
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBidirectional photoswitchability in an iron(III) spin crossover complex: symmetry-breaking and solvent effects†
Raúl
Díaz-Torres‡
 a,
Guillaume
Chastanet
b,
Eric
Collet
c,
Elzbieta
Trzop
c,
Phimphaka
Harding‡
*d and
David J.
Harding‡
*d
a,
Guillaume
Chastanet
b,
Eric
Collet
c,
Elzbieta
Trzop
c,
Phimphaka
Harding‡
*d and
David J.
Harding‡
*d
aThammasat University Research Unit in Multifunctional Crystalline Materials and Applications (TU-MCMA), Faculty of Science and Technology, Thammasat University, Pathum Thani 12121, Thailand
bUniversité de Bordeaux, ICMCB, 87 Avenue du Dr A. Schweitzer, Pessac, F-33608, France
cUniv Rennes, CNRS, IPR (Institut de Physique de Rennes) – UMR 6251, F-35000 Rennes, France
dSchool of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand. E-mail: david@g.sut.ac.th; phimphaka@g.sut.ac.th
First published on 1st June 2023
Abstract
The impact of solvent on spin crossover (SCO) behaviour is reported in two solvates [Fe(qsal-I)2]NO3·2ROH (qsal-I = 4-iodo-2-[(8-quinolylimino)methyl]phenolate; R = Me 1 or Et 2) which undergo abrupt and gradual SCO, respectively. A symmetry-breaking phase transition due to spin-state ordering from a [HS] to [HS-LS] state occurs at 210 K in 1, while T1/2 = 250 K for the EtOH solvate, where complete SCO occurs. The MeOH solvate exhibits LIESST and reverse-LIESST from the [HS-LS] state, revealing a hidden [LS] state. Moreover, photocrystallographic studies on 1 at 10 K reveal re-entrant photoinduced phase transitions to a high symmetry [HS] phase when irradiated at 980 nm or a high symmetry [LS] phase after irradiation at 660 nm. This study represents the first example of bidirectional photoswitchability and subsequent symmetry-breaking from a [HS-LS] state in an iron(III) SCO material.
Introduction
The search for bistable molecular systems is driven by their possible applications in molecular electronic devices.1,2 Spin crossover (SCO) complexes are one group of bistable materials that have been utilised as functional components in a range of devices.3–5 The interest lies in the appreciable changes in the magnetic and physical properties upon transition between the low spin (LS) and high spin (HS) state, induced by an external perturbation (temperature, pressure, light irradiation or magnetic field). In the majority of SCO complexes, these changes are gradual making them less attractive for applications. However, SCO complexes with abrupt transitions result in dramatic changes in their properties, and are excellent prototypes for use in molecular sensors, switches, data storage or spintronics.6,7 To achieve abrupt SCO, strong interactions between the SCO centres, the so-called cooperativity, are required with two different strategies employed: the polymeric approach, where covalent bonds are used to directly connect the spin sites,8,9 and the supramolecular approach, where intermolecular interactions are employed (π–π, hydrogen and halogen bonds), which have the advantage of being more elastic.10–12 These interactions are responsible for transmitting structural distortions along the crystal as the spin transition occurs. In most cases, this results in a single-step spin transition, sometimes with hysteresis. However, multi-step transitions can also occur13,14 often due to the presence of inequivalent spin centres that undergo SCO at different temperatures. More rarely, previously equivalent sites become inequivalent upon SCO in a process termed symmetry-breaking (SB).15–19 The first example of SB in FeIII was reported by Morgan and co-workers20 in [FeIII(3,5-OMe-sal2bapen)]ClO4 {3,5-OMe-sal2bapen = N,N′-bis(3,5-dimethoxysalicylidene)-1,5,8,12-tetraazadodecane} where incomplete SCO occurs in three steps from HS → [2HS-LS] → [HS-2LS]. Since then a number of SB SCO systems have been discovered with [FeIII(H-5-Br-thsa)(5-Br-thsa)]·H2O (H2-5-Br-thsa = 5-bromo-2-hydroxy(benzylidene)hydrazinecarbothioamide) exhibiting a remarkable 5-step SCO.21Quinolylsalicylaldiminate complexes are particularly prevalent in iron(III) SCO chemistry22–25 with three SB examples now known. [Fe(qsal-Br)2]NO3·2MeOH (qsal-Br = 4-bromo-2-[(8-quinolylimino)methyl]phenolate),26 exhibits SB coupled to a complete two-step SCO through HS (1 centre) → [HS-LS] (2 centres) → LS (1 centre), showing an intermediate phase (IP) [HS-LS] plateau of 96 K. [Fe(qsal)2][(C6F3I3)I] also shows SB from HS (1 centre) → [HS-LS] (2 centres), with the LS state thermally inaccessible.27 Lastly, [Fe(qsal-4-F)2]NO3·0.91MeOH·0.57H2O (qsal-4-F = 5-fluoro-2-[(8-quinolylimino)methyl]phenolate) exhibits SCO in two steps, but this time from HS → [HS-LS] → [LS-LS], with no re-entrant LS phase.28 Recent studies suggest that symmetry-breaking and SCO phenomena are coupled through volume strain.29,30 This behaviour emerges through the appearance of competing short/long range elastic interactions (e.g. H-bonds, π–π interactions) in the crystal.31 In molecular systems, symmetry-breaking most often occurs when previously equivalent sites become ordered into patterns of HS and LS states driven in part by these elastic interactions. However, a lack of systematic studies, especially for FeIII, that probe the influence of each factor on symmetry-breaking has made it difficult to improve these systems.
