
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum carbonyl assisted reductive tetramerization of CO by activated magnesium(I) compounds: squarate dianion vs. metallo-ketene formation†
K.
Yuvaraj‡
 a,
Jeremy C.
Mullins
a,
Thayalan
Rajeshkumar
b,
Iskander
Douair
b,
Laurent
Maron
*b and
Cameron
Jones
*a
a,
Jeremy C.
Mullins
a,
Thayalan
Rajeshkumar
b,
Iskander
Douair
b,
Laurent
Maron
*b and
Cameron
Jones
*a
aSchool of Chemistry, Monash University, PO Box 23, VIC, 3800, Australia. Web: http://www.monash.edu/science/research-groups/chemistry/jonesgroupE-mail: cameron.jones@monash.edu; Web: https://twitter.com/Jones_Research
bUniversité de Toulouse et CNRS, INSA, UPS, LPCNO, UMR 5215, 135 Avenue de Rangueil, F-31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 24th April 2023
Abstract
Reactions of a dimagnesium(I) compound, [{(DipNacnac)Mg}2] (DipNacnac = [HC(MeCNDip)2]−, Dip = 2,6-diisopropylphenyl), pre-activated by coordination with simple Lewis bases (4-dimethylaminopyridine, DMAP; or TMC, :C(MeNCMe)2), with 1 atmosphere of CO in the presence of one equivalent of Mo(CO)6 at room temperature, led to the reductive tetramerisation of the diatomic molecule. When the reactions were carried out at room temperature, there is an apparent competition between the formation of magnesium squarate, [{(DipNacnac)Mg}{cyclo-(κ4-C4O4)}{μ-Mg(DipNacnac)}]2, and magnesium metallo-ketene products, [{(DipNacnac)Mg}[μ-O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC{Mo(CO)5}C(O)CO2]{Mg(D)(DipNacnac)}], which are not inter-convertible. Repeating the reactions at 80 °C led to the selective formation of the magnesium squarate, implying that this is the thermodynamic product. In an analogous reaction, in which THF is the Lewis base, only the metallo-ketene complex, [{(DipNacnac)Mg}[μ-OCC{Mo(CO)5}C(O)CO2]{Mg(THF)(DipNacnac)}] is formed at room temperature, while a complex product mixture is obtained at elevated temperature. In contrast, treatment of a 1
CC{Mo(CO)5}C(O)CO2]{Mg(D)(DipNacnac)}], which are not inter-convertible. Repeating the reactions at 80 °C led to the selective formation of the magnesium squarate, implying that this is the thermodynamic product. In an analogous reaction, in which THF is the Lewis base, only the metallo-ketene complex, [{(DipNacnac)Mg}[μ-OCC{Mo(CO)5}C(O)CO2]{Mg(THF)(DipNacnac)}] is formed at room temperature, while a complex product mixture is obtained at elevated temperature. In contrast, treatment of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the guanidinato magnesium(I) complex, [(Priso)Mg–Mg(Priso)] (Priso = [Pri2NC(NDip)2]−), and Mo(CO)6, with CO gas in a benzene/THF solution, gave a low yield of the squarate complex, [{(Priso)(THF)Mg}{cyclo-(κ4-C4O4)}{μ-Mg(THF)(Priso)}]2, at 80 °C. Computational analyses of the electronic structure of squarate and metallo-ketene product types corroborate the bonding proposed from experimental data, for the C4O4 fragments of these systems.
:1 mixture of the guanidinato magnesium(I) complex, [(Priso)Mg–Mg(Priso)] (Priso = [Pri2NC(NDip)2]−), and Mo(CO)6, with CO gas in a benzene/THF solution, gave a low yield of the squarate complex, [{(Priso)(THF)Mg}{cyclo-(κ4-C4O4)}{μ-Mg(THF)(Priso)}]2, at 80 °C. Computational analyses of the electronic structure of squarate and metallo-ketene product types corroborate the bonding proposed from experimental data, for the C4O4 fragments of these systems.
