
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBicyclic (alkyl)(amino)carbene (BICAAC) in a dual role: activation of primary amides and CO2 towards catalytic N-methylation†
Nimisha
Gautam
 a,
Ratan
Logdi
a,
Sreejyothi
P.
a,
Antara
Roy
b,
Ashwani K.
Tiwari
*a and
Swadhin K.
Mandal
*a
a,
Ratan
Logdi
a,
Sreejyothi
P.
a,
Antara
Roy
b,
Ashwani K.
Tiwari
*a and
Swadhin K.
Mandal
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur Campus, Nadia, 741246, West Bengal, India. E-mail: swadhin.mandal@iiserkol.ac.in; ashwani@iiserkol.ac.in
bDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur, 721302, West Bengal, India
First published on 20th April 2023
Abstract
Herein, we report the first catalytic methylation of primary amides using CO2 as a C1 source. A bicyclic (alkyl)(amino)carbene (BICAAC) exhibits dual role by activating both primary amide and CO2 to carry out this catalytic transformation which enables the formation of a new C–N bond in the presence of pinacolborane. This protocol was applicable to a wide range of substrate scopes, including aromatic, heteroaromatic, and aliphatic amides. We successfully used this procedure in the diversification of drug and bioactive molecules. Moreover, this method was explored for isotope labelling using 13CO2 for a few biologically important molecules. A detailed study of the mechanism was carried out with the help of spectroscopic studies and DFT calculations.
Introduction
The global average CO2 emissions have climbed significantly over the last two decades, despite the several restrictions on human activities imposed by the COVID-19 pandemic.1 To address this issue, a novel concept known as carbon capture and utilisation (CCU) is gaining increasing attention. CCU involves the conversion of CO2 into fuels and chemicals while adding value, which can offset the cost of CO2 capture.2 CO2 is regarded as an appealing carbon feedstock in chemical synthesis since it is non-toxic and it is becoming gradually plentiful.3 However, the utilisation of CO2 as a C1 source is rather limited because of its high thermodynamic and kinetic stability,4 and in recent years, the use of metal-free catalysis is gaining popularity for CO2 activation.5 For example, the concept of CO2 activation and its catalytic reduction of CO2 to methanol employing a metal-free N-heterocyclic carbene (NHC) as a catalyst was introduced by Ying and co-workers.6 The other approach of hydroborative reduction of CO2 to methanol with aNHC7 (abnormal N-heterocyclic carbene) was reported by our group in 2016.8 However, the raw materials for fine chemical synthesis cannot be satisfied simply by reducing or functionalising CO2. In this aspect, Cantat coined a fascinating term, “diagonal transformation”, which means simultaneous reduction and functionalisation.9 This innovative concept of combining the reduction and functionalisation of CO2 enables the formation of new C–C, C–N, and C–O bonds, and it can broaden the range of chemical synthesis using CO2.9 This concept has successfully functionalised amines producing formamides, aminals and methylamines, which are the 2e, 4e, and 6e reduced products of CO2, respectively, along with the formation of a new C–N bond.5 However, such diagonal transformation was mostly limited to amines till 2018, when the first catalytic formylation of primary amides was reported by our group.10 Compared to amines, the functionalisation of primary amides with CO2 is profoundly challenging as amides are extremely weak bases making them chemically inert. As a result of the electronic conjugation of nitrogen lone pair of electrons with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionality of the amide, they are less reactive to nucleophilic attack, which makes them difficult to functionalise.11 Consequently, the aminals were prepared from amine by CO2 under reductive conditions using metal-free catalysts,12–14 but corresponding methylenated products of amides could not be prepared using CO2 until recently when we successfully designed mesoionic N-heterocyclic imines (mNHIs) and utilised them for catalytic amide methylenation using CO2.15 Despite such development, methylation of primary amides using CO2 as a reagent has not been reported, although corresponding N-methylation of amines is abundant in the current literature.16–20 We thus became interested in exploring the N-methylation of primary amides using CO2 as a C1 source as a part of our ongoing research in developing a metal-free catalyst for CCU.5,10,15,20,21 In this regard, we resorted to an ambiphilic singlet carbene named bicyclic (alkyl)(amino)carbene (BICAAC), which was introduced by Bertrand and co-workers in 2017.22 It is a storable carbene with high σ-donating and π-accepting properties.22 Since its discovery, the use of BICAAC as a ligand has received gradually increasing attention in the field of organometallic catalysis.23–26 Recently, we reported BICAAC as a catalyst for the reduction of nitriles in the presence of a borane.27 As BICAAC successfully reduced the polar C
O functionality of the amide, they are less reactive to nucleophilic attack, which makes them difficult to functionalise.11 Consequently, the aminals were prepared from amine by CO2 under reductive conditions using metal-free catalysts,12–14 but corresponding methylenated products of amides could not be prepared using CO2 until recently when we successfully designed mesoionic N-heterocyclic imines (mNHIs) and utilised them for catalytic amide methylenation using CO2.15 Despite such development, methylation of primary amides using CO2 as a reagent has not been reported, although corresponding N-methylation of amines is abundant in the current literature.16–20 We thus became interested in exploring the N-methylation of primary amides using CO2 as a C1 source as a part of our ongoing research in developing a metal-free catalyst for CCU.5,10,15,20,21 In this regard, we resorted to an ambiphilic singlet carbene named bicyclic (alkyl)(amino)carbene (BICAAC), which was introduced by Bertrand and co-workers in 2017.22 It is a storable carbene with high σ-donating and π-accepting properties.22 Since its discovery, the use of BICAAC as a ligand has received gradually increasing attention in the field of organometallic catalysis.23–26 Recently, we reported BICAAC as a catalyst for the reduction of nitriles in the presence of a borane.27 As BICAAC successfully reduced the polar C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of nitriles having a high bond dissociation energy of 179.3 kcal mol−1, we were thus curious if it could activate the CO bond of CO2.28 Moreover, there are reports of activation of CO2 and B–H bond of boranes by similar monocyclic (alkyl)(amino)carbenes (CAACs).29–31
N bond of nitriles having a high bond dissociation energy of 179.3 kcal mol−1, we were thus curious if it could activate the CO bond of CO2.28 Moreover, there are reports of activation of CO2 and B–H bond of boranes by similar monocyclic (alkyl)(amino)carbenes (CAACs).29–31
In particular, our primary focus has been the reductive functionalisation of CO2 for the methylation of amides. N-Methyl amides are the core moiety in various marketed drugs (Scheme 1a). Traditionally, N-methyl amide compounds are synthesised by coupling carboxylic acids with amines in the presence of stoichiometric amounts of toxic reagents such as SOCl2, N,N′-diisopropylcarbodiimide (DIC), N,N′-dicyclohexane carbodiimide (DCC) and acid chloride, etc. (Scheme 1b).32,33 However, such processes are non-catalytic. The other less popular approach to the N-methylation of amides is using methanol as a methylating agent in the presence of a transition metal-based catalyst (Scheme 1b).34,35 At present, N-methylation of amides using CO2 is not known. Therefore, developing a protocol for N-methylation of amides using CO2 as a C1 source and avoiding metal-based catalysts or toxic methylating reagents is highly desirable. With this vision, in this work, we report catalytic N-methylation of a diverse range of primary amides using CO2 under metal-free catalytic conditions (Scheme 1c). A detailed mechanistic investigation using control experiments and DFT calculations unravelled that the BICAAC plays a dual role in integrating primary amide activation by N-borylation and CO2 activation by forming boron formate, which then recombines in delivering the N-methylated amides.