Light-Induced Excited Spin-State Trapping or LIESST involves light activation of a SCO material, typically from a LS to a HS state. The vast majority of reports concern FeII complexes,32–36 with comparatively few known for FeIII.37–39 The first FeIII complex known to exhibit LIESST was [Fe(pap)2]ClO4·H2O (pap = 2-[(pyrid-2-yl)methyleneamino]phenolate) reported by Sato and co-workers40 in 2000. Subsequent work seems to indicate that large distortions at the metal centre are key to the observation of LIESST in FeIII systems.41 Interestingly, for both FeII and FeIII it is also possible to use a different wavelength of light to switch from the metastable HS state back to the LS state, a process known as reverse-LIESST (Fig. 1a). Moreover, light irradiation sometimes permits access to hidden spin states that are not accessible thermally (Fig. 1b). While common in coordination polymers,42–47 we are aware of only one mononuclear complex where this has been observed.38 In this work we present a study of [Fe(qsal-I)2]NO3·2ROH (qsal-I = 4-iodo-2-[(8-quinolylimino)methyl]phenolate; R = Me 1, Et 2) where the different alcohols result in symmetry-breaking SCO or non-symmetry breaking and gradual SCO, respectively and compare it with bromo- analog [Fe(qsal-Br)2]NO3·2MeOH (3).26 Remarkably, 1 exhibits bidirectional switching from the low symmetry [HS-LS] state, allowing access to a hidden LS state or a HS state, both of higher symmetry.
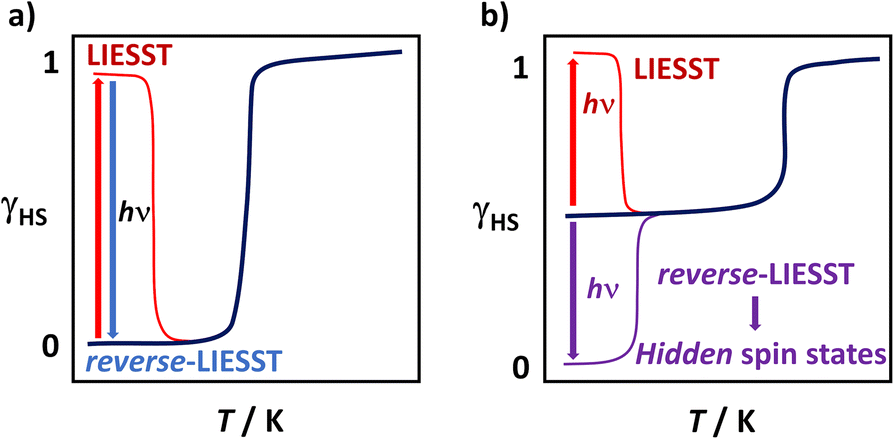 | ||
| Fig. 1 Visual representation of (a) LIESST and reverse-LIESST and (b) light activation to access hidden (not easily accessible) spin states. | ||
Results and discussion
Synthesis
Both compounds were prepared by layering a solution of Fe(NO3)3·9H2O in MeOH (1) or EtOH (2) over a solution of Hqsal-I in CH2Cl2 in which NEt3 had been added. Full spectroscopic and Experimental details can be found in the ESI† and are consistent with the proposed formulae.Structural descriptions
The crystal structure of 2 was collected at 300 and 150 K with the asymmetric unit showing one [Fe(qsal-I)2]+ cation, a nitrate anion and two ethanol molecules at both temperatures (Fig. S1†). For 1, symmetry-breaking is observed, showing one independent FeIII centre at high temperature (300, 240 K), and two centres (Fe1 and Fe2) at low temperature (150 and 100 K; Table S1†). Both compounds crystallize in triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a pseudo-octahedral FeIII metal centre and two meridionally coordinated qsal-I ligands (Fig. 2). In 1, the Fe-Nav distances and octahedral distortion parameters48 indicate HS FeIII at 300 K (ca. davFe1–N = 2.132 Å and Θ = 235, respectively),49 while at 240 K they shrink slightly to ca. davFe1 = 2.082 Å and Θ = 187, consistent with ca. 80% HS. Further cooling to 150 K results in doubling of the unit cell volume (V300 K = 1699 Å3; V150 K = 3316 Å3), indicating symmetry-breaking and resulting in two [Fe(qsal-I)2]+ cations in the doubled asymmetric unit (Fig. 1). At this temperature, the Fe-Nav distances are Fe1–N = 1.96 Å and Fe2–N = 2.13 Å respectively, showing that Fe1 is LS while Fe2 is HS.49 This long-range ordering of HS and LS molecular states corresponds to the formation of a spin-state concentration wave, SSCW.29 No significant changes are observed between 150 K and 100 K (Table S2†).
with a pseudo-octahedral FeIII metal centre and two meridionally coordinated qsal-I ligands (Fig. 2). In 1, the Fe-Nav distances and octahedral distortion parameters48 indicate HS FeIII at 300 K (ca. davFe1–N = 2.132 Å and Θ = 235, respectively),49 while at 240 K they shrink slightly to ca. davFe1 = 2.082 Å and Θ = 187, consistent with ca. 80% HS. Further cooling to 150 K results in doubling of the unit cell volume (V300 K = 1699 Å3; V150 K = 3316 Å3), indicating symmetry-breaking and resulting in two [Fe(qsal-I)2]+ cations in the doubled asymmetric unit (Fig. 1). At this temperature, the Fe-Nav distances are Fe1–N = 1.96 Å and Fe2–N = 2.13 Å respectively, showing that Fe1 is LS while Fe2 is HS.49 This long-range ordering of HS and LS molecular states corresponds to the formation of a spin-state concentration wave, SSCW.29 No significant changes are observed between 150 K and 100 K (Table S2†).