Introduction
Carbon monoxide is produced on an industrial scale by processes including the partial oxidation of methane, and the gasification of coal or biomass.1 The gas is used as a C1 feedstock in a variety of processes, which produce value-added commodity chemicals on mega-tonne scales per annum. Of most relevance to this study is the Fischer–Tropsch process (FT), which transforms mixtures of CO and H2 (syngas) into liquid hydrocarbons and oxygenates, typically in the presence of heterogeneous transition metal catalysts, and under elevated temperatures and pressures.2 While little success has been had catalysing FT under homogeneous conditions, a great deal of work has centred on the use of organometallic compounds to study the fundamental steps of this process in solution.3 Such studies have led to a growing understanding of how the very strong bond in CO (BDE = 257 kcal mol−1)4 can activated and functionalized by reactive metal centres.With respect to the study of C–C bond forming processes, the reductive homologation of CO into a good number of cyclic and acyclic C–C coupled oxo-carbon anions, [CmOm]n− (m, n > 1), has been achieved by reaction of the gas with low-valent complexes involving electropositive elements from the p, d- and f-blocks of the periodic table.5 In 2019 we extended this work to the s-block for the first time, with the reductive trimerization of CO to the deltate complex 1 (Fig. 1), by dimagnesium(I) complexes [(DipNacnac)Mg–Mg(D)(DipNacnac)] ((DipNacnac) = [HC(MeCNDip)2]−, Dip = 2,6-diisopropylphenyl; D = Lewis base), which were activated by coordination of one Mg center with a simple Lewis base.6,7 It is of note that in the absence of Lewis bases (D), three-coordinate dimagnesium(I) compounds, [LMg–MgL] (L = β-diketiminate or guanidinate), are unreactive towards CO under ambient conditions.6 The only other report of deltate formation from CO reduction came from Cloke and co-workers, who used a uranium(III) complex as the reductant.8 They, and others, later showed that ethynediolate and squarate ([CmOm]2−, m = 2 and 4 respectively) complexes can also be selectively accessed by altering the steric profile of the f-block organometallic reductant.9 In a similar vein, when we reacted the less bulky dimagnesium(I) compounds [(ArNacnac)Mg–Mg(DMAP)(ArNacnac)] (Ar = mesityl (Mes) or 2,6-xylyl (Xyl); DMAP = 4-dimethylaminopyridine) with CO, reductive dimerization of the gas to [C2O2]2−, followed by C–H activation of DMAP, yielded the ethenediolates, 2.10,11
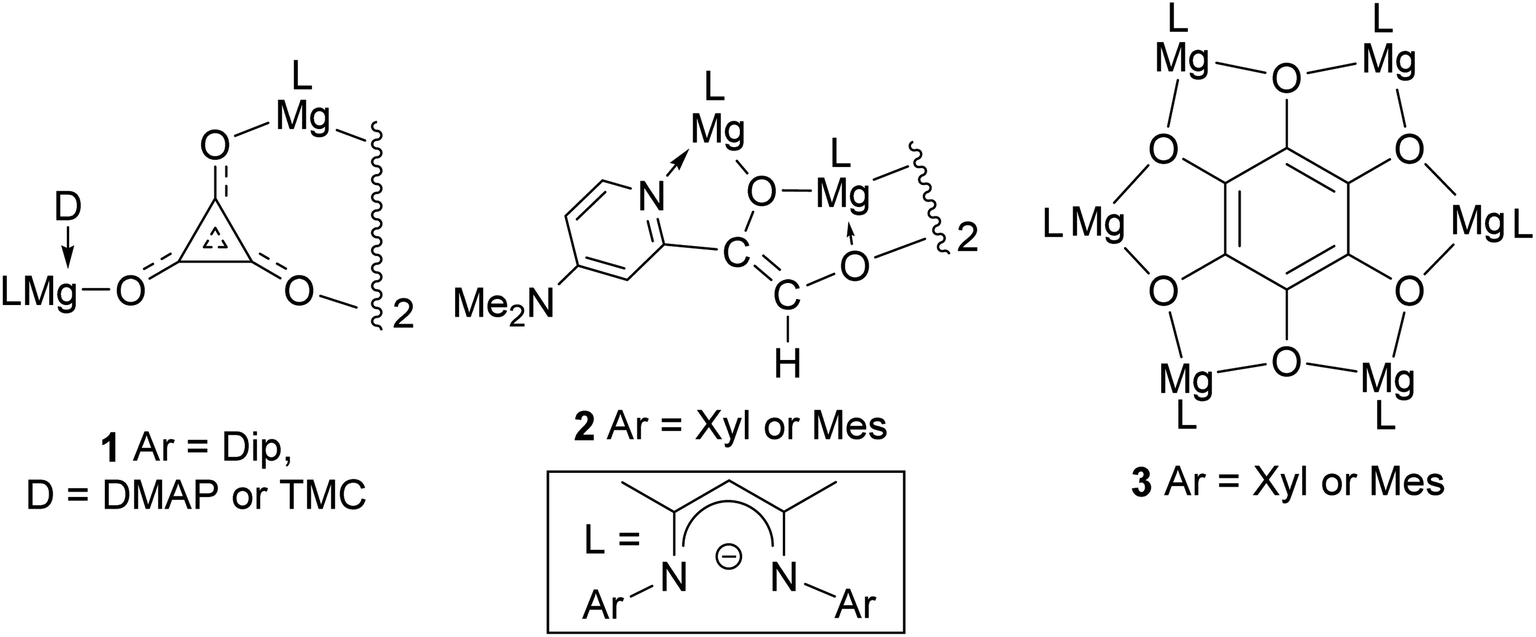 | ||
| Fig. 1 Previously described products derived from the reductive homologation of CO by neutral dimagnesium(I) compounds. | ||
As an alternative to Lewis base activation of dimagnesium(I) compounds, we have shown that cooperative heterobimetallic strategies for CO homologation are effective. For example, treating [(ArNacnac)Mg–Mg(ArNacnac)] (Ar = Xyl or Mes) with CO in the presence of catalytic Mo(CO)6 led to reductive hexamerization of the gas, and formation of the benzenehexolate complexes, 3.12 These represented the first well-defined benzenehexolate complexes derived from CO, though it is noteworthy that K6C6O6 was proposed by Liebig to have been formed in the reaction between molten potassium and CO, as long ago as 1834.13 Given our success using dimagnesium(I) compounds for the selective reductive dimerization, trimerization and hexamerization of CO, we were keen to explore routes to other C–C coupled oligomers of the diatomic molecule. We were especially interested in squarate formation, [C4O4]2−, given the results of a recent study that showed MgCl− to reduce CO to squarate in the gas phase.14 Here, we reveal that by combining dimagnesium(I) activation by simple Lewis base coordination, and a cooperative heterobimetallic strategy, selective reductive tetramerizations of CO can, indeed, be achieved. The two CO derived product types obtained using this approach, magnesium squarates and magnesium metallo-glyoxylatoketenes (hereafter, on occasion, abbreviated as metallo-ketenes), are unprecedented as isolated species. Moreover, the distribution of the two products can be readily altered by manipulating the reaction conditions. The electronic structure of the products of these reactions has been explored using DFT calculations.
