DOI:
10.1039/D3SC01295F
(Edge Article)
Chem. Sci., 2023,
14, 6087-6094
From closed-shell edge-extended kekulenes to open-shell carbonylated cycloarene diradicaloid†
Received
10th March 2023
, Accepted 13th May 2023
First published on 15th May 2023
Abstract
The precise synthesis of cycloarenes remains a challenging topic in both organic chemistry and materials science due to their unique fully fused macrocyclic π-conjugated structure. Herein, a series of alkoxyl- and aryl-cosubstituted cycloarenes (kekulene and edge-extended kekulene derivatives, K1–K3) were conveniently synthesized and an unexpected transformation of the anthryl-containing cycloarene K3 into a carbonylated cycloarene derivative K3-R was disclosed by controlling the temperature and gas atmosphere of the Bi(OTf)3-catalyzed cyclization reaction. All their molecular structures were confirmed by single-crystal X-ray analysis. The crystallographic data, NMR measurements, and theoretical calculations reveal their rigid quasi-planar skeletons, dominant local aromaticities, and decreasing intermolecular π–π stacking distance with extension of the two opposite edges. The much lower oxidation potential for K3 by cyclic voltammetry explains its unique reactivity. Moreover, carbonylated cycloarene derivative K3-R shows a remarkable stability, large diradical character, a small singlet–triplet energy gap (ΔES–T = −1.81 kcal mol−1), and weak intramolecular spin–spin coupling. Most importantly, it represents the first example of carbonylated cycloarene diradicaloids as well as the first example of radical-acceptor cycloarenes and will shed some light on synthesis of extended kekulenes and conjugated macrocyclic diradicaloids and polyradicaloids.
Introduction
Cycloarenes are fully fused macrocycles with an enclosed cavity, by the combination of annellations of benzene units.1 In the beginning, many theoretical studies focused on the question of their electron structure: whether π-electrons in cycloarenes are localized on sextets or delocalized throughout the whole conjugated skeleton,2 until the first cycloarene kekulene, a regular hexagonal macrocycle consisting of 12 benzene rings, was successfully synthesized by Staab and Diederich in 1978.3 The deshielded inner protons by 1H NMR measurement suggest that π-electrons are localized on individual benzenoid rings, which supports the hypothesis proposed by McWheeny2b and the model described by Clar.2c In the following three decades, the single synthesis strategy and the poor solubility of products limited the development of cycloarenes,4 and only a contracted kekulene cyclo[d,e,d,e,e,d,e,d,e,e]decakisbenzene was reported.5 Recently, kekulene's seven-sided cousin septulene synthesized by ring-closing metathesis of olefins6 and the alkoxy substituted octagonal cousin octulene prepared by the fold-in method were reported in 2012 and 2016, respectively.7 Subsequently we and Wu's group developed an efficient synthetic strategy for aryl-substituted cycloarenes and alkoxy substituted heterocycloarenes through Bi(OTf)3-catalyzed cyclization from a macrocyclic methoxyethenylated precursor.8 In 2021, based on this strategy, Wu's group reported a series of core-expanded kekulenes, more than two edges of which are expanded.9 Moreover, there are some peripherally fused kekulene analogues obtained by on-surface synthesis10 or Diels–Alder cycloaddition in solution.11 Although some effective synthetic strategies have been explored over the years, only a few cycloarenes were reported,12 and the poor molecular diversity severely limited their further development.
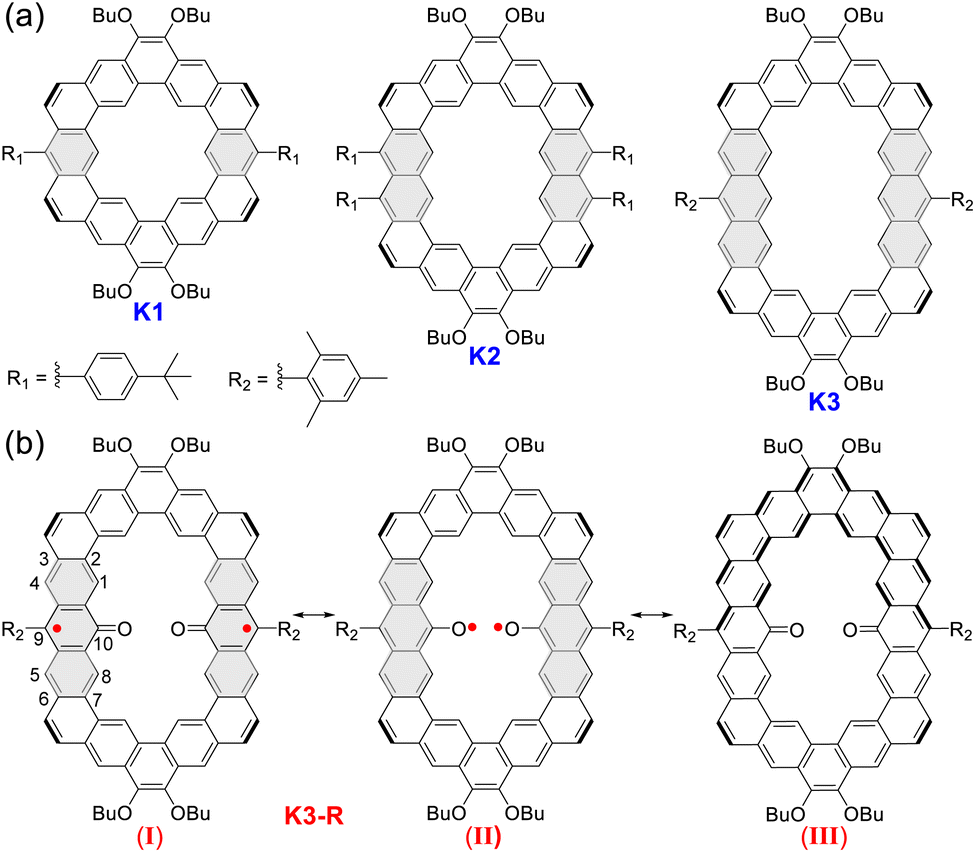
Herein, according to the extension of phenyl building blocks from benzene to anthracene, we have successfully designed and synthesized an alkoxyl- and aryl-cosubstituted kekulene (K1) and its edge-extended homologues (K2 and K3) by the Suzuki coupling reaction followed by Bi(OTf)3-catalyzed cyclization of vinyl ethers at room temperature. Interestingly, a carbonylated cycloarene derivative K3-R with two anthroxyl radicals was obtained when we raised the temperature of the cyclization reaction to 80 °C under an air atmosphere, which displays open-shell singlet ground diradical character with a small singlet–triplet energy gap (ΔES–T = −1.81 kcal mol−1) verified by X-ray crystallography and variable-temperature electron spin resonance (VT ESR) measurements. According to X-ray single crystal analysis, optical spectroscopy and theoretical calculations, we have systematically investigated the size-dependent molecular overlap model, electronic structure and aromaticity of these three closed-shell cycloarenes and the open-shell carbonylated cycloarene diradicaloid. The low oxidation potential for K3 measured by cyclic voltammetry (CV) supports reasonable unique reactivity of anthryl-based cycloarene and facilitates the formation of diradical product K3-R. Meanwhile, K3-R can be drawn in at least three different resonance forms (Fig. 1):13 an open-shell diradical form with the unpaired electron localized at the 9-position (I); an open-shell diradical form with the unpaired electron localized at oxygen atoms (II); and a closed-shell form containing a quinoidal and an aromatic dibenzo[a,o]pentaphene (III). Thus, the electronic structure, aromaticity, and radical character of open-shell anthroxyl-based macrocycle diradical K3-R are of great interest due to the contributions of multiple resonance forms. The crystallographic data reveals the dominant contribution of resonance structure I, and the theoretical calculations show its large radical character and weak intramolecular spin–spin coupling between the two neighboring spins.
