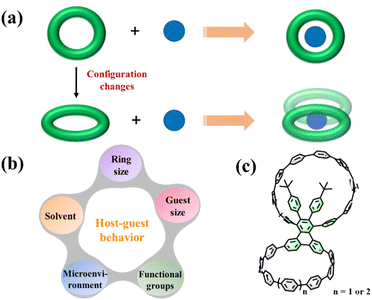 |
| | Fig. 1 (a) Tuning supramolecular characteristics of conjugated macrocycles to construct an unconventional host–guest system. (b) Several factors affecting host–guest behaviors. (c) Molecular structures of bismacrocycles 1a and 1b. | |
The hosting of fullerene molecules in the cavity is mainly influenced by the ring size, geometric configuration, microenvironment, functional groups, fullerene size, and even solvent (Fig. 1b). To date, previous studies showed that only modifying a single macrocycle cannot fully achieve the abovementioned goals.20–22 Recently, the development of cycloparaphenylene bismacrocycles enriches the family of fullerene-containing supramolecular systems with a high stoichiometry ratio and tunable cavity shapes.23–26 For example, Cong and coworkers employed a cyclooctatetrathiophene unit as the linker to build a figure-of-eight bismacrocycle whose adaptive cavities enable the formation of peanut-like 1:2 host–guest complexes with C60 or C70.25 Notably, these bismacrocycles often have two macrocycles with the same sizes, shapes, and supramolecular properties because there are no other aryl groups at the linker that can affect the microenvironment of the bismacrocycle. Based on this, we envision that the geometric configurations could be finely tuned when these macrocycles are formed with different microenvironments, resulting in different and interesting supramolecular properties.
Herein, we report the design and synthesis of two novel dimeric macrocycles 1a and 1b, in which two cycloparaphenylene macrocycles with tunable sizes are anchored onto a triphenylene derivative (Fig. 1c). Interestingly, 1a exhibits a very weak host ability and can only capture one C60 or C70 molecule to form 2:1 host–guest complexes by cooperating with another bismacrocycle. By changing the ring size, another bismacrocycle 1b can interact with three C60 molecules to construct a 2:3 host–guest complex, demonstrating the first 2:3 host–guest complex in the supramolecular chemistry of bismacrocycles.
Results and discussion
Molecular design and synthesis
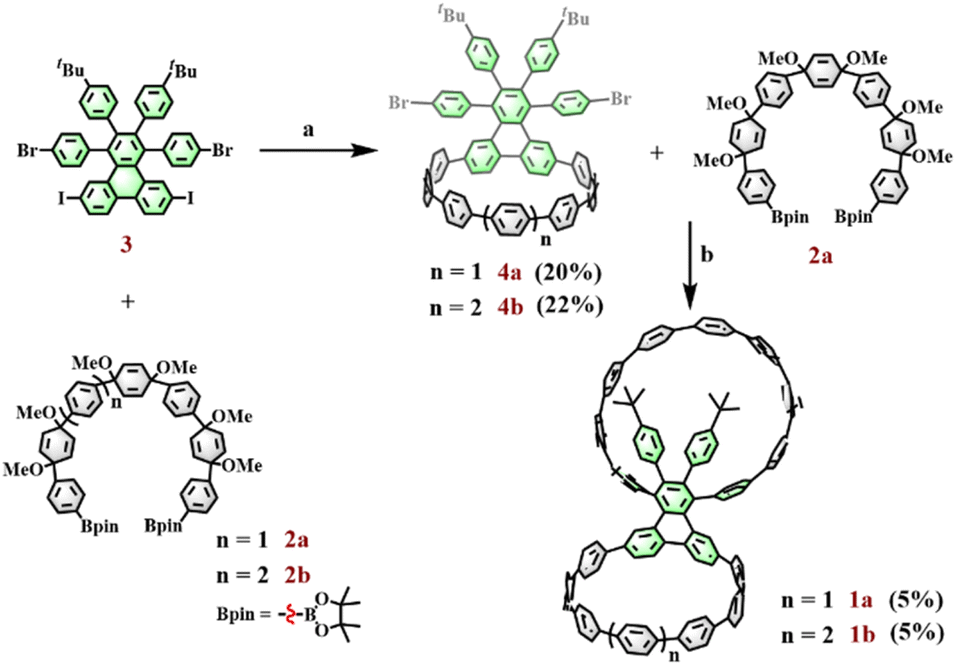
To compare the effect of microenvironments on a host–guest system, it is important to rationally select the appropriate building blocks and synthesis strategy to construct a bismacrocycle with different geometric configurations. In this work, a triphenylene derivative, 1,4-bis(4-bromophenyl)-2,3-bis(4-tert-butylphenyl)-6,11-diiodotriphenylene, is used; it is a suitable precursor since these halogen groups can provide different reactive sites for subsequent macrocycle formations, whereas the peripheral 4-tert-butylphenyl groups can influence the geometrical environment of the macrocycle to which it is connected. A prior study showed that a cyclic fragment can selectively react with the iodine groups within this precursor to form a bifunctional macrocyclic structure.27 Inspired by this report, we can choose different curved synthones to react with this triphenylene derivative via the Suzuki coupling reaction and aromatization reaction to give target bismacrocycles with different sizes.
