
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric rhodium-catalyzed click cycloaddition to access C–N axially chiral N-triazolyl indoles†
Li
Zhou‡
a,
Yankun
Li‡
a,
Shunian
Li
a,
Zhenwei
Shi
a,
Xue
Zhang
a,
Chen-Ho
Tung
 a and
Zhenghu
Xu
*abc
a and
Zhenghu
Xu
*abc
aKey Lab for Colloid and Interface Chemistry of Education Ministry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: xuzh@sdu.edu.cn
bState Key Laboratory of Organometallic Chemistry Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, PR China
cKey Laboratory of Precise Synthesis of Functional Molecules of Zhejiang Province, School of Science, Westlake University, No. 18 Shilongshan Road, Hangzhou 310024, China
First published on 20th April 2023
Abstract
The copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is regarded as a prime example of “click chemistry”, but the asymmetric click cycloaddition of internal alkynes still remains challenging. A new asymmetric Rh-catalyzed click cycloaddition of N-alkynylindoles with azides was developed, providing atroposelective access to C–N axially chiral triazolyl indoles, a new type of heterobiaryl, with excellent yields and enantioselectivity. This asymmetric approach is efficient, mild, robust and atom-economic, and features very broad substrate scope with easily available Tol-BINAP ligands.
Introduction
Axially chiral heterobiaryls are common in natural products and bioactive molecules and also as crucial scaffolds of chiral ligands or catalysts.1 Catalytic asymmetric construction of these axially chiral molecules has become one of the most challenging yet fascinating issues in contemporary organic synthesis.2 Compared to the well-developed C–C axial chirality construction, the catalytic atroposelective construction of C–N axial chirality3 has been much less developed until recently. In particular, C–N atropisomeric indole derivatives,4,5 a type of privileged 5-membered heterocycle, widely exist in bioactive natural products such as polybrominated bisindoles and murrastifoline F and in chiral ligands (Scheme 1a).6 However, due to the lower rotation barrier caused by high rotational freedom of the C–N axis and shrinked size of the five-membered ring, catalytic atroposelective synthesis of such C–N axially chiral indole frameworks remains a formidable challenge and only limited examples have been reported to date. Atroposelective de novo construction of indole rings via a palladium-catalyzed or organocatalytic asymmetric intramolecular hydroamination of 2-alkynylaniline has been developed by the Kitagawa group5a in 2010 and the Ye group5b in 2022 (Scheme 1b). Very recently, Tan and coworkers reported a chiral phosphoric acid-catalyzed atroposelective construction of axially chiral N-aryl indoles via direct C–N bond formation,5c and a palladium-catalyzed desymmetrical C–H alkenylation of pre-existing C–N axial has also been developed (Scheme 1b).5d From the retro-analysis of N-aryl axially chiral indoles, it was found that the de novo construction of an N-attached aromatic or heteroaromatic ring is an attractive synthetic method, yet this hasn't been reported to date. Herein we report the first example of such a reaction, which applied a chiral-rhodium catalyzed click cycloaddition reaction (Scheme 1c).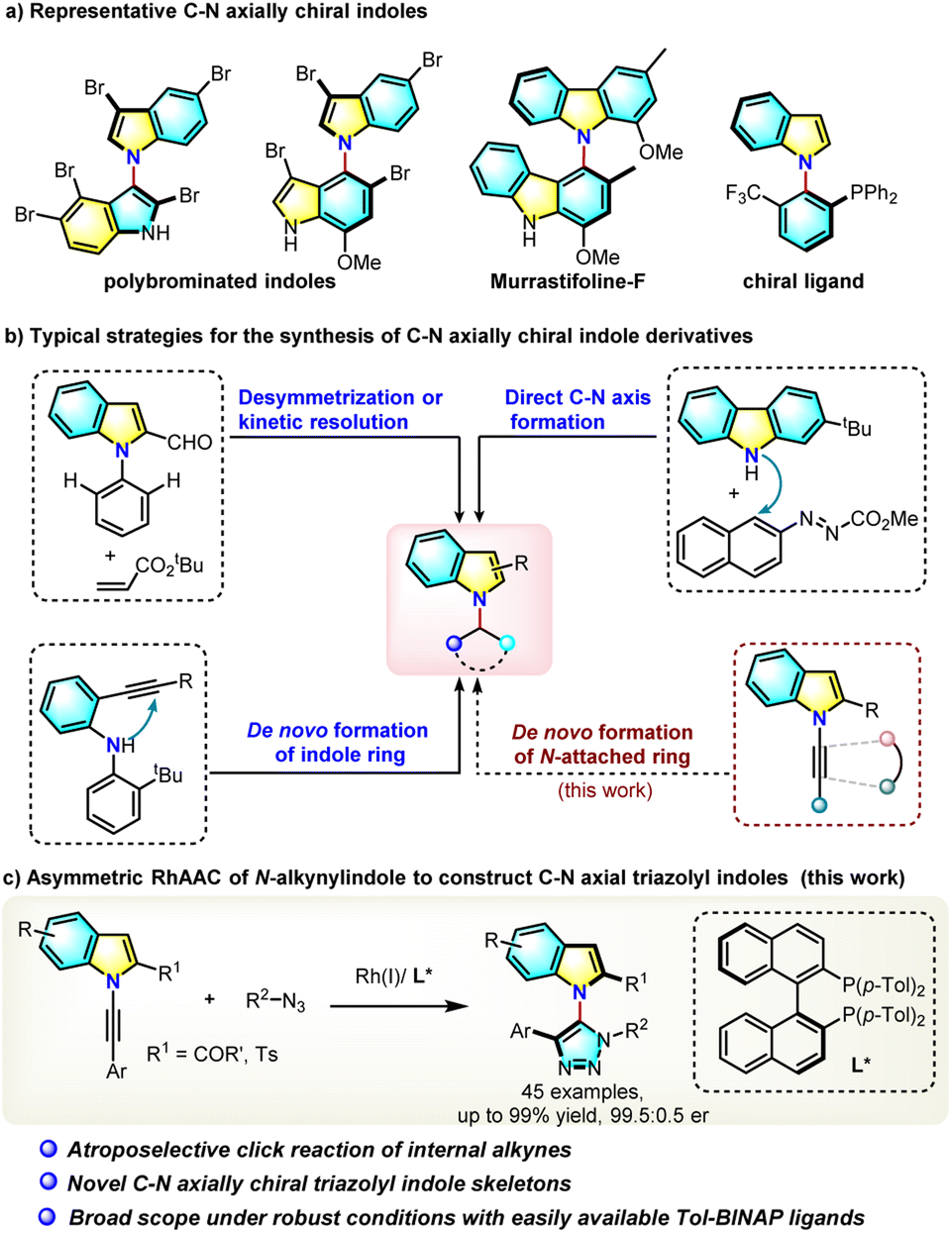 | ||