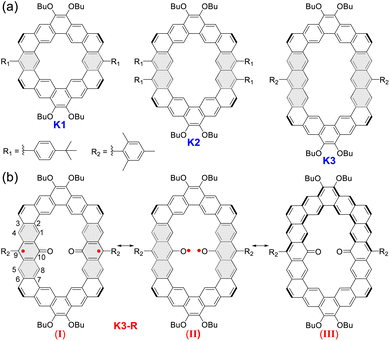 |
| | Fig. 1 (a) Structures of alkoxyl- and aryl-cosubstituted kekulene K1 and its edge-extended homologues K2 and K3. (b) The representative resonance forms of carbonylated cycloarene diradical K3-R. | |
Results and discussion
Synthesis
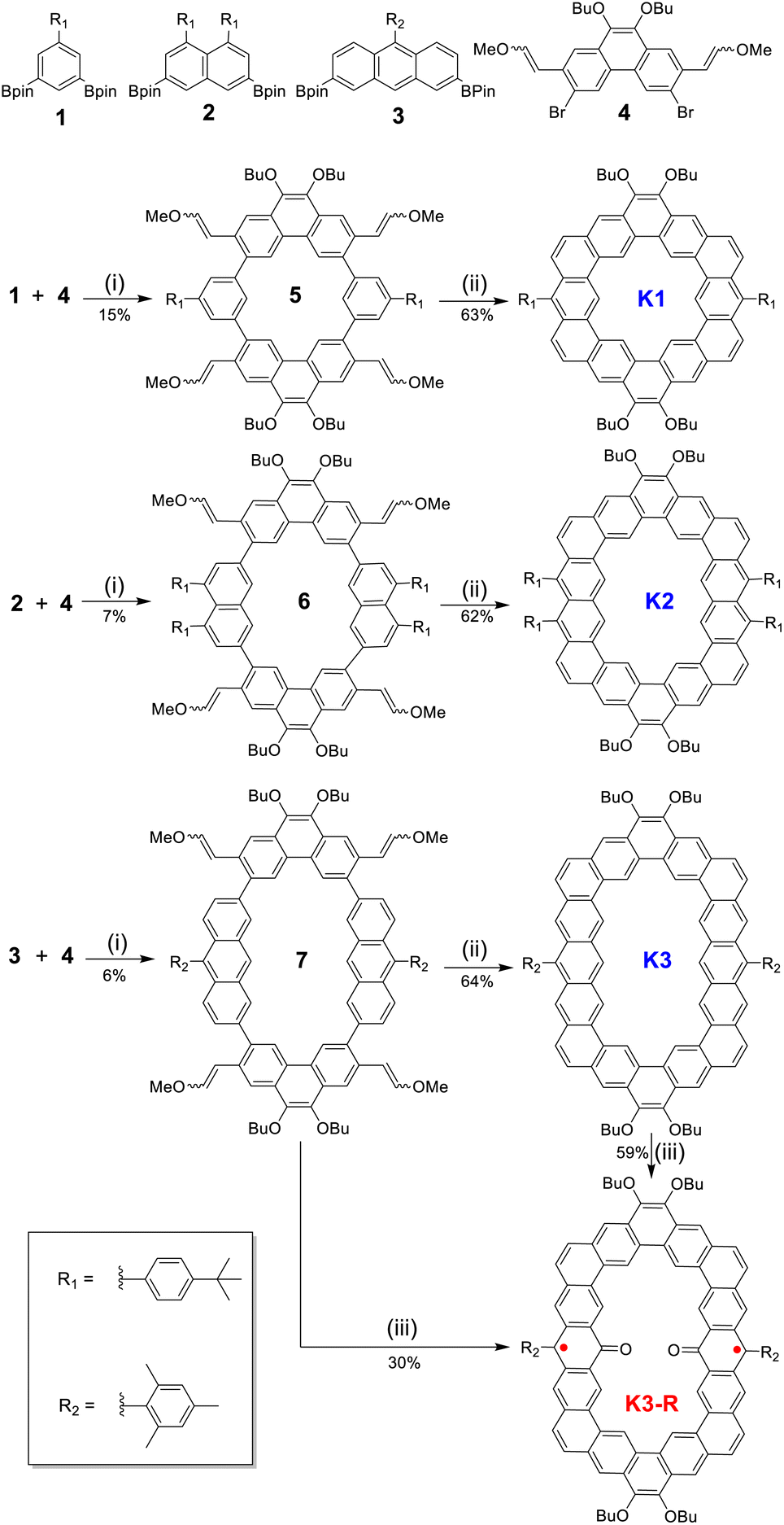
The synthesis of alkoxyl- and aryl-cosubstituted kekulene K1 and its edge-extended homologues K2 and K3 was carried out as shown in Scheme 1. K1 can be regarded as an aryl- and alkoxy-cosubstituted kekulene, and its tetramethoxyethenylated macrocyclic precursor 5 was synthesized by the Suzuki coupling reaction using XPhos Pd G2 as a catalyst14 in 14.7% isolated yield, with benzene and phenanthrene units as building blocks. Then, treatment of 5 with Bi(OTf)3 catalyzed cyclization in 1,2-dichloroethane (DCE) at room temperature gave the fully fused product K1 in 63% yield.8,14,15 By the replacement of benzene units with naphthalene and anthracene units, the higher homologues K2 and K3 were synthesized as yellow and orange solids, respectively. All new products were characterized by NMR spectroscopy and high-resolution mass spectroscopy.
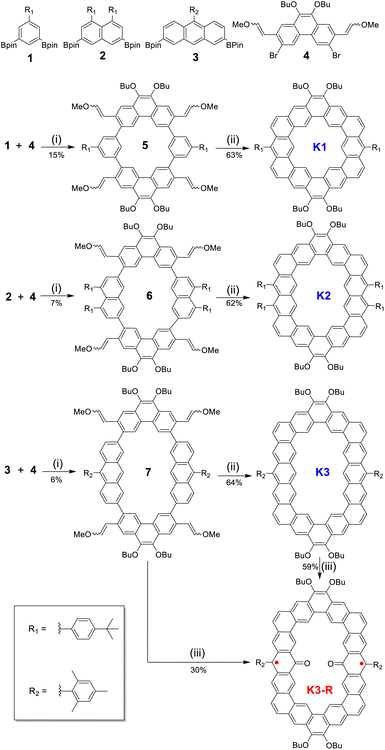 |
| | Scheme 1 Synthetic route of alkoxyl- and aryl-cosubstituted cycloarenes K1–K3 and carbonylated cycloarene diradical K3-R: (i) XPhos Pd G2, K3PO4, THF/H2O, 50 °C; (ii) Bi(OTf)3, CH2ClCH2Cl, rt; (iii) Bi(OTf)3, air (O2), CH2ClCH2Cl, 80 °C. | |
Interestingly, when heating a mixture of anthryl-based tetramethoxyethenylated macrocyclic precursor 7 and Bi(OTf)3 in DCE at 80 °C under an air atmosphere, we obtain an anthroxyl-based diradical product K3-R as a black solid, which can be regarded as a carbonylated cycloarene derivative. It is stable enough to be purified over conventional flash silica gel column chromatography (dichloromethane as an eluent). In the NMR spectrum, although sharp signals corresponding to the methyl protons and some CH2 protons of the butoxyl group were observed, a broad resonance signal was observed in the aromatic region at room temperature, which suggests the paramagnetic character of K3-R. Even at −90 °C, the 1H NMR signal of K3-R was still broad (Fig. S5†) indicating appreciable triplet character.16 This product was identified by high-resolution mass spectrum measurement (Fig. S37†), single crystal X-ray diffraction analysis (Fig. 2) and ESR spectroscopy (Fig. S6† and 6a). It is worth noting that the diradical product K3-R can also be obtained directly by treating cycloarene K3 under the same heating conditions, indicating that the transformation from 7 to K3-R involves both cyclization and oxidation. Generally, the oxidation of anthracene to an anthroxyl radical requires high reaction temperature (200–300 °C).17 Meanwhile, in the absence of bismuth triflate, the oxidation of 7 or K3 to diradical K3-R did not proceed, even under an air (O2) atmosphere, which indicates that Bi(OTf)3 may significantly reduce the activation energy of oxidation and the oxidation is the result of the combination of the bismuth(III) catalyst and O2 as the oxidant.18 Additionally, this oxidation was not observed in the benzene- or naphthalene-based macrocyclic system, indicating that this is the intrinsic nature of anthryl-based cycloarene.