The rate of oxidative addition in a Suzuki–Miyaura reaction is largely controlled by the bond dissociation energies (BDEs) of C–X (X = I, Br, Cl) bonds.28,29 The key point in this strategy is to take advantage of the difference in BDEs between Ar–I and Ar–Br bonds (Ar–I > Ar–Br) to allow for a selective reaction in the ring formations. The synthetic strategy for 1a and 1b is shown in Fig. 2. Starting materials 2a, 2b, and 3 were prepared according to the reported literature.27,30,31 Initially, diboronate 2a was selectively reacted with the iodine groups in compound 3 to prepare bifunctional macrocycle 4a under relatively weak reaction conditions (catalyst: Pd(PPh3)4; base: K2CO3) and subsequent reductive aromatization with H2SnCl4. With this key fragment 4a in hand, the second intermolecular cyclization between 4a and 2a was performed under strong Suzuki coupling reaction conditions (catalyst: Pd2(dba)3, S-Phos; base: Cs2CO3) and subsequent reductive aromatization with H2SnCl4 to afford the target bismacrocycle 1a. It is worth noting that the two-step macrocyclization strategy can significantly improve the yield of the target bismacrocycle when compared to the one-pot method. By applying this strategy, another bismacrocycle 1b could also be prepared by replacing compound 2a with its 8-ring partner 2b. Following a similar procedure to that of 1a, diboronate 2b coupled with 3 to give key macrocycle 4b, which then underwent the second Suzuki macrocyclization with 2a and subsequent reductive aromatization to deliver bismacrocycle 1b.
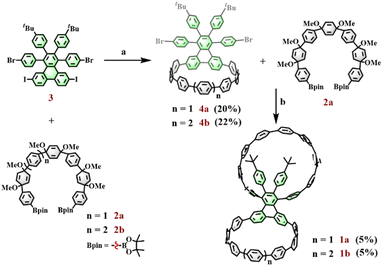 |
| | Fig. 2 Reaction conditions: (a) (1) Pd(PPh3)4, K2CO3, THF, H2O, 68 °C; (2) H2SnCl4, THF, rt. (b) (1) Pd2(dba)3, S-Phos, Cs2CO3, toluene, MeOH, H2O, 100 °C; (2) H2SnCl4, THF, rt. | |
Physical characterizations
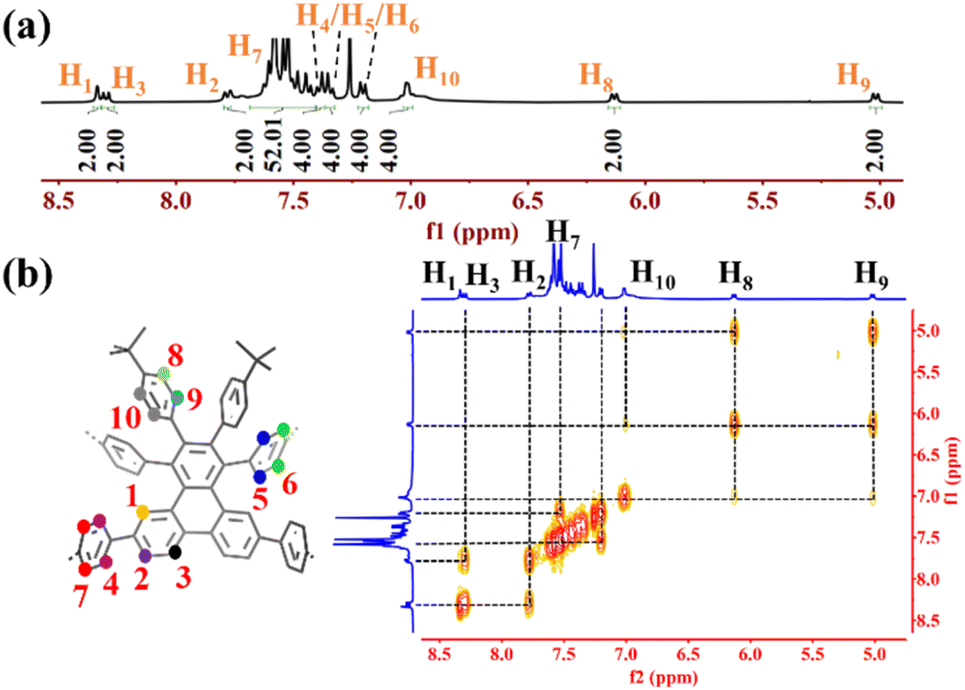
The successful synthesis of 1a and 1b was confirmed by a combination of MALDI-TOF mass spectrometry, nuclear magnetic resonance (NMR), and X-ray crystallography. The peaks found at m/z 1705.7546 and 1781.7811 (calculated for 1a: C134H96 [M]+ 1705.7501; 1b: C140H100 [M]+ 1781.7859) suggested successful synthesis of the target compounds. Further characterization by 1H NMR spectroscopies also confirmed the successful synthesis of 1a and 1b. As shown in Fig. 3a, the characteristic singlet at δ = 8.34 ppm can be assigned to H1 because its vicinal positions are all quaternary carbons. The multiplets at δ = 7.43–7.63 ppm (H7) are mainly from the protons in the cycloparaphenylene moieties according to the overlapped peak shape, chemical shifts and number of integral protons. To fully assign all the protons in 1a, we recorded the 2D 1H–1H COSY NMR spectrum (Fig. 3b). The doublet at δ = 7.78 ppm is correlated with the signals at δ = 8.34 ppm and 8.30 ppm, suggesting that the doublets at δ = 7.78 ppm and 8.30 ppm can be assigned to H2 and H3, respectively. Three doublets at δ = 7.20 ppm, 7.34 ppm, and 7.39 ppm are strongly correlated with the signals at δ = 7.43–7.63 ppm, confirming that those signals belong to H4, H5, and H6. Another three signals at δ = 5.02 ppm, 6.13 ppm, and 7.00 ppm show no interactions with the protons of cycloparaphenylene moieties, implying that they may originate from phenyl groups connected to the tert-butyl groups. Combined with the crystal data, protons H8 and H9 could be located in the shielding region induced by the macrocyclic ring current, forcing their chemical shifts to the high field. Therefore, the doublets at δ = 5.02 ppm and 6.13 ppm can be ascribed to H8 and H9, respectively, and the signal at δ = 7.00 ppm can be assigned to H10. Similar assignments can be made for the protons in 1b.
