
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid and column-chromatography-free peptide chain elongation via a one-flow, three-component coupling approach†
Naoto
Sugisawa
,
Akira
Ando
and
Shinichiro
Fuse
 *
*
Department of Basic Medicinal Sciences, Graduate School of Pharmaceutical Sciences, Nagoya University, Nagoya, 464-8601, Japan. E-mail: fuse@ps.nagoya-u.ac.jp
First published on 12th June 2023
Abstract
Short peptides are extremely important as drugs and building blocks for the syntheses of longer peptides. Both solid- and liquid-phase peptide syntheses suffer from a large number of synthetic steps, high cost, and/or tedious purification. Here, we developed a rapid, mild, inexpensive, and column-chromatography-free peptide chain elongation via a one-flow, three-component coupling (3CC) approach that is the first to use α-amino acid N-carboxy anhydrides (α-NCAs) both as electrophiles and nucleophiles. We demonstrated the high-yielding and column-chromatography-free syntheses of 17 tripeptides, as well as a gram-scale synthesis of a tripeptide. The total synthesis of beefy meaty peptide was achieved by repeating the 3CC approach with the addition of only one column chromatographic purification. We also demonstrated a one-flow tripeptide synthesis via in situ preparation of α-NCA starting from three readily available protected amino acids. With this study, we achieved dramatic reductions in both time and cost compared with typical solid-phase synthesis.
Introduction
Peptide-based drugs have become increasingly important over the past several decades.1 Reportedly, ca. 80% of peptide drugs on the market are short peptides consisting of less than or equal to 10 amino acids.2 Longer peptides can be readily prepared by linking these short peptides via ligation technologies.3 Therefore, efficient and scalable preparation of short peptides is extremely important both in academia and industry. Solid-phase peptide synthesis (SPPS) is a compelling option due to its facile purification.4 Pentelute and co-workers recently demonstrated a sophisticated solid-phase automated rapid-flow peptide/protein chain elongation.5 However, SPPS usually requires the use of a large amount of materials (i.e., expensive coupling agents, additives, a solid phase, amino acids, and solvents).6 Thus, production cost becomes high even for the synthesis of short peptides. Liquid-phase peptide synthesis (LPPS) has been frequently used for the production of short peptides because it does not require the use of an excess amount of materials. However, burdensome and costly purifications are usually required for the removal of remained and/or reacted materials from the target peptides after every step.7 Another problem both in SPPS and LPPS is the requirement of deprotection steps that account for almost half of the total number of synthetic steps. Researchers have long coveted an ideal process that enables fewer synthetic steps, low cost, mild, rapid, and scalable peptide chain elongation for short peptides without tedious purification.One-pot/one-flow, multi-component coupling approaches are attractive because they skip intermediate purifications, and, thus, these can shorten the time for synthesis and reduce waste. However, as far as we could ascertain, there have been only three reports for peptide syntheses based on such an approach. Sampaio-Dias et al. used protection-free amino acid A as a scaffold in their one-pot, three-component coupling (3CC) approach (Scheme 1a).8 It is well known that the nucleophilicity of an amino group is higher than that of a carboxyl group in A under basic conditions. Therefore, amine A was coupled with activated ester B, and the remaining carboxyl group was activated by TBTU for the following coupling with amine C to afford tripeptide D. Fuller et al. used urethane-protected α-amino acid N-carboxy anhydride (UNCA) E as a scaffold