
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequencing palladium-catalyzed cycloisomerization cascades in a synthesis of the gelsemine core†
Guoduan
Liang
and
Edward A.
Anderson
 *
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: edward.anderson@chem.ox.ac.uk
First published on 5th June 2023
Abstract
Transition metal-catalyzed cycloisomerization is a powerful strategy for the construction of cyclic organic molecules, and the use of palladium catalysts can deliver a wide range of monocyclic and bicyclic products. However, applications of cycloisomerizations in complex target synthesis in which more than one cycloisomerization process is deployed in a cascade context are rare. Here we report investigations of the relative rates of two different types of ene-ynamide cycloisomerization that form fused and spirocyclic rings, and use of these results to design a sequence-controlled cascade cycloisomerization that prepares the tetracyclic core of gelsemine in a single step. Crucial to this work was an evaluation of the kinetics of each cycloisomerization in competition experiments, which revealed a key influence of the ynamide electron-withdrawing group on the cycloisomerization reaction.
Introduction
Transition metal-catalyzed cycloisomerizations have been widely explored as atom-economic processes to form cyclic organic molecules from acyclic polyunsaturated starting materials.1–4 Among many metals, palladium catalysts are well-established as versatile promoters of such transformations.5–12 When substrates feature multiple unsaturated bonds, elegant cascade processes have been developed whereby intermediate alkyl- or alkenylpalladium species undergo multiple C–C bond forming events in a single step;13 one pioneering example from the Trost group is shown in Scheme 1a (1 → 2).14 Pd-catalyzed cycloisomerizations have also been widely used in total synthesis,1,15–22 however applications of cascade cycloisomerizations are rare in this context. Sequential cycloisomerizations have been reported, such as an elegant approach to dendrobine described by the Chen group in which palladium- and rhodium-catalyzed cycloisomerizations were implemented in consecutive steps to construct the bicyclic pyrrolidine 3 from dienyne 4.23 A palladium-catalyzed cycloisomerization cascade was described by Trost et al. in which dienyne 5 underwent cyclization to triene 6 in the synthesis of various terpenoid natural products.24 However, to our knowledge, no examples of cascade cycloisomerizations have been reported in which a transition metal catalyst operates twice on a reaction substrate, where the order of events is critical for reaction success.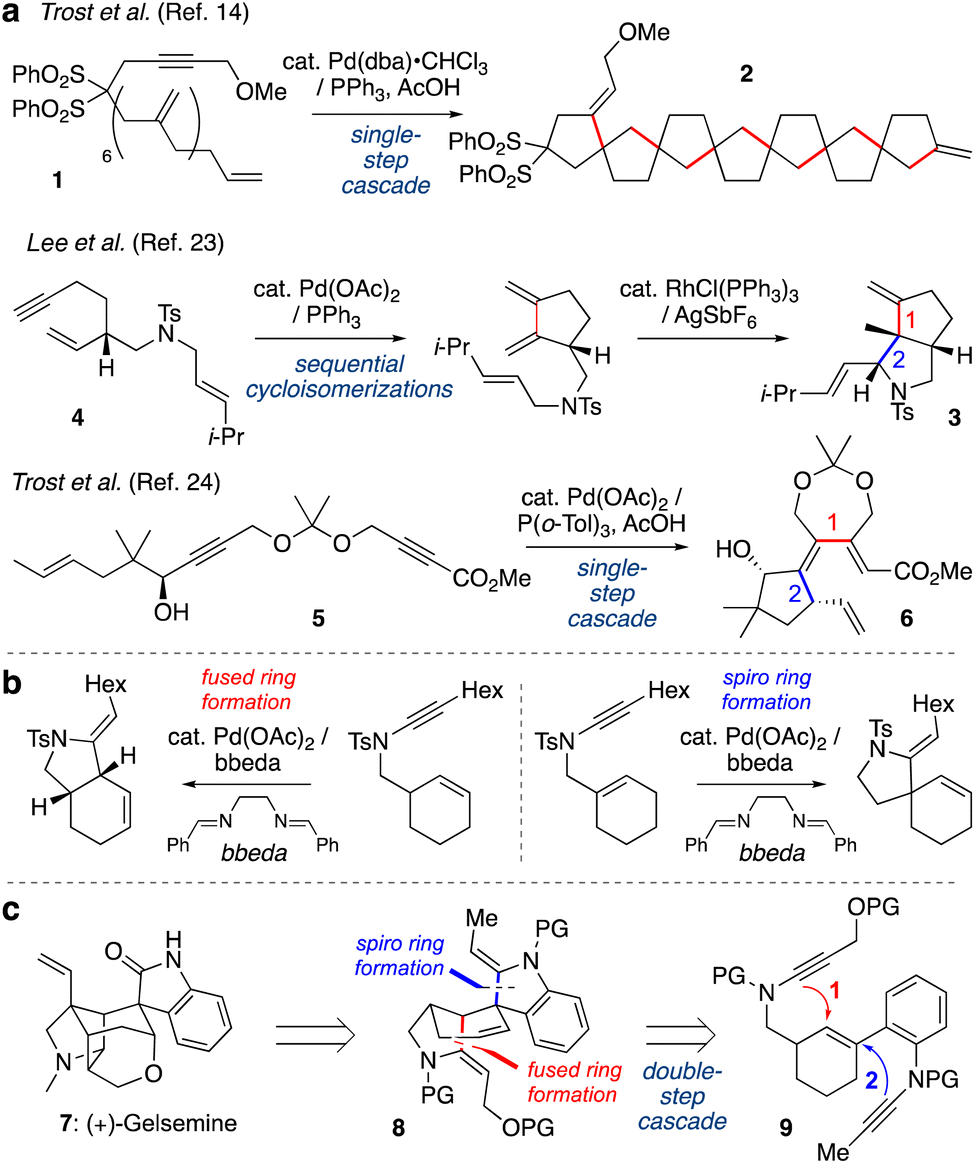 | ||
| Scheme 1 (a) Cascade or sequenced cycloisomerizations. (b) Previous work from our group on palladium-catalyzed ynamide cycloisomerization. (c) This work: time-resolved palladium-catalyzed cascade cycloisomerization towards the synthesis of the gelsemine core. | ||