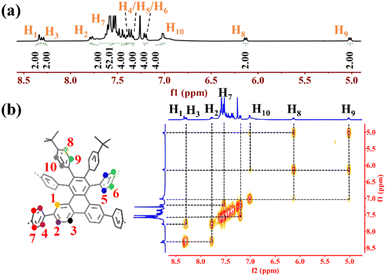 |
| | Fig. 3 (a) Aromatic regions of the 1H NMR spectrum of 1a. (b) Expanded 2D 1H–1H COSY NMR spectrum of 1a. | |
Photophysical properties
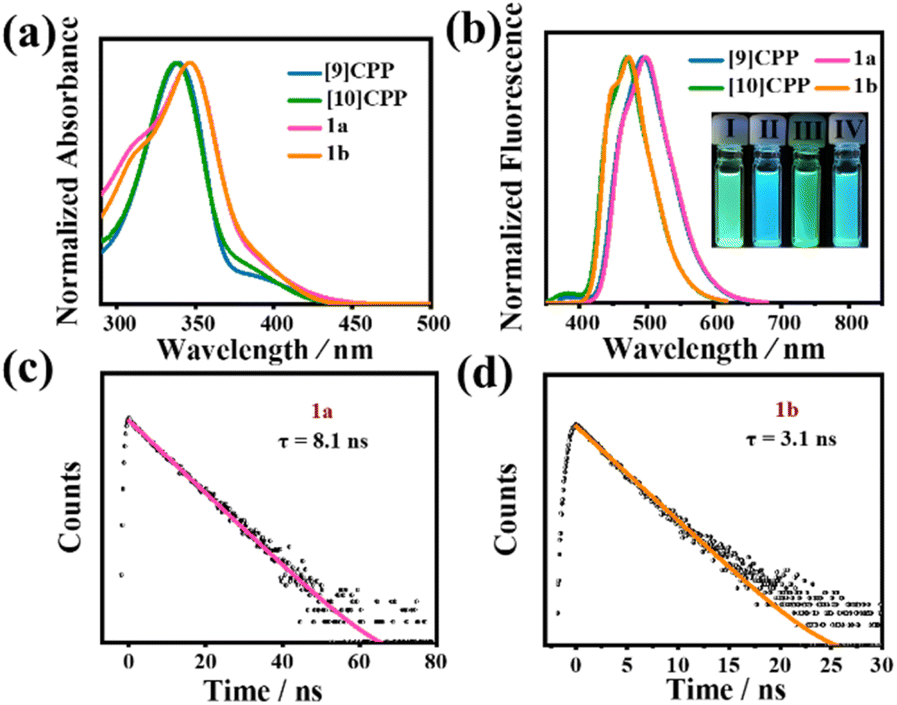
The photophysical properties of 1a and 1b were studied by UV-vis absorption spectroscopy, steady-state fluorescence spectroscopy, and time-resolved fluorescence decay. Interestingly, the absorption and fluorescence spectra of 1a and 1b both reveal unique features of this type of bismacrocycle compared with [9]CPP and [10]CPP (Fig. 4a and b). No significant difference is found in the absorption spectra of 1a and 1b; their maximum absorption peaks are located at 346.5 nm and 346 nm, respectively (molecular absorption coefficients ε1a = 1.54 × 104 M−1 cm−1, ε1b = 9.01 × 104 M−1 cm−1), which are slightly redshifted compared to [n]CPPs.32 To identify the difference in optical properties, time-dependent density functional theory (TD-DFT) with PBE0/6-31G(d,p)//PCM was performed. The calculation results reveal that the observed experimental peak at 346.5 nm in 1a mainly corresponds to the HOMO−2 → LUMO and HOMO → LUMO+2 electronic transitions, while the peak at 346 nm in 1b corresponds to the HOMO → LUMO+1 and HOMO−4 → LUMO transitions. In the fluorescence spectra, 1a is nearly identical to that of [9]CPP with no traces of the fluorescence peaks that originated from [10]CPP. Similarly, 1b also only exhibits the fluorescence features of [10]CPP, whereas the addition of another macrocycle has no effect on its fluorescence properties. The fluorescence quantum yields (ΦF) of 1a and 1b were estimated to be approximately 12.9% and 7.5%, respectively. Under irradiation by a hand-held UV lamp at λ = 365 nm, the photoluminescence of the diluted solution (1.0 × 10−5 M) of 1a and 1b, i.e., green and cyan, respectively, are also the same as those of [9]CPP and [10]CPP, which are consistent with the results of the fluorescence spectroscopy. The luminescence lifetimes (τs) of 1a and 1b were measured by time-resolved fluorescence decay using the time-resolved photoluminescence (TRPL) technique (Fig. 4c and d). When excited at 390 nm, the τs of 1a and 1b were determined to be approximately 8.1 ns at 499 nm and 3.1 ns at 475 nm, respectively, using single-exponential decay fitting.