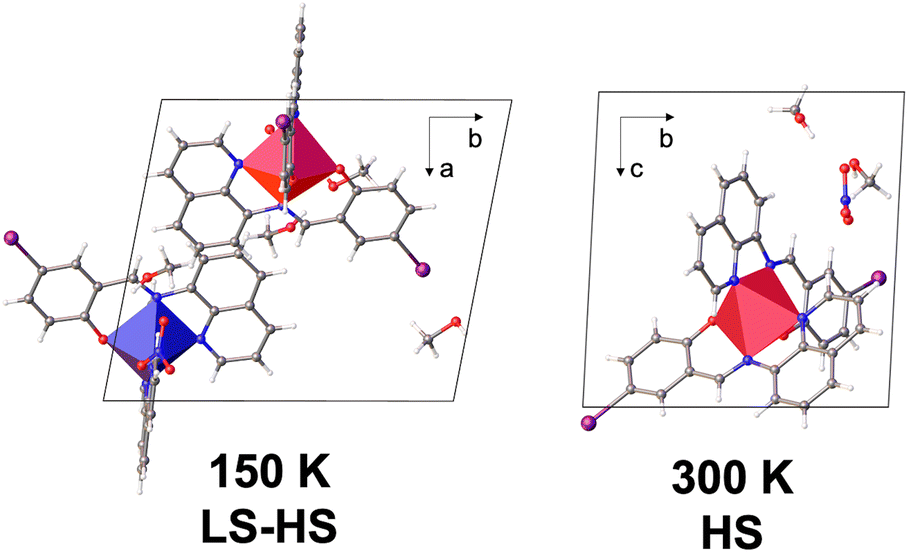 | ||
| Fig. 2 Unit cell components of 1 at 150 K (left) and 300 K (right). The a, b and c axis at 300 K and 150 K correspond to different directions. | ||
Heating back to 300 K, reveals reversible spin conversion and an increase in symmetry with a single FeIII centre in the asymmetric unit. The reversibility of symmetry-breaking SCO was confirmed by recooling to 150 K with the structure again showing two independent FeIII centres. This behaviour is similar to [Fe(qsal-Br)2]NO3·2MeOH 3,26 but the iodo group seems to prevent access to the full LS state, presumably because of its steric bulk that favours stronger antiferroelastic interactions between the FeIII centres stabilizing [HS-LS] order.22 There are comparatively few FeIII systems that undergo symmetry-breaking SCO18,20,50–53 and this is only the third example from a [HS] to an ordered [HS-LS] SSCW (Table S3†).54
The ethanol complex, 2 has simpler behaviour, with Fe–N/O distances (Fe-Nav = 2.12, Fe-Oav = 1.90 Å) and octahedral parameters (Σ = 59 and Θ = 204) at 300 K consistent with HS FeIII.49 Cooling to 150 K, results in a decrease in the Fe–N/O bond distances (ΔFe-Nav = 0.137 and ΔFe-Oav = 0.025) and the octahedral parameters (ΔΣ = 15 and ΔΘ = 87) indicating a HS to LS transition (Table S4†). The lack of symmetry-breaking in 2 suggests that the methanol solvent is primarily responsible, modifying elastic interactions between the FeIII centres, mirroring results reported for [Fe(1-bpp-SiPr)2][BF4]2·sol (1-bpp-SiPr = 1-bpp-SiPr = 2,6-di{pyrazol-1-yl}-4-isopropylthiopyridine).55
Crystal packing
The packing in the structures consist of 1D chains along the c axis (Fig. 3 and S2†) where the FeIII cations are connected via two sets of perpendicular π–π interactions (Type A and Type B) supported by C–H⋯O interactions. In the case of 1 at 150 K, symmetry-breaking does not alter the packing of the FeIII cations with each Fe centre in separate 1D chains (Fe1–Fe1 and Fe2–Fe2) as shown in Fig. 3. The π–π distances in 1 and 2 are similar, with Type B being slightly stronger (by about 0.12 Å) compared to Type A. One of the most significant differences between 1 and 2 is the shorter Fe–Fe distance within the 1D chain for 1, by about 0.3 Å. This feature is also observed in the P4AE (parallel fourfold aryl embraces)56 and C–H⋯I interactions, that connect neighbouring 1D chains and form a 2D sheet in the (ac) plane (Fig. 3 and S3†). In both cases, the interactions are much shorter for 1 (by about 0.3 Å) compared to 2. The final 3D structure is formed by C–H⋯I interactions connecting the 2D planes along the b axis (Fig. 3). This higher dimensional structure is also supported by halogen bonds (I⋯I) in the case of 1 (Fig. S4 and S5†). | ||
| Fig. 3 Schematic illustration of the packing from the asymmetric unit to the final supramolecular 3D structure for 1 at 150 K (blue Fe1 centres, red Fe2 centres). This figure is intended as a guide to the discussion only. | ||
The nitrate and solvent molecules (MeOH for 1 and EtOH for 2) are mainly located in the space between the chains (Fig. S6†). These molecules interact by C–H⋯O, O–H⋯O and O⋯I contacts and aid in the formation of the 2D structure by connecting the qsal-I ligands from different chains. This results in the formation of a supramolecular square (Fig. S6†). In the case of 2, the halogen bond (N–O⋯I) is slightly stronger (3.37 Å) than in 1 (3.51 Å). In all cases, small changes are observed with temperature.