Our group has developed a variety of metal-catalyzed cycloisomerizations of alkenyl ynamides (e.g., Scheme 1b).25–27 These compounds are readily accessed by alkynylation of appropriate carbamate and sulfonamide precursors,28–36 and undergo Pd-catalyzed cyclizations to azacycles under mild reaction conditions. We further studied the mechanism of these cycloisomerizations, where a series of deuterium-labelling studies demonstrated that the so-called ‘ligand’ bis(benzylidene)ethylenediamine (bbeda) in fact also serves a source of a palladium(II) hydride species that initiates the catalysis.37 Building from this work, we questioned whether we could develop a cycloisomerization cascade in which two discrete cycloisomerizations would be sequenced in a time-resolved manner, thus generating products of greater complexity with higher efficiency.
We were particularly attracted to the challenge of differentiating between fused-ring and spiro-ring formation (as shown in Scheme 1b), as we recognized that a sequence of these reactions could generate the core scaffold of the indole alkaloid gelsemine (7, Scheme 1c). This natural product features a hexacyclic core with seven contiguous stereocenters, including two quaternary carbons, whose intriguing structure has attracted the attention of many chemists.38–41 We questioned whether the challenging tetracyclic spirooxindole core of gelsemine could be obtained using a one-pot cascade polycyclization sequence. Specifically, tetracycle 8 might derive from cyclization of bis-ynamide 9 by initial fused-ring formation (Step 1), and then spiro-ring formation (Step 2), ‘walking’ the double bond of the linking cyclohexene around the six-membered ring. To achieve this time-resolved process, an understanding of the relative rates of each process would be critical, as if Step 2 occurs before Step 1 then a totally different skeleton would be formed. We were aware that the relative rates of these processes would likely depend not only on the conformational demands of the cyclizations, and the relative rates of the elementary steps of the cycloisomerization, but also on the nature of the ynamide electron-withdrawing groups, which had not previously been studied. Here we describe the exploration of these factors, and the successful execution of this cascade cycloisomerization, which to our knowledge represents the first example of the sequencing of two independent ring-forming events in palladium-catalyzed cycloisomerization chemistry.
Results and discussion
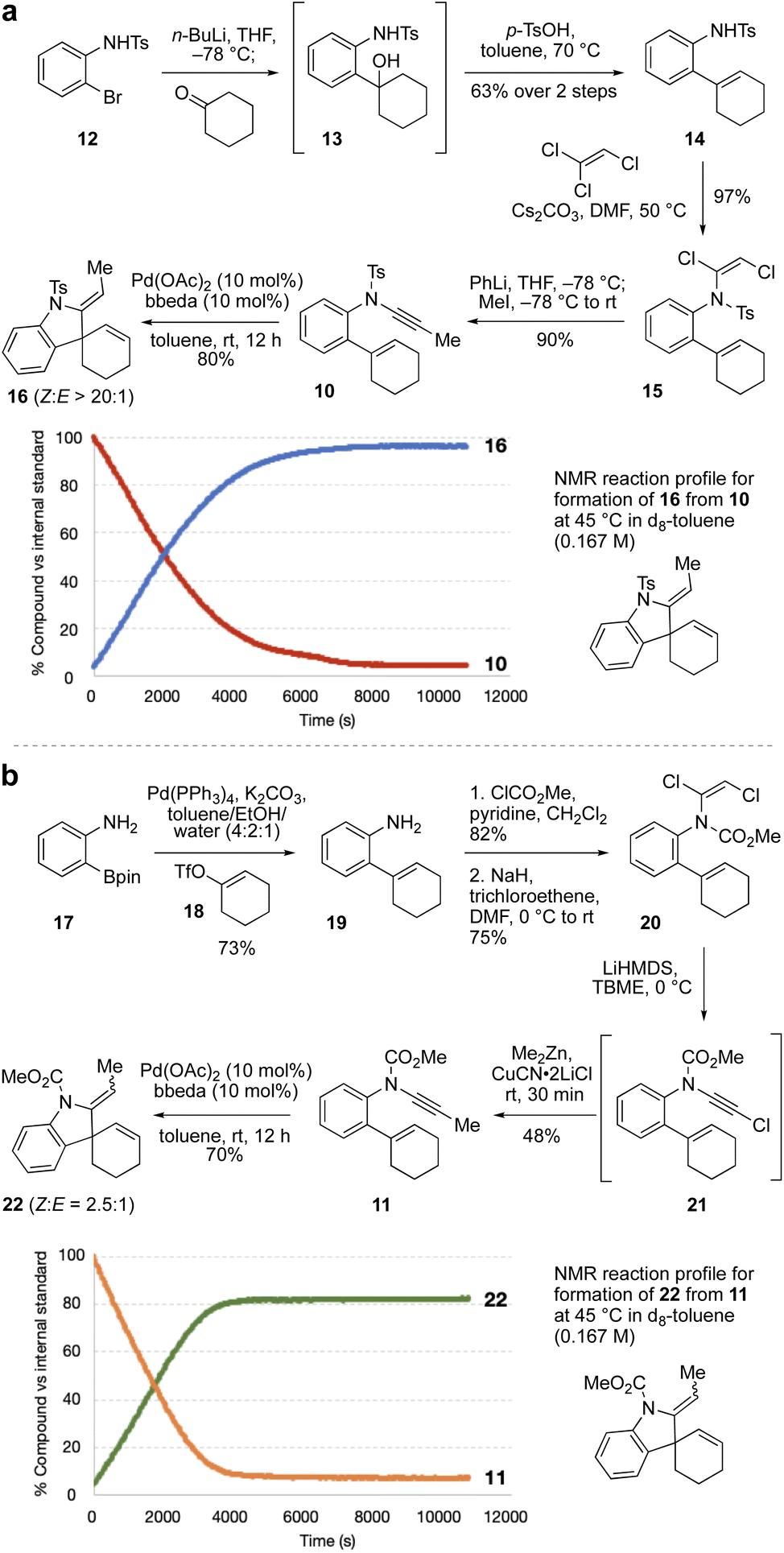