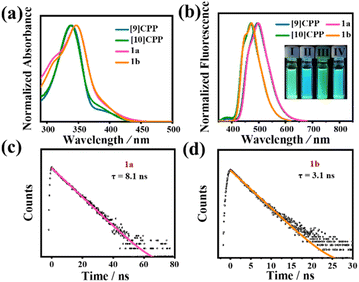 |
| | Fig. 4 (a) UV-vis absorption of CH2Cl2 solutions of 1a, 1b, [9]CPP and [10]CPP. (b) Fluorescence spectra of the CH2Cl2 solutions of 1a, 1b, [9]CPP and [10]CPP. Inset: photographs showing the fluorescence for 1a (I), 1b (II), [9]CPP(III), and [10]CPP(IV) in CH2Cl2 under a UV lamp at λ = 365 nm. (c) Emission lifetime for 1a. (d) Emission lifetime for 1b. | |
Supramolecular chemistry in the solid-state and solution
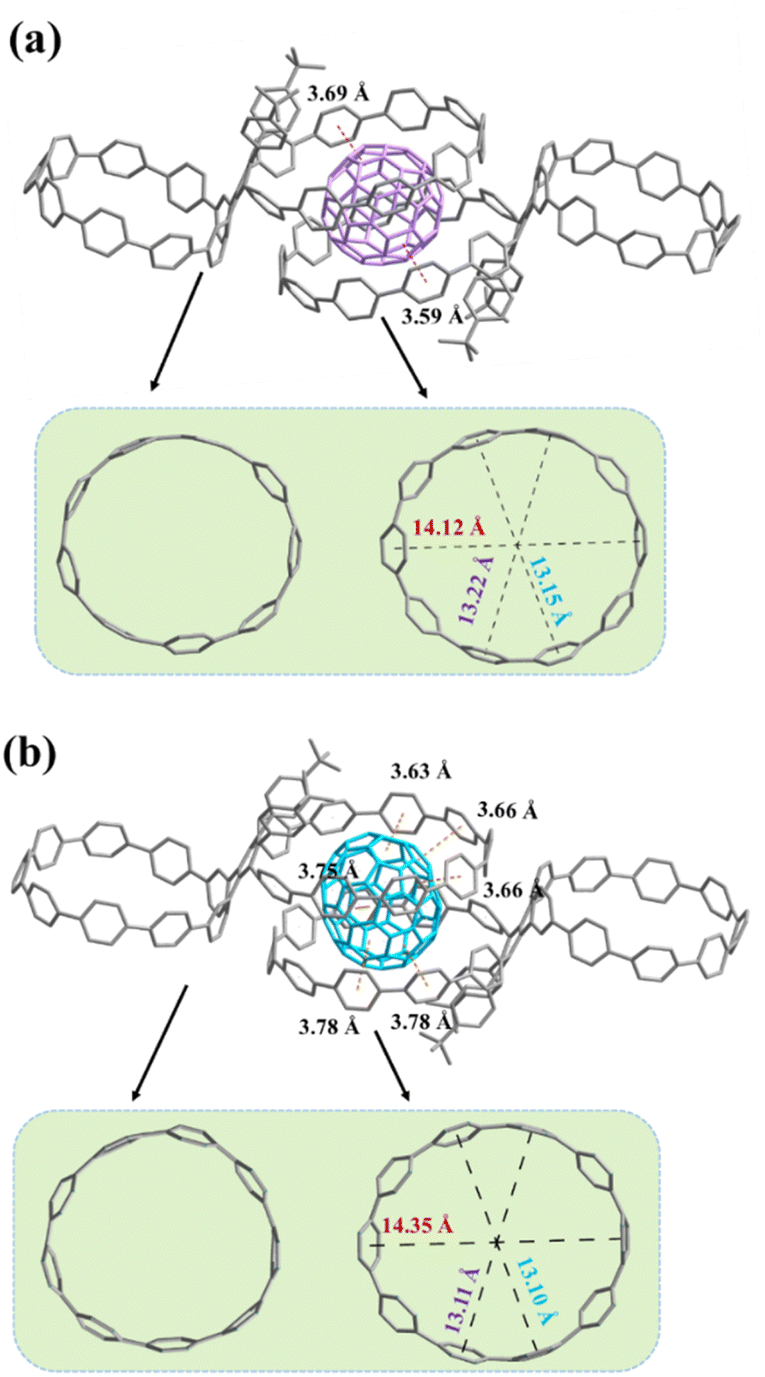
Analogous to the fullerene hosting properties of [10]CPP,17,33,34 slow liquid diffusion of hexane into a chlorobenzene solution containing 1a and excess C60 or C70 afforded black single crystals suitable for single-crystal X-ray measurements. The crystal structure not only unambiguously validates the composition and stereochemistry of 1a but also reveals intriguing and unexpected host–guest properties. Notably, two 1a molecules are bridged by a C60 or C70 molecule to build the 2:1 host–guest systems, in which the C60 or C70 molecule is shared by two [10]CPP moieties from different 1a molecules. As shown in the crystal structure of C60@(1a)2 (Fig. 5a), the host moiety exhibits an oval shape with long and short axes of 14.12 Å and 13.15 Å, respectively. The interfacial distances between the host moieties and C60, defined as the distance between the centroid of a benzene unit of the macrocycle and the nearest centroid of a hexagon or pentagon of C60, are mostly greater than 4.0 Å with only a few parts less than 3.8 Å (highlighted in red, more detailed information see in Fig. S12†), implying that only a few aromatic rings participate in the convex–concave π–π stacking interactions and that the supramolecular interactions are quite weak. Therefore, a single oval-shaped macrocycle cannot capture a C60 molecule but rather can only bind that molecule by cooperating with the same oval-shaped macrocycle from another bismacrocycle. This behavior is probably because the oval-shaped macrocycle configuration and the steric hindrance of tert-butylphenyl groups change the supramolecular interactions between the conjugated macrocycle