The distance between the chains (termed dchain) is affected by the solvent's size, being shorter for 1 and 3 (12.0 Å) compared to 2 (12.3 Å) at 300 K and consistent with the more compact structure for 1 (Fig. 4, S7 and Table S5†). In the case of 2, dchain does not show any variation with temperature, while for 1 and 3, after symmetry breaking, the distances become longer and shorter by ca. 0.25 Å for Fe1 HS and Fe2 LS chains, respectively. Interestingly, in 3, dchain in the LS state is slightly higher than that in the HS state. In contrast, the distance between the planes (termed dplane) is slightly higher for 1, 12.5 Å, compared to 12.3 Å for 2 and 3. The similarity in dplane in 2 and 3 is unexpected but may be a factor in the thermal inaccessibility of the LS state in 1, as the iodo substituents are located between the planes. Overall, the shorter Fe–Fe distances within the chains and the lower dchain values that reflect the distance between the chains indicate that the more compact structure in 1 enhances elastic interactions between the FeIII centres and the antiferroelastic interactions that favour [HS-LS] ordering and long-range ordered SSCW.29,30,57
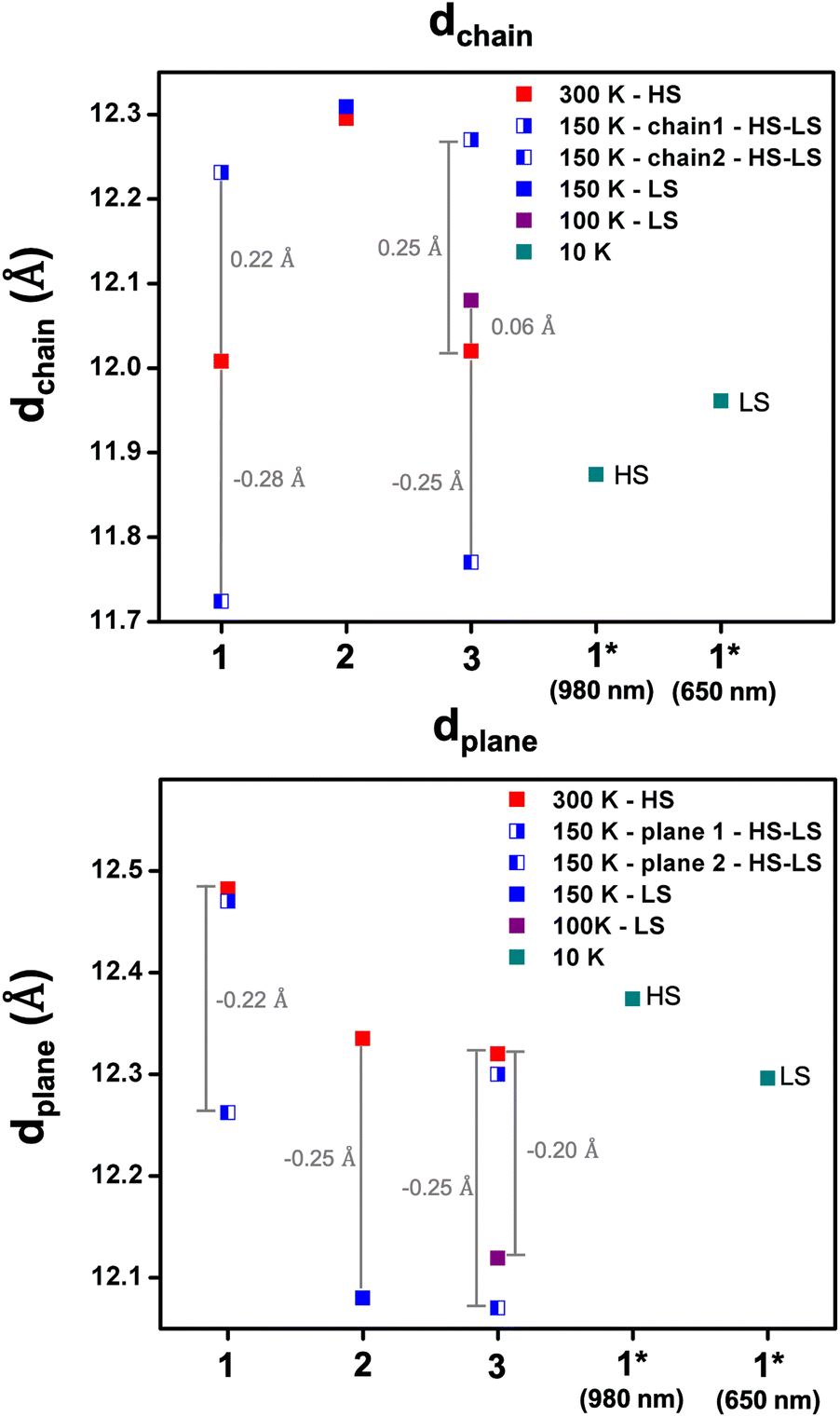 | ||
| Fig. 4 Plots of the variation of the dchain (top) and dplane (bottom) for 1, 2, and 3 at different temperatures. The changes in these values are indicated by the grey bars. *indicates irradiated samples. | ||
To better quantify the supramolecular interactions present in 1–3 we undertook Hirshfeld analysis. This reveals no significant differences between the compounds. The contributions, as well as their strength, are broadly similar in all systems (Table S8, Fig. S8 and S9†). The one exception is the O⋯H/H⋯O interactions, which are significantly stronger for the MeOH complexes (1.7 and 2.0 Å, for 1 and 3, respectively) compared to 2 (2.5 Å) at 300 K. The reason for this difference is that in 2 at 300 K one of the ethanol molecules is disordered. However, upon cooling, the hydrogen bonds in 2 become much stronger (2.05 Å), whereas for 1 weaker hydrogen bonding is observed (2.25 Å). The fact that this change is not observed in 3, where the values remain almost unchanged, strongly suggests that the SB process is unlikely to be related to these changes in the hydrogen bonding contacts.
Magnetic studies
Magnetic susceptibility data recorded on polycrystalline samples of 1 and 2 between 5–300 K, are shown in Fig. 5 and S10–S13.† At 300 K, the χMT values for 1 and 2 are ca. 3.90–4.10 cm3 K mol−1 consistent with the HS state.49 Cooling of 1 shows a moderately abrupt SCO (ΔT80 = 70 K; defined as the temperature range over which the SCO is 80% complete)58 with T1/2 = 210 K. At 150 K, χMT = 2.40 cm3 K mol−1 and consistent with the [HS-LS] state observed crystallographically. Interestingly, a further decrease in χMT is observed around 80 K reaching a value of 2.15 cm3 K mol−1. It is important to note that this behaviour is repeatable over several measurements and the additional step at 80 K does not depend on kinetics (see Fig. S10†). Moreover, a structure collected at 10 K shows no changes in the Fe–N/O bond lengths despite this small decrease in χMT (Table S2†). For 2, cooling results in a more gradual change (ΔT80 = 88 K) in χMT, reaching a value of 0.60 cm3 K mol−1 at 150 K, indicative of the full LS state (Fig. 5 and S11†). In this case T1/2 is around 250 K, 40 K higher than in 1 suggesting that the slight differences in the packing in the EtOH solvate modifies the ligand field thereby favouring the LS state in 2. At high temperatures, the SCO profiles change markedly due to desolvation (Fig. S12–S14†), in agreement with TGA analysis (Fig. S15†). After heating to 350 K, both samples exhibit slightly different profiles (Fig. S14†), probably due to differing degrees of incomplete desolvation. However, in both cases a much more gradual SCO (ΔT80 = 178 K) than either of the solvates are observed. The trapping of 1 in the [HS-LS] state contrasts with [Fe(qsal-Br)2]NO3·2MeOH where the full LS state is accessible.26 It would appear that the small increase in the van der Waal radius between the bromo (1.86 Å) and iodo atoms (2.04 Å) is sufficient to make the LS state thermally inaccessible.59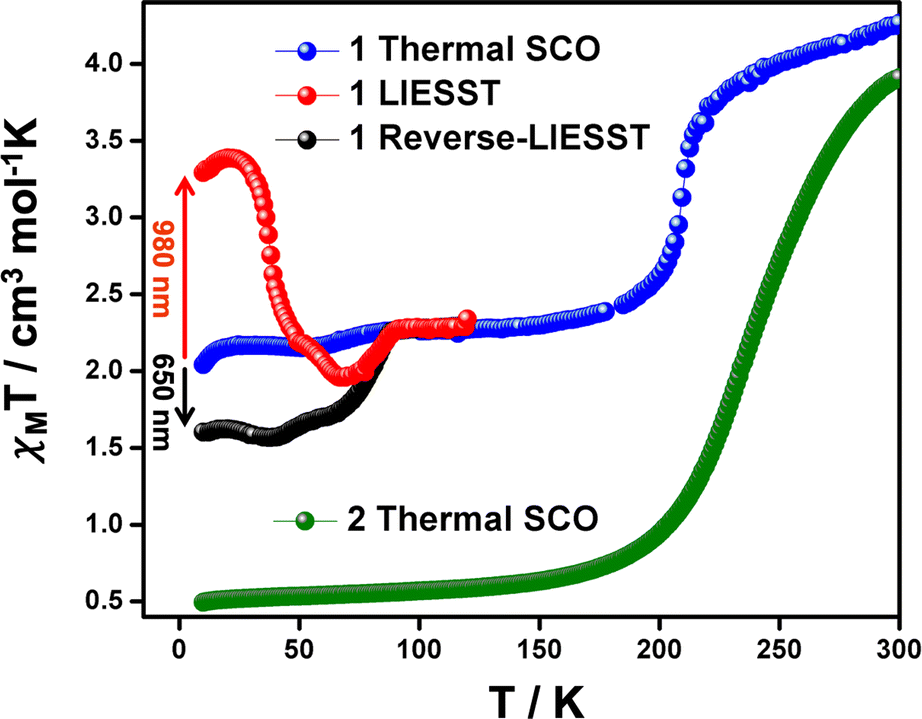 | ||