| Scheme 1 Representatives of C–N axially chiral indole derivatives and synthetic methods. | ||
The copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is one of the most powerful catalyzed reactions developed in the past twenty years and has been widely used in medicinal chemistry, polymer chemistry and materials science.7 The click reaction product, a 1,2,3-triazole, represents a very important pharmacophore building block in medicinal chemistry.8 Recently, the Zhou group developed an elegant desymmetric CuAAC reaction for the construction of chiral quaternary carbon centers,9 and Topczewski et al. reported a highly enantioselective CuAAC reaction by dynamic kinetic resolution of allyl azides.10 These enantioselective CuAAC reactions are limited to terminal alkynes building point-chiral triazoles. Such a reaction of internal alkynes is much more challenging and couldn't be realized by current CuAAC reactions,11 because the two substituents at both ends of an alkyne bond increase its steric hindrance, lower its reactivity, and more importantly, increase the difficulty of regiocontrol. Although the direct enantioselective click cycloaddition of an azide with a sterically hindered internal alkyne is a concise and straightforward approach to atropisomeric triazoles, no relevant reports appeared before 2022. Very recently, Li,12 Cui13 and our group14 independently reported the first atropoenantioselective AAC reaction building axially chiral aryltriazoles. However, these reactions leading to C–C axial naphthyltriazoles are limited to only one type of substrate, 2-hydroxynaphthylalkynes. Development of practical asymmetric click reactions of other internal alkynes is highly desirable, and no example of an asymmetric click reaction producing C–N axial chirality has been reported.
In an effort to understand the challenging asymmetric click reaction of internal alkynes, a N-alkynyl indole (1) was selected as a substrate for the following reasons: (1) the resulting indole-based structure represents one of the most important classes of heterobiaryls in organic and natural product chemistry; (2) the electronically biased alkynyl amine (1) is more reactive in transition-metal coordination than simple internal alkynes, as evidenced by the rich ynamide chemistry; (3) the ortho sulfonyl group is not only a bulky and sterically hindering moiety which blocks rotation, but could also serve as an important directing group to help chiral induction.
Results and discussion
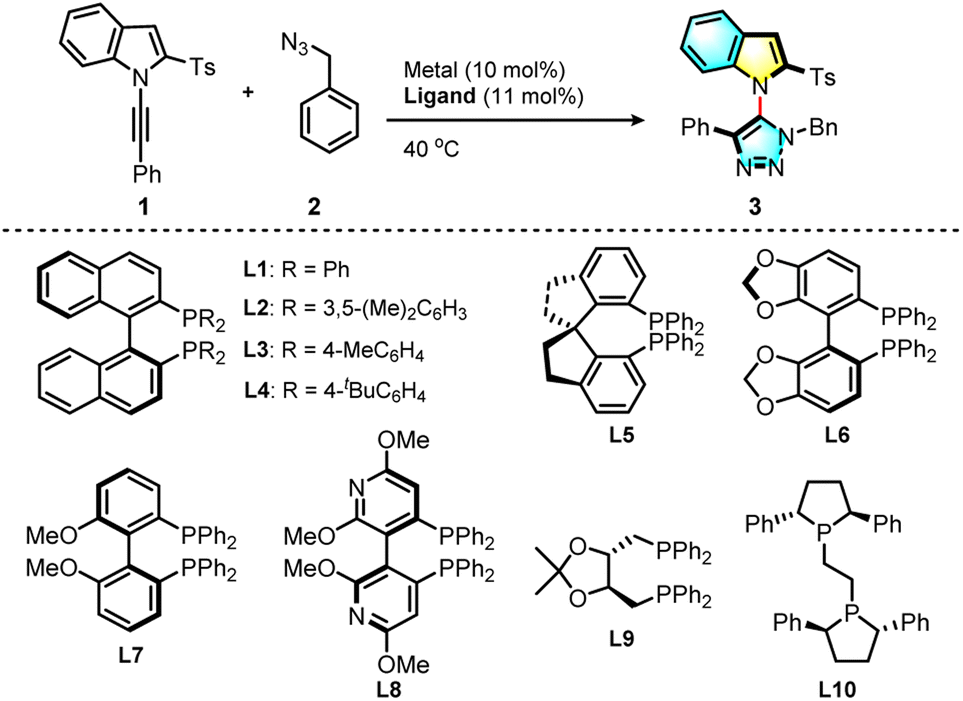
We explored the click cycloaddition of a N-alkynylindole (1) and benzyl azide (2) in the presence of transition-metal catalysts (Table 1, for details see the ESI†). Unsurprisingly, the use of copper(I) catalysts such as CuI did not lead to any product (Table 1, entry 1). When [Rh(CO)2Cl]2 was used as the catalyst, however, the desired triazolyl indole (3) was isolated in 55% yield as a single regioisomer (entry 2), suggesting that the biased electronic effect determined the regioselectivity. We explored the asymmetric version of the reaction with a combination of a Rh(I) catalyst with various chiral phosphine ligands. We found that when the reaction was performed in the presence of cationic Rh(COD)2BF4 with commercially available (S)-BINAP (L1) in the solution of 1,2-dichloroethane (DCE), the product (3) was obtained in 21% yield with a good 89.