As the relative rate of spiro ring formation (Step 2 in Scheme 1c) compared to fused ring formation was expected to be crucial for the success of the cascade, we first synthesized two model ‘spiro’ enynamide substrates 10 and 11 (Scheme 2a and b respectively), differing in the nature of the ynamide electron-withdrawing group, which as noted above could make a significant difference to the rate of cyclization. The synthesis of N-aryl ynamides can be challenging, and we selected an approach based on dichloroenamide precursors developed previously by our group.42,43 Towards sulfonamide ynamide 10, lithiation of bromide 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 followed by addition to cyclohexanone gave tertiary alcohol 13 which, without purification, underwent dehydration to afford alkene 14 (63% over two steps). Treatment of 14 with trichloroethene and Cs2CO3 gave dichloroenamide 15 (97%), which was converted to N-tosyl ynamide 10 on treatment with PhLi, then iodomethane (90%). Subjection of 10 to Pd(OAc)2/bbeda (10 mol%) at room temperature for 12 h gave the spiroindoline 16 in 80% yield as a single alkene diastereomer. Other catalyst systems tested (e.g. Pd(OAc)2/2PPh3, or Pd(OAc)2 alone) also proceeded efficiently.45 An NMR reaction profile obtained at 45 °C revealed that the reaction reached completion after around 2 hours at that temperature.
44 followed by addition to cyclohexanone gave tertiary alcohol 13 which, without purification, underwent dehydration to afford alkene 14 (63% over two steps). Treatment of 14 with trichloroethene and Cs2CO3 gave dichloroenamide 15 (97%), which was converted to N-tosyl ynamide 10 on treatment with PhLi, then iodomethane (90%). Subjection of 10 to Pd(OAc)2/bbeda (10 mol%) at room temperature for 12 h gave the spiroindoline 16 in 80% yield as a single alkene diastereomer. Other catalyst systems tested (e.g. Pd(OAc)2/2PPh3, or Pd(OAc)2 alone) also proceeded efficiently.45 An NMR reaction profile obtained at 45 °C revealed that the reaction reached completion after around 2 hours at that temperature.
 | ||
| Scheme 2 Model spiro-ring synthesis (a), model fused-ring synthesis (b), and reaction kinetics. | ||
Synthesis of the equivalent N-carbamate ynamide 11 was attempted using a similar route, but proved unsuccessful due to formation of a cyclic carbamate in the first step (by cyclization of the tertiary alcohol onto the N-carbamate in the equivalent of intermediate 13).45 Instead, Suzuki coupling of boronic ester 1746 and enol triflate 18 afforded aniline 19 in 73% yield, which was converted to dichloroenamide 20 in two steps (62%). Treatment of 20 with LiHMDS generated an intermediate chloroynamide 21, which underwent in situ copper-catalyzed cross-coupling with Me2Zn to give ynamide 11 in 48% yield. Pd(OAc)2/bbeda-catalyzed cyclization of 11 at room temperature afforded the spirocyclic product 22 in 70% yield, as a 2.5:1 (Z:E) mixture of diastereomers. Interestingly, the two cycloisomerizations (of 10 and 11) appeared to proceed at quite similar reaction rates; an NMR reaction profile obtained at 45 °C revealed the reaction of 11 reached completion after around 1 hour.