| Fig. 5 χ M T versus T plot (recorded at 0.5 K min−1) of 1 showing the thermal behaviour in the dark without any previous light irradiation (blue), after LIESST effect at 980 nm (red) and after reverse-LIESST effect at 650 nm (black) and thermal behaviour of 2 (green). | ||
Photomagnetic studies
Photomagnetic studies on 1 reveal that at 10 K irradiation at 980 nm causes an increase in χMT reaching 3.38 cm3 K mol−1 at 20 K and indicative of LIESST. The photoconversion efficiency is ca. 67% and one of the most efficient yet reported for FeIII (Table S6 in the ESI†). The derivative of χMT vs. T reveals that T(LIESST) is 38 K. Interestingly, above 60 K the χMT values dip below the thermal SCO profile with an additional decrease of χMT at the same temperature then a small jump in the SCO curve. The reason for this remains unclear, but similar observations have been noted in [Fe(qsal)2][(C6F3I3)I].27 One possible explanation is that relaxation from the high-symmetry HS state proceeds initially to a metastable LS state of the same symmetry, before further warming results in SB to the long-range ordered [HS-LS] SSCW state. No LIESST effect was observed in 2, mirroring the results found in 3. Given how similar the supramolecular contacts are in 1 and 2 (see Hirshfeld analysis above) it seems likely that the SB that occurs in 1, but not in 2 is responsible. Symmetry-breaking is also not required in 3 as the LS state is of high symmetry and may explain the lack of LIESST in this system. This is consistent with the surprisingly different T(LIESST) values in the MeCN and MeNO2 solvates of [Fe(1-bpp-SiPr)2][BF4]2·sol.55 The MeCN solvate shows no SB and is observed on the normal T(LIESST) line for this class of compound. In contrast, the MeNO2 solvate exhibits SB from a LS (P21/c, Z = 12) phase to a HS (P21, Z = 4) phase, upon light irradiation, and a lower than expected T(LIESST) value. In the case of FeIII, where relaxation is generally more rapid,37,60 perhaps SB permits observation of LIESST in compounds where it is not normally observed. The presence of efficient LIESST in [FeIII(psalpm-Cl)2]PTFB (psalpm-Cl = 4-chloro-(R,S)-((phenyl(2-pyridyl)methylimino)methyl)phenolate; PTFB = phenyltrifluoroborate)54 and [Fe(qsal)2][(C6F3I3)I].27 would seem to support this hypothesis. However, no structural data is available for either compound in the photoinduced HS state and further examples will be needed to see if this is a more general trend. Overall, these observations support the fact that the lifetime of the photo-induced state does not depend exclusively on the coordination sphere parameters but is also influenced by all the additional energies associated with symmetry changes, such as SB and order-disorder transitions.The strong reverse-LIESST exhibited by [Fe(naphBzen)2]I38 encouraged us to irradiate 1 with 650 nm light. While the reverse-LIESST is far from complete, there is a clear drop in χMT, which reaches 1.60 cm3 K mol−1 at 10 K. This value is maintained until 45 K, after which it increases gradually, before a more rapid rise to rejoin the thermal SCO profile at 90 K. The presence of significant LIESST and reverse-LIESST from a [HS-LS] state, formed upon symmetry-breaking, is to the best of our knowledge unique in FeIII SCO chemistry. Interestingly, [Fe(qsal)2][(C6F3I3)I] when irradiated at 650 nm shows relaxation from the meta-stable HS state to the [HS-LS] state with no evidence of a hidden LS state.27 Moreover, it appears that the multistep character of the thermal SCO is maintained in the curves recorded after LIESST and reverse-LIESST effects.