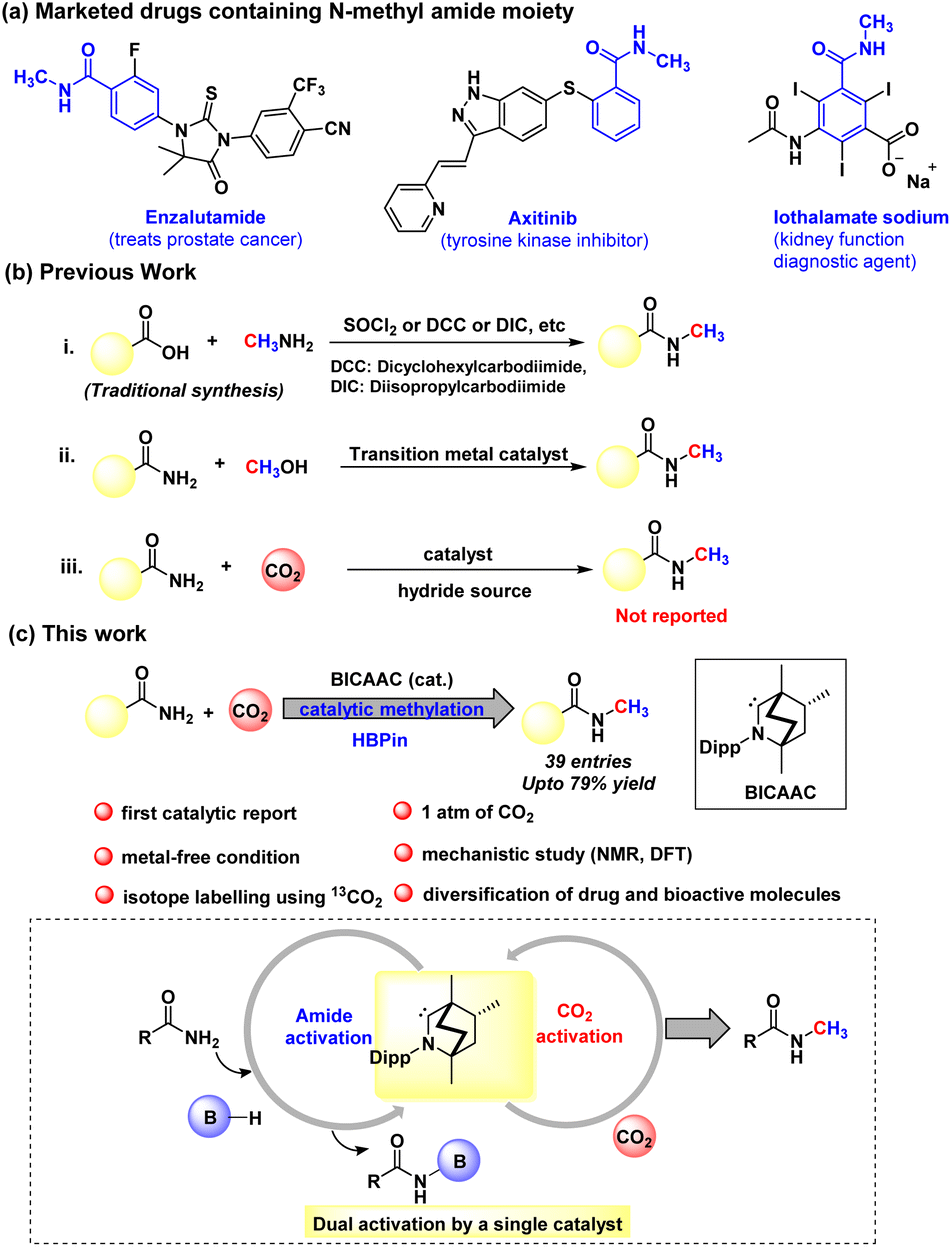 | ||
| Scheme 1 (a) Examples of N-methyl amides in pharmaceuticals. (b) Previous strategy for N-methyl amide synthesis. (c) This work represents the first catalytic methylation of amides using CO2. | ||
Results and discussion
The current investigation was initiated by optimising the methylation of a primary amide, 4-methyl benzamide (2a), as a model substrate using a catalytic amount of BICAAC (1), and borane as a reducing agent in the presence of 1 atm of CO2. BICAAC (1) was prepared according to the method reported in the literature.22 In the absence of the catalyst, the product was observed in trace amounts (entry 1, Table 1). After several trials, it was found that BICAAC functions as an efficient catalyst for the N-methylation of primary amides. Initially, we were delighted to observe that BICAAC in the presence of a borane (HBpin) in dioxane can afford N-methylated product with NMR conversion of 15% at 50 °C within 24 h (entry 3, Table 1), which was increased to 70% by raising the reaction temperature to 100 °C within 12 h (entry 4, Table 1). Under such conditions, other solvents, such as tetrahydrofuran (THF) and acetonitrile (ACN), were screened (entries 9 and 10, Table 1). This study establishes that dioxane is the best solvent for this transformation. Next, we screened several other boranes, such as borane dimethyl sulfide complex (BH3·SMe2) and catechol borane (HBcat), however, the reaction did not give the desired product (entries 11 and 12, Table 1). Moreover, we screened the reaction with a commercially available base, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), simple carbenes such as 1,3-bis(2,6-diisopropyl)phenyl-4,5-dihydroimidazol-2-ylidene (SIPr), and 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr), (entries 13–15, Table 1). Also, we tested a mesoionic carbene, aNHC (entry 16, Table 1). Treatment of catalytic amount of BICAAC salt (10 mol%) with equivalent amount of base can result in situ generated free BICAAC which results in the 72% conversion to the desired N-methylated amide (entry 17, Table 1). This result indicates that air stable BICAAC salt can be used for such catalytic conversion.|
|
|||||
|---|---|---|---|---|---|
| Entry | Borane | Catalyst (mol%) | Solvent | T (°C) | Conversion (%) |
| a All reactions were carried out with amide (0.2 mmol), borane (0.8 mmol), dry solvent (1.0 mL) and 1 atm CO2. b 12 h. c 36 h. d Isolated yield. e In the absence of 1 atm of CO2. | |||||
| 1 | HBpin | — | Dioxane | 120 | Trace |
| 2 | — | 1 (10) | Dioxane | 120 | — |
| 3 | HBpin | 1 (10) | Dioxane | 50 | 15 |
| 4 | HBpin | 1 (10) | Dioxane | 100 | 70b |
| 5 | HBpin | 1 (10) | Dioxane | 100 | 77c |
| 6 | HBpin | 1 (10) | Dioxane | 120 | 87 |
| 7 | HBpin | 1 (5) | Dioxane | 100 | 60 |
| 8 | HBpin | 1 (10) | Dioxane | 100 | 69d |
| 9 | HBpin | 1 (10) | THF | 100 | 54d |
| 10 | HBpin | 1 (10) | ACN | 100 | 52d |
| 11 | BH3·SMe2 | 1 (10) | Dioxane | 100 | — |
| 12 | HBcat | 1 (10) | Dioxane | 100 | — |
| 13 | HBpin | DBU (10) | Dioxane | 120 | 21d |
| 14 | HBpin | SIPr (10) | Dioxane | 100 | — |
| 15 | HBpin | IPr (10) | Dioxane | 100 | Trace |
| 16 | HBpin | aNHC (10) | Dioxane | 40 | 16 |
| 17 | HBpin | 1 salt + KHMDS (10) | Dioxane | 120 | 72 |
| 18 | HBpin | 1 (10) | Dioxane | 120 | —e |
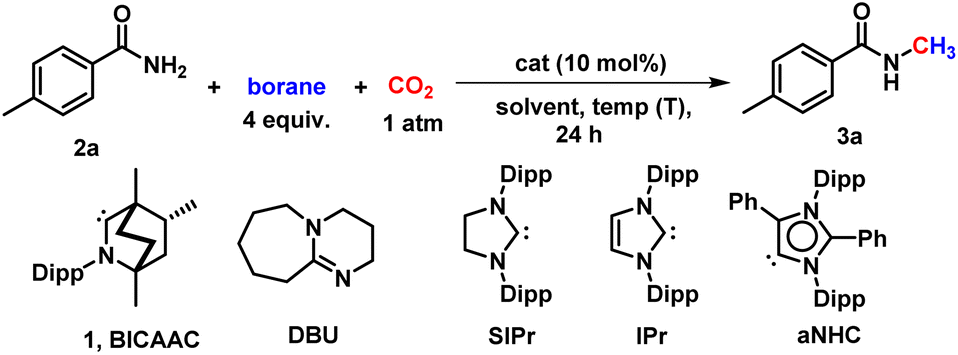
From this optimisation study, it can be concluded that 10 mol% of BICAAC with HBpin as a hydride source at 120 °C under 1 atm CO2 gave the best result improving the NMR yield up to 87% (entry 6, Table 1).
After having the optimal catalytic conditions in hand, we next explored the scope of the reaction for the N-methylation of various primary amides under an atmospheric pressure of CO2 (Table 2). Various commercially accessible amides comprising aromatic, aliphatic and also heterocycles were attempted. The protocol offered a good to a very good isolated yield of the N-methylated amides.
| a All reactions were carried out with amide (0.2 mmol), borane (0.8 mmol), dry solvent (1.0 mL) and 1 atm CO2. Isolated yields (average yield calculated from the yields obtained from two independent catalytic runs, shown in the parenthesis). b NMR conversion based on the unreacted substrate. c NMR conversion using hexamethylbenzene as an internal standard. |
|---|
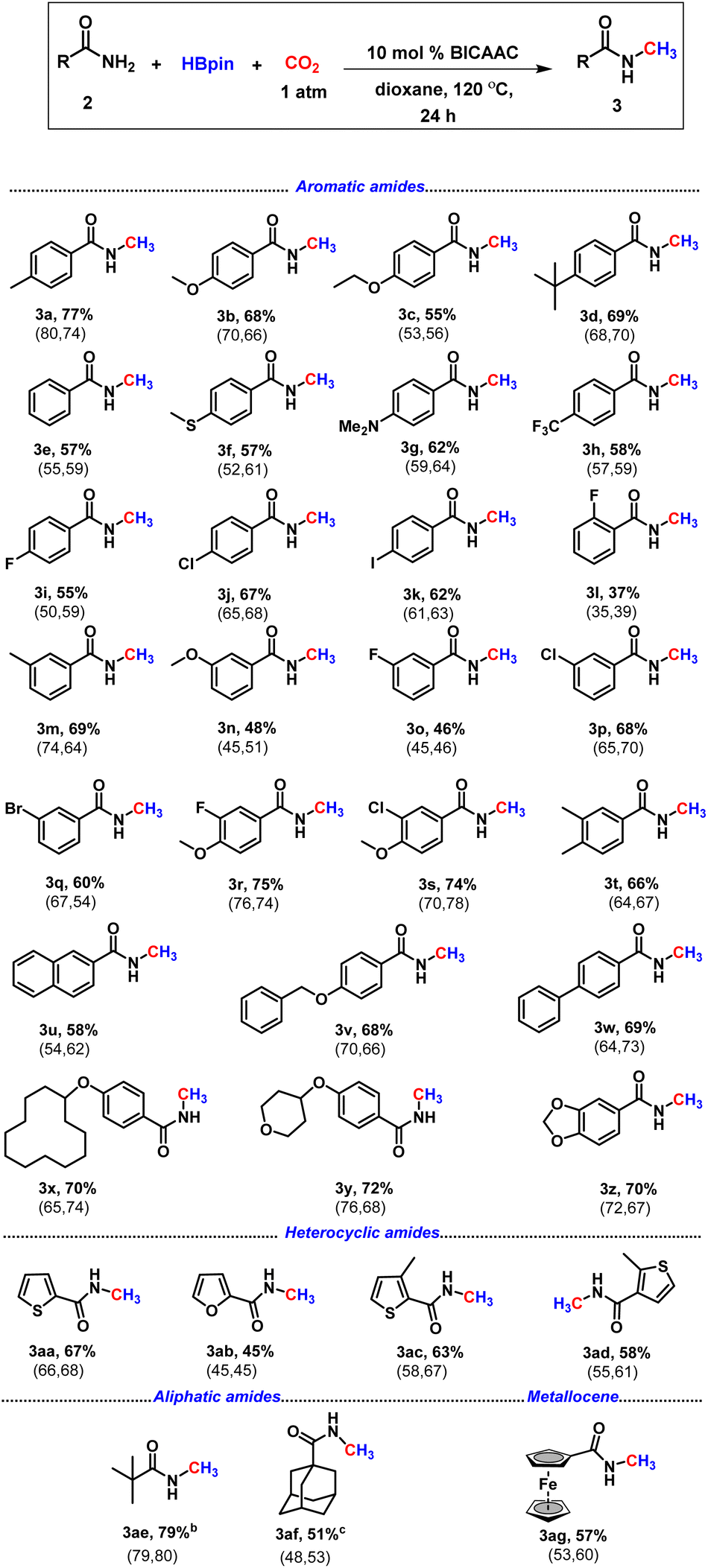
|
Amides containing electron-donating groups such as methyl and tert-butyl at the benzene ring gave the N-methylated product 3a (77%), 3d (69%), and 3m (69%) in very good isolated yield (Table 2). Aromatic amides bearing highly electron-rich groups such as –SMe and –NMe2 also delivered the N-methyl derivatives in good yield, 3f (57%) and 3g (62%). Substrates with electron-withdrawing groups such as trifluoromethyl, fluoro, and phenyl exhibited low to very good reactivity (3h–i, 3l, 3o, 3r and 3w, Table 2). Under the optimised reaction conditions, methylation of the N–H bond of several amides bearing electron-withdrawing groups was performed, delivering isolated yields in the range of 37–75% (3h–3l, 3o–3s, Table 2). The reactivity difference between electron-deficient and electron-rich amides might have originated from the availability of electron density on the nitrogen center of amide as supported by our NBO calculations (see the ESI, Fig. S129†). The meta-substituted primary amides appeared to be good candidates affording the desired product in 46% to 69% isolated yields (3m–3q). However, the ortho-substituted amide produced a low yield of 2-fluoro-N-methyl benzamide, 3l (37%). 2-Naphthamide displayed good reactivity resulting in a 58% yield (3u). In the presence of macrocyclic substitution (3x) and a cyclic ether, i.e. tetrahydropyran (3y) at the para position, the reaction proceeded smoothly with yields of 70% and 72%, respectively. Next, we explored its reactivity towards heteroaryl amides, which afforded N-methylated carboxamides in good yields (3aa–3ad, 45–67%). Heteroaryl motifs are crucial for the manufacture of pharmaceutical and agricultural products.36 Also, we were pleased to observe that aliphatic amides underwent N-methylation resulting in 3ae (79%) and 3af (51%). We next attempted this protocol with a metallocene containing primary amide, ferrocenecarboxamide, which yielded the desired product (3ag) in 57% yield.
Next, we checked the applicability of our protocol towards diversification of bioactive molecules. Three bioactive N-methyl amides were synthesised from (−)-borneol, (1R,2S,5R)-(−) menthol and estrone furnishing isolated yields of 71%, 67% and 59%, respectively (3ah–3aj, Scheme 2). Moreover, we were also successful in functionalising derivatives of three drug molecules, probenecid (treats gout), adapalene (a third-generation topical retinoid) and (±)-α-tocopherol, i.e., vitamin E (3ak–3am, Scheme 2). Furthermore, we perceive that our protocol might act as an efficient method for isotopic labelling using 13CO2. Such isotopic labelling could pave the way to radioactive labelling using 14CO2, which has immense potential in the metabolic tracking of drug candidates required for regulatory approval.37 These studies thus offer the importance of isotope labelling in pharmacokinetic investigations. Therefore, we next proceeded to perform our catalytic protocol using 13CO2 for a few biologically and medicinally relevant substrates. To our delight, very good isolated yields of 13C labelled N-methylated bioactive molecules were obtained (3ah′–3al′, Scheme 3) when we used 13CO2.
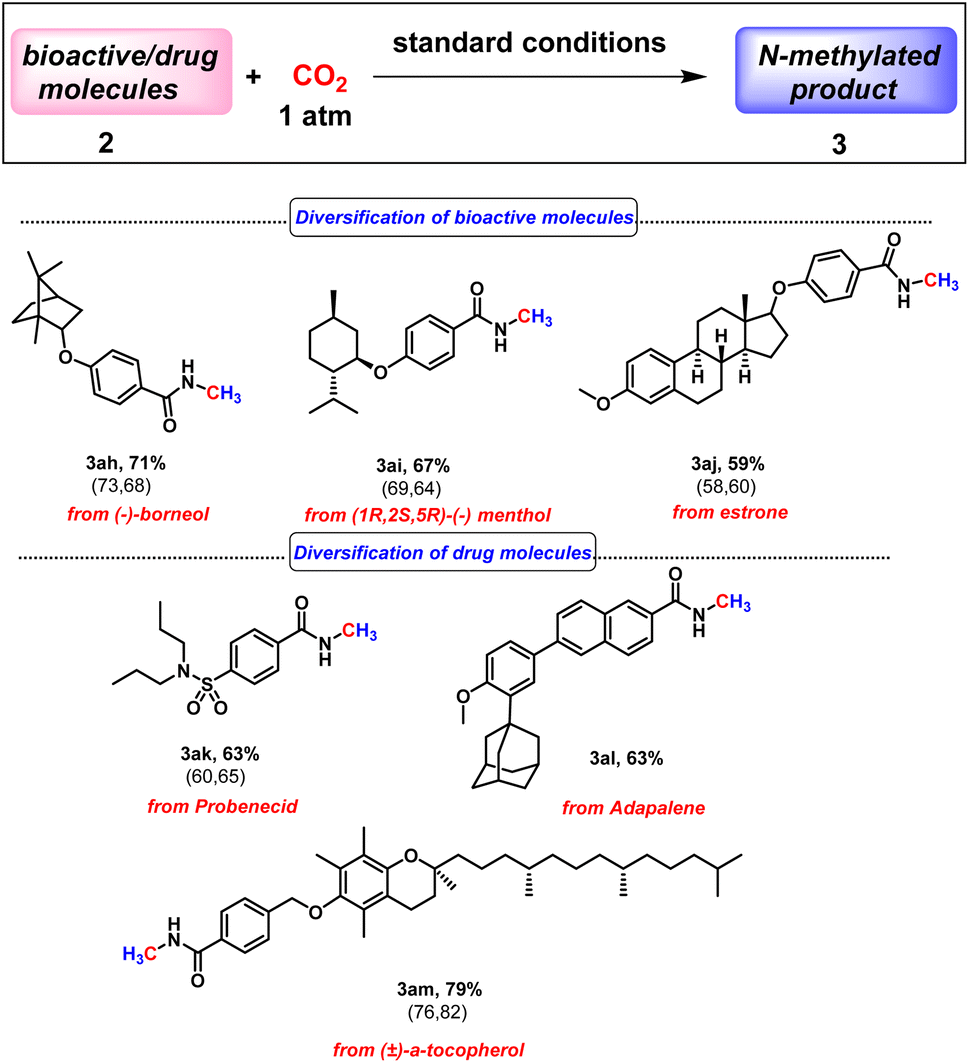 | ||
| Scheme 2 Applications of BICAAC catalysed N-methylation for the diversification of compounds with biological importance. Isolated yields (average yield calculated from the yields obtained from two independent catalytic runs, shown in the parenthesis). | ||
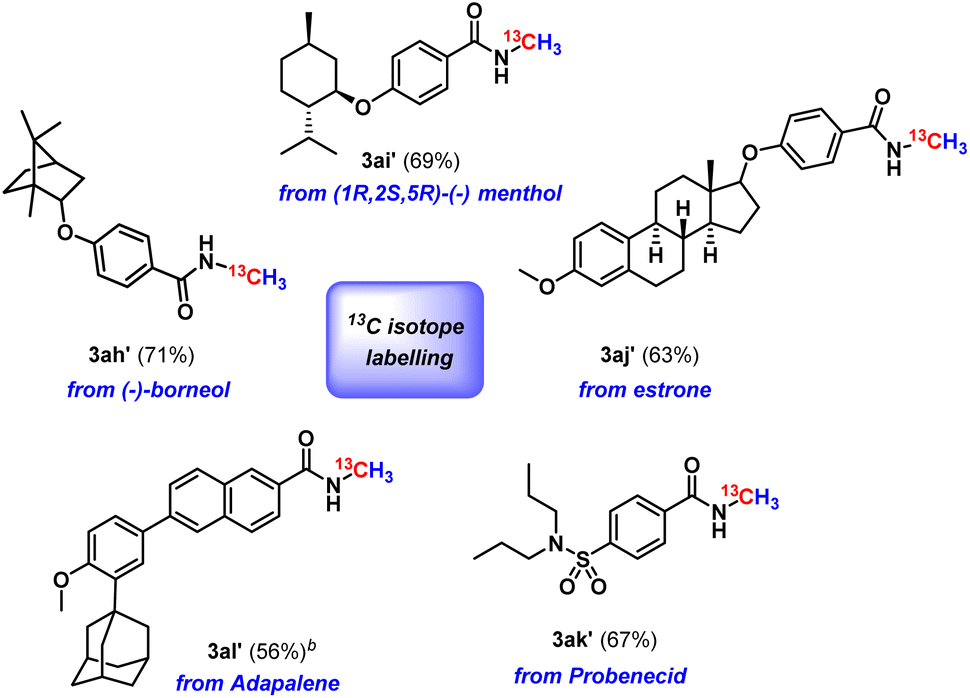 | ||
| Scheme 3 13C isotope labelling of biologically and medicinally important molecules using 13CO2a. aAll reactions were carried out with amide (0.2 mmol), borane (0.8 mmol), dry solvent (1.0 mL) and 1 atm 13CO2. Isolated yields are given in parentheses. bPerformed with a 0.1 mmol scale. | ||
Next, a series of control experiments were performed to gain insight into the mechanism of this transformation. From our previous study, we are aware that a stoichiometric treatment of BICAAC with HBPin results in the BICAAC B–H insertion product (1A) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture with respect to the bridgehead of BICAAC.27 As in this study, CO2 is another reagent, so to unveil the reactivity of BICAAC (1) towards these reagents, the reaction between BICAAC and CO2 was performed in ether separately which afforded a white solid, BICAAC–CO2 adduct (1B) after removal of solvent. The 13C NMR spectrum of BICAAC–CO2 adduct in CDCl3 displayed a peak at δ 193.1 ppm, which was assigned to CO of the BICAAC–CO2 adduct.26 Next, 1A or 1B was separately used as a catalyst in the methylation of 4-methyl benzamide (2a) under a CO2 atmosphere. To our surprise, both 1A and 1B furnished the desired N-methylated amide (Fig. 1a). This result indicated that 1A and 1B under a CO2 atmosphere could lead to the catalytic N-methylation, indicating they might interconvert in the presence of CO2 and borane. This made us curious to understand the reaction further, and towards this direction, frontier molecular orbital analysis was carried out with the help of density functional theory (DFT) calculations. The DFT calculations were performed employing M06-2X/6-311++G**//M06-2X/6-31G* methodology with CPCM solvent model (dioxane as solvent). A closer look at the HOMO of 1 shows that it contains a σ orbital at the carbene carbon centre, and LUMO contains a delocalised π orbital at carbene carbon (see the ESI, Fig. S125†). Then, the HOMO–LUMO of BICAAC–HBpin (1A), CO2, HBpin and BICAAC–CO2 adduct (1B) were computed. It is clear from the diagram (Fig. 1b) that the HOMO of CO2 is centred on the p orbital of oxygen, which can interact with the LUMO of 1A (13.1 eV). In the case of 1B, the HOMO is raised by 4.4 eV compared to free CO2; this can interact with the LUMO of HBpin (10.0 eV). This analysis indicates that the catalytic reaction is feasible for 1A and 1B. Gibbs's free energy calculations were conducted for both approaches to comprehend further how these species are capable of catalysing this process. For the pathway involving 1A, the route was found to be energetically demanding with ΔG = 67.7 kcal mol−1 (see the ESI, Fig. S126†), and next, we proceeded to the pathway involving 1B (Fig. 1c). In this pathway (involving 1B as a catalyst), at first, catalyst 1 interacts with CO2, where the HOMO of carbene interacts with the carbon centre of CO2, which is its LUMO forming 1BviaTS3 with an activation energy barrier of 1.3 kcal mol−1. In 1B, the electron density is located on the oxygen centre, which then reacts with the electron-deficient boron centre of pinacolborane to give VIviaTS4 having a 6.5 kcal mol−1 activation energy barrier. Here, a hydride shift takes place to produce 4. This step requires a very small barrier (1.4 kcal mol−1); hence, from this computed energy profile diagram, we can conclude that the transformation of 1B to 4 is highly feasible.
:1 diastereomeric mixture with respect to the bridgehead of BICAAC.27 As in this study, CO2 is another reagent, so to unveil the reactivity of BICAAC (1) towards these reagents, the reaction between BICAAC and CO2 was performed in ether separately which afforded a white solid, BICAAC–CO2 adduct (1B) after removal of solvent. The 13C NMR spectrum of BICAAC–CO2 adduct in CDCl3 displayed a peak at δ 193.1 ppm, which was assigned to CO of the BICAAC–CO2 adduct.26 Next, 1A or 1B was separately used as a catalyst in the methylation of 4-methyl benzamide (2a) under a CO2 atmosphere. To our surprise, both 1A and 1B furnished the desired N-methylated amide (Fig. 1a). This result indicated that 1A and 1B under a CO2 atmosphere could lead to the catalytic N-methylation, indicating they might interconvert in the presence of CO2 and borane. This made us curious to understand the reaction further, and towards this direction, frontier molecular orbital analysis was carried out with the help of density functional theory (DFT) calculations. The DFT calculations were performed employing M06-2X/6-311++G**//M06-2X/6-31G* methodology with CPCM solvent model (dioxane as solvent). A closer look at the HOMO of 1 shows that it contains a σ orbital at the carbene carbon centre, and LUMO contains a delocalised π orbital at carbene carbon (see the ESI, Fig. S125†). Then, the HOMO–LUMO of BICAAC–HBpin (1A), CO2, HBpin and BICAAC–CO2 adduct (1B) were computed. It is clear from the diagram (Fig. 1b) that the HOMO of CO2 is centred on the p orbital of oxygen, which can interact with the LUMO of 1A (13.1 eV). In the case of 1B, the HOMO is raised by 4.4 eV compared to free CO2; this can interact with the LUMO of HBpin (10.0 eV). This analysis indicates that the catalytic reaction is feasible for 1A and 1B. Gibbs's free energy calculations were conducted for both approaches to comprehend further how these species are capable of catalysing this process. For the pathway involving 1A, the route was found to be energetically demanding with ΔG = 67.7 kcal mol−1 (see the ESI, Fig. S126†), and next, we proceeded to the pathway involving 1B (Fig. 1c). In this pathway (involving 1B as a catalyst), at first, catalyst 1 interacts with CO2, where the HOMO of carbene interacts with the carbon centre of CO2, which is its LUMO forming 1BviaTS3 with an activation energy barrier of 1.3 kcal mol−1. In 1B, the electron density is located on the oxygen centre, which then reacts with the electron-deficient boron centre of pinacolborane to give VIviaTS4 having a 6.5 kcal mol−1 activation energy barrier. Here, a hydride shift takes place to produce 4. This step requires a very small barrier (1.4 kcal mol−1); hence, from this computed energy profile diagram, we can conclude that the transformation of 1B to 4 is highly feasible.
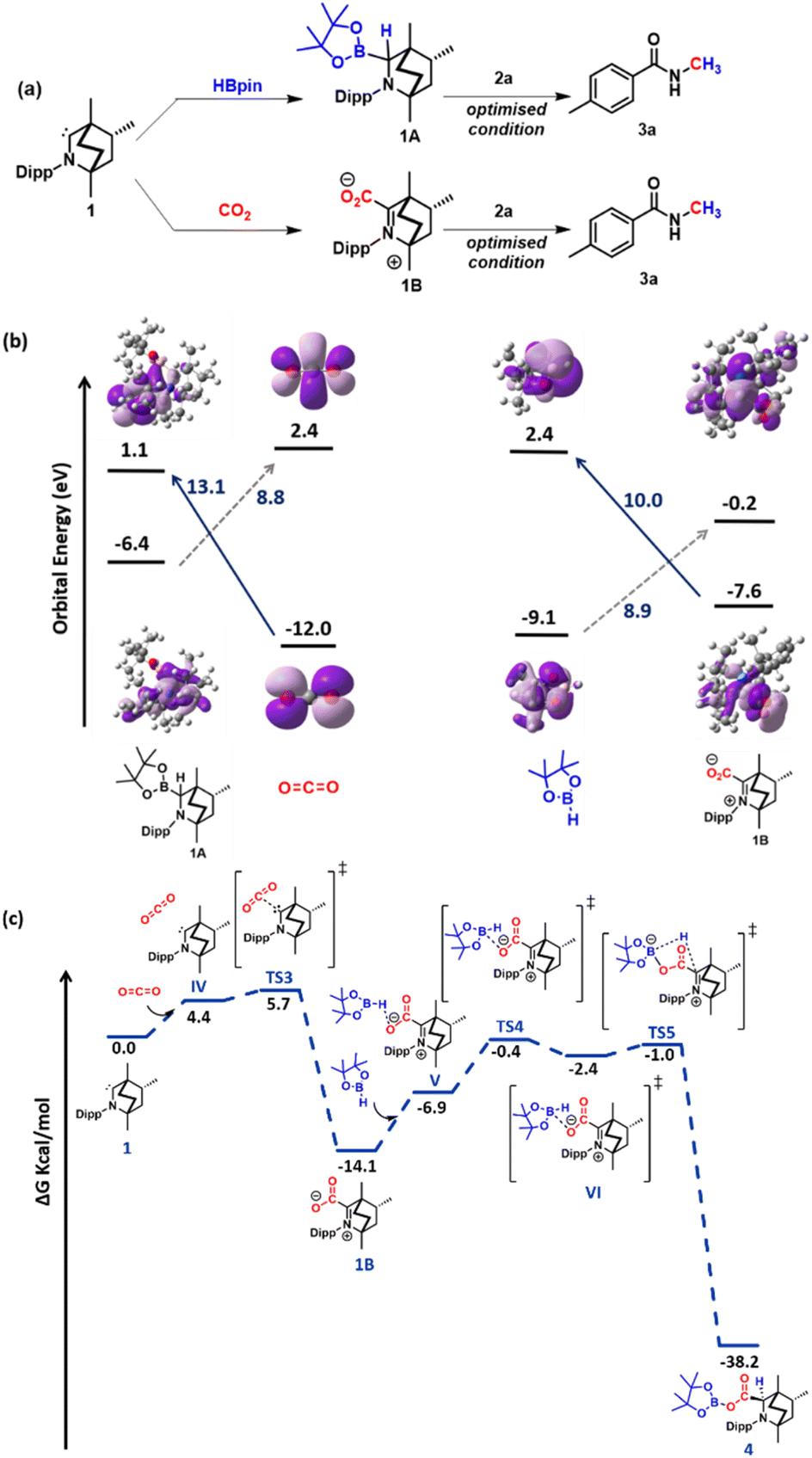 | ||
| Fig. 1 (a) Testing 1A and 1B as catalysts. (b) Frontier molecular orbital analysis disclosing the possibility of interaction of 1A with CO2 or 1B with HBpin in contrast to HBpin alone. (c) Computed Gibbs free energy profile for pathway involving 1B enabled by the interaction of 1 with CO2 leading to compound 4. | ||
To check if 4 can be traced experimentally, a stoichiometric reaction of BICAAC–CO2 adduct with HBpin in the presence of CO2 in CD3CN in a J. Young valve NMR tube was performed (Scheme 4a), which rendered two singlets at δ 4.04 and 4.30 ppm indicating the formation of a pair of 1:1 diastereomeric mixture of 4, as characterised by 1H NMR spectroscopy. The 11B{1H} NMR spectrum showed two peaks nearly merged at δ 21.2 and 20.6 ppm in the reaction mixture.
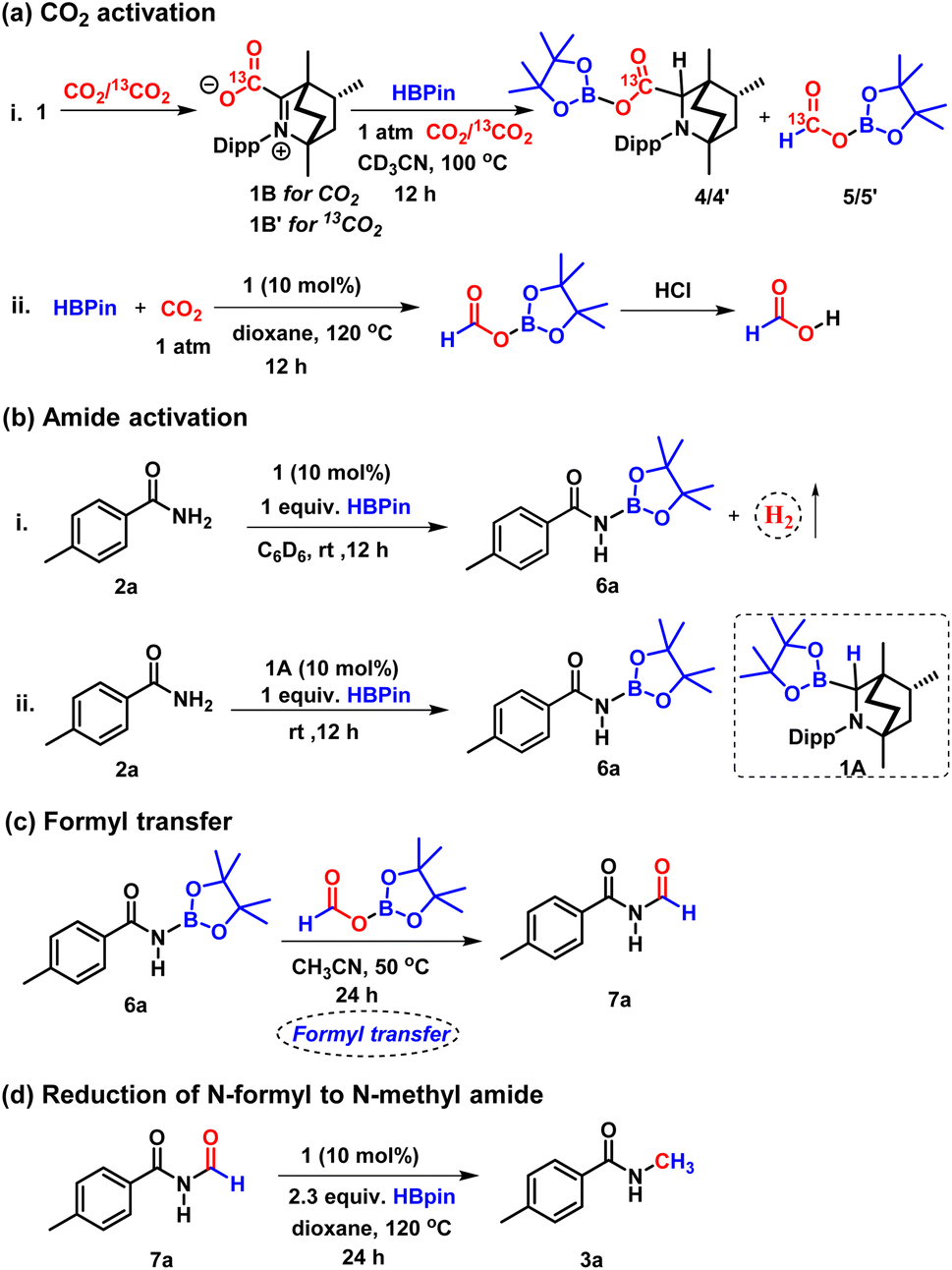 | ||
| Scheme 4 Control experiments: (a) CO2 activation. (b) Amide activation. (c) Formyl transfer reaction. (d) Reduction of N-formyl amide to N-methyl amide. | ||
In addition to 4, we have detected the formation of another compound displaying a peak at δ 8.19 ppm in the 1H NMR spectrum, indicating the formation of boron formate 5.38 Further, to confirm the formation of 4 and 5, the reaction was performed in the presence of 13C-labelled CO2, and the 13C{1H} NMR spectrum of the reaction mixture in CD3CN displayed two peaks at δ 172.2 and 171.3 ppm, which were assigned to the two diastereomers of 4′ (see the ESI, Fig. S7†) while a peak at δ (13C NMR) = 164.7 ppm was assigned to 13C labelled boron formate, 5′. Moreover, in the 1H NMR spectrum of 13C-labelled 4 (4′), two singlets at δ 4.04 and 4.30 ppm appeared as two doublets with 2JC–H = 3.5 and 1.5 Hz.39,40 Compound 4′ was also detected by mass spectrometry, m/z calculated for 13CC28H47BNO4+ [M + H]+: 485.3671, found 485.3638.
Also, in the 1H NMR spectrum, the singlet at δ 8.19 ppm corresponding to boron formate (5) was transformed into a doublet with 1JC–H = 206.5 Hz15 due to very strong one bond coupling with the 13C nucleus. The formation of 5 was further confirmed by executing catalytic reduction of CO2 with HBPin using 10 mol% of BICAAC in dioxane at 120 °C leading to the formation of formic acid (δ 8.31 ppm in 1H NMR spectrum in CDCl3) by treatment with aqueous HCl at the end of the reaction (Scheme 4a).41,42 These experiments and DFT calculations established the CO2 activation pathway by BICAAC.
Next, for the amide activation, we monitored the reaction of 4-methyl benzamide (2a) and HBpin in a 1:1 ratio in the absence of 1; the reactants stayed unreacted after performing the reaction at 25 °C for 12 h. However, in the presence of 10 mol% 1, when methyl benzamide (2a) and HBpin were mixed in a 1:1 ratio, gas evolution was noticed at 25 °C and was monitored by 1H NMR spectroscopy in a screw cap NMR tube in C6D6 (Scheme 4b). The gas was detected as dihydrogen by the 1H NMR spectroscopic peak at δ 4.47 ppm and was further verified by GC-MS analysis.20 Subsequently, we performed a reaction of 4-methyl benzamide (2a) and HBpin in a 1:1 ratio in the presence of 10 mol% 1 in CD3CN at room temperature for 12 h. 1H and 11B NMR spectroscopy of the reaction mixture confirmed the formation of N-borylated amide (6) with δ (11B NMR) = 24.2 ppm, which is in accordance with the reported literature.43 We also performed this N-borylation reaction with 1A as a catalyst, which rendered a similar result, indicating 1A to be the active species for the activation of primary amide (Scheme 4b). The transformation of amide to N-borylated amide catalysed by 1A may proceed through a concerted pathway (see the ESI, Fig. S127†).10
The above experiments established that BICAAC plays a dual role in activating CO2 to boron formate 5, and it can catalytically activate primary amide to N-borylated amide, such as 6a. Following this, we performed another control experiment between boron formate 5 and N-borylated amide 6a (Scheme 4c). A doublet at δ 9.37 ppm was observed in the 1H NMR spectrum of the reaction mixture recorded in CDCl3, indicating the formation of an N-formylated product 7a.10 This observation indicates that the N-formylated product is the intermediate for this N-methylation reaction. To further establish that N-methylation proceeds via N-formylation of amide, we also performed the reduction of N-formyl-4-methylbenzamide (7a) with only 2.3 equiv. of HBpin (instead of 4 equiv. HBpin) in the presence of a catalytic amount of 1 (Scheme 4d). The formation of N-methylated product in 67% isolated yield confirms that the given reaction proceeds through N-formylation. Moreover, a white solid was produced as a by-product of the catalytic reaction under the optimised condition that was identified as BpinOBpin by 1H and 11B NMR spectroscopy (11B in C6D6, δ 21.8 ppm).44
On the basis of these control reactions and theoretical studies, the following plausible mechanistic cycle is proposed (Scheme 5). In the first step, BICAAC activates the B–H bond of pinacolborane to yield the BICAAC B–H insertion product (1A), which has been previously reported.27 This activates the amide by borylation of one of its N–H bonds producing 6 along with the liberation of dihydrogen and regenerating 1. Amide activation can occur at 25 °C, as noticed from the H2 evolution on mixing the reagents, as also established from the characterisation of H2 gas by 1H NMR and GC-MS studies. The BICAAC (1) also can react with CO2 to produce BICAAC–CO2 zwitterionic adduct (1B).261B then reacts with the pinacolborane, followed by a hydride transfer to the carbene centre viaTS5, generating 4, which can produce boron formate 5 in the presence of CO2, regenerating the catalyst 1. The formation of 4 and 5 was established from our control experiments described above. The N-borylated amide (6) reacts with boron formate (5) when the formyl transfer reaction takes place, leading to N-formylated product (7) and (Bpin)2O (8) as a byproduct. The control experiment in Scheme 4c supports such a proposition. 7 then undergoes successive hydroboration of the carbonyl centre,20 leading to N-methylated amide, along with the formation of (Bpin)2O (8) as a byproduct.20 Thus, in total 4 equiv. of reducing agent is required. Amide and CO2 activation require two equiv. of HBpin, while the rest 2 equiv. is needed for reducing N–CHO to N–CH3.
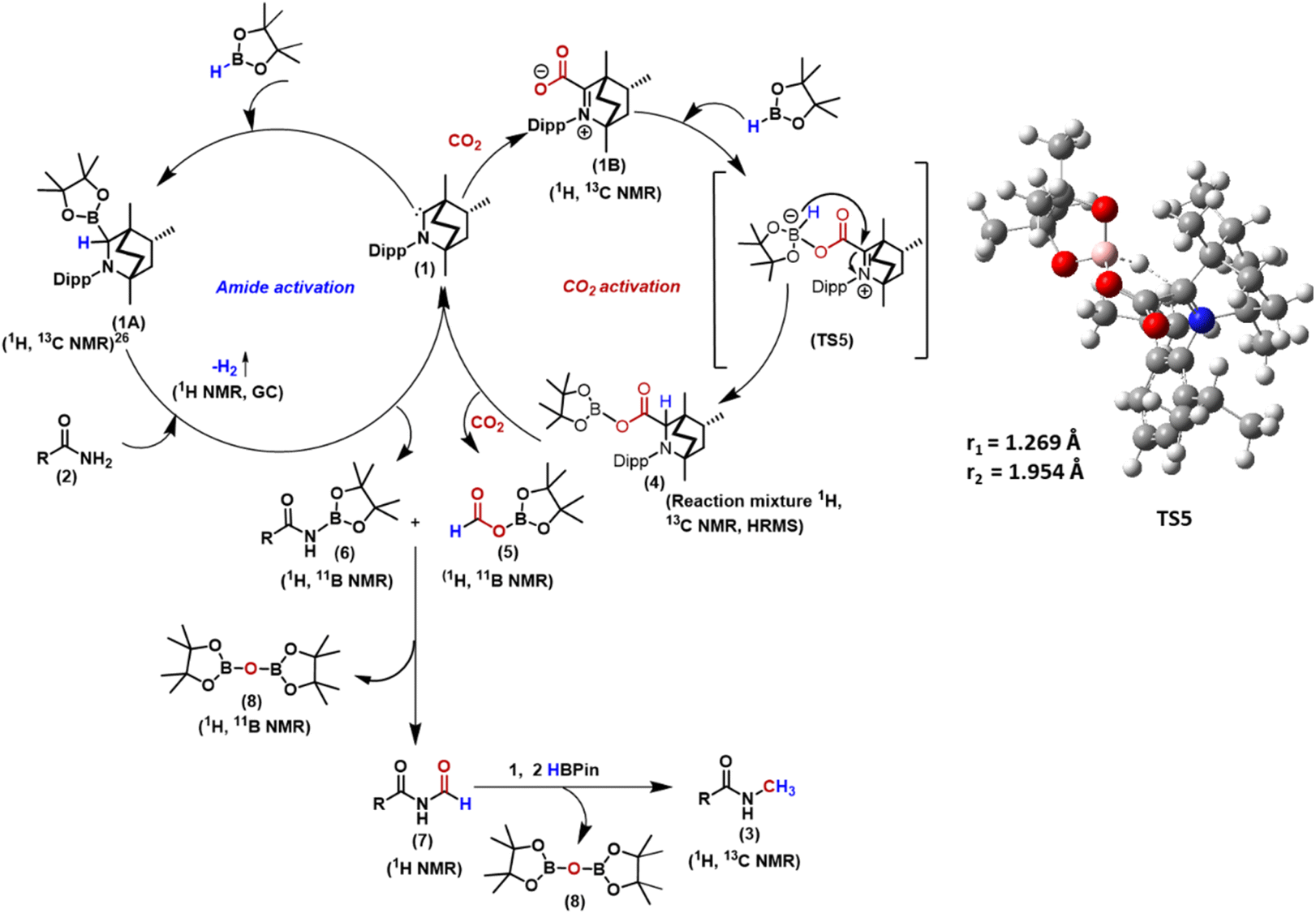 | ||
| Scheme 5 Proposed mechanistic cycle for N-methylation of amide catalysed by BICAAC. | ||
Conclusions
Although amine methylation was known using CO2, the current literature lacked a similar process with chemically inert amides. The present study reports the first N-methylation of primary amides utilising a greenhouse gas such as CO2 as a C1 source under metal-free conditions. This mechanistic investigation discloses that BICAAC plays a dual role in activating CO2 as well as amide to accomplish the methylation of various aromatic, aliphatic and heteroaromatic primary amides with HBpin as the hydride source. Furthermore, the present protocol was successful towards the diversification of drug and bioactive molecules. A few biologically important molecules were isotope-labelled using 13CO2. Considering our earlier efforts, with this study, we have been able to complete the 2e (formylation),104e (methylenation)15 and 6e reductive functionalisation (present study) of amides under metal-free conditions using CO2 as a reagent.Data availability
All experimental procedures, characterization details, copies of NMR spectra for all compounds, and computational data related to this article have been uploaded as part of the ESI.†Author contributions
SKM and NG conceived the idea of this work. SKM and NG planned the experiments. NG carried out the catalytic reactions and control experiments. RL conducted all the theoretical calculations and AKT analyzed the data. SP contributed to the mechanistic study. AR carried out the starting material synthesis. SKM supervised the work. SKM and NG analyzed the data and prepared the manuscript with the help of AKT and RL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKM and AKT thank the STARS Program (Grant no. MoE-STARS/STARS-1/473) of India for financial support. N. G., R. L., S. P. thank IISER-Kolkata, CSIR and DST-Inspire Program, respectively, for their research fellowships.Notes and references
- R. Lindsay, Climate Change: Atmospheric Carbon Dioxide, https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide, accessed 16/02/2023 Search PubMed.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. Schneider, H. F. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- P. Sreejyothi and S. K. Mandal, Chem. Sci., 2020, 11, 10571–10593 RSC.
- S. N. Riduan, Y. G. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556–559 CrossRef CAS PubMed.
- S. Sau, R. Bhattacharjee, P. Vardhanapu, G. Vijaykumar, A. Datta and S. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef CAS PubMed.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed.
- P. K. Hota, S. C. Sau and S. K. Mandal, ACS Catal., 2018, 8, 11999–12003 CrossRef CAS.
- C. R. Kemnitz and M. J. Loewen, J. Am. Chem. Soc., 2007, 129, 2521–2528 CrossRef CAS PubMed.
- X. Frogneux, E. Blondiaux, P. Thuéry and T. Cantat, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS.
- C. Xie, J. Song, H. Wu, B. Zhou, C. Wu and B. Han, ACS Sustainable Chem. Eng., 2017, 5, 7086–7092 CrossRef CAS.
- X.-Y. Li, S.-S. Zheng, X.-F. Liu, Z.-W. Yang, T.-Y. Tan, A. Yu and L.-N. He, ACS Sustainable Chem. Eng., 2018, 6, 8130–8135 CrossRef CAS.
- A. Das, P. Sarkar, S. Maji, S. K. Pati and S. K. Mandal, Angew. Chem., Int. Ed., 2022, 61, e202213614 CAS.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186–12190 CrossRef CAS PubMed.
- W.-C. Chen, J.-S. Shen, T. Jurca, C.-J. Peng, Y.-H. Lin, Y.-P. Wang, W.-C. Shih, G. P. A. Yap and T.-G. Ong, Angew. Chem., Int. Ed., 2015, 54, 15207–15212 CrossRef CAS PubMed.
- S. Maji, A. Das and S. K. Mandal, Chem. Sci., 2021, 12, 12174–12180 RSC.
- S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC.
- E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef PubMed.
- S. Yazdani, G. P. Junor, J. L. Peltier, M. Gembicky, R. Jazzar, D. B. Grotjahn and G. Bertrand, ACS Catal., 2020, 10, 5190–5201 CrossRef CAS.
- S. Chakrabortty, M. Kaur, M. Adhikari, K. K. Manar and S. Singh, Inorg. Chem., 2021, 60, 6209–6217 CrossRef CAS PubMed.
- Y. Gao, S. Yazdani, A. Kendrick IV, G. P. Junor, T. Kang, D. B. Grotjahn, G. Bertrand, R. Jazzar and K. M. Engle, Angew. Chem., Int. Ed., 2021, 60, 19871–19878 CrossRef CAS PubMed.
- N. M. Rajendran, N. Gautam, P. Sarkar, J. Ahmed, A. Das, S. Das, S. K. Pati and S. K. Mandal, Chem. Commun., 2021, 57, 5282–5285 RSC.
- N. Gautam, R. Logdi, P. Sreejyothi, N. M. Rajendran, A. K. Tiwari and S. K. Mandal, Chem. Commun., 2022, 58, 3047–3050 RSC.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- G. Kuchenbeiser, M. Soleilhavoup, B. Donnadieu and G. Bertrand, Chem.–Asian J., 2009, 4, 1745–1750 CrossRef CAS PubMed.
- G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444–9447 CrossRef CAS PubMed.
- S. Würtemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 22, 13032–13036 CrossRef PubMed.
- J. J. Li, Name Reactions: A Collection of Detailed Reaction Mechanisms, Springer, Berlin, 2006 Search PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- B. Paul, D. Panja and S. Kundu, Org. Lett., 2019, 21, 5843–5847 CrossRef CAS PubMed.
- B. Paul, M. Maji, D. Panja and S. Kundu, Asian J. Org. Chem., 2022, 11, e202100678 CAS.
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed.
- C. S. Elmore and R. A. Bragg, Bioorg. Med. Chem. Lett., 2015, 25, 167–171 CrossRef CAS PubMed.
- X. Wang, K. Chang and X. Xu, Dalton Trans., 2020, 49, 7324–7327 RSC.
- G. J. Karabatsos, J. Am. Chem. Soc., 1961, 83, 1230–1232 CrossRef CAS.
- P. Mohanakrishnan and K. R. K. Easwaran, Chem. Phys., 1986, 104, 409–414 CrossRef CAS.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459–2462 CrossRef CAS.
- Y. Tang, Y. Li, V. Fung, D. E. Jiang, W. Huang, S. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Zhang, A. I. Frenkel and F. F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed.
- C. Yu, C. Guo, L. Jiang, M. Gong and Y. Luo, Organometallics, 2021, 40, 1201–1206 CrossRef CAS.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01216f |
| This journal is © The Royal Society of Chemistry 2023 |