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.5 e.r. The solvent effect was examined (entries 3–6) and it was found that the target product (3) was not obtained from the reaction in n-hexane (entry 5), probably due to the poor solubility of n-hexane. When a mixed solvent of n-hexane and DCE (4/1) was used for this reaction, the product (3) was obtained in 52% yield and in 91.5:8.5 e.r. (entry 6). The identity of the counterion is very important with this catalyst, and when using 11 mol% of sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]-borate (NaBArF4) as the additive, the yield increased dramatically to 81% without any erosion in the enantioselectivity (entry 7). When the reaction was performed in a mixed (4/1) solvent of cyclohexane and DCE, the enantioselectivity increased to 93.5:6.5 e.r. albeit with slightly decreased yield (entry 8). Nonpolar solvents favored the enantioselectivity of the reaction, but decreased the reaction rate. After extension of the reaction time to 48 h, 3 was obtained in 95% yield with 93.5:6.5 e.r. (entry 9). Finally, various chiral bisphosphine ligands were screened (entries 8–17). Among all the biphosphine ligands tested, (S)-Tol-BINAP ((S)-(−)-2,2′-p-tolyl-phosphino)-1,1′-binaphthyl), (L3) gave 93% yield with 95:5 e.r. (entry 11), and the reaction with ligand L8 gave 59% yield with the highest 96.5:3.5 e.r. (entry 16). With a balance of both reactivity and enantioselectivity, the more easily available ligand (L3) was chosen for further study.
:10.5 e.r. The solvent effect was examined (entries 3–6) and it was found that the target product (3) was not obtained from the reaction in n-hexane (entry 5), probably due to the poor solubility of n-hexane. When a mixed solvent of n-hexane and DCE (4/1) was used for this reaction, the product (3) was obtained in 52% yield and in 91.5:8.5 e.r. (entry 6). The identity of the counterion is very important with this catalyst, and when using 11 mol% of sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]-borate (NaBArF4) as the additive, the yield increased dramatically to 81% without any erosion in the enantioselectivity (entry 7). When the reaction was performed in a mixed (4/1) solvent of cyclohexane and DCE, the enantioselectivity increased to 93.5:6.5 e.r. albeit with slightly decreased yield (entry 8). Nonpolar solvents favored the enantioselectivity of the reaction, but decreased the reaction rate. After extension of the reaction time to 48 h, 3 was obtained in 95% yield with 93.5:6.5 e.r. (entry 9). Finally, various chiral bisphosphine ligands were screened (entries 8–17). Among all the biphosphine ligands tested, (S)-Tol-BINAP ((S)-(−)-2,2′-p-tolyl-phosphino)-1,1′-binaphthyl), (L3) gave 93% yield with 95:5 e.r. (entry 11), and the reaction with ligand L8 gave 59% yield with the highest 96.5:3.5 e.r. (entry 16). With a balance of both reactivity and enantioselectivity, the more easily available ligand (L3) was chosen for further study.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal | Ligand | Solvent | Yield/%b | e.r.c |