and C60. Due to the unique ellipsoid shape, the interfacial distances between C70 and the host moieties are relatively smaller than those of C60 but also show the weak supramolecular interactions (Fig. 5b, the distances less than 3.8 Å are highlighted in orange and more detailed information see in Fig. S15†). Interestingly, C60@(1a)2 is aligned in a linear manner, while C70@(1a)2 exhibits a herringbone alignment style (Fig. S14 and S17†). It should be mentioned that there are no significant intermolecular interactions between C60@(1a)2 and C70@(1a)2 molecules.
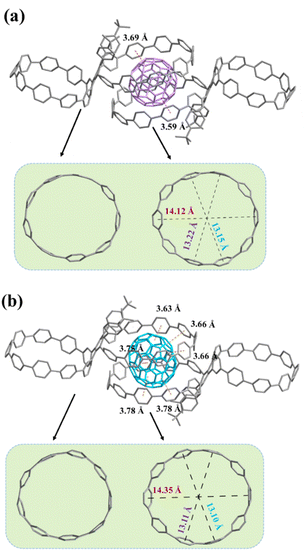 |
| | Fig. 5 (a) Crystal structure of C60@(1a)2. (b) Crystal structure of C70@(1a)2. Hydrogen atoms and disordered solvent molecules are omitted for clarity. | |
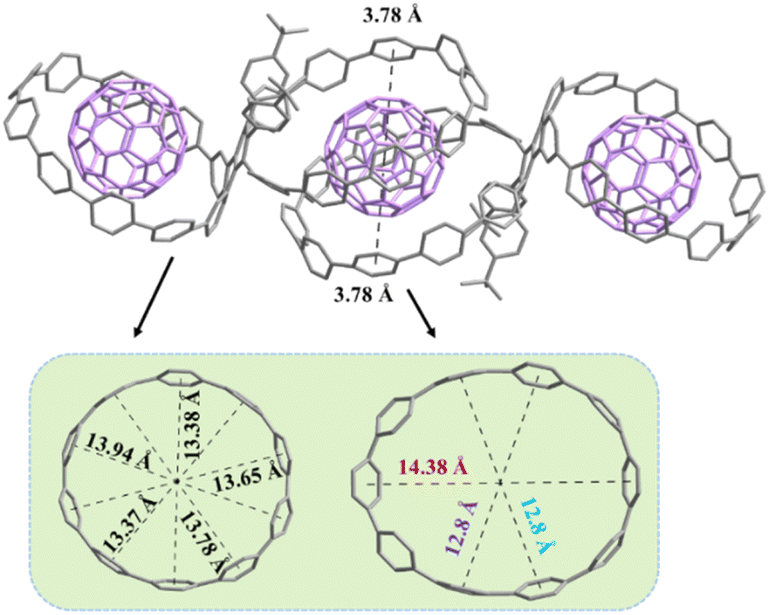
Encouraged by these results, we are able to build the first 2:3 host–guest system between 1b and C60. By slow evaporation of toluene solutions containing 1b and excess C60, black single crystals suitable for X-ray measurements can be obtained. As shown in Fig. 6, the two macrocycles within 1b show different geometric shapes: one is approximately circular with an inner diameter of (13.66 ± 0.28) Å, and the other is oval with long and short axes of 14.38 Å and 12.8 Å, respectively. The crystal structure reveals that two 1b molecules interact with three C60 molecules to form a five-membered complex, with each circular macrocycle binding one C60 molecule and two oval-shaped macrocycles just sharing one. The interfacial distances between C60 and the circular macrocycle are found to be within the range of 3.58–3.91 Å (only three are more than 3.80 Å), demonstrating strong π–π stacking interactions. However, the interfacial distances between C60 and the oval-shaped macrocycle are mostly greater than 4.0 Å, indicating a weak π–π stacking interaction (the distances less than 3.8 Å are highlighted in black and more detailed information see in Fig. S18†). Similarly, 3C60@(1b)2 molecules align in a herringbone manner, and no significant intermolecular interactions have been observed between 3C60@(1b)2 molecules (Fig. S20†).