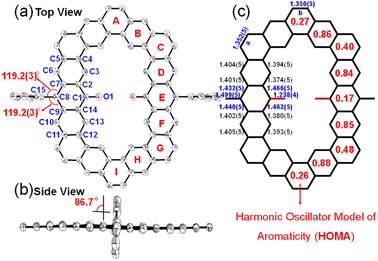 |
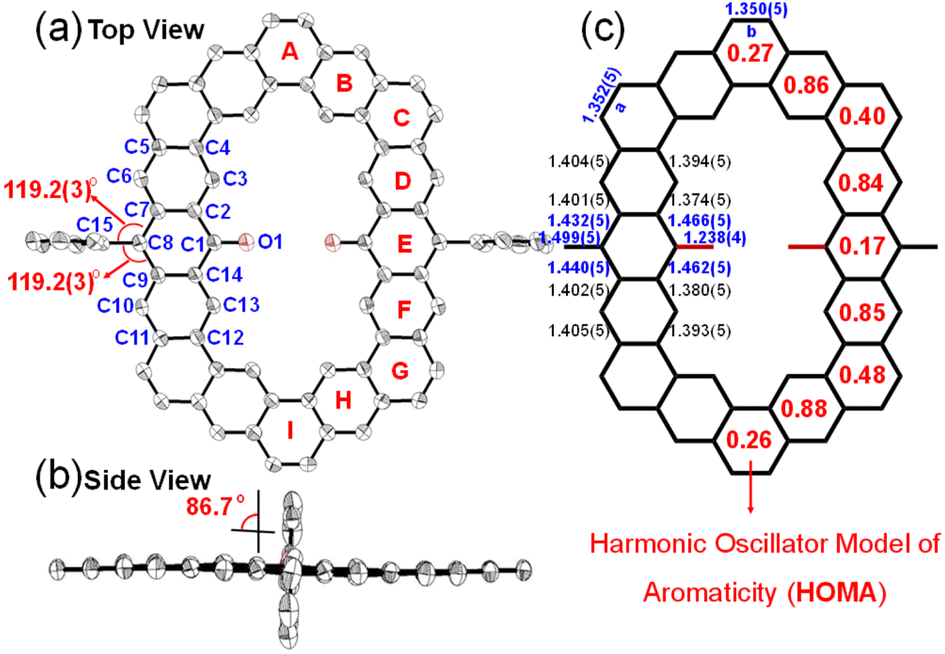
| | Fig. 2 X-ray crystallographic structure of K3-R: (a) top view with labels; (b) side view; (c) selected bond lengths (in Å) of the backbone. The numbers in parentheses are the estimated standard deviations (esds). The red numbers in rings A–I are the calculated HOMA values. Hydrogen atoms, alkyl, and alkoxy substituents are omitted for clarity. | |
X-ray crystallographic structure and aromaticity
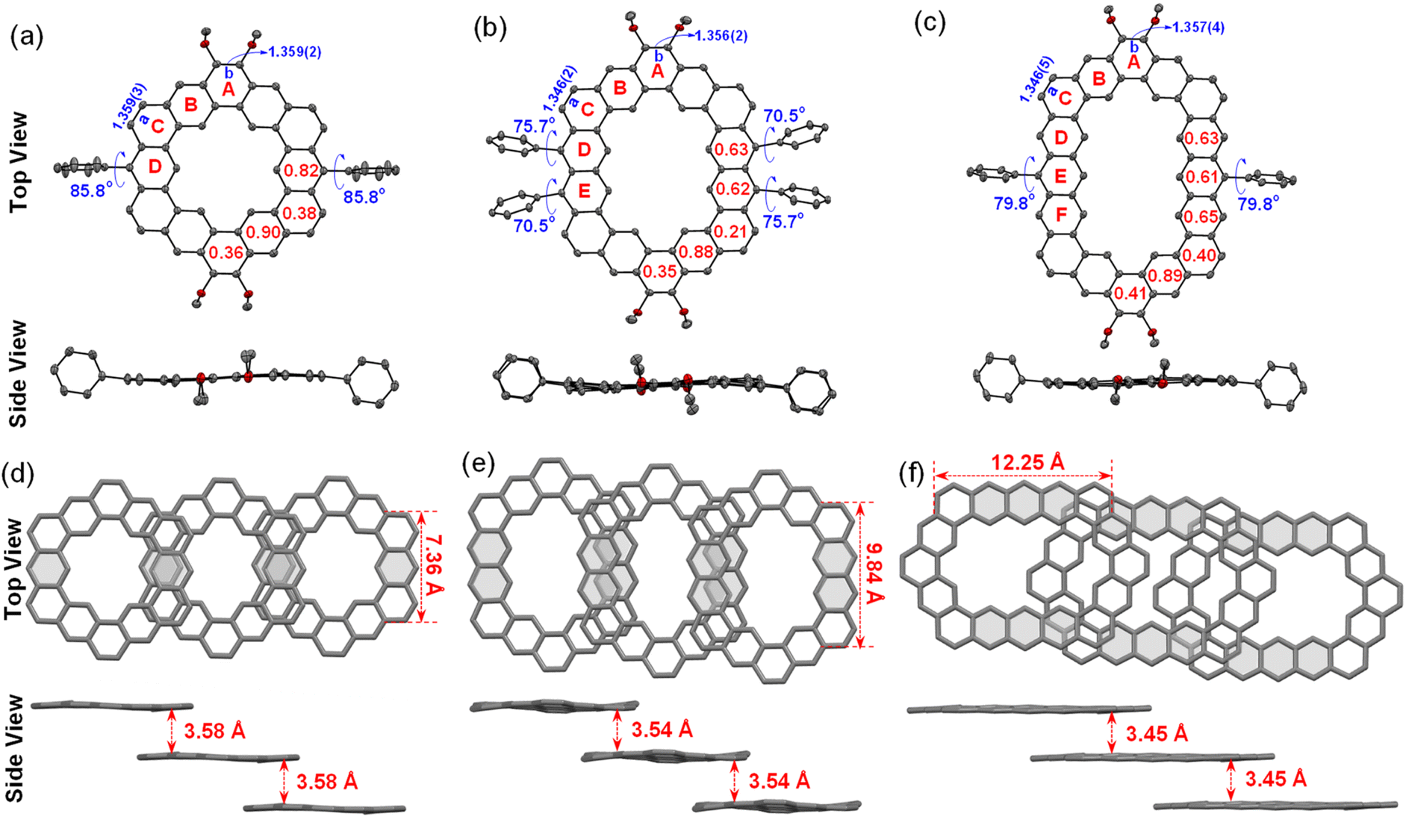
The single crystals of three alkoxyl- and aryl-cosubstituted cycloarenes K1–K3 were grown by slow diffusion of n-hexane into the carbon disulfide solution under ambient conditions. All three crystals are aligned in triclinic unit cells with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Their crystallographic structures are shown in Fig. 3, which clearly reveal slightly distorted coplanar π-conjugated skeletons. The hexagonal skeleton is stretched with the extension of the building block from benzene to anthracene, and the side length of zigzag edges connected to aryl-substituted groups is calculated to be 7.36 Å for K1, 9.84 Å for K2 and 12.25 Å for K3 (Fig. 3). Different from the herringbone pattern of kekulene with no substituent group,19 alkoxyl- and aryl-cosubstituted K1–K3 adopt a one-dimensional (1D) slipped stacking, and the interplanar distance decreases with extension of the two opposite edges. By creating planes based on all of the C atoms in skeletons, the interplanar distance was measured to be 3.58 Å for K1, 3.54 Å for K2 and 3.45 Å for K3 (Fig. 3). Meanwhile, with the replacement of the t-butylphenyl group with a larger steric hindrance mesityl group, the overlap of adjacent molecules concentrates from the aryl-substituted sides for K1/K2 to the alkoxy-substituted sides for K3, which may facilitate tighter packing.