| a Reaction conditions: a mixture of 1 (0.1 mmol), 2 (0.2 mmol), metal (10 mol%), ligand (11 mol%), and solvent (2.5 mL) under N2 and at 40 °C for 36 hours. b Isolated yields. c Determined by HPLC analysis using a chiral stationary phase. d NaBArF4 (11 mol%). e 48 h. | |||||
| 1 | CUI | — | DCE | 0 | — |
| 2 | [Rh(CO)2Cl]2 | — | CH3CN | 55 | — |
| 3 | Rh(COD)2BF4 | L1 | DCE | 21 | 89.5:10.5 |
| 4 | Rh(COD)2BF4 | L1 | Toluene | 19 | 90:10 |
| 5 | Rh(COD)2BF4 | L1 | Hexane | 0 | — |
| 6 | Rh(COD)2BF4 | L1 | Hexane:DCE = 4:1 |
52 | 91.5:8.5 |
| 7d | Rh(COD)2BF4 | L1 | Hexane:DCE = 4:1 |
81 | 91.5:8.5 |
| 8d | Rh(COD)2BF4 | L1 | Cyclohexane:DCE = 4:1 |
62 | 93.5:6.5 |
| 9d,e | Rh(COD)2BF4 | L1 | Cyclohexane:DCE = 4:1 |
95 | 93.5:6.5 |
| 10d,e | Rh(COD)2BF4 | L2 | Cyclohexane:DCE = 4:1 |
89 | 91.5:8.5 |
| 11d,e | Rh(COD)2BF4 | L3 | Cyclohexane:DCE = 4:1 |
93 | 95:5 |
| 12d,e | Rh(COD)2BF4 | L4 | Cyclohexane:DCE = 4:1 |
84 | 94.5:5.5 |
| 13d,e | Rh(COD)2BF4 | L5 | Cyclohexane:DCE = 4:1 |
47 | 30:70 |
| 14d,e | Rh(COD)2BF4 | L6 | Cyclohexane:DCE = 4:1 |
67 | 94.5:5.5 |
| 15d,e | Rh(COD)2BF4 | L7 | Cyclohexane:DCE = 4:1 |
48 | 90:10 |
| 16d,e | Rh(COD)2BF4 | L8 | Cyclohexane:DCE = 4:1 |
59 | 3.5:96.5 |
| 17d,e | Rh(COD)2BF4 | L9 | Cyclohexane:DCE = 4:1 |
<10 | — |
| 18d,e | Rh(COD)2BF4 | L10 | Cyclohexane:DCE = 4:1 |
56 | 49:51 |
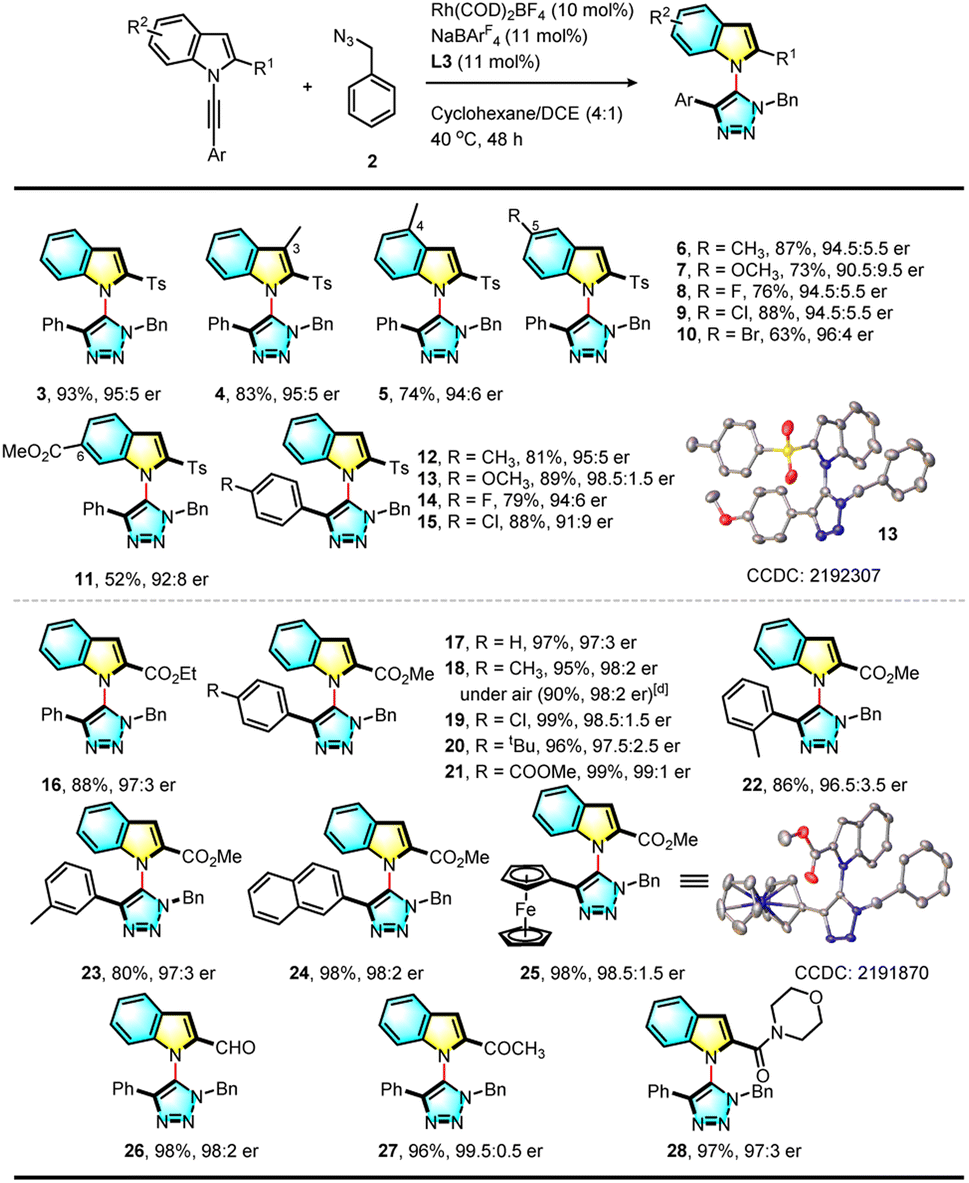
With the optimal conditions established for this asymmetric RhAAC reaction, the scope of substrates was further investigated. First, the reactivities of various N-alkynyl indole precursors were examined (Table 2). Various substituents such as methyl, fluoro, chloro, and methoxyl groups at the 3, 4, 5 or 6-position of the indole ring failed to affect this reaction (3–15). Interestingly, when the ortho Ts group was replaced by an ethyl ester group, the reaction was equally successful, and desired triazolyl indole (16) was isolated in 88% yield with an even higher 97:3 e.r. Using the smaller methyl ester led to a higher (97%) yield with 97:3 e.r. (17). Substrates bearing various electron-donating or withdrawing substitutions at the para position of the terminal aromatic ring or a methyl group at the meta or ortho position were all viable for this reaction, giving the corresponding C–N axially chiral triazolyl indoles in both excellent yields and excellent enantioselectivity. A ferrocenyl internal alkyne led to 98% yield and 98.5:1.5 e.r. (25). The structures of 13 and 25 were confirmed by X-ray single crystal diffraction, and the absolute configuration was established as (aR). More importantly, the ester could be further replaced by an aldehyde (26), acetyl (27), or amide group (28), and their reactions were successful and formed the corresponding functionalized products. Further study indicated that the reaction proceeded well under an air atmosphere without decreasing enantioselectivity, showing the robust nature of this click reaction.