Photocrystallographic studies
Photocrystallographic studies were conducted on 1 at 10 K by irradiating a crystal at 980 and 660 nm. In both cases, the structures retain triclinic symmetry but now contain a single FeIII centre with the same lattice periodicity as the high temperature HS phase, as the initially different [HS-LS] sites become equivalent in the fully [HS] or [LS] states. It is likely that this high symmetry recovery, which corresponds to a photoinduced re-entrant phenomena, is responsible for the relatively strong LIESST and reverse-LIESST that is observed in 1,55 due to the elastic coupling between the spin state and symmetry changes.29 In the 980 nm structure, the bond lengths are indicative of HS FeIII, while the Fe-Nave distances in the 660 nm structure are 1.98 Å and typical for LS FeIII, Fig. 6. This is supported by the octahedral distortion parameters which are Σ = 61 and Θ = 228 (HS-980 nm) and Σ = 41 and Θ = 118 (LS-660 nm). We note that dchain and dplane in the photoinduced HS and LS structures show relatively small changes upon SCO by 0.09 and 0.08 Å, respectively (Table S5† and Fig. 4). As observed in 3, dchain is shorter in the HS structure, while dplane is shorter in the LS structure. However, 3 (HS and LS structures only) shows a smaller change in dchain (0.06 Å), but a much larger change in dplane (0.21 Å). The complete SCO observed in the single crystal contrasts with the incomplete LIESST and reverse-LIESST observed in the magnetic studies and probably reflects the extremely dark colour of the crystals which limits light penetration in the bulk.34,61,62 The HS structure at 300 K and that of the photoinduced HS structure at 10 K are almost identical. Moreover, the packing in the photoinduced HS and LS structures closely mirrors that at 300 K but with much shorter supramolecular contacts (see ESI Table S7†). An interesting difference is that the O(nitrate)⋯I halogen bond is lost in the LS structure (Fig. S16†).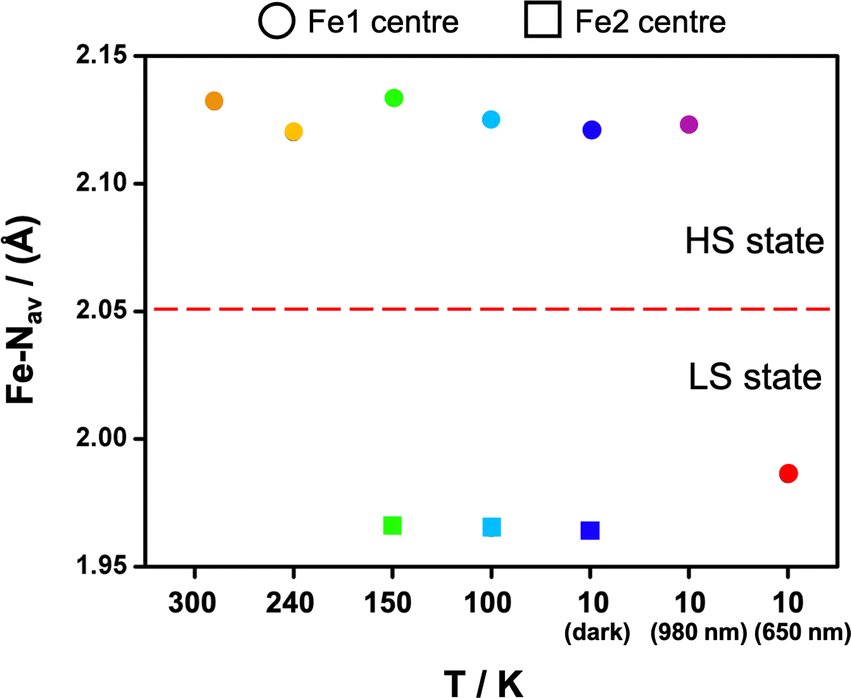 | ||
| Fig. 6 Plot of the Fe-Nav distance for 1 at 300 K (orange), 240 K (yellow), 150 K (green), 100 K (light blue), 10 K (blue), 10 K (980 nm) (purple) and 10 K (660 nm) (red). Circles represent the Fe1 centre and squares the Fe2 centre. | ||
Comparing 1 with existing SCO systems it is important to note that the thermal SCO and SB are clearly coupled. This is not the case in previously studied examples where a ‘hidden’ LS state is present.38,43,63 Hence, in 1 at 10 K where there are two distinct FeIII centres, one HS and the other LS, a change in both symmetry (2 FeIII centres to 1 FeIII centre) and spin state is required to access the LS state. In this case the phase transition is reconstructive as the fully LS phase is of higher symmetry than the [HS-LS] phase and therefore the phase transition is discontinuous and associated with latent heat. Recalling that the low symmetry [HS-LS] state is stabilized through the elastic coupling of the SB to the volume strain,29 there is an elastic cost to reach the high symmetry fully LS state, which makes it inaccessible at low temperature following a thermal equilibrium path. However, in the case of relaxation from the LIESST HS state, a relaxation towards the fully LS state is made easier because there is no symmetry change between these states, and therefore no elastic cost related to symmetry-breaking.
Conclusions
In summary, we have explored the impact of two alcohol solvents on SCO behaviour in an FeIII SCO complex. While the EtOH solvate exhibits a gradual, complete and single step SCO, the MeOH solvate shows a rare symmetry-breaking spin transition to a long-range ordered [HS-LS] SSCW state combined with a re-entrant photoinduced phase transition towards HS or hidden LS states under irradiation with visible or near-IR light. The results demonstrate how solvent subtly alters the packing in the structure, allowing for chemical tuning of the supramolecular interactions between the active SCO centres and favouring here incomplete symmetry-breaking SCO in 1, which in turn permits bidirectional switching in an FeIII SCO material. Moreover, the results suggest that SB seems to favour LIESST in FeIII, although further structural studies will be needed to determine how applicable the observations in 1 are to other FeIII complexes.Data availability
Magnetic data for this article are available upon request.Author contributions
Raúl Díaz-Torres: investigation, formal analysis, data curation, visualization, writing – original draft. Guillaume Chastanet: investigation, formal analysis, data curation, writing – review & editing. Eric Collet: investigation, data curation, writing – review & editing. Elzbieta Trzop: methodology, investigation, formal analysis, data curation. Phimphaka Harding: conceptualization, resources, writing – review & editing, supervision. David J. Harding: conceptualization, resources, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Research Council of Thailand (NRCT) grant number NRCT5-RSA63019-02 for funding. Walailak University is thanked for a postdoctoral research fellowship to R. D. T. The National Science Technology and Innovation Policy Office for Integrated Research and Innovation Plan (Grant No. 256113A3050001) is thanked for funds to purchase an X-ray diffractometer.Notes and references
- M. Ratner, Nat. Nanotechnol., 2013, 8, 378–381 CrossRef CAS PubMed.
- H. Song, M. A. Reed and T. Lee, Adv. Mater., 2011, 23, 1583–1608 CrossRef CAS PubMed.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1–23 CrossRef PubMed.
- J.-F. Létard, P. Guionneau and L. Goux-Capes, in Spin Crossover in Transition Metal Compounds III, Springer-Verlag, Berlin/Heidelberg, 2004, pp. 221–249 Search PubMed.