We next targeted a model fused-ring system (23, Scheme 3). An N-tosyl electron-withdrawing group was selected due to the efficiency of ynamide formation for aliphatic amides with this group, compared to carbamates. This synthesis commenced with an Ac2O-promoted Diels–Alder reaction47 between commercially-available aldehyde 24 and acrylonitrile (60%). Reduction of the cyclohexene (H2, Pd/C) and the nitrile (LiAlH4) followed by tosylation of the resulting amine afforded 26 (53% over three steps). Elimination of the alcohol using the Burgess reagent, and base-promoted isomerization of the alkene48 gave 27 exclusively, with the alkene in conjugation with the aromatic ring. Sulfonamide 27 was then coupled33 with bromoalkyne 28 to afford ynamide 29 (86%). Cycloisomerization of 29 using Pd(OAc)2/bbeda did not proceed at room temperature, but on heating to 45 °C for 3 h gave the desired fused-ring bicyclic product 23 in 50% yield. Surprisingly, use of Pd(OAc)2 alone6 proved similarly effective, generating 23 in 54% yield. An NMR reaction profile obtained at 45 °C revealed the reaction of 29 reached completion at around 3 hours.
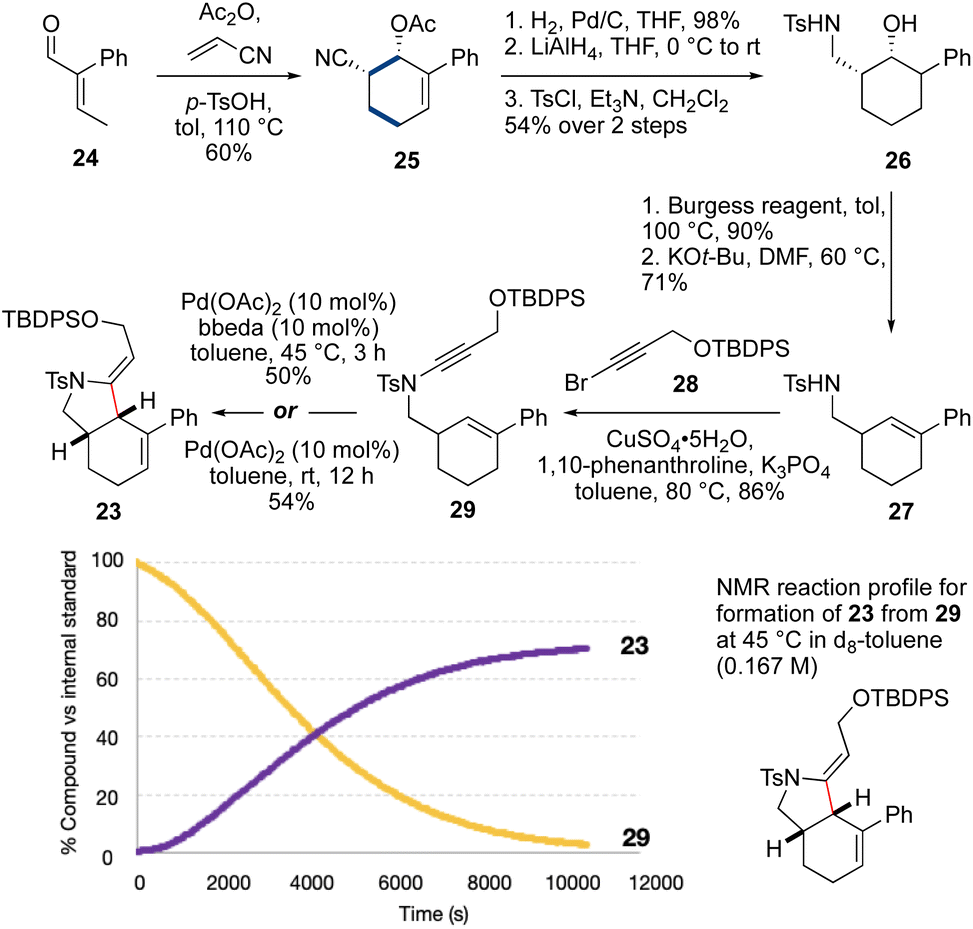 | ||
| Scheme 3 Pd-catalyzed cycloisomerization to fused-ring model 23. | ||
The individual NMR timecourse experiments (at 45 °C) suggested that spiro-ring synthesis should be favoured over fused-ring synthesis, which is contrary to our synthetic design. To further explore this, competition experiments were carried out between ynamides 10 and 29, and ynamides 11 and 29, each in a 1:1 ratio (Fig. 1). We first compared the reactivity of the two sulfonamide-substituted ynamides 10 and 29 (Fig. 1a). This revealed somewhat similar rates of ynamide consumption and product formation for the two substrates. However, to our surprise, the equivalent competition between N-carbamate ynamide 11 and N-tosyl ynamide 29 (Fig. 1b) resulted in quite distinct reaction profiles, in which sulfonamide-substituted fused-ring formation outcompeted carbamate-substituted spiro-ring formation, in spite of the significantly higher rate of reaction of the latter when conducted in isolation (compare Schemes 2b and 3). In a competition setting, this appears to suggest preferential complexation of the sulfonamide-substituted ynamide to the palladium(II) catalyst over the carbamate-substituted ynamide. Cycloisomerization of the former may proceed via a low-energy intermediate (i.e. a catalyst resting state) that retards the overall observed rate of reaction for both compounds.