Results and discussion
At the outset of this study the reaction between [(DipNacnac)Mg–Mg(DMAP)(DipNacnac)] and CO, in the presence of 25 mol% Mo(CO)6, was carried out at room temperature. This led to isolation of low yields of the colourless squarate and orange metallo-ketene complexes, 4 and 5 respectively, in addition to a trace of magnesium deltate 1 (D = DMAP) (Scheme 1). This result shows that, unlike for the synthesis of benzenehexolate 3, Mo(CO)6 is largely consumed in the reaction and does not act as a catalyst, at least in the formation of 5. As a result, the reaction was repeated, but using one equivalent of Mo(CO)6. This led to increased isolated yields of 4 and 5 (and no 1), with 5 being the predominant product. Similar reactions were subsequently carried out with TMC (:C(MeNCMe)2) and THF adducts of the dimagnesium(I) compound.15 The former gave a mixture of squarate 4 and metallo-ketene 6, whereas only the metallo-ketene 7 was isolated from the latter reaction, and no squarate 4 was observed in the reaction mixture (as determined by an 1H NMR spectroscopic analysis). In order to determine if the metallo-ketene products, 5–7, are intermediates in the formation of squarate, 4, benzene solutions of the compounds were heated to 80 °C under atmospheres of CO. In no case was squarate 4 formed, suggesting a competition exists between distinct reaction pathways to the squarate and metallo-ketene products. That the metallo-ketenes do not convert to squarate 4 is perhaps not surprising, considering that the formation of the former must involve at least one C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond cleavage. Transformation to 4 would require O-migration through formal C–O bond cleavage and formation steps, ring closure through formation of a C–C bond, and loss of the Mo(CO)5 Lewis base fragment.
O bond cleavage. Transformation to 4 would require O-migration through formal C–O bond cleavage and formation steps, ring closure through formation of a C–C bond, and loss of the Mo(CO)5 Lewis base fragment.
 | ||
| Scheme 1 Synthesis of compounds 4–7 at either room temperature or 80 °C (isolated yields given in parentheses). | ||
So as to determine if the product distributions obtained from the reactions involving DMAP and TMC activated systems are affected by reaction temperature, they were repeated, but with heating at 80 °C overnight. In both cases squarate 4 was isolated in higher yields than obtained for the corresponding room temperature reactions, and no metallo-ketene products were recovered. This may indicate that squarate 4 is the thermodynamic product, whereas metallo-ketenes 5 and 6 are kinetic products. In addition, the fate of the molybdenum carbonyl, and DMAP and TMC Lewis bases, in the formation of 4 appears to be generation of the cis-adducts [Mo(CO)4(D)2] (D = DMAP or TMC), which were isolated from the respective reactions, and crystallographically characterised. These compounds were subsequently rationally synthesized and spectroscopically characterised, which allowed confirmation that they are also low yield products in the associated reactions carried out at room temperature. Moreover, the formation of stable cis-[Mo(CO)4(D)2] seemingly shuts down any catalytic activity of Mo(CO)6, as was important in the formation of magnesium benzenehexolate 3.12
It is interesting that no squarate complex was formed in the reaction that gave 7. Indeed, when that reaction was repeated at elevated temperature, squarate 4 was again not isolated from the reaction mixture, which an 1H NMR spectroscopic analysis showed to contain a complex mixture of products. The differences with the reactions involving DMAP or TMC activated dimagnesium(I) compounds might stem from the fact that cis-[Mo(CO)4(THF)2] is not a stable compound (see below), whereas the stability of cis-[Mo(CO)4(D)2] (D = DMAP or TMC) might help drive formation of squarate 4. To explore this further, [(DipNacnac)Mg–Mg(DipNacnac)] was substituted with the related guanidinate coordinated system [(Priso)Mg–Mg(Priso)] (Priso = [Pri2NC(NDip)2]−)16 in a potentially THF activated reaction.17 Although a reaction did occur at room temperature, it was very slow, with only a fraction of the dimagnesium(I) compound being consumed overnight. However, repeating the reaction at 80 °C overnight, afforded a low yield (17%) of the colourless THF coordinated squarate compound 8, upon work-up (Scheme 2). It is not known why squarate formation occurs, whereas it does not in the reaction involving [(DipNacnac)Mg–Mg(DipNacnac)] in the presence of THF. The fact that the Mg centres in 8 are THF coordinated is likely a result of them being more sterically accessible than those in squarate complex 4.