. Their crystallographic structures are shown in Fig. 3, which clearly reveal slightly distorted coplanar π-conjugated skeletons. The hexagonal skeleton is stretched with the extension of the building block from benzene to anthracene, and the side length of zigzag edges connected to aryl-substituted groups is calculated to be 7.36 Å for K1, 9.84 Å for K2 and 12.25 Å for K3 (Fig. 3). Different from the herringbone pattern of kekulene with no substituent group,19 alkoxyl- and aryl-cosubstituted K1–K3 adopt a one-dimensional (1D) slipped stacking, and the interplanar distance decreases with extension of the two opposite edges. By creating planes based on all of the C atoms in skeletons, the interplanar distance was measured to be 3.58 Å for K1, 3.54 Å for K2 and 3.45 Å for K3 (Fig. 3). Meanwhile, with the replacement of the t-butylphenyl group with a larger steric hindrance mesityl group, the overlap of adjacent molecules concentrates from the aryl-substituted sides for K1/K2 to the alkoxy-substituted sides for K3, which may facilitate tighter packing.
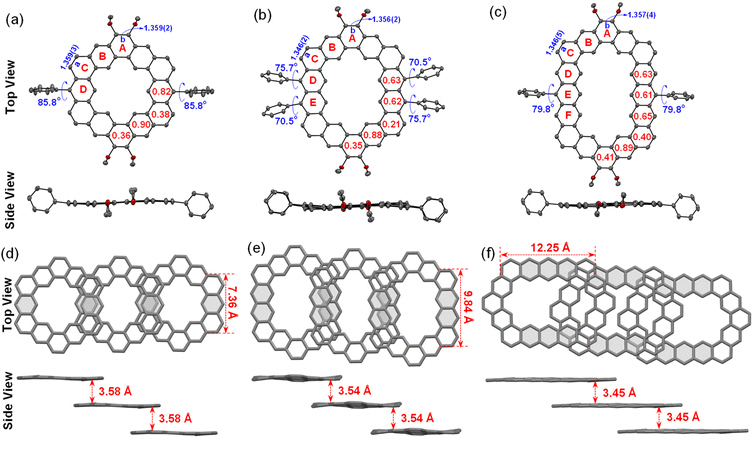 |
| | Fig. 3 Single-crystal X-ray diffraction structures (hydrogen atoms and alkyl substituents are omitted and the long alkoxy substituents are shortened to methoxy substituents for clarity), selected bond lengths (Å), calculated HOMA values, and crystal framework packing structures of (a and d) K1, (b and e) K2, and (c and f) K3. | |
The single crystal of carbonylated cycloarene derivative K3-R was grown by slow diffusion of methanol into a CHCl3 solution at room temperature and is shown in Fig. 2. The crystal of K3-R is aligned in a triclinic unit cell and there are two molecules of K3-R in each asymmetric unit, which is similar to its closed-shell precursor K3. The molecular backbone is rigid and slightly distorted and the molecular packing exhibits 1D parallel stacking with an intermolecular distance of 3.48 Å, which is a little wider than that of K3. The bond length of CO bonds at the 10-position of anthracene units of K3-R is 1.238(4) Å, which is almost as short as the ketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length in benzophenone (1.23 Å),20 and indicates their doubled bond characters. The C7–C8 (1.432(5) Å) and C8–C9 (1.440(5) Å) are longer than the C(sp2)–C(sp2) bond (1.39 Å) in benzene, and close to the typical C(sp2)–C(sp2) single-bond (1.46 Å),20 which suggests its single-bond character. Moreover, the bond angles of ∠C15–C8–C7 (119.2(3)o) and ∠C15–C8–C9 (119.2(3)o) are near the sp2 hybridization bond angle (120o), which verifies that the atom C8 is an sp2 hybrid. The above crystallographic analysis suggests its carbonylated cycloarene structure containing two anthroxyl radicals. In addition, the lack of both significant bond length alternation and NMR signal at room temperature implies that the major resonance contribution of open-shell K3-R is form I as shown in Fig. 1.
O bond length in benzophenone (1.23 Å),20 and indicates their doubled bond characters. The C7–C8 (1.432(5) Å) and C8–C9 (1.440(5) Å) are longer than the C(sp2)–C(sp2) bond (1.39 Å) in benzene, and close to the typical C(sp2)–C(sp2) single-bond (1.46 Å),20 which suggests its single-bond character. Moreover, the bond angles of ∠C15–C8–C7 (119.2(3)o) and ∠C15–C8–C9 (119.2(3)o) are near the sp2 hybridization bond angle (120o), which verifies that the atom C8 is an sp2 hybrid. The above crystallographic analysis suggests its carbonylated cycloarene structure containing two anthroxyl radicals. In addition, the lack of both significant bond length alternation and NMR signal at room temperature implies that the major resonance contribution of open-shell K3-R is form I as shown in Fig. 1.