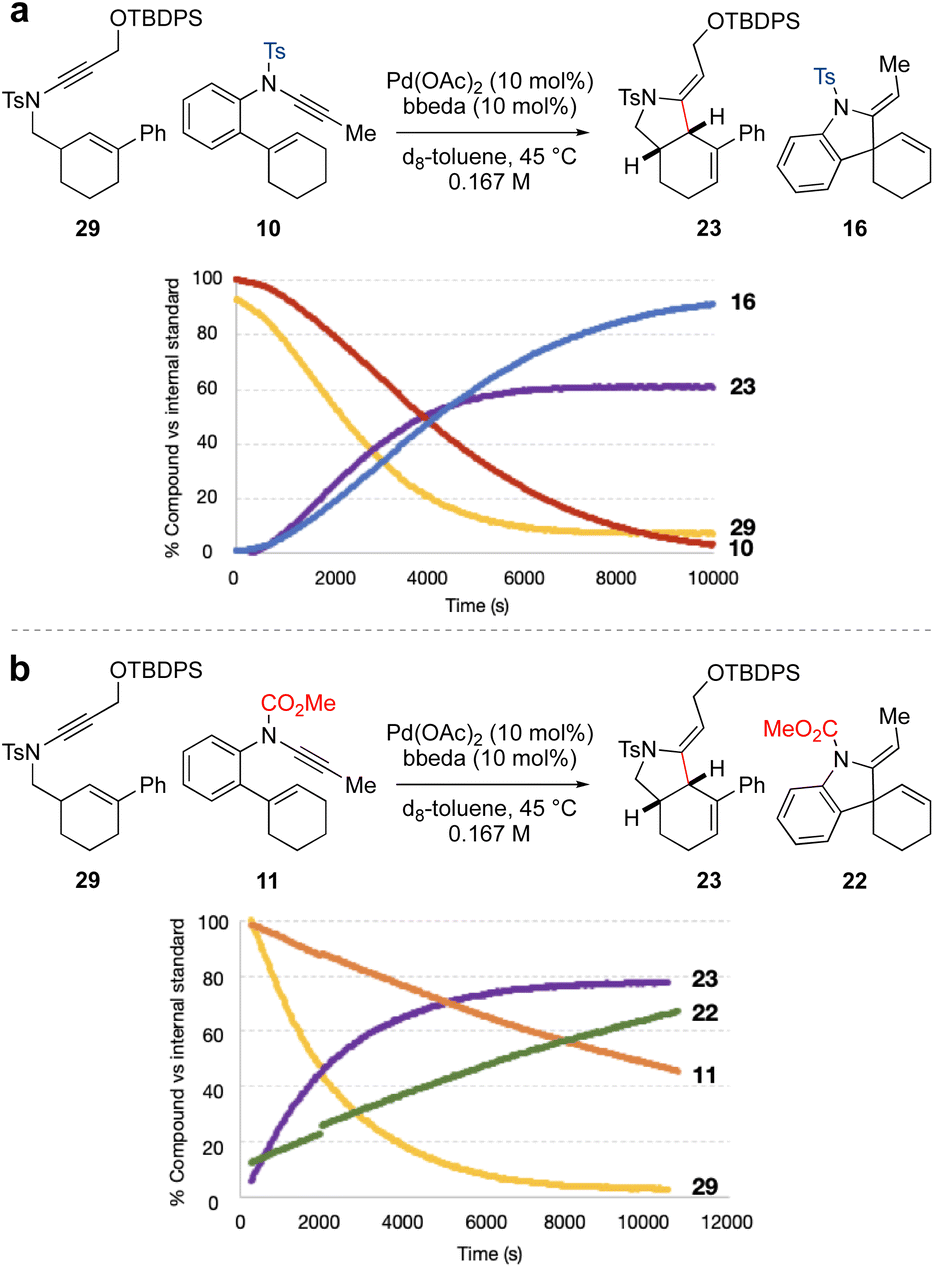 | ||
| Fig. 1 Competition experiments for Pd-catalyzed cycloisomerization of 10 and 11vs.29. | ||
This fortunate finding set the stage for execution of the double cycloisomerization cascade, in which the spiro-ring ynamide would feature a carbamate, and the fused-ring ynamide a sulfonamide (i.e., bis-ynamide 30, Scheme 4). The synthesis of 30 began with a Heck reaction49 of 2-iodoaniline with cyclohexenone, which after carbamoylation of the aniline and Wittig olefination of the ketone gave diene 31 (19% yield over three steps). Regioselective hydroboration/oxidation of 31 and subsequent Mitsunobu reaction gave 32 (57% yield over two steps). After selective carbamate deprotection of the sulfonamide-bearing nitrogen, the resulting sulfonamide was coupled33 with bromoalkyne 28 to obtain 33 (78% yield over two steps). In preparation for installation of the second ynamide, dichloroenamide 34 was first formed;42,43 however, treatment of 34 under ynamide-forming conditions (using LiHMDS) led only to unexpected cleavage of the sulfonamide ynamide,45 with no formation of the desired bis-ynamide 30 being observed.