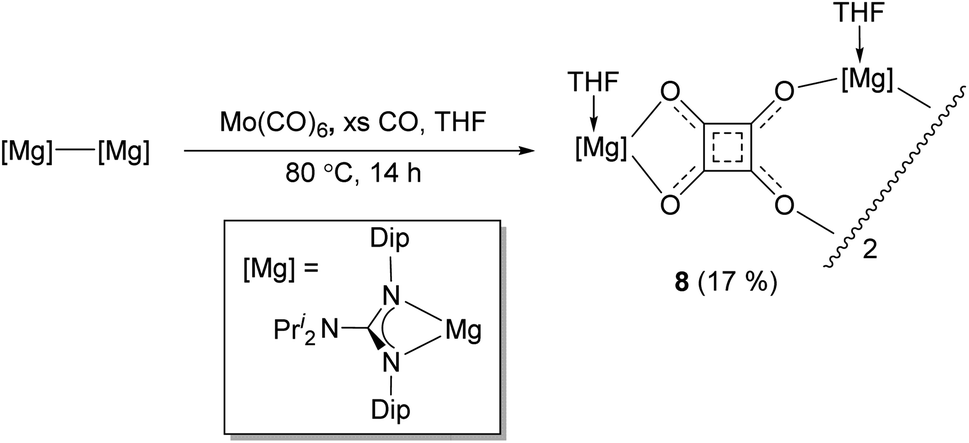 | ||
| Scheme 2 Synthesis of compound 8 (isolated yield given in parentheses). | ||
Once crystallised, squarate 4 is poorly soluble in non-coordinating deuterated solvents, and decomposes upon attempts to dissolve it in neat d8-THF. As a result, meaningful solution state 13C{1H} NMR spectroscopic data could not be obtained. Despite the 1H NMR spectrum of the compound in d6-benzene displaying very broad signals, the spectral pattern is consistent with its solid-state structure. Similarly, the guanidinato-magnesium squarate complex 8 is poorly soluble in d6-benzene and d8-toluene, though 1H and 13C{1H} NMR spectra of the compound, contaminated with significant amounts of the soluble guanidine decomposition product, PrisoH,16 were obtained and tentatively assigned. No signals for the squarate carbon centres were observed in the 13C{1H} NMR spectrum, so the compound was prepared under an atmosphere of 13CO, and the spectrum acquired before work-up. This showed two signals at δ 231.4 and 237.7 ppm, which were assigned to two inequivalent sets of [C4O4]2− carbon centres, as would be expected if the compound retained its solid-state structure in solution. It is noteworthy that the only molybdenum carbonyl signal observed in this spectrum was for Mo(CO)6 (δ = 201.3 ppm), though the presence of other insoluble molybdenum carbonyl products in the reaction suspension cannot be ruled out (e.g. as resulting from the decomposition of the transiently generated, and unstable by-product Mo(CO)4(THF)2, cf. stable [Mo(CO)4(D)2]; D = DMAP or TMC; isolated when 4 was prepared in the presence of DMAP or TMC).
As was the case for 4, the 1H NMR spectra of the metallo-ketene complexes 5–7, each exhibit sets of signals for two chemically inequivalent DipNacnac ligands, as would be expected for the solid-state structures of the compounds being preserved in solution. The poor solubility of the compounds hampered the acquisition of 13C{1H} NMR spectra of sufficient quality to enable the assignment of signals for the carbon centres of the central C4O4 fragment. Accordingly, compound 7 was prepared under an atmosphere of 13CO, and its 13C{1H} NMR spectrum recorded as a d6-benzene solution. That enabled doublet signals at δ 167.6 and 190.4 ppm (1JCC = 61 Hz) to be assigned to the backbone carbons of the C2O3 glyoxylato fragment (C(1) and C(2) in the solid-state structure, see ESI†), while the ketene carbon, CCO, was tentatively assigned as a singlet δ 170.3 ppm. No resonance was observed for the Mo coordinated ketenyl carbon (MoC = CO), perhaps due to signal broadening. It is noteworthy that the doublet signals were superimposed on singlet resonances, of similar intensity, at their centre point. This confirms that the two carbons giving rise to those signals can originate from either 13CO or Mo(12CO)6. This is likely also the case for the other carbons of the C4O4 unit. Further to this, the intensity of the 13C NMR signals for the coordinated Mo(CO)5 unit reveal that appreciable 13CO for 12CO substitution within that unit has occurred during the reaction.
The X-ray crystal structures of squarate complexes 4 and 8 were determined, and their molecular structures are depicted in Fig. 2 and 3, respectively. They are both dimers in which dianionic squarate units are bridged by four-coordinate (DipNacnac)Mg or five-coordinate (Priso)(THF)Mg cations, giving rise to non-planar C4O4Mg2 ten-membered rings. In addition, each squarate fragment chelates another magnesium centre. The C–C and C–O bond lengths within each of the planar squarate moieties are similar, and lie between those normally seen for single and double bonds. This strongly suggests significant delocalisation over the [C4O4]2− units, as has been proposed by Cloke and co-workers for related uranium(IV) squarates, e.g. [[U(η-C8H6{SiPri3}2-1,4)(η-C5Me4H)]2(μ-κ2:κ2-C4O4)].9a