in their one-pot 3CC approach (Scheme 1b).9 The UNCA E has an activated C-terminal moiety and a protected N-terminal moiety; thus, a coupling with amine C and removal of the Cbz group by Pd/C and H2 afforded an amine. Coupling of the amine with F afforded the desired tripeptide D. Yamamoto et al. used silyl-protected cyclic dipeptide G as a scaffold in their one-pot, 3CC approach (Scheme 1c).10 The N-terminus of G was selectively deprotected by n-Bu4NF and coupled with an activated ester that was derived from carboxylic acid H, EDCI, and HOBt. Then, the C-terminus of the resultant dipeptide was again activated by EDCI and HOBt, which was followed by coupling with amine C′ to afford the tetrapeptide I. All the reported approaches demonstrated oligopeptide synthesis without intermediate purifications. These approaches, however, require activation and/or deprotection steps using expensive reagents and additives as well as tedious column chromatographic purifications. In addition, although one-pot, multi-component Ugi reactions for peptide synthesis have been reported, these afford a diastereomeric mixture of peptides.11 Hirschmann et al. reported a one-pot, protecting group-free synthesis of short peptides (≤6 residues) using α-amino acid N-carboxy anhydride (α-NCA) J and J′ under carefully controlled conditions (pH = 10.2, 0–2 °C, high sheer mixing) and achieved an epoch for ribonuclease S synthesis by connecting the short peptides (Scheme 1d).12 Although α-NCA-based peptide chain elongation is ideal because it requires neither activation nor deprotection steps and emits only CO2 during the coupling step, but the process has rarely been used due to difficulties in suppressing undesired reactions. In fact, Blacker et al. reported the synthesis of di- and tri-peptides using α-NCA J in 2017.13 Although they achieved peptide synthesis using α-NCAs in a continuous stirred tank reactor, they pointed out the difficulties in purifying the obtained peptides due to the generation of undesired products. α-Dehydroamino acid NCAs are much more stable than α-NCAs14 and have been used for peptide chain elongation via a one-pot, 3CC approach because the risk of undesired reactions is low.15
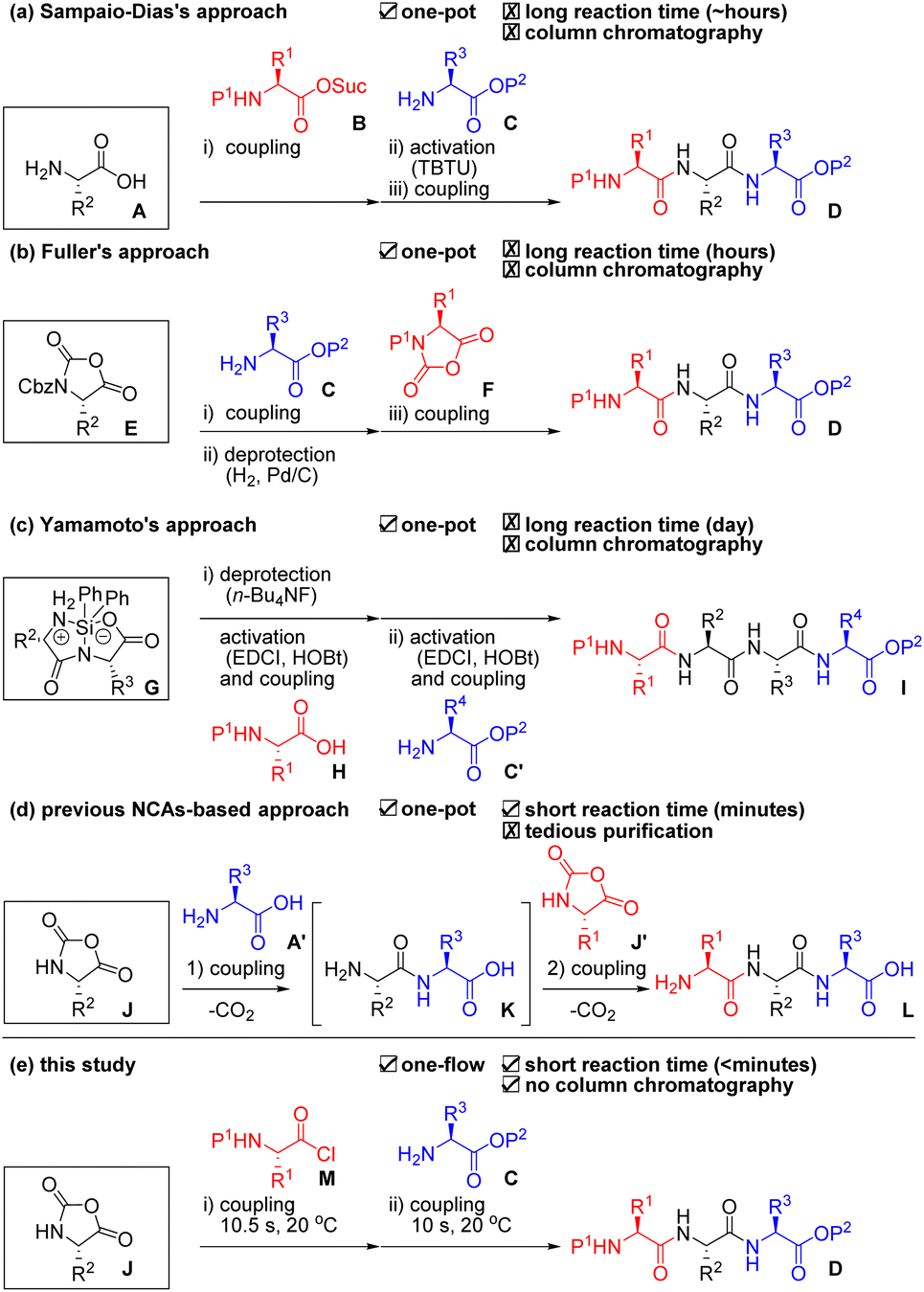 | ||
| Scheme 1 Previously reported one-pot/one-flow, three-component coupling (3CC) approaches for peptide chain elongation and their problems. (a) Sampaio-Dias's one-pot approach using an unprotected amino acid A as a scaffold. (b) Fuller's one-pot approach using an UNCA E as a scaffold. (c) Yamamoto's one-pot approach using a silyl protected cyclic dipeptide G as a scaffold. (d) Previously reported one-pot approach using α-NCA J as an electrophile. (e) Our developed one-flow approach using the J both as a nucleophile and an electrophile. P = protecting group; Suc = succinimide; TBTU = 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate; Cbz = benzyloxycarbonyl; Ph = phenyl; HOBt = 1-hydroxybenzotriazole; EDCI = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride. | ||
Micro-flow technologies allow precise control of both the reaction time on a short scale (<1 s) by rapid mixing (<milli-seconds) and reaction temperature.16 We reported a micro-flow peptide chain elongation using highly electrophilic species,17 as well as a one-step micro-flow synthesis of NCAs18 and the derivatizations.19 Herein, we report an efficient peptide chain elongation based on a one-flow, 3CC approach using α-NCA J (Scheme 1e). Although all the previously reported α-NCA-based approaches used α-NCA only as an electrophile (Scheme 1d), our approach is the first to use α-NCA J as both a nucleophile and an electrophile. The developed approach for the synthesis of various short peptides enables a reduction in the number of synthetic steps. The process also is mild (20 °C), rapid (<minutes), scalable, high-yielding, and column-chromatography-free.