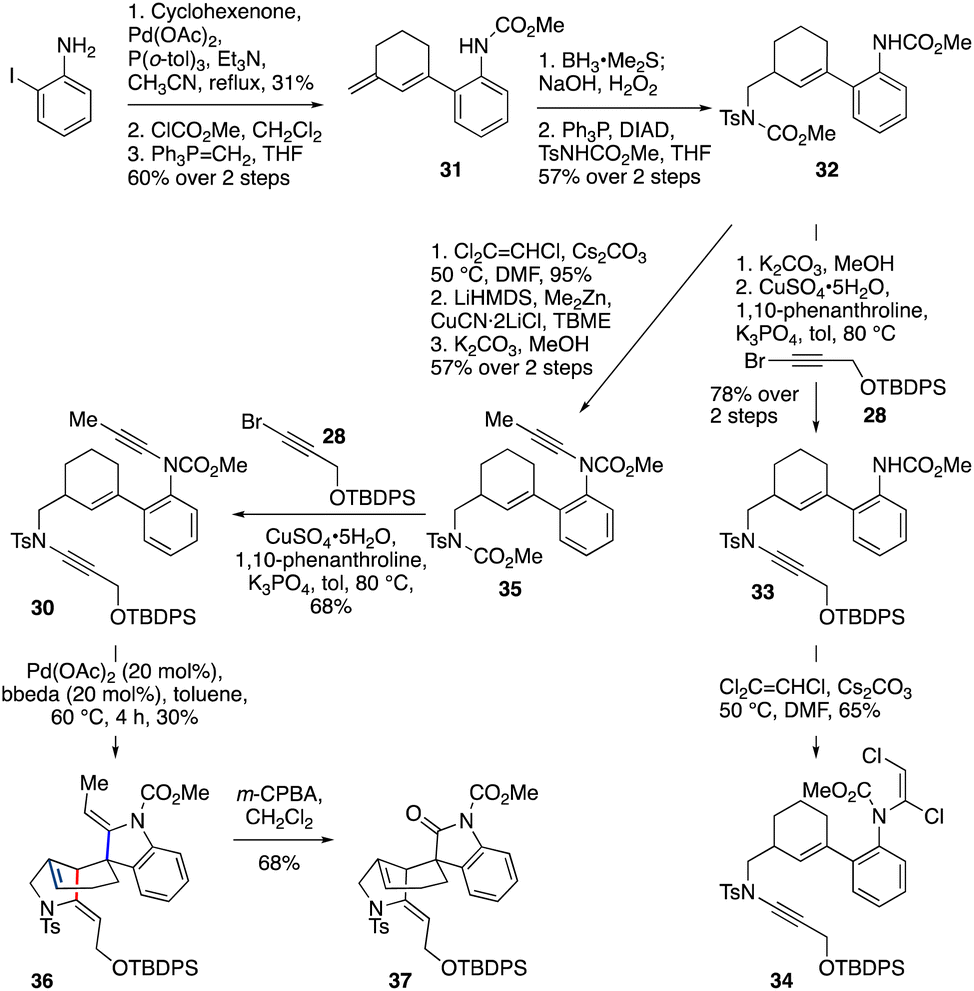 | ||
| Scheme 4 Pd-catalyzed cascade cycloisomerization to the tetracyclic core of gelsemine, 36. | ||
Pleasingly, this obstacle could be overcome by switching the order of ynamide formation. Thus, dichloroenamide synthesis from 32 (95%) was followed by conversion to the methyl ynamide 35via elimination (with LiHMDS), copper-catalyzed cross-coupling of the intermediate chloroalkyne with dimethylzinc, and cleavage of the ‘sulfonamide’ carbamate. Coupling of 35 with bromoalkyne 28 successfully afforded bis-ynamide 30. To our delight, subjection of 30 to Pd(OAc)2/bbeda-catalyzed cyclization conditions gave tetracyclic compound 36 – the gelsemine core – in 30% yield. Surprisingly, this cascade was accompanied by migration of the double bond of the six membered ring to the (more-substituted) ring junction, presumably by chain-walking of the palladium(II) hydride species.50–52 This yield is comparable with that of the two individual cyclizations (50% and 70% for 29 and 11 respectively); based on observations with the model system 29 we believe the low yield is mainly impacted by substrate degradation during the first fused-ring cycloisomerization. Finally, we demonstrated that differentiation of the two enamides in this product could be achieved on reaction with m-CPBA,53 which led to selective oxidative cleavage of the indoline enamide double bond, furnishing the spirooxindole 37 in 68% yield, as required in the gelsemine framework.