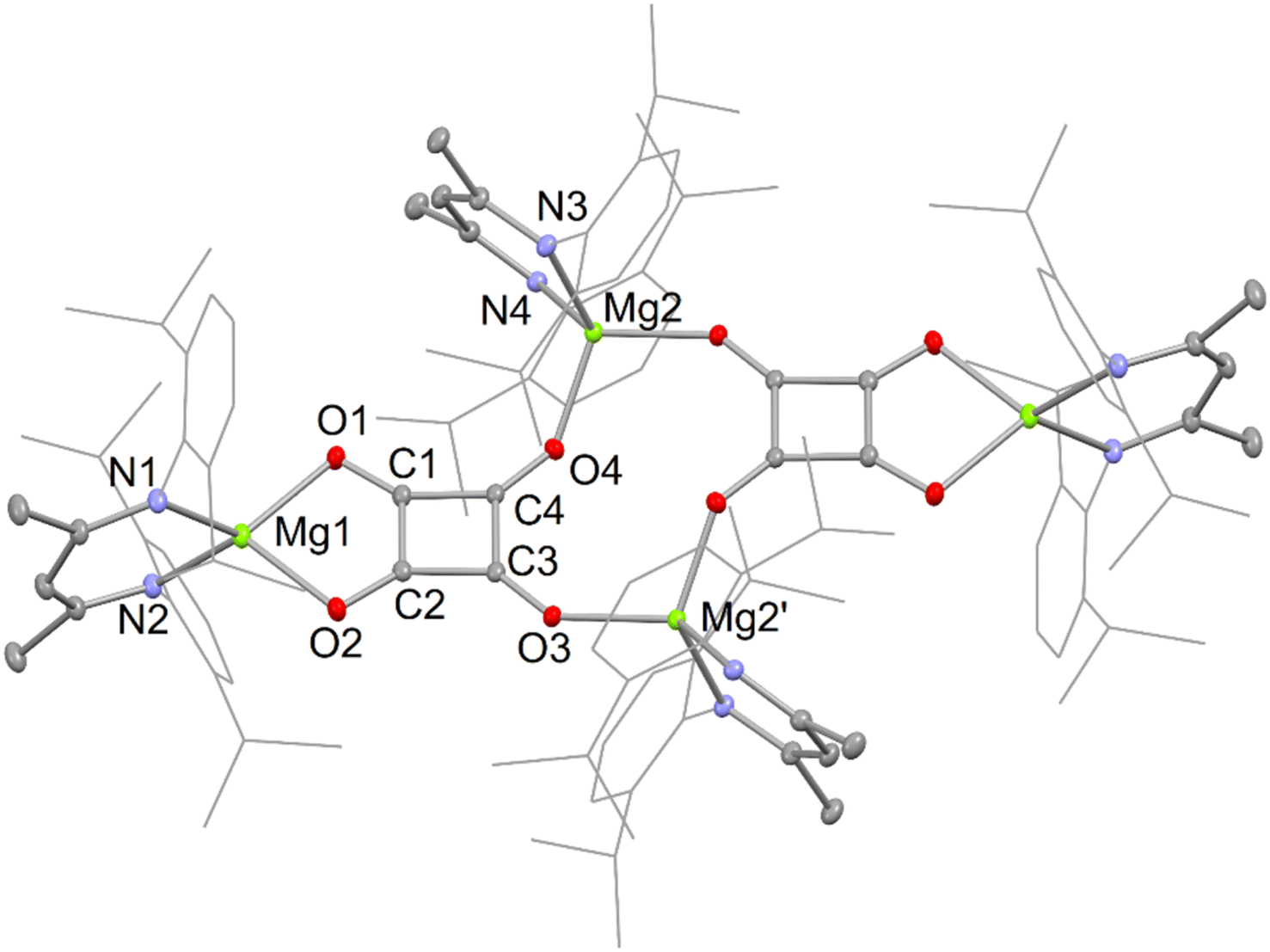 | ||
| Fig. 2 Molecular structure of 4 (25% thermal ellipsoids; hydrogen atoms omitted; Dip substituents shown as wire-frame for sake of clarity). Selected bond lengths (Å) and angles (°): Mg(1)–O(2) 2.056(3), Mg(1)–O(1) 2.079(2), O(1)–C(1) 1.259(4), C(1)–C(2) 1.430(5), C(1)–C(4) 1.464(4), Mg(2)–O(4) 1.935(2), Mg(2)′–O(3) 1.953(2), O(2)–C(2) 1.259(4), C(2)–C(3) 1.463(4), O(3)–C(3) 1.244(4), C(3)–C(4) 1.473(4), O(4)–C(4) 1.244(4), O(2)–Mg(1)–O(1) 88.16(9), C(3)–O(3)–Mg(2)′ 138.5(2), C(4)–O(4)–Mg(2) 143.2(2). | ||
 | ||
| Fig. 3 Molecular structure of 8 (25% thermal ellipsoids; hydrogen atoms omitted; Dip, THF, and isopropyl substituents shown as wire-frame for sake of clarity). Selected bond lengths (Å) and angles (°): Mg(1)–O(5) 2.0285(12), Mg(1)–O(2) 2.0853(12), Mg(1)–O(1) 2.1468(12), O(1)–C(1) 1.2606(17), C(1)–C(2) 1.4384(19), C(1)–C(4) 1.465(2), Mg(2)–O(3) 1.9378(12), Mg(2)′–O(4) 1.9535(12), O(2)–C(2) 1.2616(17), C(2)–C(3) 1.461(2), O(3)–C(4) 1.2484(17), C(3)–O(4) 1.2445(18), C(3)–C(4) 1.475(2), O(2)–Mg(1)–O(1) 86.92(5), C(4)–O(3)–Mg(2) 168.16(10), C(3)–O(4)–Mg(2)′ 155.73(10). | ||
In the solid state, compounds 5–7 are essentially isostructural. Accordingly, only the molecular structure of one of these, 6, is depicted in Fig. 4, while those of the other two can be found in the ESI.† The compounds include a central C4O4 fragment which is κ1-coordinated to Mo(CO)5 and (DipNacnac)(D)Mg (D = DMAP, TMC or THF) units, while also chelating a (DipNacnac)Mg cation. We are unaware of any structurally characterised C4O4 units, similar to those in 5–7, which can be considered as glyoxylato-ketenes. While unique, they are closely related to several f-block carboxylato-ketene complexes formed by reductive trimerisation of CO, e.g. [(CptttTm)2(μ–O2C–CCO)] (Cpttt = [C5H2But3-1,2,4]−).18 Indeed, the glyoxylatoketene unit in 5–7 can be viewed as the product of a formal insertion of CO into the C–C single bond of the carboxylato-ketene fragment, as found in [(CptttTm)2(μ–O2C–CCO)]. An examination of the bond lengths within the C4O4 fragment is instructive. There appears to be significant electronic delocalisation over the C(1)O2 unit, while the C(1)–C(2) bond is clearly single, signifying carboxylate-character within that C2O2 moiety. Interestingly, the bond lengths within the C3O2 fragment containing C(3) are indicative of some charge delocalisation over that fragment, which could be viewed as a metallo-ketene (as depicted in Scheme 1), or as having partial carbenic character at C(3), with a build up of negative charge at O(3) [cf. Crimmin's {(DipNacnac)Al}2{(μ-C4O4)M(CO)5}], M = Mo or W,19 complexes derived from AlI induced CO tetramerisation). In this respect, the C–Mo distance in 6 is at the upper end of the range for reported (CO)5Mo–C(3-coord) bond lengths.20
 | ||