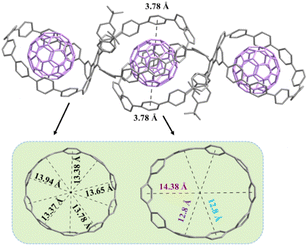 |
| | Fig. 6 Crystal structure of 3C60@(1b)2. Hydrogen atoms and disordered solvent molecules are omitted for clarity. | |
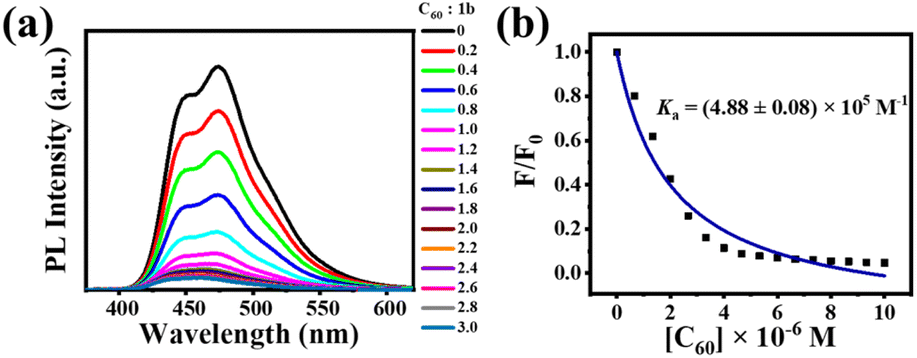
Although crystallographic data can accurately characterize a host–guest complex, the crystalline structure does not mean it will adopt the same conformation in solution or other phases.35 Supramolecular chemistry in dilute solution differs from the supramolecular assembly behavior of 1a and 1b with C60 or C70 in the solid-state in that weak interactions are influenced by both the molecular structure and the solvent. According to the MALDI-TOF mass spectrum and fluorescence quenching measurements, no supramolecular interaction was observed between 1a and C60 or C70 in solution. The reason for this phenomenon may be that the very weak supramolecular interaction between [10]CPP moieties within 1a and fullerenes dissociated by the solvent molecule. However, the interaction between 1b and C60 can be clearly observed when C60 was added to a solution of 1b, resulting in a distinct color change from cyan to brown and a reduced fluorescence intensity. The MALDI-TOF mass spectrum displayed a peak at m/z 2502.7812 (calculated for C200H100 [C60@1b]+: 2502.7892), indicating the formation of a 1:1 complex (Fig. S21†). This apparent contradictory host–guest ratio has also been observed in other fullerene-based supramolecular complexes.36,37 Since there is only a size difference between these two bismacrocycles, we speculate that the supramolecular interaction mainly occurs between the circular macrocycle within 1b and C60, so that only 1:1 complex can be detected by mass spectrum. In order to quantitatively investigate the supramolecular assembly of 1b with C60, we carried out fluorescence measurements with an excitation wavelength of 320 nm. Upon gradual addition of C60 to the solution of 1b, the fluorescence intensity of 1b dramatically decreased, as shown in Fig. 7a. Based on the quenching results, the binding constant was calculated to be approximately Ka = (4.88 ± 0.08) × 105 M−1 (Fig. 7b). The deviation between the fitted isotherm and the experimental data could be attributed to the very weak interaction between the oval-shaped macrocycle and C60 in solution. Furthermore, experimental data points were fitted to a standard 1:1 stoichiometry and different types of 1:2 stoichiometries according to the reported methods,38–40 but all the fitting errors were large (the minimum error is greater than 13%, more detailed information see in Fig. S22–S26†), indicating that the supramolecular interaction between 1b and C60 is difficult to achieve perfect equilibrium of 1:1 or 1:2 in solution. Recently, it has been reported that a chrysenylene-based carbon macrocycle can interact with C70 in 1:1, 1:2, and 2:1 stoichiometries in solution, which is significantly different from crystal structures.41 Even for a simple CPP macrocycle, von Delius and coworkers demonstrated complicated supramolecular interactions between [10]CPP and fullerenes, where 1:1 and 2:1 complexes of [10]CPP and C60/C70 were detected in solution.42 Therefore, after many attempts, we still failed to obtain the accurate supramolecular complexation stoichiometry of 1b and C60 in solution, indicating a similar complicated complexation in solution and a huge challenging determination of stoichiometry. Fortunately, we have successfully obtained the crystal structures of the complexes of these two bismacrocycles and fullerenes, which allowed us to precisely determine the stoichiometries of these complexes in the solid state.