Conclusions
In summary, we have demonstrated a time-resolved palladium-catalyzed cycloisomerization cascade, in which sequential fused- and then spiro-ring formation accessed the core of the natural product gelsemine. Critical to this chemistry was an understanding of the relative reactivity of the different ynamides involved, where kinetics studies revealed a preferential (but slower) reaction of a sulfonamide-substituted ynamide over a carbamate-substituted ynamide, perhaps suggesting this reaction may proceed via an intermediate resting state that acts as a catalyst reservoir. Application of these findings enabled a 10 step synthesis of the tetracyclic gelsemine core, but perhaps more significantly opens up opportunities for further invention and exploitation of cycloisomerization cascades in synthesis.Data availability
All data for experimental procedures and compound characterization are available in the paper and its ESI files.†Author contributions
G. L. and E. A. A. conceived the work. G. L. performed the experiments and analysed the results. E. A. A. directed the project. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EAA thanks the EPSRC for support (EP/S013172/1).Notes and references
- Y. Hu, M. Bai, Y. Yang and Q. Zhou, Org. Chem. Front., 2017, 4, 2256–2275 RSC.
- Y. Yamamoto, Chem. Rev., 2012, 112, 4736–4769 CrossRef CAS PubMed.
- V. Michelet, P. Y. Toullec and J. P. Genet, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed.
- C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813–834 CrossRef CAS PubMed.
- B. M. Trost, G. J. Tanoury, M. Lautens, C. Chan and D. T. Macpherson, J. Am. Chem. Soc., 1994, 116, 4255–4267 CrossRef CAS.
- B. M. Trost, D. L. Romero and F. Rise, J. Am. Chem. Soc., 1994, 116, 4268–4278 CrossRef CAS.
- J. Nugent, E. Matoušová, M. G. Banwell and A. C. Willis, J. Org. Chem., 2017, 82, 12569–12589 CrossRef CAS PubMed.
- B. M. Trost, Acc. Chem. Res., 1990, 23, 34–42 CrossRef CAS.
- S. Yadav and S. S. V. Ramasastry, Chem.–Asian J., 2020, 15, 2764–2774 CrossRef CAS PubMed.
- S. Mondal, T. Ballav, K. Biswas, S. Ghosh and V. Ganesh, Eur. J. Org. Chem., 2021, 4566–4602 CrossRef CAS.
- M. Lanzi, T. Cañeque, L. Marchiò, R. Maggi, F. Bigi, M. Malacria and G. Maestri, ACS Catal., 2018, 8, 144–147 CrossRef CAS.
- J. Biemolt and E. Ruijter, Adv. Synth. Catal., 2018, 360, 3821–3871 CrossRef CAS.
- B. Gabriele, R. Mancuso, L. Veltri, I. Ziccarelli and N. Della Ca, Eur. J. Org. Chem., 2019, 5073–5092 CrossRef CAS.
- B. M. Trost and Y. Shi, J. Am. Chem. Soc., 1993, 115, 9421–9438 CrossRef CAS.
- N. Gao, M. G. Banwell and A. C. Willis, Org. Lett., 2017, 19, 162–165 CrossRef CAS PubMed.
- X. Ma, N. Gao, M. G. Banwell, P. D. Carr and A. C. Willis, J. Org. Chem., 2017, 82, 4336–4341 CrossRef CAS PubMed.
- J. Nugent, E. Matoušová and M. G. Banwell, Eur. J. Org. Chem., 2015, 3771–3778 CrossRef CAS.
- L. Zilke and D. G. Hall, Eur. J. Org. Chem., 2012, 4153–4163 CrossRef CAS.
- P. A. Peixoto, R. Severin, C.-C. Tseng and D. Y. K. Chen, Angew. Chem., Int. Ed., 2011, 50, 3013–3016 CrossRef CAS PubMed.
- B. M. Trost, L. Dong and G. M. Schroeder, J. Am. Chem. Soc., 2005, 127, 10259–10268 CrossRef CAS PubMed.
- B. M. Trost and Y. Li, J. Am. Chem. Soc., 1996, 118, 6625–6633 CrossRef CAS.
- B. M. Trost, P. A. Hipskind, J. Y. L. Chung and C. Chan, Angew. Chem., Int. Ed., 1989, 28, 1502–1504 CrossRef.
- Y. Lee, E. M. Rochette, J. Kim and D. Y. K. Chen, Angew. Chem., Int. Ed., 2017, 56, 12250–12254 CrossRef CAS PubMed.