| a Reaction conditions: a mixture of N-alkynyl indole (0.1 mmol), 2 (0.2 mmol), Rh(COD)2BF4 (10 mol%), L3 (11 mol%), NaBArF4 (11 mol%), cyclohexane (2.0 mL) and DCE (0.5 mL) under N2 and at 40 °C for 48 hours. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. d Reaction was conducted under an air atmosphere. |
|---|
|
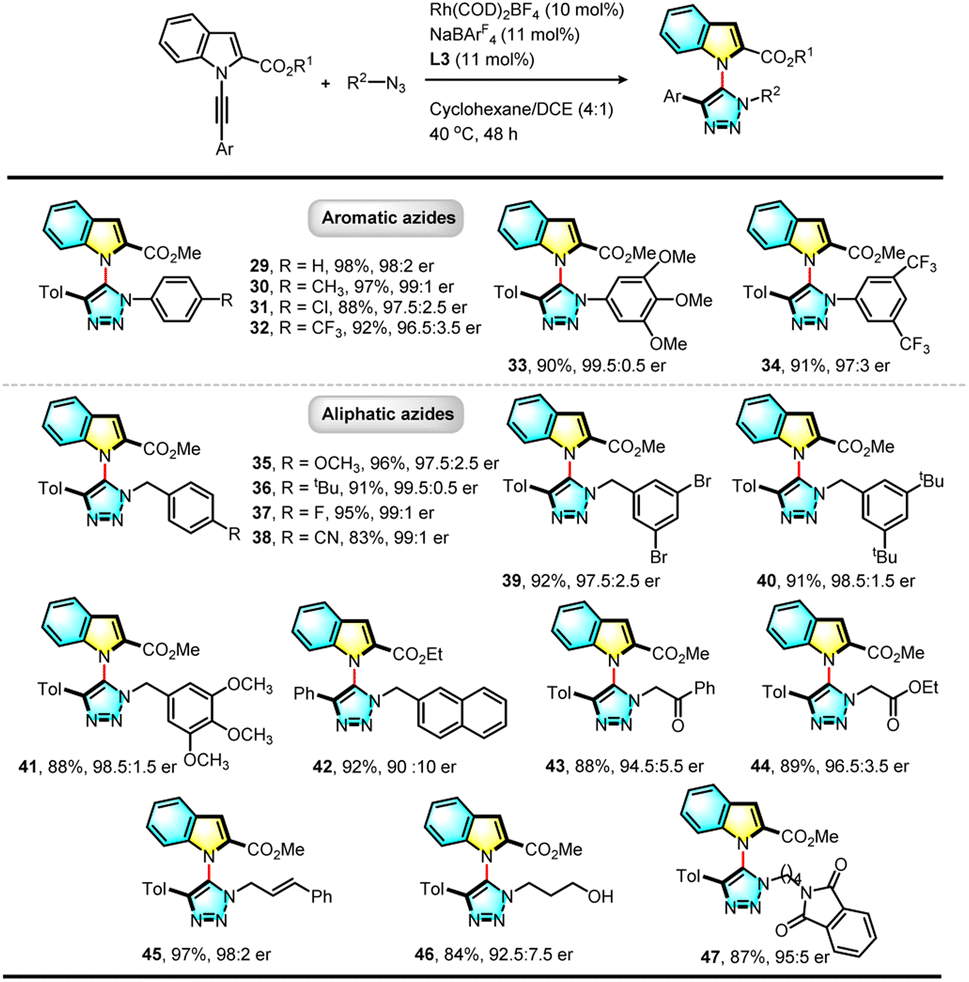
The substrate scope of azides was investigated extensively. As shown in Table 3, both aromatic and aliphatic azides are suitable substrates and a large variety of chiral triazolyl indoles (29–47) were synthesized in high yields with excellent enantioselectivity. Electron-rich or poor aromatic azides are all viable, and substrates containing three methoxyl groups (33) or two trifluoromethyl groups (34) reacted efficiently to give the corresponding triazolyl indoles in excellent yields with high enantioselectivity. Aliphatic azides bearing different functional groups such as ketone (43), ester (44), cinnamyl (45), free linear alcohol (46) and phthalyl amine (47) proceeded efficiently, demonstrating the compatibility and generality of the transformation.