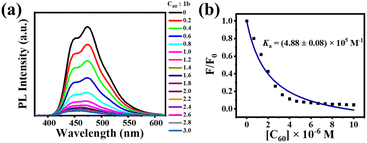 |
| | Fig. 7 (a) Fluorescence spectra of 1b (3.33 × 10−6 M) in the presence of C60 in toluene. The concentrations of C60 ranged from 0 to 1.0 × 10−5 M. (b) Plot of the absorption changes at 474 nm versus [C60] in toluene for calculating the binding constant. | |
Theoretical calculations
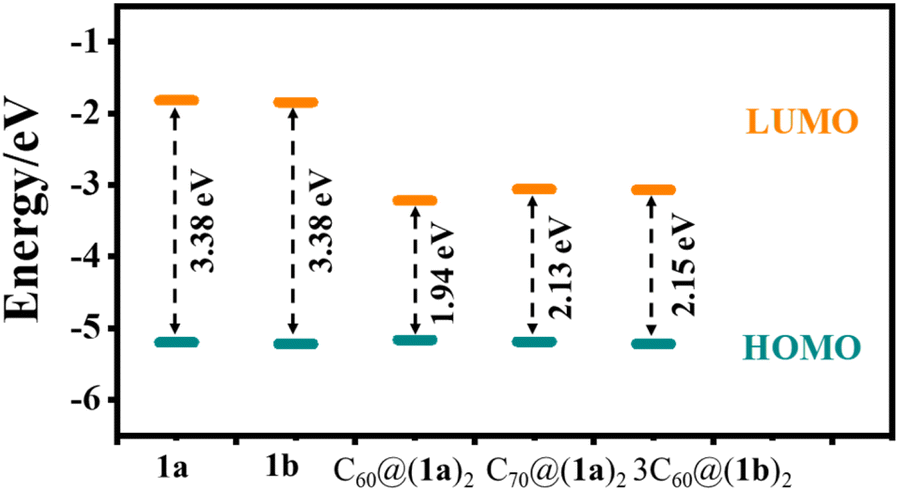
Geometrical optimization was carried out at the theoretical level of B3LYP/6-31G(d,p), where DFT-D3(BJ) and polarizable continuum model (PCM) methodologies can be used to correct the dispersion energy and solvent effect of dichloromethane.43,44 The calculation results show that two tert-butylbenzene groups lie at an average angle of 73.28° for 1a and 74.79° for 1b with a bridging phenyl in the [10]CPP moiety. Unexpectedly, the strain energies of 1a and 1b are as high as 103.12 kcal mol−1 and 103.19 kcal mol−1, respectively. Compared with [9]CPP (64.71 kcal mol−1) and [10]CPP (59.47 kcal mol−1), the strain energies of 1a and 1b increase dramatically, which can be attributed to the unique topological structure. The frontier molecular orbitals of 1a, 1b, C60@(1a)2, C70@(1a)2, and 3C60@(1b)2 are shown in Fig. S28–S32.† Due to the structural similarity, 1a and 1b possess almost the same occupied molecular orbitals and unoccupied molecular orbitals and thus have similar HOMO–LUMO gaps (∼3.38 eV). However, their inclusion complexes of C60 or C70 feature a downshift of the unoccupied molecular orbital and an upshift of the occupied molecular orbitals, and the HOMO–LUMO gaps decreases (Fig. 8).
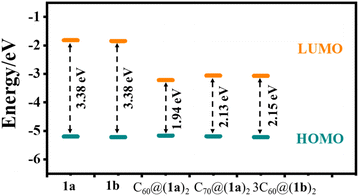 |
| | Fig. 8 Schematic diagram illustrating the frontier orbital energy levels of 1a, 1b, C60@(1a)2, C70@(1a)2, and 3C60@(1b)2. | |
Conclusions
In summary, we have designed and synthesized two dimeric macrocycles by anchoring two cycloparaphenylene macrocycles on a triphenylene derivative. These two bismacrocycles show interesting photophysical properties: the absorption spectra of 1a and 1b are almost identical, whereas the fluorescence spectra of 1a and 1b are quite different. The unique structure of the triphenylene derivative allows two macrocycles to be under different microenvironments, which in turn generate varying geometric configurations and supramolecular properties. Notably, the macrocycles within the bismacrocycles show supramolecular behaviors distinct from those of pristine units. In the solid-state, two 1a molecules cooperate to bind a C60 or C70 molecule to build the 2:1 host–guest complexes, while two 1b molecules and three C60 molecules can form a 2:3 host–guest complex. Interestingly, 1b can binds one C60 molecule in a 1:1 ratio to build another host–guest system in solution due to the weak host ability of the oval-shaped macrocycle. All these results show that the supramolecular properties of dimeric macrocycles can be finely tuned by their configurations, microenvironments, and even solvents.
Data availability
Experimental procedures, characterization data, titration experiments, and computational data are available in the ESI.†
Author contributions
P. D. conceived and designed this research with the help of M. C. and P. F. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22225108, 21971229, U1932214, 52172053).
Notes and references
- I. Hiroshi, H. Kiyoshi, A. Tsuyoshi, A. Masanori, T. Seiji, O. Tadashi, S. Masahiro and S. Yoshiteru, Chem. Phys. Lett., 1996, 263, 545–550 CrossRef.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- D. Canevet, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248–9259 CrossRef CAS.
- F. Diederich and M. Gómez-López, Chem. Soc. Rev., 1999, 28, 263–277 RSC.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC.
- C. García-Simón, M. Costas and X. Ribas, Chem. Soc. Rev., 2016, 45, 40–62 RSC.
- J. Effing, U. Jonas, L. Jullien, T. Plesnivy, H. Ringsdorf, F. Diederich, C. Thilgen and D. Weinstein, Angew. Chem., Int. Ed. Engl., 1992, 31, 1599–1602 CrossRef.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229–231 CrossRef CAS.
- J. Calbo, A. de Juan, J. Aragó, J. Villalva, N. Martín, E. M. Pérez and E. Ortí, Phys. Chem. Chem. Phys., 2019, 21, 11670–11675 RSC.
- H. Li, Q. Chen, C. Schönbeck and B.-H. Han, RSC Adv., 2015, 5, 19041–19047 RSC.
- Y. Xu, S. Gsänger, M. B. Minameyer, I. Imaz, D. Maspoch, O. Shyshov, F. Schwer, X. Ribas, T. Drewello, B. Meyer and M. von Delius, J. Am. Chem. Soc., 2019, 141, 18500–18507 CrossRef CAS PubMed.
- Q. Shi, X. Wang, B. Liu, P. Qiao, J. Li and L. Wang, Chem. Commun., 2021, 57, 12379–12405 RSC.
- W. Li, F. Qu, L. Liu, Z. Zhang, J. Liang, Y. Lu, J. Zhang, L. Wang, C. Wang and T. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116854 CAS.
- D. Lu, G. Zhuang, H. Wu, S. Wang, S. Yang and P. Du, Angew. Chem., Int. Ed., 2017, 56, 158–162 CrossRef CAS PubMed.
- S. Wang, X. Li, X. Zhang, P. Huang, P. Fang, J. Wang, S. Yang, K. Wu and P. Du, Chem. Sci., 2021, 12, 10506–10513 RSC.
- J. Xia, J. W. Bacon and R. Jasti, Chem. Sci., 2012, 3, 3018–3021 RSC.
- Z. Xia, S. H. Pun, H. Chen and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 10311–10318 CrossRef CAS PubMed.
- M. Takeda, S. Hiroto, H. Yokoi, S. Lee, D. Kim and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 6336–6342 CrossRef CAS PubMed.
- Q. Huang, Y. Wu, Y. Zhou, H. Liu, J. Wang, S. Wang and P. Du, Synthesis, 2020, 52, 2535–2540 CrossRef CAS.
- Q. Huang, G. Zhuang, H. Jia, M. Qian, S. Cui, S. Yang and P. Du, Angew. Chem., Int. Ed., 2019, 58, 6244–6249 CrossRef CAS PubMed.
- Y. Xu, B. Wang, R. Kaur, M. B. Minameyer, M. Bothe, T. Drewello, D. M. Guldi and M. von Delius, Angew. Chem., Int. Ed., 2018, 57, 11549–11553 CrossRef CAS.
- K. Li, Z. Xu, H. Deng, Z. Zhou, Y. Dang and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 7649–7653 CrossRef CAS PubMed.
- Y. Yang, S. Huangfu, S. Sato and M. Juríček, Org. Lett., 2021, 23, 7943–7948 CrossRef CAS.
- L. Zhan, C. Dai, G. Zhang, J. Zhu, S. Zhang, H. Wang, Y. Zeng, C.-H. Tung, L.-Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2022, 61, e202113334 CAS.
- X. Zhang, H. Shi, G. Zhuang, S. Wang, J. Wang, S. Yang, X. Shao and P. Du, Angew. Chem., Int. Ed., 2021, 60, 17368–17372 CrossRef CAS PubMed.
- S. Wang, X. Li, G. Zhuang, M. Chen, P. Huang, S. Yang and P. Du, Chem. Commun., 2021, 57, 9104–9107 RSC.
- J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40–62 RSC.
- C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082–10093 CrossRef CAS PubMed.
- M. Gille, A. Viertel, S. Weidner and S. Hecht, Synlett, 2013, 24, 259–263 CrossRef CAS.
- X. Zhang, H. Liu, G. Zhuang, S. Yang and P. Du, Nat. Commun., 2022, 13, 3543 CrossRef CAS PubMed.
- E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, H. Takaya, T. Haino, N. Yasuda and S. Yamago, Chem.–Eur. J., 2013, 19, 14061–14068 CrossRef CAS PubMed.
- N. Geue, R. E. P. Winpenny and P. E. Barran, Chem. Soc. Rev., 2022, 51, 8–27 RSC.
- D. Felder, B. Heinrich, D. Guillon, J.-F. Nicoud and J.-F. Nierengarten, Chem.–Eur. J., 2000, 6, 3501–3507 CrossRef CAS PubMed.
- Y.-Y. Xu, H.-R. Tian, S.-H. Li, Z.-C. Chen, Y.-R. Yao, S.-S. Wang, X. Zhang, Z.-Z. Zhu, S.-L. Deng, Q. Zhang, S. Yang, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Nat. Commun., 2019, 10, 485 CrossRef CAS PubMed.
-
http://supramolecular.org
.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- K. Ikemoto, K. Takahashi, T. Ozawa and H. Isobe, Angew. Chem., Int. Ed., 2023, 62, e202219059 CrossRef CAS.
- M. Freiberger, M. B. Minameyer, I. Solymosi, S. Frühwald, M. Krug, Y. Xu, A. Hirsch, T. Clark, D. M. Guldi, M. von Delius, K. Amsharov, A. Görling, M. E. Pérez-Ojeda and T. Drewello, Chem.–Eur. J., 2023, 29, e202203734 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comp. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Nan
Yin
b,
Guilin
Zhuang
*ab,
Nan
Yin
b,
Guilin
Zhuang