Results and discussion
To the best of our knowledge, there has been no report of using α-NCA as a nucleophile in peptide bond formation. Therefore, we began by examining acylation of the H–Phe–NCA (1a). The initial examinations revealed that the use of acid chloride as an electrophile was suitable for the acylation, whereas the use of acid anhydride did not afford the desired amide product in a satisfactory yield (details are not shown). Thus, readily available pivaloyl chloride was used as a model for amino acid chloride. We connected two T-shaped mixers with Teflon tubing and immersed them in a water bath. A solution of pivaloyl chloride and 1a in CH2Cl2 was injected into the first mixer via syringe pump A. A solution of base in CH2Cl2 was injected into the first mixer via syringe pump B, which resulted in a nucleophilic acyl substitution. A solution of isopropylamine in CH2Cl2 was injected into the second mixer via syringe pump C to convert to the desired amino acid derivative 2. The resultant mixture including 2 was collected in test tubes and the results were evaluated via HPLC-UV analysis. The bases were examined (Table 1). The use of NMI, NMM, Me2NBn, Et3N, and i-Pr2NEt afforded the desired 2 in low yields (entries 1–5). The sole use of highly basic Et3N or i-Pr2NEt generated white precipitates probably due to the undesired oligomerization/polymerization of 1a. The combined use of pyridine and i-Pr2NEt afforded 2 in a low yield (entry 6), but the yield was slightly higher than that from the sole use of i-Pr2NEt (entry 5). To our delight, the combined use of nucleophilic NMI or DMAP and i-Pr2NEt or Me2NBn afforded the desired 2 in high yields (entries 7–9). In particular, the combined use of nucleophilic NMI and highly basic i-Pr2NEt afforded the desired 2 in an excellent yield (entry 7). This is probably because the nucleophilic NMI rapidly activated pivaloyl chloride via acyl N-methylimidazolium ion formation and the highly basic i-Pr2NEt rapidly deprotonate 1a to enhance its nucleophilicity. We previously reported urethane formation from α-NCAs and alkyl chloroformate, and the combined use of pyridine and tertiary amine afforded the best results.19b Interestingly, the optimal combination of bases (NMI and i-Pr2NEt) for amidation in this study was different from that for urethane formation in the previous study.|
|
||
|---|---|---|
| Entry | Base (pKaH)a | Yieldb (%) |
| a pKa of conjugated acid. b Yields were determined via HPLC-UV analysis. c The combined use of bases (1.0 equiv. + 1.0 equiv.). d Isolated yield. Bn = benzyl, NMI = N-methylimidazole; NMM = N-methylmorpholine; Me = methyl; Et = ethyl; i-Pr = isopropyl. | ||
| 1 | NMI (7.0)20a | 35 |
| 2 | NMM (7.4)20b | 2 |
| 3 | Me2NBn (8.9)20b | <1 |
| 4 | Et3N (10.7)20b | 8 |
| 5 | i-Pr2NEt (11.4)20b | <1 |
| 6c | i-Pr2NEt + pyridine (5.2)20a | 10 |
| 7c | i-Pr2NEt + NMI | >99 (>99)d |
| 8c | i-Pr2NEt + DMAP (9.7)20c | 99 |
| 9c | Me2NBn + NMI | 89 |
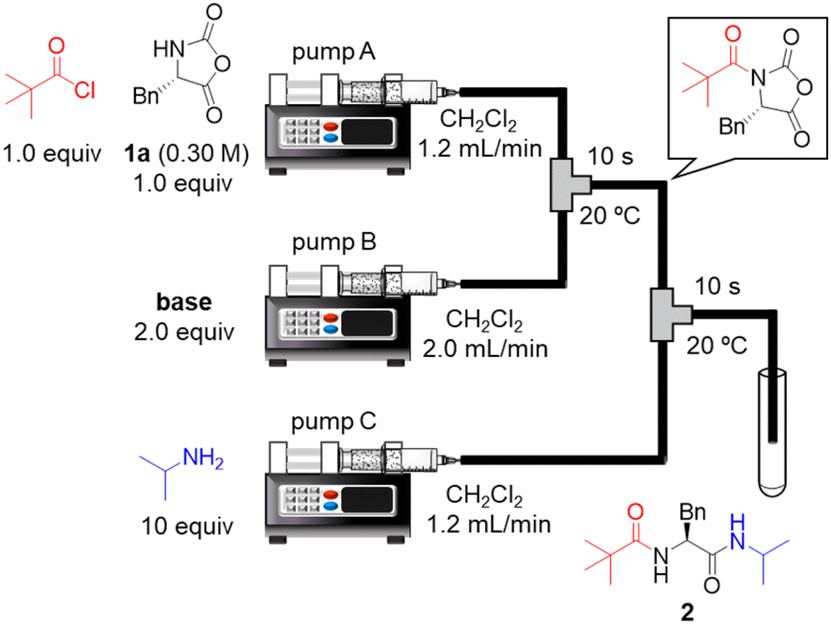
Next, we examined a one-flow 3CC for tripeptide synthesis based on the developed conditions (Fig. 1). In order to avoid the isolation of a labile acid chloride, Fmoc-protected amino acid chloride 4a was in situ prepared from Fmoc-protected amino acid 3a and SOCl2 in the presence of a base (for details of the optimization of bases, solvents and reaction times for acid chloride formation, see ESI Table S2†), which was used for the following coupling with α-NCA 1a. The coupling generated an intermediate 5a that was used for the subsequent coupling with amino acid ester 6a (for details of optimization of bases, amounts of substrate, and reaction times in the coupling between 5a and 6a, see ESI Table S3†) that afforded the desired tripeptide 7a. We successfully obtained the desired 7a in an 86% yield (30.5 s, 20 °C, 3 steps, Fig. 1). We were amazed that a rather pure peptide could be obtained after a simple aqueous work-up, and a simple recrystallization afforded a highly pure peptide without the need for column chromatographic purification. In addition, gram-scale (3.0 g) preparation of 7a was successfully achieved by simply extending the pumping time to 14 min 30 s (productivity: 12.3 g h−1). To verify the importance of the micro-flow conditions, batch reactions were examined under the same reaction conditions with the exception of the residence time (0.5 s) of amino acid chloride 4a and α-NCA 1a (for details, see ESI Table S4†). Although the reaction mixture was vigorously stirred during the experiments, reproducible results were not observed due to batch-to-batch differences in mixing efficiency. Observed yields under batch conditions were ca. 15% lower than those under flow conditions. In particular, this reaction involves the generation of toxic gas such as sulfur dioxide and hydrogen chloride, and, therefore, scale-up of this reaction in a batch reactor should be avoided.