The configurational stability of 18 at various temperatures was studied (Scheme 2a), and it was found that this new heterobiaryl retains very good thermal stability at temperatures below 100 °C, whereas the ee value of 18 slumped to 62% in 12 h at 120 °C and decreased to 40% in 24 h. On account of these results, the ΔG‡ and the half-life period of 18 at 120 °C were computed, respectively to be 32.9 kcal mol−1 and 23 h (Scheme 2b), demonstrating a good thermal stability of C–N axially chiral triazolyl indoles (for more details, see the ESI†).
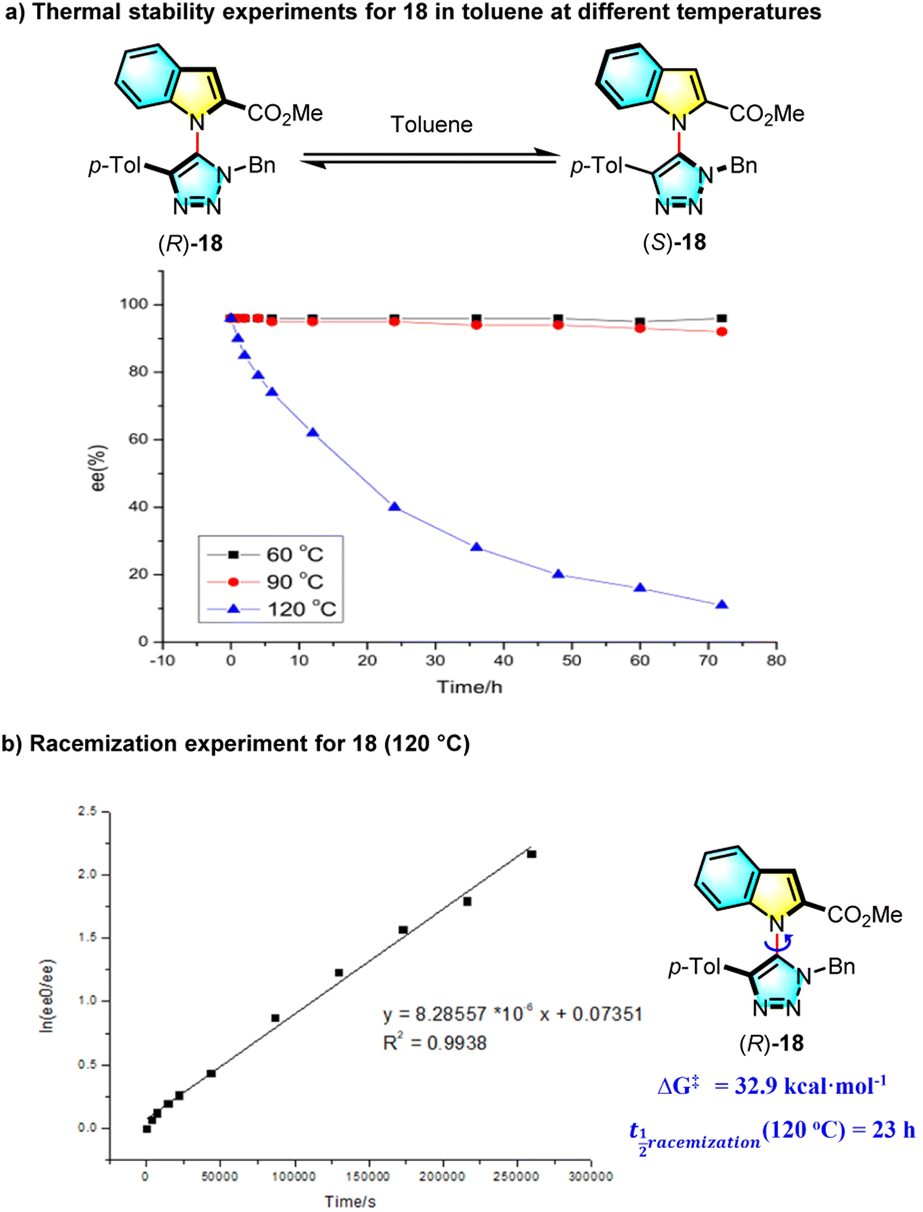 | ||
| Scheme 2 Thermal racemization experiments. | ||
Demonstrating the utility of the current protocol, a gram-scale experiment gave triazole (18) in 84% yield and 99:1 e.r. under standard conditions. The indole ring and ester group are useful synthetic handles for further transformations (Scheme 3). Treatment of 18 with Grignard reagents led to tertiary alcohols (48, 49) in 76% and 43% yield respectively. Aminolysis of 18 smoothly generated an amide (50) in 78% yield and with 98.5:1.5 e.r., and this could be further transformed into a tricyclic product (51) in 67% yield with 96.5:3.5 e.r. through a palladium-catalyzed C–H/N–H oxidative annulation. Compound 18 was efficiently converted to the 3-iodo product (52) with a 98% yield and 98:2 e.r., which could be transformed into 53 (92%, 98:2 e.r.), 54 (93%, 98:2 e.r.) and 55 (79%, 97:3 e.r.) through palladium-catalyzed cross-coupling reactions. A variety of novel axially chiral heterobiaryls, which cannot be produced with existing synthetic methods, were obtained with little or no erosion of the enantioselectivity.