| Fig. 4 Molecular structure of 6 (25% thermal ellipsoids are shown; hydrogen atoms omitted; Dip substituents shown as wireframe for sake of clarity). Selected bond lengths (Å) and angles (°): Mo(1)–C(3) 2.331(3), Mg(1)–C(10) 2.226(3), O(1)–C(1) 1.248(3), C(1)–O(2) 1.258(3), C(1)–C(2) 1.546(3), C(2)–O(3) 1.273(3), C(2)–C(3) 1.384(3), C(3)–C(4) 1.325(4), O(4)–C(4) 1.157(3), O(1)–C(1)–O(2) 126.2(2), O(1)–C(1)–C(2) 117.9(2), O(2)–C(1)–C(2) 115.8(2), O(3)–C(2)–C(3) 125.2(2), O(3)–C(2)–C(1) 113.9(2), C(3)–C(2)–C(1) 120.9(2), C(4)–C(3)–C(2) 121.5(2), C(4) C(3)–Mo(1) 108.32(18), C(2)–C(3)–Mo(1) 129.80(18), O(4)–C(4)–C(3) 170.8(3). | ||
Despite significant efforts, the complexity of the four-component reactions that gave 4–7 thwarted our attempts to compute cogent reaction pathways to these compounds. However, the nature of the bonding in complexes 4 and 6 was investigated at the DFT level (B3PW91), as this methodology has already been successfully applied to describe CO homologation in other metal systems.6,7,9,12 The optimized geometry of compound 4 compares well with that displayed in the solid state. For example, the C–C and C–O bonds within the squarate moiety are perfectly reproduced (1.46 and 1.24 Å). These distances are in line with a significant degree of electronic delocalisation over the C4O4 units. The latter was corroborated by a Natural Bonding Orbital (NBO) analysis. The associated Wiberg Bond Indexes (WBI) for the C–O units are 1.40, indicating a strong covalent character to those bonds. The somewhat reduced C–O WBI, relative to what would be expected for localised CO double bonds, is partially explained by some delocalization of the CO electron density onto an empty orbital on Mg, as highlighted in the second order donor–acceptor NBO level. The CO π bond is also found to be delocalized onto the C4 ring, giving rise to some C–C π-bond character. The latter is highlighted by the C–C WBI of 1.10. In addition, this delocalisation is clearly seen in the molecular orbitals associated with the squarate fragments (e.g. HOMO-17 in Fig. 5(a); see also Fig. S21 in the ESI†). The Mg–O bonds in 4 were found to possess strong donor–acceptor characteristics (while having high ionic character), with donation from the oxygen σ lone pair to an empty Mg orbital, and very little donation from the C–O π bonds. In concert, this results in very low Mg–O WBIs of ca. 0.1.
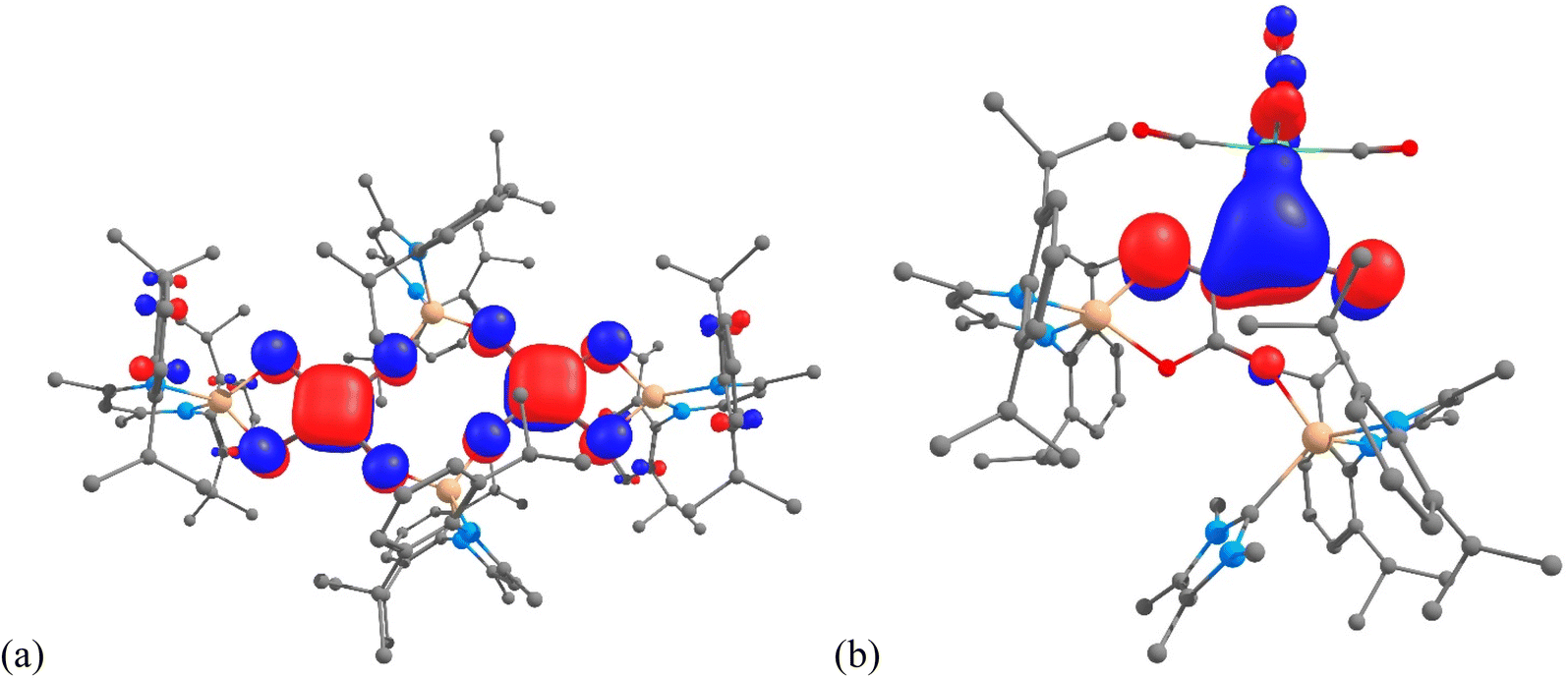 | ||
| Fig. 5 (a) HOMO-17 of 4, and (b) HOMO-13 of 6, calculated (B3PW91) for the molecules in the gas phase. | ||
A similar analysis was carried out on compound 6, which incorporates the NHC TMC as a ligand. As was the case with 4, the optimized geometry compares well with that obtained from the X-ray crystallographic analysis. For example, the Mg–C distances are within 0.04 Å, while the Mo–C separations (2.34 vs. 2.331(3) Å) are essentially identical. Moreover, all the C–C and C–O bond distances within the C4O4 core of the molecule are well reproduced, with a maximum deviation of 0.01 Å. In line with these bond distances, the NBO analysis clearly indicates the presence of a carboxylate moiety, with the two associated delocalised C–O π-bonds (WBIs = 1.37 and 1.40). The carboxylate moiety is linked to the Mo-coordinated C3O2 fragment via a C–C single bond (WBI = 0.97). Both C–C interactions within that C3O2 unit display significant double bond character (WBIs of 1.37 and 1.62), as is the case for the two C–O bonds in that unit (WBIs of 1.37 and 1.94). Therefore, the C3O2 moiety can be seen as a ketenyl unit interacting with Mo through a Mo–C bond that has a WBI of 0.29. While this interaction is not highly covalent, an inspection of the MOs clearly shows a π interaction, associated with HOMO-13 (Fig. 5(b), see also Fig. S22†), delocalised over the Mo–C–C unit. The metallo-ketene nature of the complex is also highlighted by the natural charges, which show Mo and the attached ketenyl C-centre to be negatively charged (−1.00 and −0.53, respectively).
Conclusions
In summary, reactions of a dimagnesium(I) compound, activated by simple pyridine or NHC Lewis base coordination, with CO at room temperature, in the presence of one equivalent of Mo(CO)6, led to the reductive tetramerisation of the gas. There is an apparent competition between the formation of magnesium squarate and magnesium metallo-ketene products in these reactions, which are not inter-convertible, and have no precedent in the literature. Raising the temperature of these reactions to 80 °C led to the selective formation of the magnesium squarate, implying that this is the thermodynamic product, while the metallo-ketenes are kinetic products. Computational analyses of the electronic structure of the two product types corroborates electronic delocalization within the squarate complex, as well as the metallo-ketene nature of the molybdenum complexes, as proposed from the experimental data.The results of this and prior studies show that the degree of magnesium(I) induced reductive oligomerisation of CO, can be readily controlled by altering the method(s) of magnesium(I) reagent activation, ligand steric bulk, and/or the reaction conditions. To date, this has allowed us to selectively access reduced CO dimeric, trimeric, hexameric, and now tetrameric products. With respect to the potential applicability of the latter, s-block metal squarates (cf.4) have, for example, been proposed as electro-active materials (e.g. as anode components in rechargeable batteries),21 while s-block metallo-ketenes have very recently been revealed as powerful ketenylation reagents in organic synthesis.22 We continue to explore the selective dimagnesium(I) induced homologation of CO into value-added products and materials, while gaining an understanding of the mechanisms of such reactions, and their relevance to industrial processes utilising CO as a C1 synthetic building block (e.g. FT).
Experimental section
Full synthetic, spectroscopic and crystallographic details for new compounds; and full details and references for the DFT calculations can be found in the ESI.†Data availability
Crystallographic data for compounds 4–8, cis-[Mo(CO)4(DMAP)2] and cis-[Mo(CO)4(TMC)2] have been deposited at the Cambridge Crystallographic Database under accessing numbers 2250277–2250283.Author contributions
K. Yuvaraj and J. C. Mullins performed the synthetic experimental work; K. Yuvaraj, J. C. Mullins and C. Jones interpreted the X-ray diffraction analysis; T. Rajeshkumar, I. Douair and L. Maron performed and interpreted the computational studies; C. Jones conceptualized the research, acquired funding, wrote the manuscript and supervised the work; All authors revised and edited the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
CJ thanks the Australian Research Council and the US Air Force Asian Office of Aerospace Research and Development (grant FA2386-21-1-4048). LM is a senior member of the Institut Universtaire de France and thanks the Humboldt Foundation.Notes and references
- (a) D. Unruh, K. Pabst and G. Schaub, Energy Fuels, 2010, 24, 2634–2641 CrossRef CAS; (b) N. Dahmen, E. Henrich, E. Dinjus and F. Weirich, Energy Sustain. Soc., 2012, 2, 1–44 Search PubMed; (c) S. S. Ail and S. Dasappa, Renewable Sustainable Energy Rev., 2016, 58, 267–286 CrossRef CAS.
- See for example: (a) Advances in Fischer–Tropsch Synthesis Catalysts and Catalysis, ed. B. H. Davis and M. L. Occelli, CRC, Boca Raton, FL, 2009 Search PubMed; (b) A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed; (c) C. K. Rofer-DePoorter, Chem. Rev., 1981, 81, 447–474 CrossRef CAS; (d) C. Masters, Adv. Organomet. Chem., 1979, 17, 61–103 CrossRef CAS.
- See for example: (a) L. D. Durfee and I. P. Rothwell, Chem. Rev., 1988, 88, 1059–1079 CrossRef CAS; (b) G. Erker, Acc. Chem. Res., 1984, 17, 103–109 CrossRef CAS; (c) J. A. Gladysz, Adv. Organomet. Chem., 1982, 20, 1–38 CrossRef CAS; (d) P. T. Wolczanski and J. E. Bercaw, Acc. Chem. Res., 1980, 13, 121–127 CrossRef CAS; (e) J. M. Parr and M. R. Crimmin, Angew. Chem., Int. Ed., 2023, 62, e202219203 CrossRef CAS PubMed.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2013, 117, 8981–8995 CrossRef CAS PubMed.
- (a) R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC; (b) S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- (a) K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed; (b) N.B. CO homologated products were subsequently shown to be formed in reactions between alkali metal amides and CO or syngas; see, for example, M. Xu, Z.-W. Qu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 634–638 CrossRef CAS PubMed.
- N.B. A related CO trimerization by a β-diketiminato magnesium hydride, yielding a cyclopropantriolate complex, had previously been reported by our group; see: R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829–831 CrossRef CAS PubMed.
- (a) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602–9603 CrossRef CAS PubMed; (b) A. S. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitkin, J. Am. Chem. Soc., 2008, 130, 13816–13817 CrossRef CAS PubMed; (c) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC; (d) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed; (e) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 9265–9270 CrossRef CAS PubMed; (f) N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353–1362 CrossRef CAS; (g) I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242–11243 CrossRef CAS PubMed; (h) A. J. Ryan, J. W. Ziller and W. J. Evans, Chem. Sci., 2020, 11, 2006–2014 RSC.
- K. Yuvaraj, I. Douair, D. D. L. Jones, L. Maron and C. Jones, Chem. Sci., 2020, 11, 3516–3522 RSC.
- N.B. Hill and co-workers subsequently reported the reductive dimerisation of CO to ethynediolate, by reaction with an anionic dimagnesium(I) compound; see: H.-T. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- A. Paparo, K. Yuvaraj, A. J. R. Matthews, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 630–634 CrossRef CAS PubMed.
- (a) J. Liebig, Ann. Chem. Pharmacie, 1834, 11, 182–189 CrossRef; (b) For an historical overview of this work, see: U. Rosenthal, Chem. Eur. J., 2020, 26, 14507–14511 CrossRef CAS PubMed.
- J. S. Jestilä, Z. Iker, M. J. O. Ryding and E. Uggerud, Org. Biomol. Chem., 2020, 18, 9499–9510 RSC.
- N.B. Although isolated 1:1 THF adduct, [(DipNacnac)Mg–Mg(THF)(DipNacnac)], was not used in this reaction, it is assumed to be transiently formed by loss of THF from the known 2:1 adduct, [{(DipNacnac)(THF)Mg}2], which has been reported to be facile; see
(a) S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed. It is of further note that 1:1 adducts of THF with other dimagnesium(I) compounds have been reported, see:
(b) A. J. Boutland, D. Dange, A. Stasch, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2016, 55, 9239–9243 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- Although no THF adduct of [(Priso)Mg–Mg(Priso)] has been isolated, it is assumed that a transient 1:1 THF adduct of the compound is generated in solution; c.f. [(DipNacnac)Mg–Mg(THF)(DipNacnac)].
- (a) T. Simler, K. N. McCabe, L. Maron and G. Nocton, Chem. Sci., 2022, 13, 7449–7461 RSC; (b) W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef CAS; (c) W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed; (d) R. J. Ward, I. del Rosal, S. P. Kelley, L. Maron and J. R. Walensky, Chem. Sci., 2023, 14, 2024–2032 RSC.
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed.
- As determined from a survey of the Cambridge Crystallographic Database, March, 2023 Search PubMed.
- A. Gosh and S. Mitra, ChemElectroChem, 2018, 5, 159–165 CrossRef.
- (a) M. Jorges, F. Krischer and V. H. Gessner, Science, 2022, 378, 1331–1336 CrossRef PubMed; (b) R. Wei, X.-F. Wang, D. A. Ruiz and L. L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202219211 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2250277–2250283. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01487h |
| ‡ Current address: Department of Chemistry, Indian Institute of Technology Palakkad, Palakkad-678557, Kerala, India. |
| This journal is © The Royal Society of Chemistry 2023 |