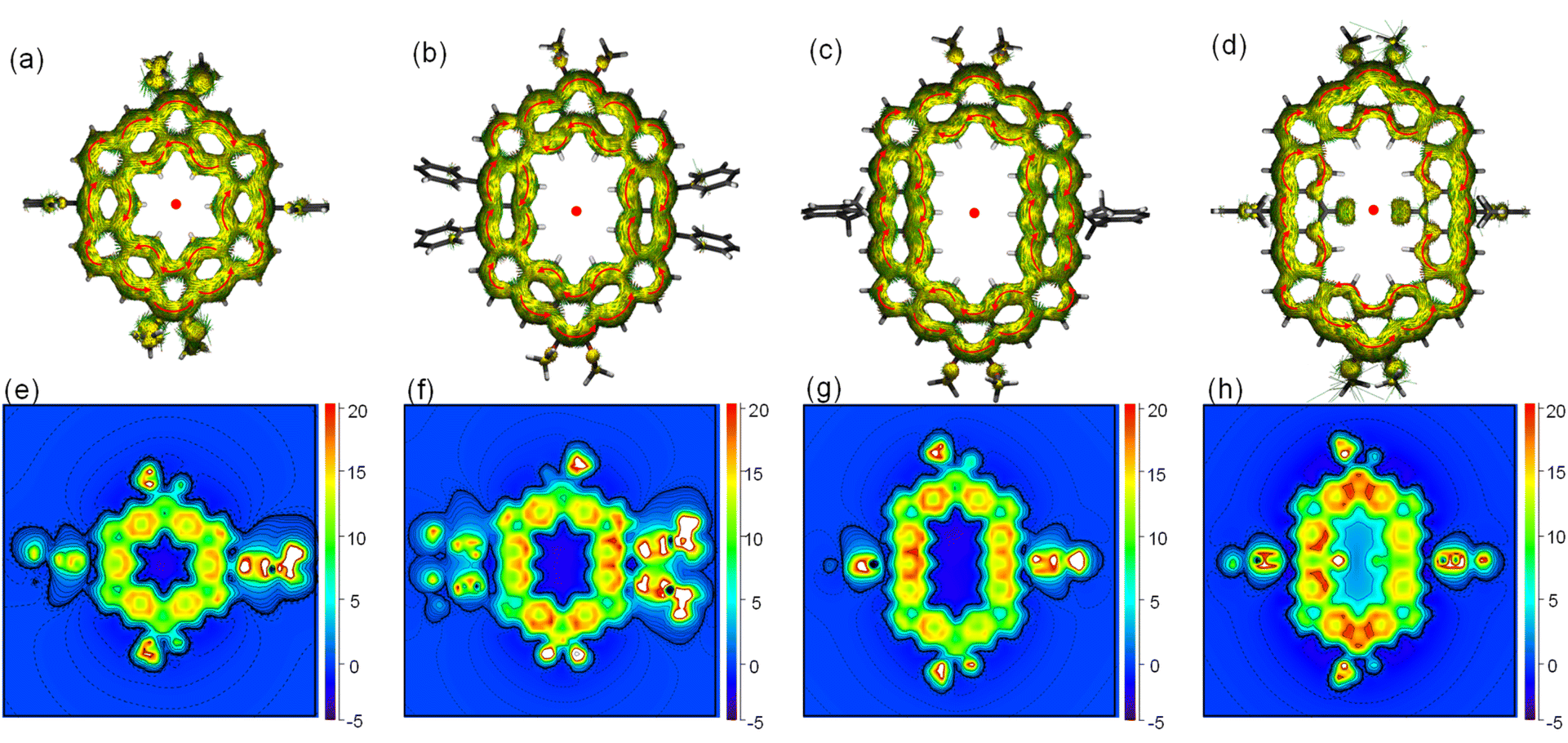
To further understand the aromaticity of three closed-shell alkoxyl- and aryl-cosubstituted cycloarenes K1–K3 and the open-shell carbonylated cycloarene derivative K3-R, the bond length analysis (Fig. 2, 3 and S1†), harmonic oscillator model of aromaticity (HOMA) calculations (Fig. 2 and 3),21 nucleus independent chemical shift (NICS) (Fig. S1†),22 anisotropy of the induced current density (ACID) (Fig. 4a–d),23 and 2D isochemical shielding surface (ICSS) (Fig. 4e–h) calculations24 were carried out. For the three closed-shell cycloarenes K1–K3, bond length analysis shows that the bond lengths of bonds a from vinyl ethers and the bonds b in phenanthrene units (1.346(2)–1.359(3) Å) are obviously shorter than that of the typical C(sp2)–C(sp2) bond (1.39 Å) in benzene20 but close to that of typical olefins (1.34 Å),25 indicating their double bond character. The small HOMA values (the numbers in red color) for individual rings also imply the weak aromaticity in the corresponding rings C and A (Fig. 3). The NICS(0) values (Fig. S1†) and ACID plots (Fig. 4a–c) display local aromatic character. The calculated 2D ICSS maps of K1–K3 (Fig. 4e–g) indicate that the building blocks benzene/naphthalene/anthracene units exhibit strong magnetic shielding. All these analysis results suggest local aromatic character of the three closed-shell cycloarenes K1–K3, like the other cycloarenes,12 and their π-electrons are mainly delocalized at the individual benzene/naphthalene/anthracene ring and the two periphery hexatomic rings of the phenanthrene unit. For the open-shell carbonylated cycloarene derivative K3-R, the bonds a and b show the same double bone character as that of its closed-shell parent K3 (Fig. 2). The NICS(0) value of the center of the macrocycle is nearly zero (−0.88 ppm) exhibiting local aromatic character. Different from the continuous ring currents over the anthracene units for K3, there is almost no ring current on the atom C1, indicating that the ring currents are mainly distributed in the outer rings of the anthroxyl structure, which is consistent with the 2D ICSS map (Fig. 4h). Meanwhile, compared with the three rings of the anthracene unit in K3 with almost equal HOMA values (0.61–0.65), the HOMA value of the centered ring E (0.17) of the anthroxyl structure in K3-R is obviously smaller than that of the lateral rings D/F (0.84–0.85) (Fig. 2), which can be attributed to the relatively long bond lengths of C7–C8 and C8–C9 due to the conjugation with the carbonyl groups. Overall, the above calculations about aromaticity verify again that form I is the dominant resonance structure of open-shell carbonylated cycloarene derivative K3-R.
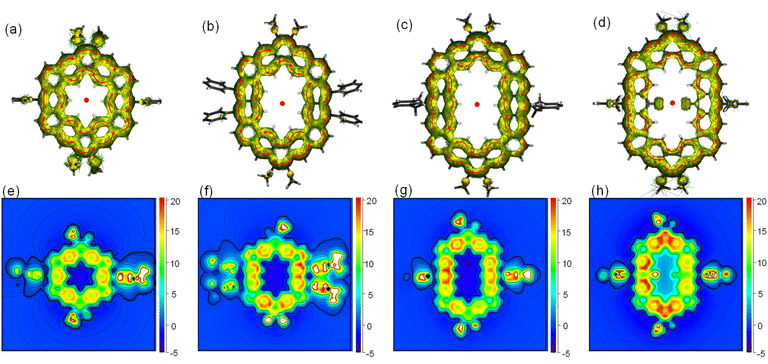 |
| | Fig. 4 Calculated ACID plots (contribution from π-electrons only) of (a) K1, (b) K2, (c) K3, and (d) K3-R. The magnetic field is perpendicular to the XY plane and points out through the paper. The red arrows indicate the clockwise (diamagnetic) current flow. Calculated 2D ICSSzz maps for (e) K1, (f) K2, (g) K3, and (h) K3-R. | |
Optical and electrochemical properties
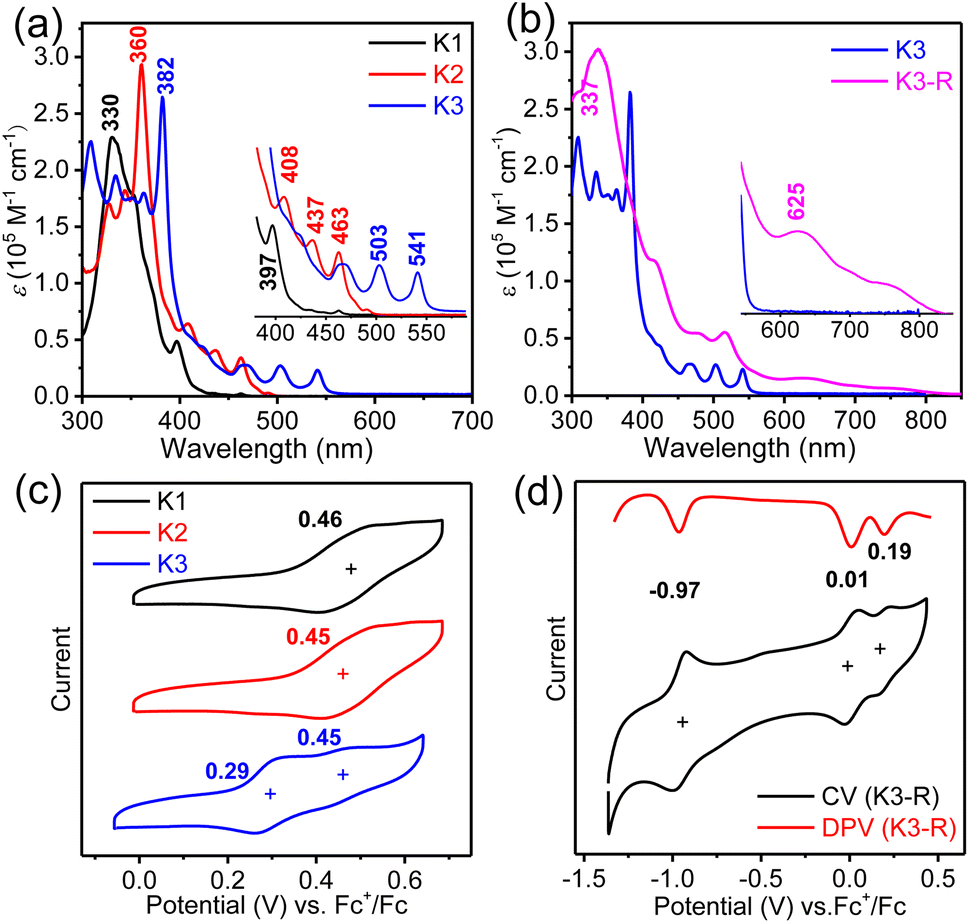