- B. M. Trost and C. Min, Nat. Chem., 2020, 12, 568–573 CrossRef CAS PubMed.
- P. R. Walker, C. D. Campbell, A. Suleman, G. Carr and E. A. Anderson, Angew. Chem., Int. Ed., 2013, 52, 9139–9143 CrossRef CAS PubMed.
- R. N. Straker, Q. Peng, A. Mekareeya, R. S. Paton and E. A. Anderson, Nat. Commun., 2016, 7, 10109 CrossRef CAS PubMed.
- S. J. Mansfield, K. E. Christensen, A. L. Thompson, K. Ma, M. W. Jones, A. Mekareeya and E. A. Anderson, Angew. Chem., Int. Ed., 2017, 56, 14428–14432 CrossRef CAS PubMed.
- L. Zhao, H. Yang, R. Li, Y. Tao, X.-F. Guo, E. A. Anderson, A. Whiting and N. Wu, J. Org. Chem., 2021, 86, 1938–1947 CrossRef CAS PubMed.
- L. Andna and L. Miesch, Org. Biomol. Chem., 2019, 17, 5688–5692 RSC.
- X. Zeng, Y. Tu, Z. Zhang, C. You, J. Wu, Z. Ye and J. Zhao, J. Org. Chem., 2019, 84, 4458–4466 CrossRef CAS PubMed.
- Y. Yang, X. Zhang and Y. Liang, Tetrahedron Lett., 2012, 53, 6557–6560 CrossRef CAS.
- A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381–4385 CrossRef CAS PubMed.
- X. Zhang, Y. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. Shen and M. R. Tracey, J. Org. Chem., 2006, 71, 4170–4177 CrossRef CAS PubMed.
- S. Hirano, R. Tanaka, H. Urabe and F. Sato, Org. Lett., 2004, 6, 727–729 CrossRef CAS PubMed.
- J. R. Dunetz and R. L. Danheiser, Org. Lett., 2003, 5, 4011–4014 CrossRef CAS PubMed.
- B. Witulski and T. Stengel, Angew. Chem., Int. Ed., 1998, 37, 489–492 CrossRef CAS.
- A. Mekareeya, P. R. Walker, A. Couce-Rios, C. D. Campbell, A. Steven, R. S. Paton and E. A. Anderson, J. Am. Chem. Soc., 2017, 139, 10104–10114 CrossRef CAS PubMed.
- A. Ghosh and R. G. Carter, Angew. Chem., Int. Ed., 2019, 58, 681–694 CrossRef CAS PubMed.
- X. Chen, S. Duan, C. Tao, H. Zhai and F. G. Qiu, Nat. Commun., 2015, 6, 7204 CrossRef PubMed.
- X. Zhou, T. Xiao, Y. Iwama and Y. Qin, Angew. Chem., Int. Ed., 2012, 51, 4909–4912 CrossRef CAS PubMed.
- H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36–51 CrossRef CAS PubMed.
- S. J. Mansfield, R. C. Smith, J. R. J. Yong, O. L. Garry and E. A. Anderson, Org. Lett., 2019, 21, 2918–2922 CrossRef CAS PubMed.
- S. J. Mansfield, C. D. Campbell, M. W. Jones and E. A. Anderson, Chem. Commun., 2015, 51, 3316–3319 RSC.
- M. R. Paleo, L. Castedo and D. Dominguez, J. Org. Chem., 1993, 58, 2763–2767 CrossRef CAS.
- See the ESI† for details.
- S. Ortgies and A. Breder, Org. Lett., 2015, 17, 2748–2751 CrossRef CAS PubMed.
- D. Strübing, A. J. von Wangelin, H. Neumann, D. Gördes, S. Hübner, S. Klaus, A. Spannenberg and M. Beller, Eur. J. Org. Chem., 2005, 107–113 CrossRef.
- M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. van Otterlo, Chem. Rev., 2015, 115, 5462–5569 CrossRef CAS PubMed.
- S. E. Reisman, J. M. Ready, M. M. Weiss, A. Hasuoka, M. Hirata, K. Tamaki, T. V. Ovaska, C. J. Smith and J. L. Wood, J. Am. Chem. Soc., 2008, 130, 2087–2100 CrossRef CAS PubMed.
- H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS PubMed.
- T. Kochi, T. Hamasaki, Y. Aoyama, J. Kawasaki and F. Kakiuchi, J. Am. Chem. Soc., 2012, 134, 16544–16547 CrossRef CAS PubMed.
- See also: L. A. Goj and R. A. Widenhoefer, J. Am. Chem. Soc., 2001, 123, 11133–11147 CrossRef CAS PubMed.
- Q. Wang, L. Zhang, J. Yao, G. Qiu, X. Li and H. Zhou, J. Org. Chem., 2018, 83, 4092–4098 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of reaction optimization, copies of 1H and 13C NMR spectra (pdf). See DOI: https://doi.org/10.1039/d3sc01353g |
| This journal is © The Royal Society of Chemistry 2023 |