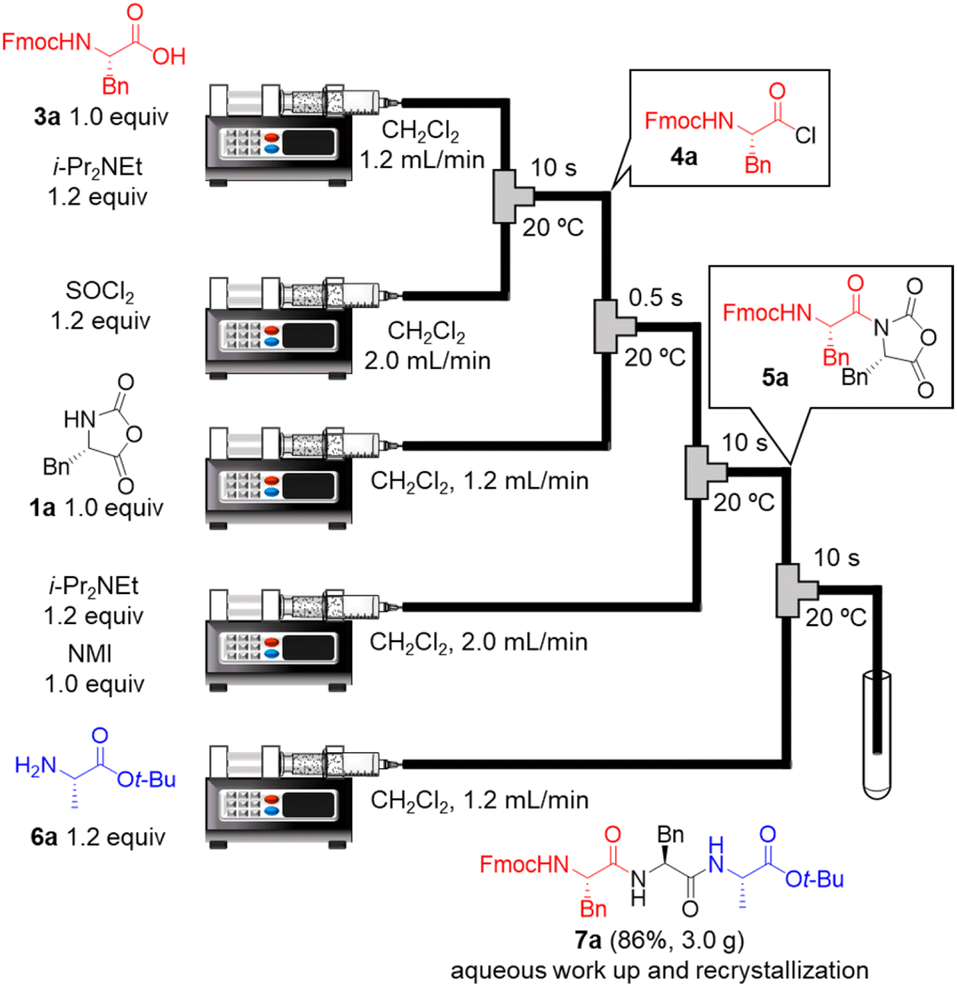 | ||
| Fig. 1 One-flow, 3CC approach for synthesis of tripeptide. Fmoc = 9-fluorenylmethyloxycarbonyl; t-Bu; tertiarybutyl. | ||
The substrate scope of our developed one-flow, 3CC approach was examined (Fig. 2). The use of Boc-protected amino acid was not suitable for this reaction due to α-NCA formation (for details, see ESI Table S6†),21 whereas the use of Cbz-protected amino acids afforded the desired tripeptides 7b–g in high yields (80–95%), although there also was a risk of α-NCA formation.22 The tripeptides 7h–p derived from Fmoc-protected amino acids afforded the desired products in high to excellent yields (83–94%). To our delight, the tripeptides 7h and 7i derived from sterically hindered Fmoc–Val–OH and Fmoc–Thr(Bn)–OH were obtained in high yields (86% and 87%). The tripeptides 7g and 7j derived from racemizable H–Cys(Bn)–NCA and Fmoc–Cys(t-Bu)–OH were also obtained in high yields (82% and 83%) without detectable racemization (<0.1%). Notoriously, none of the syntheses required column chromatographic purification, and the pure peptides were readily obtained by simple aqueous work-up with a subsequent recrystallization. A challenging synthesis of tripeptide 7q containing N-methyl amino acid residue was also examined (for details of optimization of the amounts of N-methyl amino acid, reaction times, and temperatures for the coupling between 5a and H–MePhe–OMe, see ESI Table S5†), and 7q was obtained in an acceptable yield (51%).
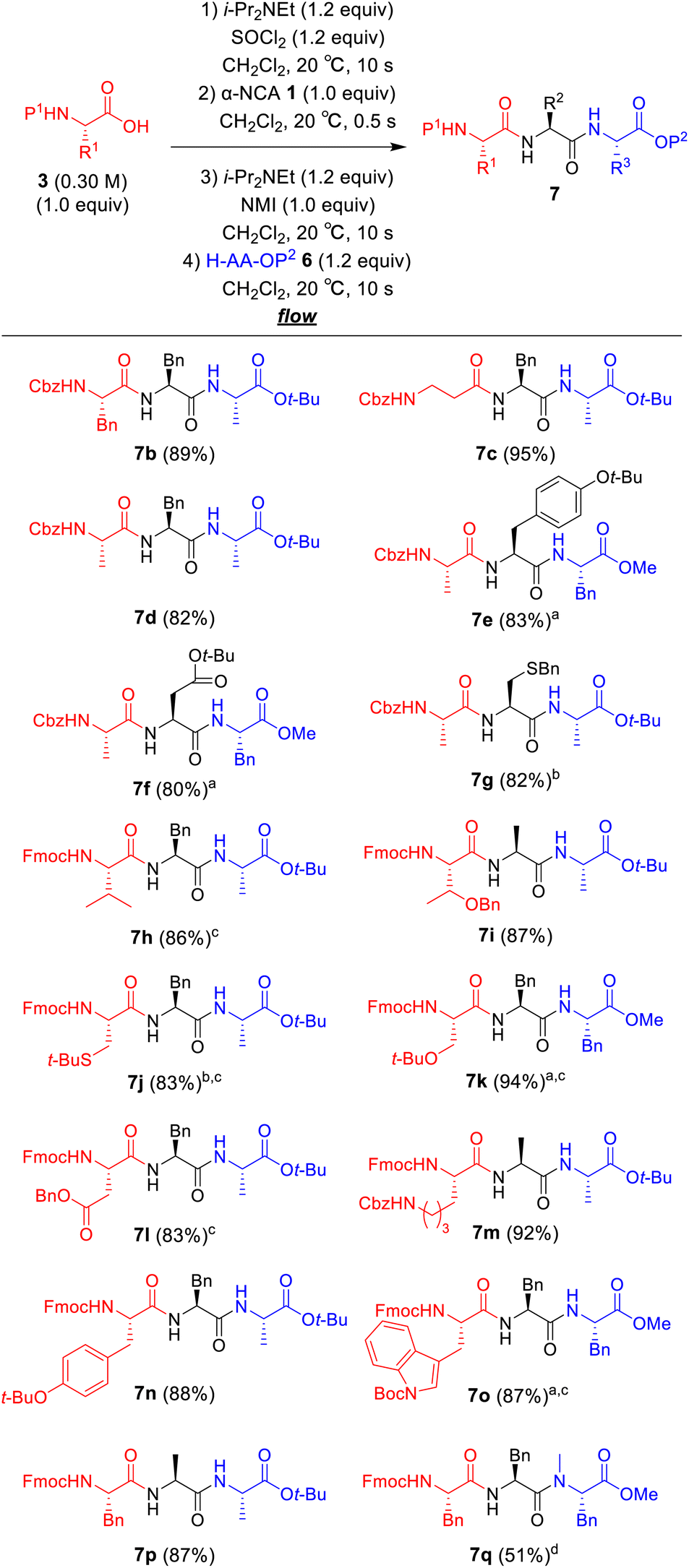 | ||
| Fig. 2 Substrate scope for the one-flow, 3CC approach. aReaction time for the coupling of N-acylated α-NCA 5 with amino acid ester 6 was 10 s (flow) + 1 min (batch). bRacemization was not detected by HPLC-UV analysis. cReaction time for the coupling of amino acid chloride 4 with α-NCA 1 was 15 s. dReaction time for the coupling of 5 with H–MePhe–OMe (1.5 equiv.) was 3 h (batch). | ||
We applied our developed approach for synthesis of the octapeptide 10b, which was shown in Scheme 2, is a beefy meaty peptide (H–Lys–Gly–Asp–Glu–Glu–Ser–Leu–Ala–OH).23 The preparation of amino acid chlorides from 3b–d and their couplings with α-NCA 1b–d was rapid (total 20.5 s). Thus, the process was carried out under micro-flow conditions. On the other hand, the following couplings with 6b, 7s, and 8b required a longer time (1–10 min), and, therefore, these were carried out under batch conditions. The first 3CC of 3b with 1b and 6b afforded crude tripeptide 7r, and the subsequent simple recrystallization afforded sufficiently pure 7r in 82% yield (HPLC purity 94%). We compared both the time and the cost of our synthesis of N-terminus protected tripeptide 7r with those of Wassenaar's solid-phase synthesis of Fmoc–Ser(t-Bu)–Leu–Ala–HMP resin (7t) that has the same peptide sequence (for details, see ESI†).24 Dramatic reductions were achieved in both time (5.2 min mmol−1vs. 158 min mmol−1) and cost (787 JPY per mmol vs. ca. 5820 JPY per mmol). The batch removal of the Cbz group in 7r afforded N-terminal-free tripeptide 7s in a quantitative yield. The second 3CC of 3c with 1c and 7s (64% yield, HPLC purity 93%) and batch removal of the Cbz group afforded N-terminal-free pentapeptide 8b (92% yield). The third 3CC of 3d with 1d and 8b (64% yield, HPLC purity 90%) and the batch removal of the Cbz group in 9a afforded N-terminal-free heptapeptide 9b (91% yield). The formation of a mixed carbonic anhydride from 3e under micro-flow conditions and the subsequent batch coupling with heptapeptide 9b afforded the desired protected beefy meaty peptide 10a (55% yield, HPLC purity 86%). Amazingly, no column chromatographic purification was needed in the synthesis of 10a. The final acidic removal of t-Bu groups and Boc groups and the following reversed phase column chromatographic purification afforded beefy meaty peptide (10b) in 85% yield (overall yield 13% in 15 steps, HPLC purity 98%). The structure of 10b was confirmed via1H NMR, 13C NMR (for details, see ESI,† NMR spectra section), and HRMS. The observed spectra were well consistent with those of commercially purchased 10b.
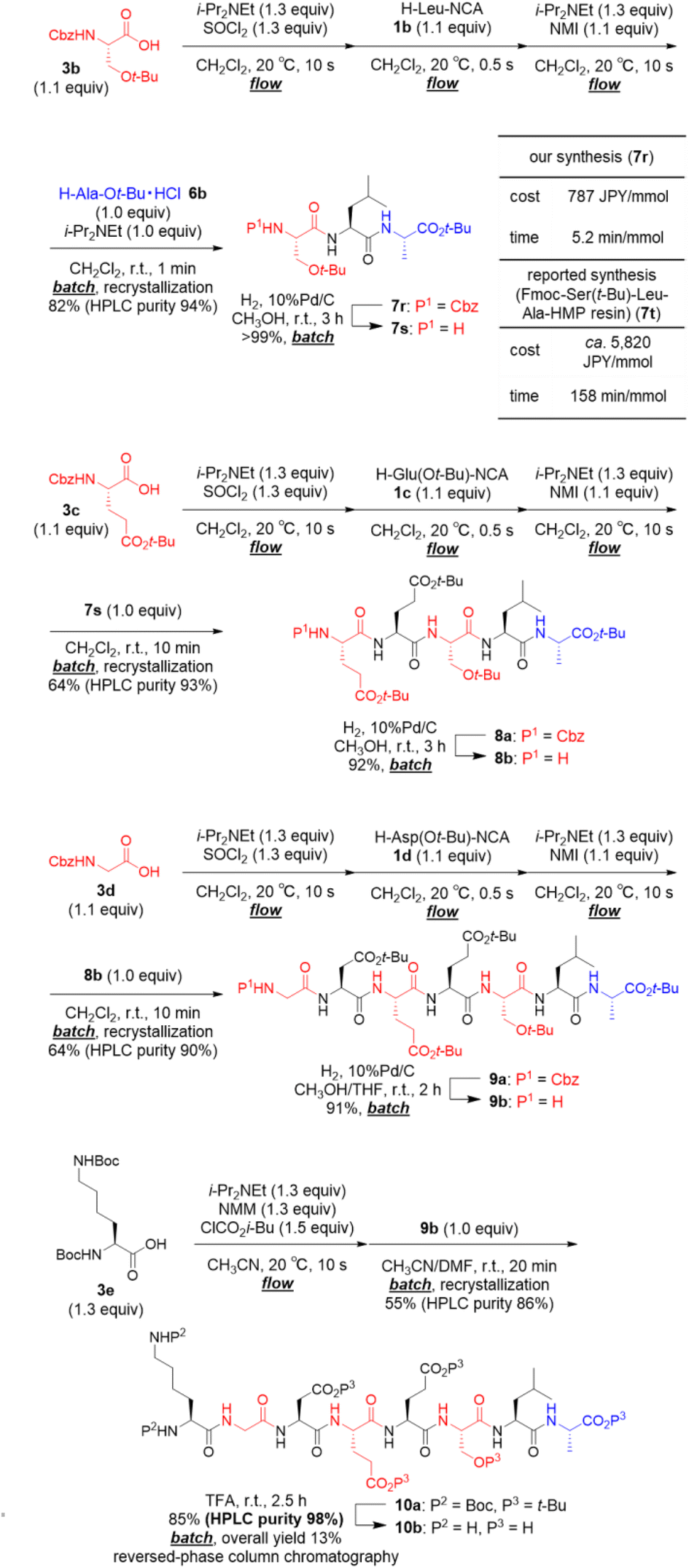 | ||
| Scheme 2 Total synthesis of beefy meaty peptide (10b). | ||
Finally, in order to achieve a one-flow synthesis of tripeptide by using only amino acid building blocks that were readily and commercially available, we examined an in situ preparation of α-NCA from Boc-protected amino acid 3f and SOCl2 in the presence of a base (for details, see ESI Table S6†). We also established the optimized amounts of reagents and reaction times for the formation of 1a as well as that for amidation between 4a and 1a (for details, see ESI Table S7†). The optimized conditions afforded the desired tripeptide 7a in 80% yield using simple silica-gel column chromatography and recrystallization (HPLC purity 95%, Fig. 3). Racemization was not detected in HPLC-UV analysis. It should be noted that the tripeptide was rapidly prepared via a one-flow 3CC approach from protected amino acids 3a, 3f, and 6b, which are inexpensive and both readily and commercially available.
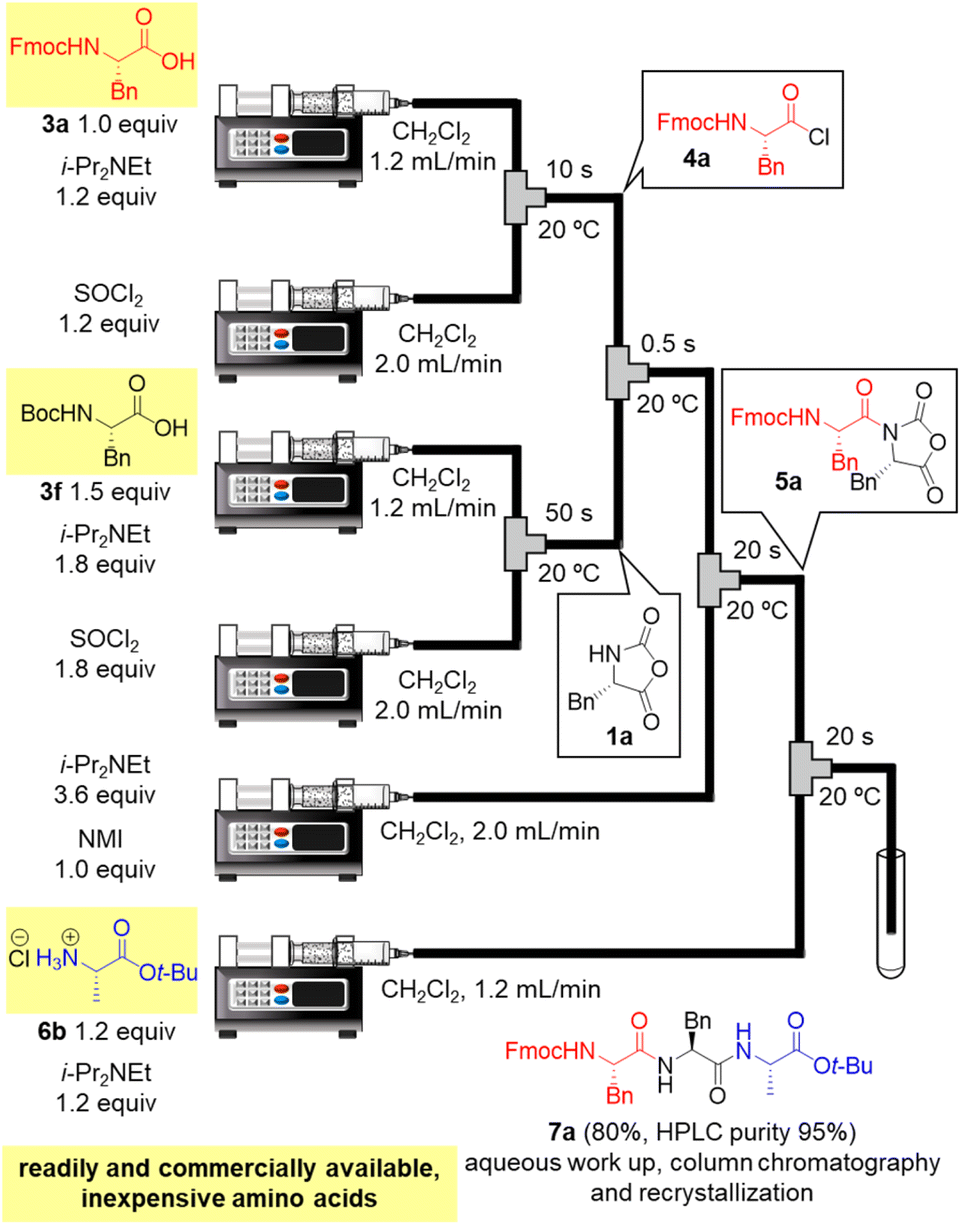 | ||
| Fig. 3 One-flow tripeptide synthesis from readily and commercially available, inexpensive amino acids 3a, 3f, and 6b. | ||
Conclusions
We developed a one-flow 3CC approach to peptide chain elongation that is both rapid and mild. The proposed approach is the first to use α-NCAs as both electrophiles and nucleophiles. The key to success was the rapid generation of acid chloride and its rapid coupling with α-NCA in the presence of NMI and i-Pr2NEt that suppressed undesired reactions of α-NCAs. The developed approach uses only readily removable reagents and substrates, which allows a simple aqueous work-up and recrystallization to afford the desired peptides in high yields and purities without tedious column chromatographic purifications. A scaled-up synthesis could be safely and readily achieved in a highly reproducible manner via simply extending the pumping time. In addition, we achieved a total synthesis of octapeptide, beefy meaty peptide, by repeating our developed 3CC approach with only the addition of one column chromatographic purification as the final step. The reductions in both time (ca. 1/31) and cost (ca. 1/7) were dramatic compared with that of typical solid-phase synthesis. Moreover, a one-flow tripeptide synthesis including in situ preparation of α-NCA was realized, which allowed a tripeptide to be rapidly prepared from readily and commercially available protected amino acid building blocks. The developed approach potentially produces longer peptides in one-flow fashion by injecting multiple NCAs. Although we challenged this powerful synthesis, we have not succeeded yet. The developed approach offers a novel and powerful option for the preparation of short peptides and should dramatically accelerate drug/material developments that are based on peptides/proteins.Data availability
The synthetic procedures, characterization, and spectral data supporting this article have been uploaded as part of the ESI.†Author contributions
Naoto Sugisawa: conceptualization, data curation, formal analysis, methodology, investigation. Akira Ando: data curation, formal analysis, investigation. Shinichiro Fuse: conceptualization, data curation, project administration and supervision. All authors: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Grant-in-Aid for Transformative Research Area (B) (21H05081), and the Research Support Project for Life Science and Drug Discovery [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from the Japan Agency for Medical Research and Development (AMED) under Grant Number JP22ama121044, and JSPS Fellows (22J12988 to N. Sugisawa). Pd/C was generously provided by Kawaken Fine Chemicals, Japan.Notes and references
- (a) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed; (b) L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 1–27 CrossRef PubMed.
- G. B. Santos, A. Ganesan and F. S. Emery, ChemMedChem, 2016, 11, 2245–2251 CrossRef CAS PubMed.
- V. Agouridas, O. E. Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- (a) R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS; (b) H. Yan and F.-E. Chen, Adv. Synth. Catal., 2022, 364, 1934–1961 CrossRef CAS.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 CrossRef CAS PubMed.
- V. Martin, P. H. Egelund, H. Johansson, S. T. Le Quement, F. Wojcik and D. S. Pedersen, RSC Adv., 2020, 10, 42457–42492 RSC.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- I. E. Sampaio-Dias, C. A. D. Sousa, S. C. Silva-Reis, S. Ribeiro, X. García-Mera and J. E. Rodríguez-Borges, Org. Biomol. Chem., 2017, 15, 7533–7542 RSC.
- Y. Zhu and W. D. Fuller, Tetrahedron Lett., 1995, 36, 807–810 CrossRef CAS.
- T. Hattori and H. Yamamoto, J. Am. Chem. Soc., 2022, 144, 1758–1765 CrossRef CAS PubMed.
- G. M. Ziarani, R. Moradi and L. Mahammadkhani, Arkivoc, 2019, 18–40 Search PubMed.
- (a) R. G. Denkewalter, H. Schwam, R. G. Strachan, T. E. Beesley, D. F. Veber, E. F. Schoenewaldt, H. Barkemeyer, W. J. Paleveda, T. A. Jacob and R. Hirschmann, J. Am. Chem. Soc., 1966, 88, 3163–3164 CrossRef CAS; (b) R. Hirschmann, R. G. Strachan, H. Schwan, E. F. Schoenewaldt, H. Joshua, B. Barkemeyer, D. F. Veber, W. J. Paleveda Jr, T. A. Jacob, T. E. Beesley and R. G. Denkewalter, J. Org. Chem., 1967, 32, 3415–3425 CrossRef CAS PubMed; (c) R. G. Denkewalter and R. Hirschmann, Am. Sci., 1969, 57, 389–409 CAS.
- K. E. Jolley, W. Nye, C. González Niño, N. Kapur, A. Rabion, K. Rossen and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1557–1565 CrossRef CAS.
- C. Shin, Y. Yonezawa and J. Yoshimura, Chem. Lett., 1981, 1635–1638 CrossRef CAS.
- (a) Y. Yonezawa, T. Yamada and C. Shin, Chem. Lett., 1982, 1567–1568 CrossRef CAS; (b) C. Shin, Y. Yonezawa and M. Ikeda, Bull. Chem. Soc. Jpn., 1986, 59, 3573–3579 CrossRef CAS; (c) C. Shin, S. Honda, K. Morooka and Y. Yonezawa, Bull. Chem. Soc. Jpn., 1993, 66, 1844–1846 CrossRef CAS.
- (a) J.-i. Yoshida, Flash Chemistry – Fast Organic Synthesis in Micro Systems, Wiley-VCH, Weinheim, 2008 CrossRef; (b) J.-i. Yoshida, A. Nagaki and T. Yamada, Chem.–Eur. J., 2008, 14, 7450–7459 CrossRef CAS PubMed; (c) J.-i. Yoshida, Chem. Rec., 2010, 10, 332–341 CrossRef CAS PubMed; (d) J.-i. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- (a) S. Fuse, Y. Mifune and T. Takahashi, Angew. Chem., Int. Ed., 2014, 53, 851–855 CrossRef CAS PubMed; (b) S. Fuse, Y. Mifune, H. Nakamura and H. Tanaka, Nat. Commun., 2016, 7, 13491 CrossRef CAS PubMed; (c) S. Fuse, K. Masuda, Y. Otake and H. Nakamura, Chem.–Eur. J., 2019, 25, 15091–15097 CrossRef CAS PubMed; (d) Y. Otake, Y. Shibata, Y. Hayashi, S. Kawauchi, H. Nakamura and S. Fuse, Angew. Chem., Int. Ed., 2020, 59, 12925–12930 CrossRef CAS PubMed.
- (a) Y. Otake, H. Nakamura and S. Fuse, Angew. Chem., Int. Ed., 2018, 57, 11389–11393 CrossRef CAS PubMed; (b) N. Sugisawa, Y. Otake, H. Nakamura and S. Fuse, Chem.–Asian J., 2020, 15, 79–84 CrossRef CAS PubMed.
- (a) N. Sugisawa, H. Nakamura and S. Fuse, Chem. Commun., 2020, 56, 4527–4530 RSC; (b) R. Okabe, N. Sugisawa and S. Fuse, Org. Biomol. Chem., 2022, 20, 3303–3310 RSC.
- (a) M. L. Bender and B. W. Turnquest, J. Am. Chem. Soc., 1957, 79, 1656–1662 CrossRef CAS; (b) H. K. Hall Jr, J. Am. Chem. Soc., 1957, 79, 5441–5444 CrossRef; (c) C. Faltin, E. M. Fleming and S. J. Connon, J. Org. Chem., 2004, 69, 6496–6499 CrossRef CAS PubMed.
- M. Akssira, M. Boumzebra, H. Kasmi, A. Dahdouh, M.-L. Roumestant and P. Viallefont, Tetrahedron, 1994, 50, 9051–9060 CrossRef CAS.
- D. Ben-Ishai and E. Katchalski, J. Am. Chem. Soc., 1952, 74, 3688–3689 CrossRef CAS.
- Y. Yamasaki and K. Maekawa, Agric. Biol. Chem., 1978, 42, 1761–1765 CAS.
- P. D. van Wassenaar, A. H. A. van den Oord and W. M. M. Schaaper, J. Agric. Food Chem., 1995, 43, 2828–2832 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01333b |
| This journal is © The Royal Society of Chemistry 2023 |