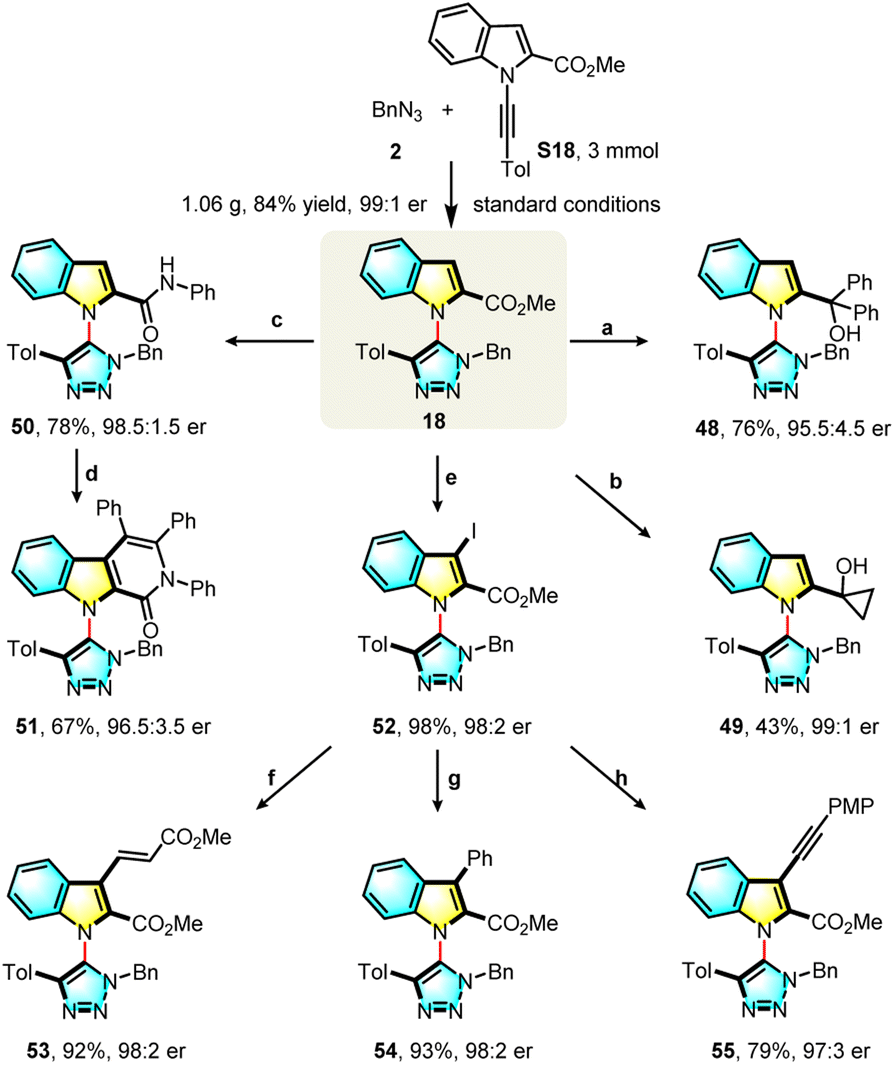 | ||
| Scheme 3 Gram-scale reaction and product elaborations (a) PhMgBr, THF, 70 °C; (b) EtMgBr, Ti(Oi-Pr)4, THF, −20 °C, (c) PhNH2, LiHMDS, toluene, rt; (d) diphenylacetylene, Cu(OAc)2, Pd(OAc)2, TBAB, 1,4-dioxane:DMSO = 9:1, 80 °C; (e) I2, AgNO3, EtOH, rt; (f) methyl acrylate, Pd2(dba)3, P(o-Tol)3, TEA, 80 °C; (g) phenylboronic acid, Pd(OAc)2, PPh3, Cs2CO3, 1,4-dioxane, 70 °C; (h)1-ethynyl-4-methoxybenzene, Pd(PPh3)2Cl2, CuI, K2CO3, THF, 65 °C. | ||
In a control experiment, a methyl or hydroxymethyl group substituted alkynyl indole was prepared and subjected to the standard reaction (Scheme 4a). The desired products were obtained in good yields but with greatly decreased enantioselectivity, possibly indicating that the neighbouring sp2 oxygen of the tosyl or carbonyl group serves as an important directing group in the Rh(I) coordination. Based on these results and previous examples, a possible mechanism was proposed and is shown in Scheme 4b. The chiral Rh(I) catalyst coordinates with both alkyne and oxygen moieties of the internal alkyne to fix the conformation, and then azide complexation, followed by asymmetric click cycloaddition forming the final product.