The UV-vis absorption spectra (Fig. 5a and b) and fluorescence emission spectra (Fig. S3†) of three closed-shell kekulene homologues K1–K3 were measured in chloroform as shown in Fig. 5a. With the major absorption at 330 nm and several shoulder peaks at 352 nm and 397 nm, the band structure of aryl- and alkoxy-cosubstituted kekulene K1 is similar to that of aryl-substituted kekulene9 and alkoxy-substituted kekulene.7 The absorption maxima (λmax) of K2 and K3 bathochromic shift to 360 nm and 382 nm, respectively, compared with that of K1 (λmax = 330 nm), which can be explained using the extended π-conjugation system. The absorption bands at 400–500 nm for K2 and 460–610 nm for K3 resemble the characteristics of the tetracene26 and pentacene moiety,27 respectively, which can be assigned a combination of HOMO → LUMO electronic transitions according to the time-dependent (TD) DFT calculations (see the ESI†). The optical energy gap (Eoptg) of K1–K3 was estimated to be 3.01 eV, 2.50 eV and 2.25 eV, respectively, from the UV-vis absorption edge (Fig. 5a). Moreover, the red-shift from the expanded conjugation was also observed in the fluorescence spectra (Fig. S3†). The absolute fluorescence quantum yields of K1–K3 in chloroform solution were measured to be 7.3%, 13.8% and 31.7%, respectively, by using the integrating sphere technique. The absorption spectrum of carbonylated cycloarene derivative K3-R shows a weak long wavelength absorption band with λmax = 625 nm along with a shoulder at 750 nm extending up to 850 nm, which is a typical character observed in many compounds with unpaired electrons.28 According to TD DFT, the NIR absorption can originate from HOMO-5 → LUMO and HOMO → LUMO+3 (Fig. S14 and Table S4†). The optical energy gap of K3-R was estimated to be 1.51 eV at the onset absorption wavelength. The stability of K3-R in CHCl3 at room temperature under ambient air and light conditions was also monitored by UV-vis measurements (Fig. S8†). The absorption intensity at 506 nm decreased by only 34% in about 97 days, indicating that K3-R is significantly more stable than other reported cycloarene-based radicals.29 Meanwhile, the superior stability of K3-R supports that the attachment of electron-acceptor groups is an efficient approach toward stable open-shell cycloarene diradicaloids and polyradicaloids. Moreover, there is no fluorescence detected for the solid or the solution of the carbonylated cycloarene derivative K3-R.
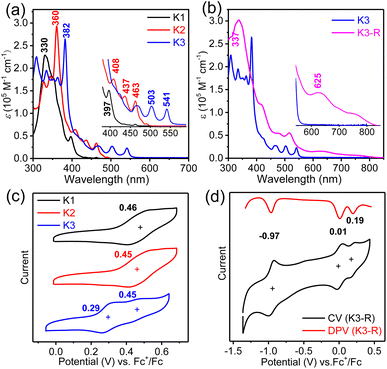 |
| | Fig. 5 UV-vis absorption spectra of (a) K1–K3 and (b) K3-R in chloroform solutions; insets show the magnified onset absorption bands. Cyclic voltammograms of (c) K1–K3 and (d) K3-R in chloroform solutions. | |
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements of K1–K3 and K3-R were performed in dry DCM by using 0.1 M tetra-n-butylammonium hexfluorophosphate as the supporting electrolyte (Fig. 5 and S4†). Similar to other cycloarenes, K1 and K2 are difficult to oxidize because of their high first oxidation potential.9 However, a low oxidation potential with a half-wave Eox1/2 of 0.29 V (vs. Fc+/Fc) of anthryl-based cycloarene K3 was observed, which can explain its unique reactivity.30 DFT calculations were performed using Gaussian 09 at the B3LYP/6-31G(d,p) level to investigate the electronic properties of K1–K3 by using simplified model molecules.31 As shown in Fig. S10,† the frontier molecular orbitals of K1–K3 were delocalized over the whole aromatic backbone, but the larger electron cloud density at five fused benzenoid rings was observed in K3, which was consistent with the observed pentacene-like band structure in the absorption spectrum. The electrochemical properties of carbonylated cycloarene derivative K3-R were investigated by the same method with the above closed-shell molecules, and two reversible oxidation waves with Eox1/2 = 0.01 and 0.19 V and one reduction wave with Ered1/2 = −0.97 V (vs. Fc+/Fc) were observed. The HOMO/LUMO energy levels of K3-R were determined to be −4.72/−3.93 eV from the onset of the first oxidation/reduction potential, and the electrochemical energy gap (EECg) was estimated to be 0.79 eV.
Magnetic properties and radical character
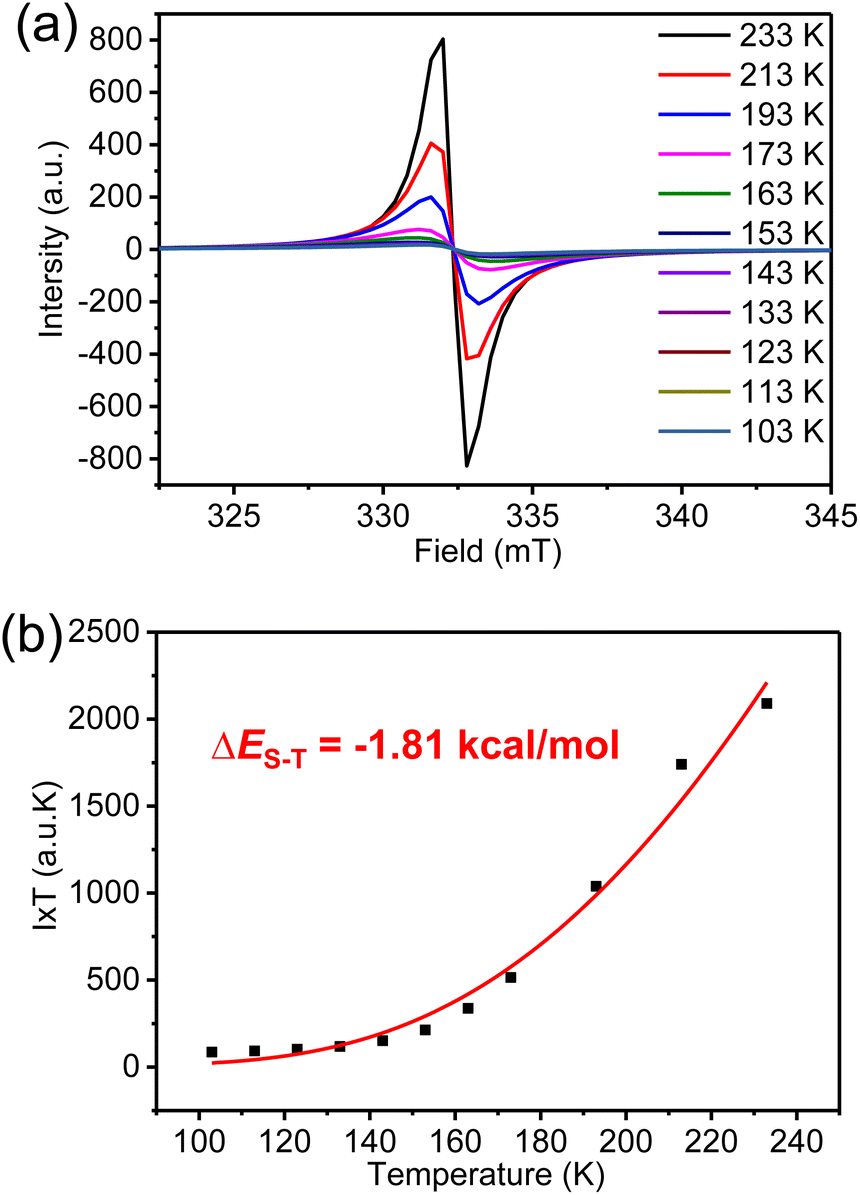
The magnetic properties of K3-R were further investigated by variable-temperature (VT) ESR measurements in a solid (Fig. 6a). The signal intensity decreased as the temperature was lowered, indicating the open-shell singlet diradical character of K3-R. By fitting the data using the Bleaney–Bowers equation (Fig. 6b),32 the singlet–triplet energy gap (ΔES–T) was calculated to be −1.81 kcal mol−1. However, the broad resonance signals in 1H NMR spectra of K3-R were observed at room temperature and even after cooling to 183 K (Fig. S5†). Indeed, the diradical character (y0)33 of K3-R was calculated to be 92% at the UCAM-B3LYP/6-31G(d,p) level, which can indicate its strong radical character and explain the broad NMR signals even at low temperature. The diradical character and excitation energies of K3-R were calculated at the unrestricted B3LYP/6-31G(d,p) level. The excitation energy from the singlet ground state to the lowest triplet was estimated to be 0.32 kcal mol−1. The natural orbital profiles and spin density distribution map of K3-R are shown in Fig. 8 and S15.† The unpaired electron density and spin density were mainly delocalized over anthroxyl units and thinly distributed in other areas of the macrocycle skeleton, suggesting the weak antiferromagnetic (AFM) coupling between the two spin centers. This weak intramolecular spin–spin coupling and the steric protection of the reactive carbon atom are effective in stabilizing anthroxyl type radicals.
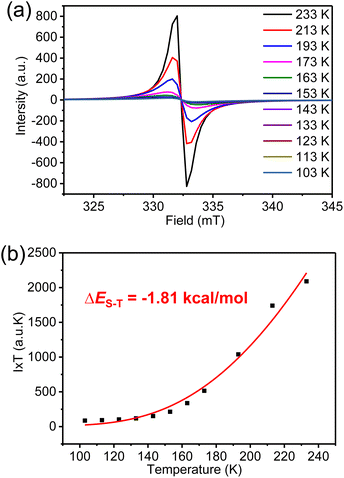 |
| | Fig. 6 (a) VT ESR spectra and (b) fitted I × T–T curves by using the Bleaney–Bowers equation of carbonylated cycloarene diradical K3-R in a solid. | |
Conclusions
In summary, an alkoxyl- and aryl-cosubstituted kekulene K1 and its homologues K2 and K3 with extensions of the two opposite edges were designed and synthesized successfully. Then, an interesting transition from the closed-shell anthryl-based cycloarene K3 to open-shell carbonylated cycloarene derivative K3-R was found via controlling the temperature and gas atmosphere of the Bi(OTf)3-catalyzed system. The unique reactivity of anthryl-based cycloarene K3 can be attributed to its low oxidation potential of 0.29 V (vs. Fc+/Fc). All their molecular structures and quasi-planar skeleton geometry were confirmed by X-ray crystallographic analysis. Clear size-dependent physical properties and crystal packing modes can be observed for this unique closed-shell cycloarene system. Most importantly, carbonylated cycloarene derivative K3-R exhibits intramolecular AFM coupling, large diradical character, small singlet–triplet energy gap, and remarkable stability and represents the first example of carbonylated cycloarene diradicaloids. Our studies disclose the distinct reactivity of alkoxyl- and aryl-cosubstituted kekulene and its edge-extended homologues, further enriching our understanding of the electronic properties of cycloarenes, and shed some light on synthesis of extended kekulenes and conjugated radical-acceptor cycloarenes.
Data availability
The crystallographic data was provided in Cambridge Structural Database, and the other necessary data of this study have been provided in the ESI.†
Author contributions
X. L. supervised the project. X. L. and D. C. designed the experiments and performed all the synthetic experiments and characterization. X. L. and J. Z. analyzed the data, performed theoretical calculations and co-wrote the manuscript. Y. S., K. C., Y. Q., and T. W. assisted in the synthesis of compounds. Y. Z. Y. L. and X. L. discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52073063, 51903052, and 61890940), the National Key R&D Program of China (2018YFA0703200), the Natural Science Foundation of Shanghai (22ZR1405800, and 23ZR1405100), and the Program for Professor of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning.
Notes and references
- H. A. Staab, F. Diederich, C. Krieger and D. Schweitzer, Chem. Ber., 1983, 116, 3504–3512 CrossRef CAS.
-
(a) L. Pauling, J. Chem. Phys., 1936, 4, 673–677 CrossRef CAS;
(b) R. McWeeny, Proc. Phys. Soc. A, 1951, 64, 921–930 CrossRef;
(c)
E. Clar, The Aromatic Sextet, J. Wiley, New York, 1972 Search PubMed.
- F. Diederich and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1978, 17, 372–374 CrossRef.
-
(a) E. F. Jenny and A. Melzer, Angew. Chem., Int. Ed. Engl., 1965, 4, 951–952 CrossRef;
(b) R. Peter and W. Jenny, Helv. Chim. Acta, 1966, 49, 2123–2135 CrossRef CAS;
(c) H. A. Staab, F. Diederich and V. Čaplar, Liebigs Ann. Chem., 1983, 1983, 2262–2273 CrossRef;
(d) H. A. Staab and M. Sauer, Liebigs Ann. Chem., 1984, 1984, 742–760 CrossRef.
- D. J. H. Funhoff and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1986, 25, 742–744 CrossRef.
- B. Kumar, R. L. Viboh, M. C. Bonifacio, W. B. Thompson, J. C. Buttrick, B. C. Westlake, M.-S. Kim, R. W. Zoellner, S. A. Varganov, P. Mörschel, J. Teteruk, M. U. Schmidt and B. T. King, Angew. Chem., Int. Ed., 2012, 51, 12795–12800 CrossRef CAS PubMed.
- M. A. Majewski, Y. Hong, T. Lis, J. Gregoliński, P. J. Chmielewski, J. Cybińska, D. Kim and M. Stępień, Angew. Chem., Int. Ed., 2016, 55, 14072–14076 CrossRef CAS PubMed.
-
(a) W. Fan, Y. Han, S. Dong, G. Li, X. Lu and J. Wu, CCS Chem., 2021, 3, 1445–1452 CrossRef CAS;
(b) M. Murai, N. Hosokawa, D. Roy and K. Takai, Org. Lett., 2014, 16, 4134–4137 CrossRef CAS PubMed;
(c) L. Yang, N. Zhang, Y. Han, Y. Zou, Y. Qiao, D. Chang, Y. Zhao, X. Lu, J. Wu and Y. Liu, Chem. Commun., 2020, 56, 9990–9993 RSC.
- W. Fan, Y. Han, X. Wang, X. Hou and J. Wu, J. Am. Chem. Soc., 2021, 143, 13908–13916 CrossRef CAS PubMed.
- M. Di Giovannantonio, X. Yao, K. Eimre, J. I. Urgel, P. Ruffieux, C. A. Pignedoli, K. Müllen, R. Fasel and A. Narita, J. Am. Chem. Soc., 2020, 142, 12046–12050 CrossRef CAS PubMed.
- U. Beser, M. Kastler, A. Maghsoumi, M. Wagner, C. Castiglioni, M. Tommasini, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 4322–4325 CrossRef CAS PubMed.
- J. C. Buttrick and B. T. King, Chem. Soc. Rev., 2017, 46, 7–20 RSC.
-
(a) T. Aotake, M. Suzuki, N. Aratani, J. Yuasa, D. Kuzuhara, H. Hayashi, H. Nakano, T. Kawai, J. Wu and H. Yamada, Chem. Commun., 2015, 51, 6734–6737 RSC;
(b) Y. Hirao, A. Konishi and T. Kubo, Org. Chem. Front., 2017, 4, 828–833 RSC;
(c) V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC;
(d) Y. Hirao, T. Saito, H. Kurata and T. Kubo, Angew. Chem., Int. Ed., 2015, 54, 2402–2405 CrossRef CAS PubMed;
(e) F. Miao, Z. L. Lim, P. Hu, S. Dong, Q. Qi, X. Zhang and J. Wu, Org. Biomol. Chem., 2017, 15, 3188–3191 RSC.
-
(a) J. Zhu, W. Li, N. Zhang, D. An, Y. Zhao, X. Lu and Y. Liu, Chem. Sci., 2022, 13, 11174–11182 RSC;
(b) N. Zhang, L. Yang, W. Li, J. Zhu, K. Chi, D. Chang, Y. Qiao, T. Wang, Y. Zhao, X. Lu and Y. Liu, J. Am. Chem. Soc., 2022, 144, 21521–21529 CrossRef CAS PubMed.
-
(a) W. Fan, T. Matsuno, Y. Han, X. Wang, Q. Zhou, H. Isobe and J. Wu, J. Am. Chem. Soc., 2021, 143, 15924–15929 CrossRef CAS PubMed;
(b) N. Zhang, J. Zhu, D. An, R. Zhang, X. Lu and Y. Liu, Org. Lett., 2022, 24, 5439–5443 CrossRef CAS PubMed.
-
(a) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed;
(b) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed;
(c) Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC;
(d) W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Z. Zeng, H. Yamada, J. Ding and J. Wu, Chem, 2017, 2, 81–92 CrossRef CAS;
(e) A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunage, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS PubMed;
(f) H. Hayashi, J. E. Barker, A. Cárdenas Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444 CrossRef CAS PubMed;
(g) A. Shimizu, T. Kubo, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Hirao, K. Matsumoto, H. Kurata, Y. Morita and K. Nakasuji, J. Am. Chem. Soc., 2010, 132, 14421 CrossRef CAS PubMed;
(h) J. J. Dressler and M. M. Haley, J. Phys. Org. Chem., 2020, 33, e4114 CAS.
- I. C. Lewis and L. S. Singer, J. Phys. Chem., 1981, 85, 354–360 CrossRef CAS.
-
(a) S. Antoniotti and E. Duñach, Eur. J. Org. Chem., 2004, 2004, 3459–3464 CrossRef;
(b) T. Zevaco, E. Duñach and M. Postel, Tetrahedron Lett., 1993, 34, 2601–2604 CrossRef CAS;
(c) K. Kallal, C. Coin, E. Dunach and M. Postel, New J. Chem., 1997, 21, 495–502 CAS.
- C. Krieger, F. Diederich, D. Schweitzer and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1979, 18, 699–701 CrossRef.
- E. Fleischer, N. Sung and S. Hawkinson, J. Phys. Chem., 1968, 72, 4311–4312 CrossRef CAS.
-
(a) J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef;
(b) T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
-
(a) D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed;
(b) T. Luo, Y. Wang, J. Hao, P. Chen, Y. Hu, B. Chen, J. Zhang, K. Yang and Z. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202214653 CAS.
-
(a) S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS;
(b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. Glaser, L. Dendi, N. Knitts and C. Barnes, Cryst. Growth Des., 2003, 3, 291–300 CrossRef CAS.
- S. A. Odom, S. R. Parkin and J. E. Anthony, Org. Lett., 2003, 5, 4245–4248 CrossRef CAS PubMed.
-
(a) Y. Kawanaka, A. Shimizu, T. Shinada, R. Tanaka and Y. Teki, Angew. Chem., Int. Ed., 2013, 52, 6643–6647 CrossRef CAS PubMed;
(b) A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980–4986 CrossRef CAS;
(c) I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS PubMed.
-
(a) X. Lu, S. Lee, J. O. Kim, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Z. Lim, Z. Zeng, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2016, 138, 13048–13058 CrossRef CAS PubMed;
(b) X. Lu, T. Y. Gopalakrishna, Y. Han, Y. Ni, Y. Zou and J. Wu, J. Am. Chem. Soc., 2019, 141, 5934–5941 CrossRef CAS PubMed.
-
(a) X. Lu, D. An, Y. Han, Y. Zou, Y. Qiao, N. Zhang, D. Chang, J. Wu and Y. Liu, Chem. Sci., 2021, 12, 3952–3957 RSC;
(b) S. Das, T. S. Herng, J. L. Zafra, P. M. Burrezo, M. Kitano, M. Ishida, T. Y. Gopalakrishna, P. Hu, A. Osuka, J. Casado, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 7782–7790 CrossRef CAS PubMed;
(c) X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052–13056 CrossRef CAS PubMed;
(d) H. Gregolińska, M. Majewski, P. J. Chmielewski, J. Gregoliński, A. Chien, J. Zhou, Y.-L. Wu, Y. J. Bae, M. R. Wasielewski, P. M. Zimmerman and M. Stępień, J. Am. Chem. Soc., 2018, 140, 14474–14480 CrossRef PubMed.
- S. H. Jang, T. B. Tai, M. K. Kim, J. W. Han, Y. H. Kim, S. C. Shin, Y. J. Yoon, S. K. Kwon and S. G. Lee, Bull. Korean Chem. Soc., 2009, 30, 618–622 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Rev. C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
-
(a) B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CrossRef CAS;
(b) B. Li, C. Yang, X. Wang, G. Li, W. Peng, W. Peng, H. Xiao, S. Luo, S. Xie, J. Wu and Z. Zeng, Angew. Chem., Int. Ed., 2021, 60, 19790 CrossRef CAS PubMed;
(c) Y. Wang, Y. Huang, T. Huang, J. Zhang, T. Luo, Y. Ni, B. Li, S. Xie and Z. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202200855 CAS.
- T. Minami and M. Nakano, J. Phys. Chem. Lett., 2012, 3, 145–150 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of all physical characterization studies and theoretical calculations and additional spectroscopic data. CCDC 2223047–2223050. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01295f |
| ‡ D. C. and J. Z. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Xuefeng
Lu
and
Xuefeng
Lu