- K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef CAS.
- Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- J. Linares, E. Codjovi and Y. Garcia, Sensors, 2012, 12, 4479–4492 CrossRef CAS PubMed.
- K. A. Zenere, S. G. Duyker, E. Trzop, E. Collet, B. Chan, P. W. Doheny, C. J. Kepert and S. M. Neville, Chem. Sci., 2018, 9, 5623–5629 RSC.
- D. J. Mondal, S. Roy, J. Yadav, M. Zeller and S. Konar, Inorg. Chem., 2020, 59, 13024–13028 CrossRef CAS PubMed.
- W. Phonsri, D. J. Harding, P. Harding, K. S. Murray, B. Moubaraki, I. A. Gass, J. D. Cashion, G. N. L. Jameson and H. Adams, Dalton Trans., 2014, 43, 17509–17518 RSC.
- A. Tsukiashi, M. Nakaya, F. Kobayashi, R. Ohtani, M. Nakamura, J. M. Harrowfield, Y. Kim and S. Hayami, Inorg. Chem., 2018, 57, 2834–2842 CrossRef CAS PubMed.
- G. Dupouy, M. Marchivie, S. Triki, J. Sala-Pala, J.-Y. Salaün, C. J. Gómez-García and P. Guionneau, Inorg. Chem., 2008, 47, 8921–8931 CrossRef CAS PubMed.
- Y. Y. Peng, S. G. Wu, Y. C. Chen, W. Liu, G. Z. Huang, Z. P. Ni and M. L. Tong, Inorg. Chem. Front., 2020, 7, 1685–1690 RSC.
- F. Fürmeyer, L. M. Carrella, V. Ksenofontov, A. Möller and E. Rentschler, Inorg. Chem., 2020, 59, 2843–2852 CrossRef PubMed.
- M. Shatruk, H. Phan, B. A. Chrisostomo and A. Suleimenova, Coord. Chem. Rev., 2015, 289–290, 62–73 CrossRef CAS.
- C. Zheng, S. Jia, Y. Dong, J. Xu, H. Sui, F. Wang and D. Li, Inorg. Chem., 2019, 58, 14316–14324 CrossRef CAS PubMed.
- Y. Meng, Q. Q. Sheng, M. N. Hoque, Y. C. Chen, S. G. Wu, J. Tucek, R. Zboril, T. Liu, Z. P. Ni and M. L. Tong, Chem.–Eur. J., 2017, 23, 10034–10037 CrossRef CAS PubMed.
- Z. Y. Li, J. W. Dai, Y. Shiota, K. Yoshizawa, S. Kanegawa and O. Sato, Chem.–Eur. J., 2013, 19, 12948–12952 CrossRef CAS PubMed.
- E. Trzop, D. Zhang, L. Piñeiro-Lopez, F. J. Valverde-Muñoz, M. Carmen Muñoz, L. Palatinus, L. Guerin, H. Cailleau, J. A. Real and E. Collet, Angew. Chem., Int. Ed., 2016, 55, 8675–8679 CrossRef CAS PubMed.
- M. Griffin, S. Shakespeare, H. J. Shepherd, C. J. Harding, J. F. Létard, C. Desplanches, A. E. Goeta, J. A. K. Howard, A. K. Powell, V. Mereacre, Y. Garcia, A. D. Naik, H. Müller-Bunz and G. G. Morgan, Angew. Chem., Int. Ed., 2011, 50, 896–900 CrossRef CAS PubMed.
- Z.-Y. Li, H. Ohtsu, T. Kojima, J.-W. Dai, T. Yoshida, B. K. Breedlove, W.-X. Zhang, H. Iguchi, O. Sato, M. Kawano and M. Yamashita, Angew. Chem., Int. Ed., 2016, 55, 5184–5189 CrossRef CAS PubMed.
- K. Takahashi, K. Yamamoto, T. Yamamoto, Y. Einaga, Y. Shiota, K. Yoshizawa and H. Mori, Crystals, 2019, 9, 81 CrossRef.
- B. J. C. Vieira, J. C. Dias, I. C. Santos, L. C. J. Pereira, V. da Gama and J. C. Waerenborgh, Inorg. Chem., 2015, 54, 1354–1362 CrossRef CAS PubMed.
- S. Hayami, Z. Z. Gu, H. Yoshiki, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2001, 123, 11644–11650 CrossRef CAS PubMed.
- B. J. C. Vieira, L. C. J. Pereira, V. da Gama, I. C. Santos, A. C. Cerdeira and J. C. Waerenborgh, Magnetochemistry, 2021, 8, 1 CrossRef.
- D. J. Harding, W. Phonsri, P. Harding, K. S. Murray, B. Moubaraki and G. N. L. Jameson, Dalton Trans., 2015, 44, 15079–15082 RSC.
- I. R. Jeon, C. Mathonière, R. Clérac, M. Rouzières, O. Jeannin, E. Trzop, E. Collet and M. Fourmigué, Chem. Commun., 2017, 53, 10283–10286 RSC.
- H. J. Sheng, C. C. Xia, X. Y. Zhang, C. C. Zhang, W. J. Ji, Y. Zhao and X. Y. Wang, Inorg. Chem., 2022, 61, 12726–12735 CrossRef CAS PubMed.
- G. Azzolina, R. Bertoni and E. Collet, J. Appl. Phys., 2021, 129, 085106 CrossRef CAS.
- E. Collet and G. Azzolina, Phys. Rev. Mater., 2021, 5, 1–11 Search PubMed.
- N. Ortega-Villar, M. Muñoz and J. Real, Magnetochemistry, 2016, 2, 16 CrossRef.
- A. Hauser, Top. Curr. Chem., 2012, 234, 155–198 CrossRef.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- G. Chastanet, M. Lorenc, R. Bertoni and C. Desplanches, C. R. Chim., 2018, 21, 1075–1094 CrossRef CAS.
- T. T. Ma, X. P. Sun, Z. S. Yao and J. Tao, Inorg. Chem. Front., 2020, 7, 1196–1204 RSC.
- V. García-López, M. Palacios-Corella, S. Cardona-Serra, M. Clemente-León and E. Coronado, Chem. Commun., 2019, 55, 12227–12230 RSC.
- M. Nakaya, R. Ohtani, L. F. Lindoy and S. Hayami, Inorg. Chem. Front., 2021, 8, 484–498 RSC.
- T. Boonprab, S. J. Lee, S. G. Telfer, K. S. Murray, W. Phonsri, G. Chastanet, E. Collet, E. Trzop, G. N. L. Jameson, P. Harding and D. J. Harding, Angew. Chem., Int. Ed., 2019, 58, 11811–11815 CrossRef CAS PubMed.
- M. Nakaya, K. Shimayama, K. Takami, K. Hirata, A. S. Alao, M. Nakamura, L. F. Lindoy and S. Hayami, Chem. Lett., 2014, 43, 1058–1060 CrossRef CAS.
- S. Hayami, Z. Gu, M. Shiro, Y. Einaga, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2000, 122, 7126–7127 CrossRef CAS.
- S. Hayami, K. Hiki, T. Kawahara, Y. Maeda, D. Urakami, K. Inoue, M. Ohama, S. Kawata and O. Sato, Chem.–Eur. J., 2009, 15, 3497–3508 CrossRef CAS PubMed.
- H. Fourati, M. Ndiaye, M. Sy, S. Triki, G. Chastanet, S. Pillet and K. Boukheddaden, Phys. Rev. B, 2022, 105, 174436 CrossRef CAS.
- K.-P. Xie, Z.-Y. Ruan, X.-X. Chen, J. Yang, S.-G. Wu, Z.-P. Ni and M.-L. Tong, Inorg. Chem. Front., 2022, 9, 1770–1776 RSC.
- M. M. Ndiaye, S. Pillet, E. E. Bendeif, M. Marchivie, G. Chastanet, K. Boukheddaden and S. Triki, Eur. J. Inorg. Chem., 2018, 2018, 305–313 CrossRef CAS.
- E. Milin, V. Patinec, S. Triki, E. E. Bendeif, S. Pillet, M. Marchivie, G. Chastanet and K. Boukheddaden, Inorg. Chem., 2016, 55, 11652–11661 CrossRef CAS PubMed.
- M. Ahmed, K. A. Zenere, N. F. Sciortino, K. S. A. Arachchige, G. F. Turner, J. Cruddas, C. Hua, J. R. Price, J. K. Clegg, F. J. Valverde-Muñoz, J. A. Real, G. Chastanet, S. A. Moggach, C. J. Kepert, B. J. Powell and S. M. Neville, Inorg. Chem., 2022, 61, 6641–6649 CrossRef CAS PubMed.
- Y. C. Chen, Y. Meng, Y. J. Dong, X. W. Song, G. Z. Huang, C. L. Zhang, Z. P. Ni, J. Navařík, O. Malina, R. Zbořil and M. L. Tong, Chem. Sci., 2020, 11, 3281–3289 RSC.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC.
- N. Phukkaphan, D. L. Cruickshank, K. S. Murray, W. Phonsri, P. Harding and D. J. Harding, Chem. Commun., 2017, 53, 9801–9804 RSC.
- B. J. C. Vieira, J. T. Coutinho, I. C. Santos, L. C. J. Pereira, J. C. Waerenborgh and V. Da Gama, Inorg. Chem., 2013, 52, 3845–3850 CrossRef CAS PubMed.
- K. D. Murnaghan, C. Carbonera, L. Toupet, M. Griffin, M. M. Dîrtu, C. Desplanches, Y. Garcia, E. Collet, J. F. Létard and G. G. Morgan, Chem.–Eur. J., 2014, 20, 5613–5618 CrossRef CAS PubMed.
- F. F. Martins, A. Joseph, H. P. Diogo, M. E. Minas da Piedade, L. P. Ferreira, M. D. Carvalho, S. Barroso, M. J. Romão, M. J. Calhorda and P. N. Martinho, Eur. J. Inorg. Chem., 2018, 2976–2983 CrossRef CAS.
- K. Miyano, T. Nishida, H. Ono, D. Hamada, T. Fujinami, N. Matsumoto and Y. Sunatsuki, Inorg. Chim. Acta, 2016, 439, 49–54 CrossRef CAS.
- Z. Liu, Z. Yao and J. Tao, Inorg. Chem., 2021, 60, 10291–10301 CrossRef CAS PubMed.
- R. Kulmaczewski, E. Trzop, L. J. Kershaw Cook, E. Collet, G. Chastanet and M. A. Halcrow, Chem. Commun., 2017, 53, 13268–13271 RSC.
- V. Russell, M. Scudder and I. Dance, J. Chem. Soc., Dalton Trans., 2001, 789–799 RSC.
- J. Cruddas and B. J. Powell, Inorg. Chem. Front., 2020, 7, 4424–4437 RSC.
- K. Jonas, A. Jean-Paul, C. Renée, C. Epiphane, K. Olivier, J. G. Haasnoot, G. Françoise, J. Charlotte, A. Bousseksou, L. Jorge, V. François and G. V. Anne, Chem. Mater., 1994, 6, 1404–1412 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- R. Bertoni, M. Lorenc, J. Laisney, A. Tissot, A. Moréac, S. F. Matar, M. L. Boillot and E. Collet, J. Mater. Chem. C, 2015, 3, 7792–7801 RSC.
- M. Gakiya-Teruya, X. Jiang, D. Le, Ö. Üngör, A. J. Durrani, J. J. Koptur-Palenchar, J. Jiang, T. Jiang, M. W. Meisel, H. Cheng, X.-G. Zhang, X. Zhang, T. S. Rahman, A. F. Hebard and M. Shatruk, J. Am. Chem. Soc., 2021, 143, 14563–14572 CrossRef CAS PubMed.
- G. Chastanet, C. Desplanches, C. Baldé, P. Rosa, M. Marchivie and P. Guionneau, Chem, 2018, 1–19 Search PubMed.
- Y. S. Ye, X. Q. Chen, Y. De Cai, B. Fei, P. Dechambenoit, M. Rouzières, C. Mathonière, R. Clérac and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 18888–18891 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallographic data, additional structural, magnetic and TGA figures. CCDC 2220036–2220047. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01495a |
| ‡ Previous address: Functional Materials and Nanotechnology Centre of Excellence, Walailak University, Thasala, Nakhon Si Thammarat, 80160, Thailand |
| This journal is © The Royal Society of Chemistry 2023 |