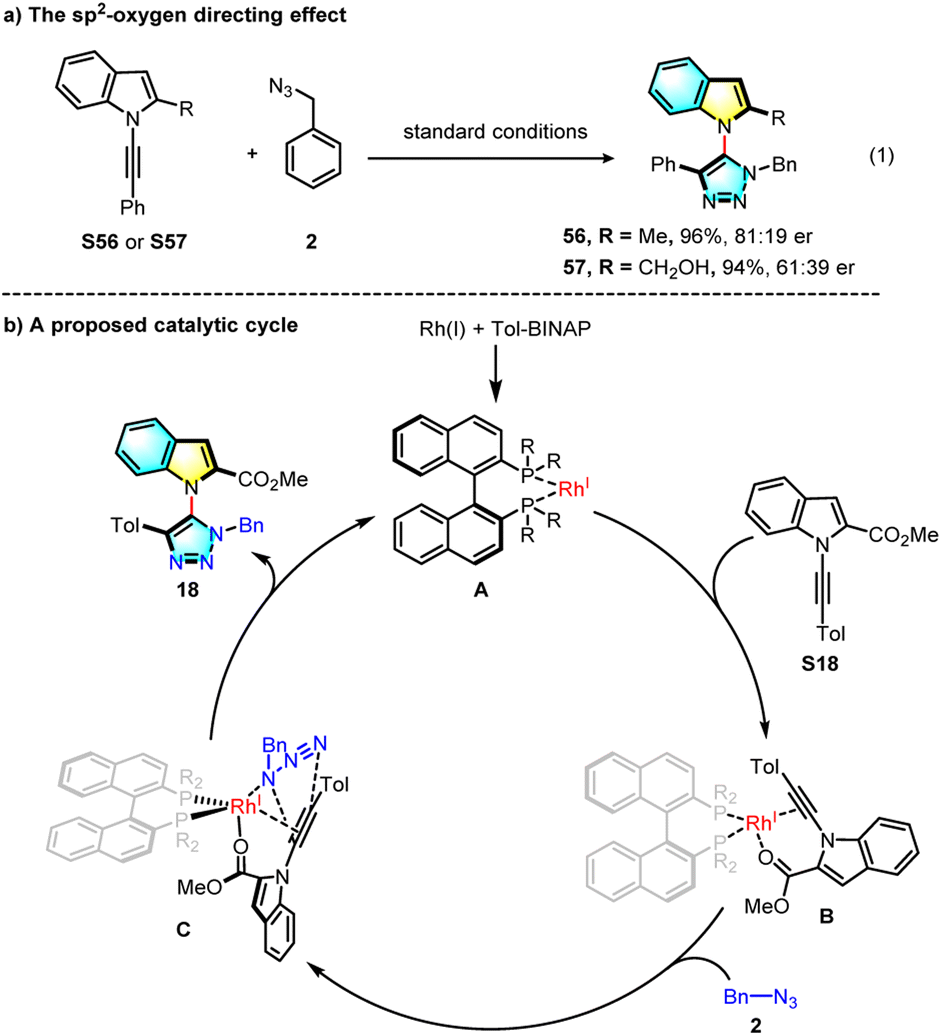 | ||
| Scheme 4 Control experiments and the proposed mechanism. | ||
Conclusions
In summary, a new Rh-catalyzed atroposelective click cycloaddition of N-alkynyl indoles with azides has been achieved. This reaction provides a robust, modular, scalable and efficient approach to C–N axially chiral triazolyl indoles in high yields with excellent regio- and enantioselctivity. These products cannot be accessed by traditional CuAAC reactions. Prominent features of this reaction include very broad substrate scope, mild reaction conditions, 100% atom economy and the use of easily available chiral Tol-BINAP ligands. We anticipate that this asymmetric RhAAC reaction could provide new insights for the application and development of other click reactions.Data availability
All experimental and characterization data including HPLC traces and NMR spectra are available in the ESI.† CCDC 2192307 (for 13) and CCDC 2191870 (for 25) contain the supplementary crystallographic data for this paper, and these data are provided free of charge by the joint Cambridge Crystallographic Data Centre.Author contributions
L. Z. and Y. L. contributed equally. Z. X. conceptualized and directed the project and drafted the manuscript with assistance from all the co-authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Natural Science Foundation of China and Shandong Province (no. 21971149, 92156007, ZR2019ZD45, and ZR2020KB005) and the Fundamental Research Funds of Shandong University.Notes and references
-
(a) N. Tajuddeen and G. Bringmann, Nat. Prod. Rep., 2021, 38, 2154–2186 RSC
; (b) H.-H. Zhang and F. Shi, Acc. Chem. Res., 2022, 55, 2562–2580 CrossRef CAS PubMed
-
(a) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed
- For selected reviews and papers on C-N axial chirality, see:
(a) J. Wang, C. Zhao and J. Wang, ACS Catal., 2021, 11, 12520–12531 CrossRef CAS
- For axially chiral indoles, see:
(a) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem. Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed
- For C-N axially chiral indoles, see:
(a) N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem. Eur. J., 2010, 16, 6752–6755 CrossRef CAS PubMed
-
(a) G. Bringmann, S. Tasler, H. Endress, J. Kraus, K. Messer, M. Wohlfarth and W. Lobin, J. Am. Chem. Soc., 2001, 123, 2703–2711 CrossRef CAS PubMed
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
-
(a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed
-
(a) F. Zhou, C. Tan and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994–10997 CrossRef CAS PubMed
-
(a) E.-C. Liu and J. J. Topczewski, J. Am. Chem. Soc., 2019, 141, 5135–5138 CrossRef CAS PubMed
-
(a) E.-C. Liu and J. J. Topczewski, J. Am. Chem. Soc., 2021, 143, 5308–5313 CrossRef CAS PubMed
- W.-T. Guo, B.-H. Zhu, Y. Chen, J. Yang, P.-C. Qian, C. Deng, L.-W. Ye and L. Li, J. Am. Chem. Soc., 2022, 144, 6981–6991 CrossRef CAS PubMed
- L. Zeng, J. Li and S. Cui, Angew. Chem., Int. Ed., 2022, 61, e202205037 CAS
- X. Zhang, S. Li, W. Yu, Y. Xie, C.-H. Tung and Z. Xu, J. Am. Chem. Soc., 2022, 144, 6200–6207 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2192307 for 13 and 2191870 for 25. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00610g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |