
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced chemo-, diastereo- and enantioselective α-C(sp3)–H functionalization of alkyl silanes†
Lili
Feng
,
Xiaofan
Chen
,
Ning
Guo
,
Yuqiao
Zhou
 ,
Lili
Lin
,
Weidi
Cao
* and
Xiaoming
Feng
*
,
Lili
Lin
,
Weidi
Cao
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: wdcao@scu.edu.cn; xmfeng@scu.edu.cn
First published on 4th April 2023
Abstract
A catalytic asymmetric α-C(sp3)–H functionalization of alkyl silanes with benzosultams was realized by merging photoredox and chiral Lewis acid catalysis. The key to success was the choice of photocatalyst with an appropriate redox potential and non-nucleophilic solvent, providing a novel entry to chiral organosilanes containing two adjacent tri- and tetra-substituted stereocenters with high to efficient diastereo- and enantioselectivity (up to 99% ee, 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 dr) under mild reaction conditions. Based on the control experiment and spectral analysis, an initial single electron transfer reduction of a benzosultam-triggered simultaneous or stepwise electron transfer/proton transfer process was proposed to rationalize the favored C(sp3)–H functionalization rather than desilylation.
:6 dr) under mild reaction conditions. Based on the control experiment and spectral analysis, an initial single electron transfer reduction of a benzosultam-triggered simultaneous or stepwise electron transfer/proton transfer process was proposed to rationalize the favored C(sp3)–H functionalization rather than desilylation.
Introduction
The direct enantioselective functionalization of C(sp3)–H is demonstrated to be an elegant, atom- and step-economic protocol to synthesize complex chiral molecules.1–3 Thanks to the successive discovery of photocatalysts4–7 in the past two decades, visible-light-promoted photocatalysis has emerged as a novel and powerful strategy for the purpose of formidable C(sp3)–H activation regardless of the high bond dissociation energy,8–12 which is complementary or even superior to that of the traditional transition metal-catalyzed C(sp3)–H insertion or using an extra quantitative oxidant. In this context, several groups have accomplished photocatalytic asymmetric C(sp3)–H functionalization of tertiary amines,13–16 ethers,17,18 sulfides,18,19 hydrocarbons and their derivatives,20–27 providing full and varied heteroatom-containing optically active compounds.Organosilanes fulfill a plethora of roles in synthetic chemistry, and have potential applications in drug discovery due to the unique physicochemical characteristics of silicon.28–30 We conceived that the direct photocatalytic asymmetric α-C(sp3)–H functionalization of silanes would offer straightforward access to chiral silicon-containing compounds. To the best of our knowledge, organic silanes have served as sought-after alkyl radical precursors in photoredox reactions by exploiting the adequately lower oxidation potential than the parent arising from the higher σ–n or σ–π interactions.31 Generally, the silanes undergo single electron transfer (SET) oxidation to form α-silyl cation radicals A, which are followed by C–Si bond or C–H bond cleavage to generate the desilylation radical species B or the α-silyl radical species C (Scheme 1a).32 The former producing non-silicon-containing products has been well developed in a variety of photoinduced enantioselective transformations (Scheme 1b);33–45 by contrast, the latter has not been explored for the synthesis of chiral silicon-containing substances. The mechanistic studies reported by Mariano32,46,47 and others48,49 revealed that the generation of desilylation and silicon-containing products greatly depended on the photoreaction solvent, and the competitive desilylation dominants by means of a nucleophile-assisted C–Si bond cleavage with the solvent acting as the nucleophile. For example, Melchiorre and co-workers elegantly established the enantioselective β-alkylation of enals with alkyl silanes through MeCN-assisted C–Si bond fragmentation.36,41 Herein, we reported the first visible-light-induced chemo-, diastereo- and enantioselective α-C(sp3)–H functionalization of silanes with benzosultams under the Ir(III)/N,N′-dioxide-Ni(II) synergetic catalysis to produce enantioenriched silicon-containing compounds (Scheme 1c); the desilylation was suppressed via the ingenious choice of a suitable photocatalyst and non-nucleophilic solvent.
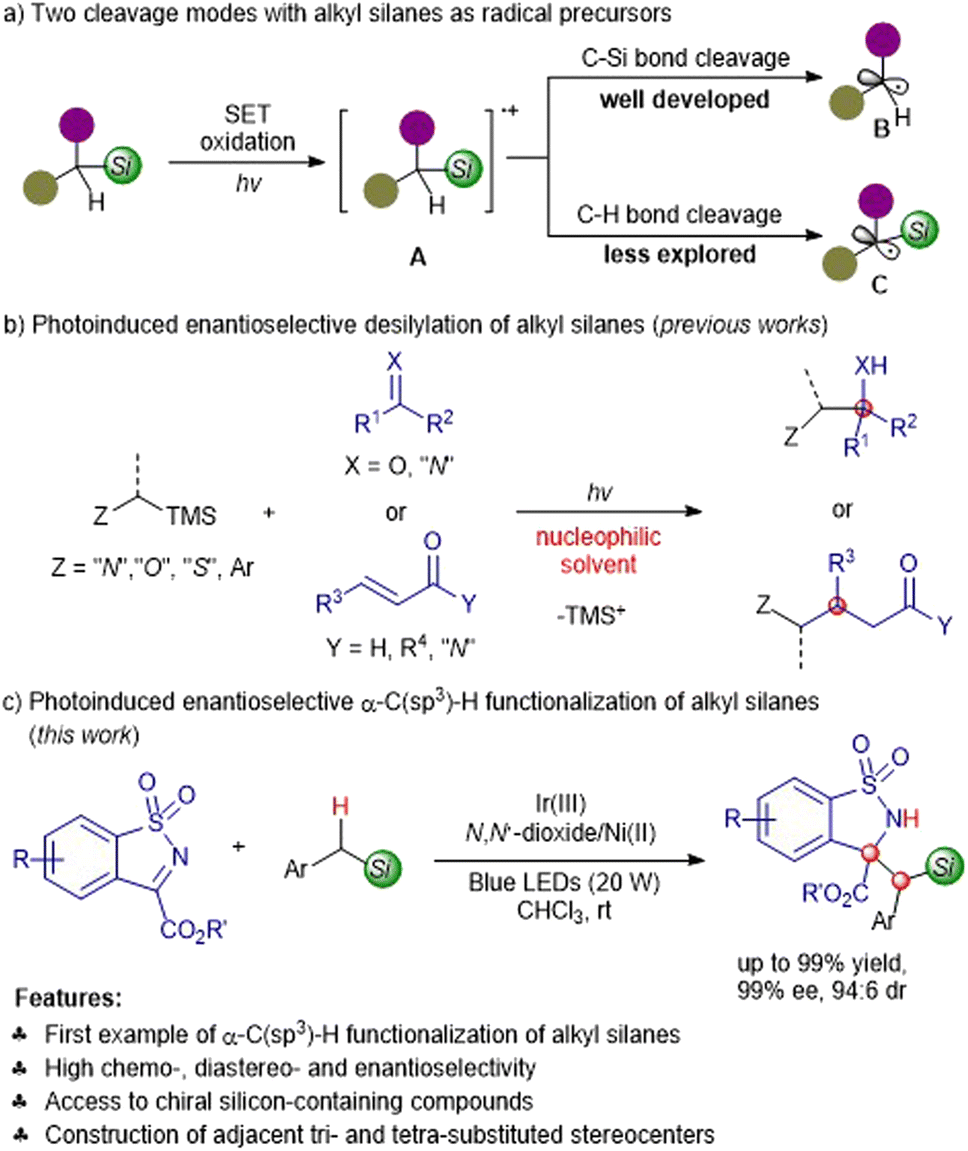 | ||
| Scheme 1 Photoinduced asymmetric radical reactions with alkyl silanes as radical precursors. | ||
Results and discussion
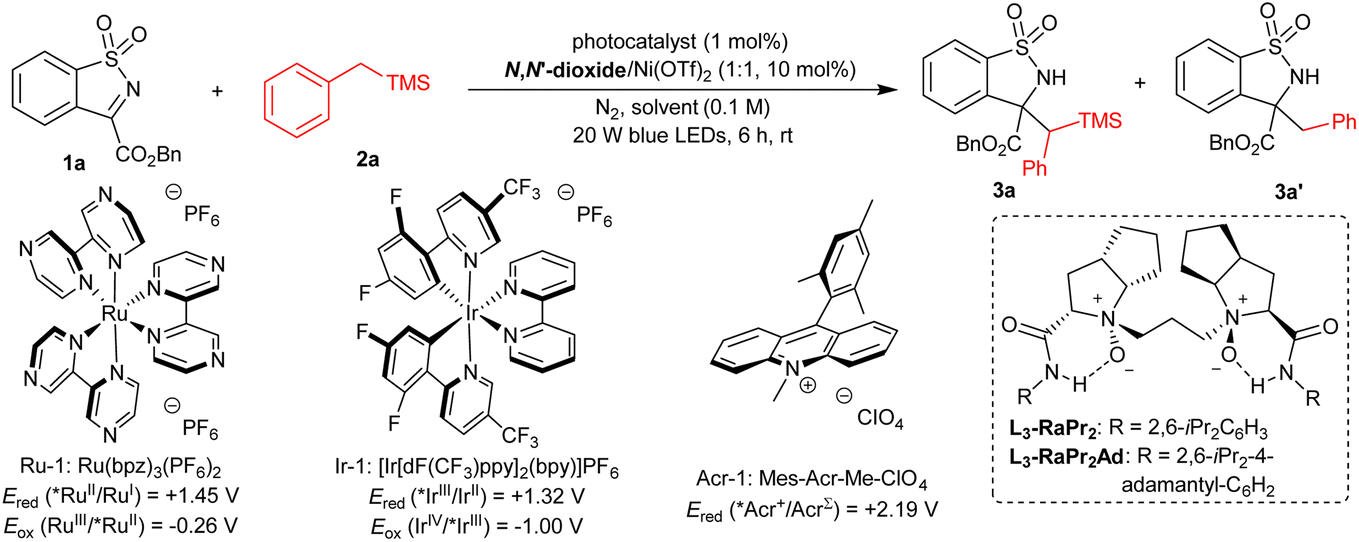
At the beginning of this study, we selected the model reaction of benzosultam 1a and benzyltrimethylsilane 2a in CHCl3. A variety of photocatalysts were investigated under the irradiation of blue LEDs (20 W) at room temperature with a L3-RaPr2/Ni(OTf)2 complex as the chiral Lewis acid catalyst (Table 1, entries 1–3, see the ESI† for details). No reaction occurred by using photocatalyst Ru-1 (Table 1, entry 1). Delightedly, the reaction proceeded smoothly to afford the desired product 3a in 69% yield, 63%/15% ee and 66:34 dr along with the formation of a trace amount of desilylation product 3a′ in the presence of Ir-1 (Table 1, entry 2). Nevertheless, the 3a′ obtained instead was catalyzed by Acr-1, which has a high exited state reduction potential (Ered (*Acr+/AcrΣ) = +2.19 V vs. SCE in CH3CN) (Table 1, entry 3).50 The screening of chiral N,N′-dioxide ligands51–61 showed that the sterically bulky L3-RaPr2Ad derived from 2,6-diisopropyl-4-adamantyl aniline promoted this reaction to give 3a in 75% yield, 86:14 dr with 94% ee (Table 1, entry 4). Increasing the dosage of 1a to 1.2 equiv. improved the yield to 94% with maintained diastereo- and enantioselectivity (Table 1, entry 5). The addition of a 4 Å molecular sieve (MS) raised the enantioselectivity slightly (95% ee, Table 1, entry 6). The solvent had an important influence on the chemoselectivity of this reaction. On switching CHCl3 to nucleophilic solvents, such as CH3OH, CH3CN and THF, 3a′ was obtained without observation of 3a (Table 1, entries 7–9), and the lower yield of 3a′ in THF was due to the competing α-C(sp3)–H functionalization of THF with 1a (Table 1, entry 9). Similarly, the reaction between toluene and 1a dominated with toluene as the solvent (Table 1, entry 10). Other reaction parameters were also examined, including a central metal salt, catalytic loading and so on, but no better result was achieved (see the ESI† for details). Additionally, control experiments were carried out to understand the synergic catalysis. No reaction occurred without light or a photocatalyst, proving the photochemical nature (Table 1, entries 11 and 12). Ir-1 could mediate this reaction itself, but afforded 3a with 48% yield and 52:48 dr, implying that the L3-RaPr2Ad/Ni(OTf)2 complex played a significant role in enhancing the reactivity and diastereoselectivity (Table 1, entry 13).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Photocatalyst | Ligand | Solvent | Yield of 3ab (%) | ee of 3ab (%) | dr (3a)c | Yield of 3a′c (%) | ee of 3a′c (%) |
| a Unless otherwise noted, all the reactions were performed with a photocatalyst (1 mol%), Ni(OTf)2 (10 mol%), ligand (10 mol%), 1a (0.10 mmol) and 2a (0.10 mmol) in solvent (1.0 mL) at room temperature under the irradiation of 20 W blue LEDs for 6 h. b Yield of the isolated product. c The ee and dr values were determined by UPCC analysis. d Acr-1 (2 mol%). e 1a (0.12 mmol). f With 4 Å MS (20 mg), 0.5 h. g Without light. h In the absence of a L3-RaPr2Ad/Ni(II) complex. n. d. = not determined. | ||||||||
| 1 | Ru-1 | L3-RaPr2 | CHCl3 | 0 | — | — | 0 | — |
| 2 | Ir-1 | L3-RaPr2 | CHCl3 | 69 | 63/15 | 66:34 |
Trace | n. d. |
| 3d | Acr-1 | L3-RaPr2 | CHCl3 | 0 | — | — | 95 | 17 |
| 4 | Ir-1 | L3-RaPr2Ad | CHCl3 | 75 | 94/47 | 86:14 |
Trace | n. d. |
| 5e | Ir-1 | L3-RaPr2Ad | CHCl3 | 94 | 93/43 | 86:14 |
Trace | n. d. |
| 6e,f | Ir-1 | L3-RaPr2Ad | CHCl3 | 95 | 95/43 | 86:14 |
Trace | n. d. |
| 7e | Ir-1 | L3-RaPr2Ad | CH3OH | 0 | — | — | 98 | 7 |
| 8e | Ir-1 | L3-RaPr2Ad | CH3CN | 0 | — | — | 99 | 36 |
| 9e | Ir-1 | L3-RaPr2Ad | THF | 0 | — | — | 11 | n. d. |
| 10e | Ir-1 | L3-RaPr2Ad | Toluene | 23 | 92/35 | 85:15 |
75 | 47 |
| 11e,g | Ir-1 | L3-RaPr2Ad | CHCl3 | 0 | — | — | 0 | — |
| 12e | — | L3-RaPr2Ad | CHCl3 | 0 | — | — | 0 | — |
| 13e,h | Ir-1 | — | CHCl3 | 48 | — | 52:48 |
0 | — |
With the optimized reaction conditions in hand (Table 1, entry 6), the substrate scope was then evaluated. As shown in Scheme 2, on changing the benzyl ester of benzosultams to a methyl ester (1b) and ethyl ester (1c), the corresponding products 3b62 (96% ee, 83:17 dr) and 3c (90% ee, 86:14 dr) were obtained smoothly. With respect to the substituents on the phenyl group, the reaction of 1f–1i bearing an electron-withdrawing group afforded 3f–3i with excellent enantioselectivities (96–99% ee), which was superior to that of electron-rich ones (3d-3e, 84–88% ee). An imine containing fused naphthyl motif was also tolerated but provided 3j in only 30% yield, 82% ee and 80:20 dr. Next, we turned our attention to the scope of silanes. Various substituted benzyl trimethylsilanes were applicable in this synergistic catalytic system to afford the α-C(sp3)–H functionalization products 3k–3s. Both the electrical properties and position of the substituents have obvious effects on this reaction. The silanes bearing electron-donating groups at the para-position of the phenyl group exhibited lower reactivity and stereoselectivity. For example, salines containing halo-substituents at the para-position, gave 3n–3p with 86–93% yield and 96% ee, while para-methyl and para-methoxyl benzyl substituted 3l-3m were obtained in 21–63% yield with lower enantioselectivity (87–93% ee). If the electron-withdrawing groups were located at the ortho- or meta-positions, decreased reactivity and stereoselectivity were observed (3q–3s, 46–68% yield, 83–89% ee, and 78:22–83:17 dr). Noteworthily, regioselective secondary C(sp3)–H functionalization adjacent to the silicon atom took place to afford 3k-3l solely without observation of the primary C(sp3)–H bond fragmentation. Trimethyl(naphthalen-2-ylmethyl)silane reacted with 1a to deliver 3t in 51% yield and 94% ee with 83:17 dr, and the desilylation product 3t′ was also isolated in 41% yield with 47% ee. Benzyltriethylsilane (BnTES), benzylic dimethyl tert-butylsilane (BnTBS), BnSiMe2CH2Cl and BnSiMe2H were also suitable substrates, successfully generating 3u–3x with 56–68% yield, 80–96% ee and 73:27–92:8 dr. It was worth mentioning that no Si–H bond cleavage product through a silyl radical intermediate was observed.63
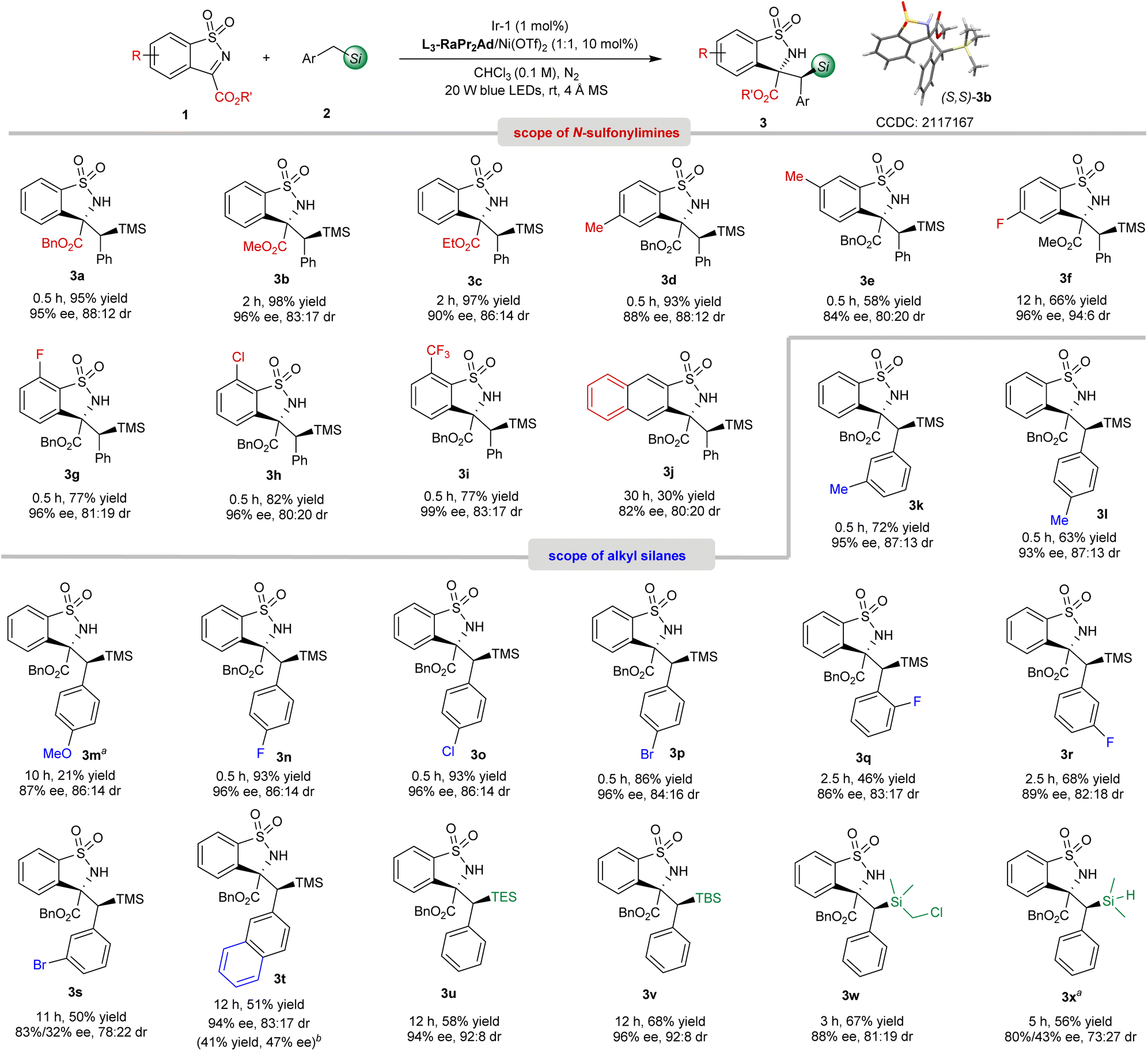 | ||
| Scheme 2 The substrate scope. Reaction conditions: Ir-1 (1 mol%), Ni(OTf)2/L3-RaPr2Ad (1:1, 10 mol%), 1 (0.12 mmol), 2 (0.10 mmol), and 4 Å MS (20 mg) in CHCl3 (1.0 mL) at rt under the irradiation of 20 W blue LEDs for the indicated time. Yield of the isolated product, and the dr value was determined by 1H NMR spectroscopy and ee value was analyzed by UPCC with a chiral column. a[Ir[dF(CF3)ppy]2(dtbbpy)]PF6 (1 mol%) instead. bThe data of desilylation product 3t′ are given in parenthesis. | ||
To get insight into the reaction process, a series of mechanistic studies were conducted. The reaction was completely inhibited with the addition of 2,2,6,6-tetramethylpiperidine-1-oxyl (see the ESI† for details). The Stern–Volmer fluorescence quenching experiments unambiguously showed that 1a quenched Ir-1 effectively, but 2a has no obvious quenching effect (Scheme 3a). Moreover, 1a underwent homocoupling to give the product 4a with 26% yield in the presence of Ir-1 (0.5 equiv.), while no reaction of 2a occurred (Scheme 3b). The observation was consistent with the redox potential between Ir-1 and 1a or 2a (Scheme 3b). The electron paramagnetic resonance (EPR) measurement was also performed with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a trapping agent. It witnessed the formation of the persistent radical 6 (g = 2.0066, AN = 12.91 G, and AH = 10.40 G) through EPR simulation (Scheme 3c),64 which was further conformed based on the detection of 6 by using the high resolution mass spectrum (HRMS) (Scheme 3c). These results strongly indicated that Ir-1 underwent oxidative quenching to trigger the generation of a radical anion 1a˙− from imine. In addition, deuterated experiments revealed that the proton source of the N–H bond in 3a came from 1a (see the ESI† for details), and a primary kinetic isotope effect (KIE) of about 1.7 and 1.47 for the competitive and parallel experiments revealed that the C(sp3)–H bond cleavage may be involved in the rate-limiting step (Scheme 3d). Next, the pathway of C(sp3)–H bond cleavage of alkyl silane was clarified: direct hydrogen atom transfer (HAT), electron transfer (ET)/proton transfer (PT) sequence or multisite proton-coupled electron transfer (MS-PCET)65–68 (see the ESI† for details)? Firstly, the photocatalysts Ir-1 and Ir-2 possessed the same triplet state energy (60.1 kcal mol−1);69 however, the latter could not facilitate this reaction (Scheme 3e), and the result was consistent with the observation of the formation of 3x rather than the Si–H bond cleavage product,63 which might exclude the direct HAT between 2a and triplet state 1a that was formed through energy transfer. Secondly, the reactions between 1a and THF or toluene proceeded smoothly (Table 1, entries 9 and 10) although the oxidation potentials of THF (>+2.4 V vs. SCE in CH3CN) and toluene (+2.26 V vs. SCE in CH3CN) are much higher than that of the Ir-1 (Eox(IrIV/III) = +1.69 V vs. SCE in CH3CN). Moreover, Ir-3 could mediate this reaction to give 3a with 32% yield (Scheme 3e), but the potential was also mismatched between Ir-3 (Ered (*IrIII/IrII) = +0.75 V, Eox (IrIV/IrIII) = +1.49 V vs. SCE in CH3CN) and 2a (Eox (2a˙+/2a) = +1.55 V vs. SCE in CH3CN, see the ESI† for details). Thus, a SET reduction of imine followed by the MS-PCET mechanism was surmised. However, 3a′ was obtained exclusively in nucleophilic solvents (Table 1, entries 7–9), indicating the existence of radical cation 2a˙+ generated through SET oxidation of 2a by Ir-1(IV), which meant that the stepwise ET/PT may be also involved. In addition, the quantum yield was found to be 0.23, suggesting that a radical chain propagation in this reaction may not be the predominant process.
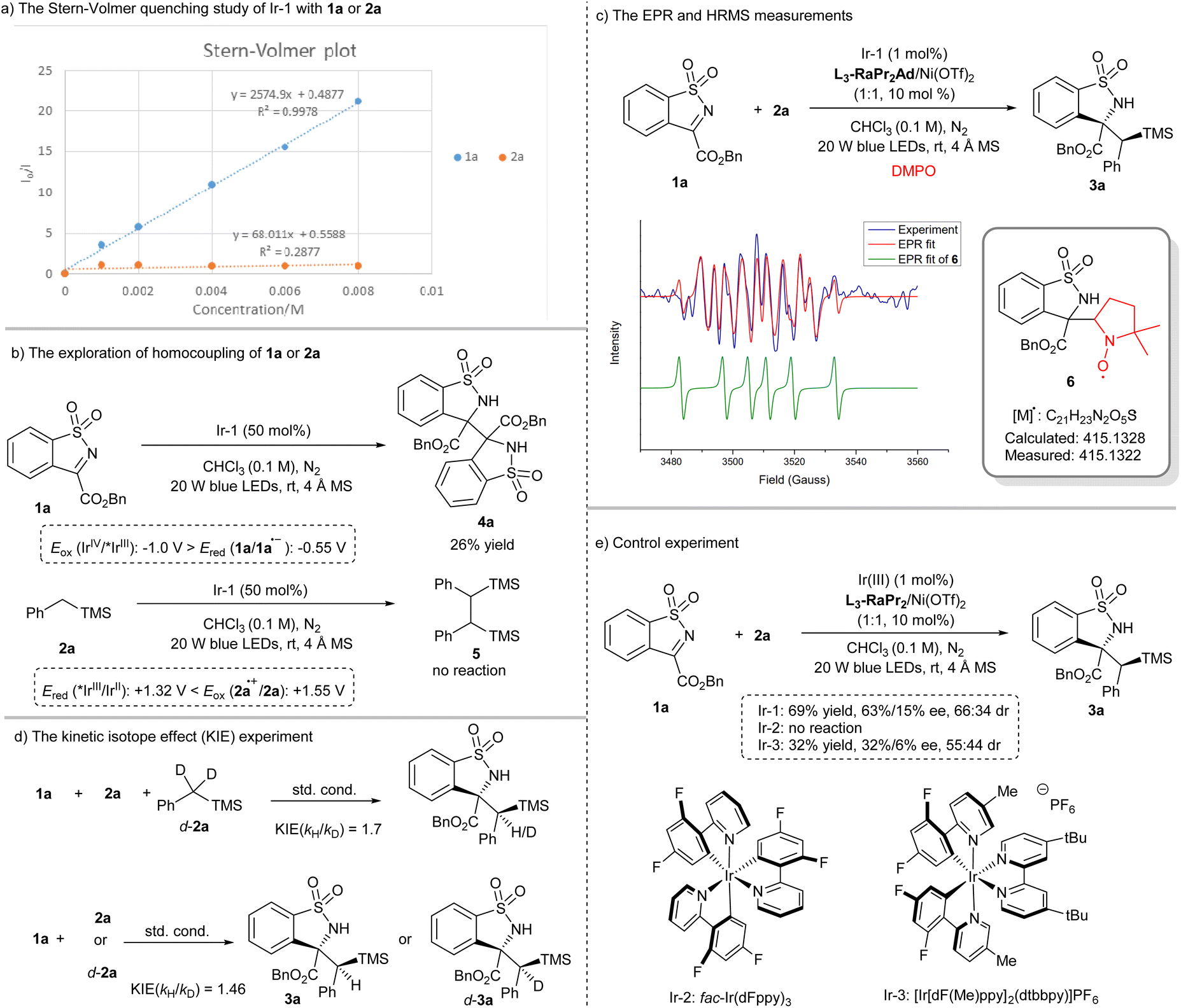 | ||
| Scheme 3 The mechanistic studies. | ||
According to above experimental results, the catalytic cycle was proposed as shown in Fig. 1. The L3-RaPr2Ad/Ni(OTf)2 complex coordinated with 1a to form the intermediate I, which underwent SET reduction with the photocatalyst [IrIII] under visible-light illumination and afforded the radical anion II; accordingly, [IrIII] went through oxidative quenching to form the [IrIV] species. The following C–H bond cleavage of 2a through MS-PCET involving electron transfer to [IrIV] (oxidant) and proton transfer to II (base)70 regenerated [IrIII] and produced a radical–radical pair III (path a). Alternatively, III was formed through a stepwise process, namely, SET between 2a and [IrIV] took place to afford the radical cation IV, followed by proton transfer to II (path b). Subsequently, III underwent stereocontrolled radical coupling to provide 3a. Additionally, an unproductive homocoupling of II proceeded to yield the byproduct 4a.
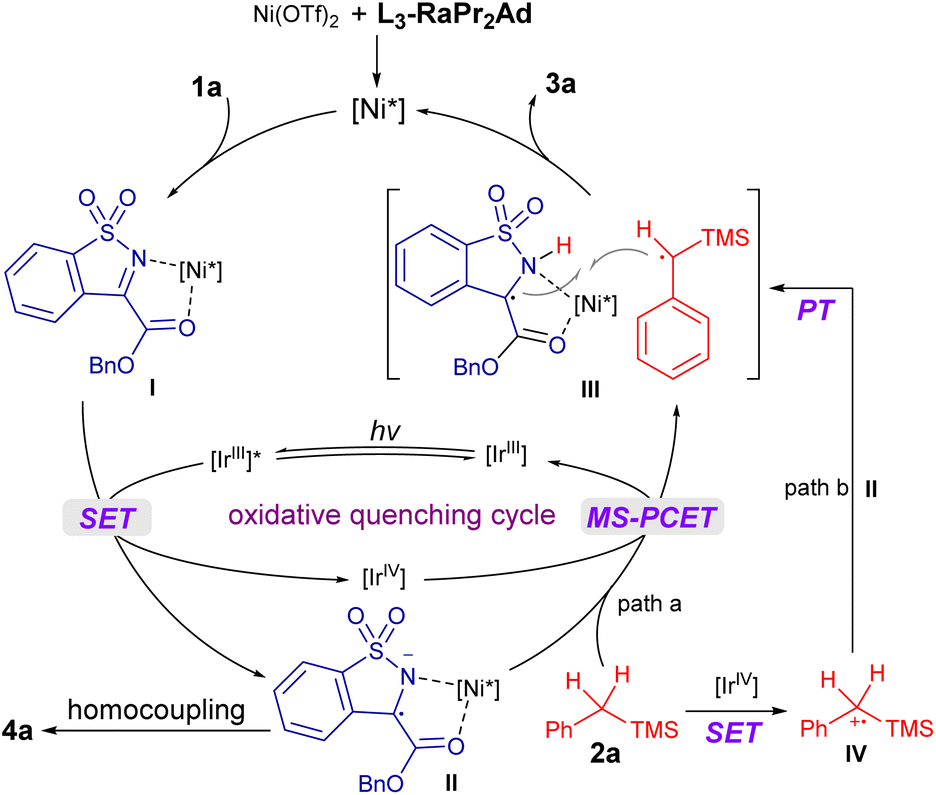 | ||
| Fig. 1 The proposed catalytic cycle. | ||
Conclusions
In summary, a bimetallic synergetic photocatalytic chemo-, diastereo- and enantioselective α-C(sp3)–H functionalization of alkyl silanes with benzosultams was discovered. Diverse experimental evidence revealed that the Ir(III) photocatalyst underwent an oxidative quenching cycle to facilitate SET reduction of benzosultam and afforded a radical anion. It triggered the following MS-PCET or stepwise ET/PT of alkyl silanes and radical coupling. This protocol provided a rapid and facile access to chiral organosilanes containing two adjacent tri- and tetra-substituted stereocenters with good to excellent yield and stereoselectivities. The extension of this methodology to synthesize chiral silanes is underway in our laboratory.Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC, CD spectra, X-ray crystallographic data for 3b are available in the ESI.†Author contributions
L. L. F. preformed the experiments. X. F. C. repeated the data. N. G. participated in the synthesis of substrates and ligands. X. M. F., L. L. L. and W. D. C. supervised the project. W. D. C. and L. L. F. co-wrote the manuscript. Y. Q. Z. performed the X-ray single crystal diffraction analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (no. 22188101, 22071160 and 92256302), and Sichuan Science and Technology Program (no. 2021YJ0562) for financial support. We thank Dr Hanjiao Chen (Sichuan University) for the assistance with the EPR experiment.Notes and references
- C. G. Newton, S. G. Wang and C. C. Oliveira, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed.
- T. G. Saint-Denis, R. Y. Zhu, G. Chen, Q. F. Wu and J. Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed.
- Q. Zhang and B. F. Shi, Chin. J. Chem., 2019, 37, 647–656 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed.
- Y. Wu, D. Kim and T. S. Teets, Synlett, 2022, 33, 1154–1179 CrossRef CAS.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- Q. Qin, H. Jiang, Z. Hu, D. Ren and S. Yu, Chem. Rec., 2017, 17, 754–774 CrossRef CAS PubMed.
- L. Revathi, L. Ravindar, W. Y. Fang, K. P. Rakesh and H. L. Qin, Adv. Synth. Catal., 2018, 360, 4652–4698 CrossRef CAS.
- M. Uygur and O. G. Mancheño, Org. Biomol. Chem., 2019, 17, 5475–5489 RSC.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768–13771 CrossRef CAS PubMed.
- C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef CAS PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- T. Y. Zhan, L. K. Yang, Q. Y. Chen, R. Weng, X. H. Liu and X. M. Feng, CCS Chem., 2022, 4 DOI:10.31635/ccschem.022.202202405.
- C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed.
- Z. D. Tan, S. B. Zhu, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202203374 CAS.
- W. Yuan, Z. Zhou, L. Gong and E. Meggers, Chem. Commun., 2017, 53, 8964–8967 RSC.
- D. Mazzarella, G. E. Crisenza and P. Melchiorre, J. Am. Chem. Soc., 2018, 140, 8439–8443 CrossRef CAS PubMed.
- Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016–1026 CrossRef CAS.
- H. Mitsunuma, S. Tanabe, H. Fuse, K. Ohkubo and M. Kanai, Chem. Sci., 2019, 10, 3459–3465 RSC.
- Z. Y. Dai, Z. S. Nong and P. S. Wang, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS.
- S. Cao, W. Hong, Z. Ye and L. Gong, Nat. Commun., 2021, 12, 2377 CrossRef CAS PubMed.
- J. Kikuchi, S. Kodama and M. Terada, Org. Chem. Front., 2021, 8, 4153–4159 RSC.
- Z. Y. Dai, Z. S. Nong, S. Song and P. S. Wang, Org. Lett., 2021, 23, 3157–3161 CrossRef CAS PubMed.
- H. Yu, T. Zhan, Y. Zhou, L. Chen, X. H. Liu and X. M. Feng, ACS Catal., 2022, 12, 5136–5144 CrossRef CAS.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668–6684 CrossRef CAS PubMed.
- J. I. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed.
- E. Hasegawa, W. Xu, P. S. Mariano, U.-C. Yoon and J.-U. Kim, J. Am. Chem. Soc., 1988, 110, 8099–8111 CrossRef CAS.
- L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef CAS PubMed.
- J. Ma, K. Harms and E. Meggers, Chem. Commun., 2016, 52, 10183–10186 RSC.
- T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953–6958 CrossRef CAS PubMed.
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed.
- Z. Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562–4568 RSC.
- B. Han, Y. Li, Y. Yu and L. Gong, Nat. Commun., 2019, 10, 3804 CrossRef PubMed.
- S. K. Pagire, N. Kumagai and M. Shibasaki, Chem. Sci., 2020, 11, 5168–5174 RSC.
- E. Le Saux, D. Ma, P. Bonilla, C. M. Holden, D. Lustosa and P. Melchiorre, Angew. Chem., Int. Ed., 2021, 60, 5357–5362 CrossRef CAS PubMed.
- X. Dong, Q. Y. Li and T. P. Yoon, Org. Lett., 2021, 23, 5703–5708 CrossRef CAS PubMed.
- J. Y. Kim, Y. S. Lee and D. H. Ryu, ACS Catal., 2021, 11, 14811–14818 CrossRef CAS.
- S. M. Cho, J. Y. Kim, S. Han and D. H. Ryu, J. Org. Chem., 2022, 87, 11196–11203 CrossRef CAS PubMed.
- L. Z. Hou, Y. Q. Zhou, H. Yu, T. Y. Zhan, W. D. Cao and X. M. Feng, J. Am. Chem. Soc., 2022, 144, 22140–22149 CrossRef CAS PubMed.
- U.-C. Yoon and P. S. Mariano, Acc. Chem. Res., 1992, 25, 233–240 CrossRef CAS.
- U. C. Yoon, P. S. Mariano, L. Cermenati, M. Freccero, P. Venturello and A. Albini, J. Am. Chem. Soc., 1995, 117, 7869–7876 CrossRef.
- J. P. Dinnocenzo, S. Farid, J. L. Goodman, I. R. Gould, S. L. Mattes and W. P. Todd, J. Am. Chem. Soc., 1989, 111, 8973–8975 CrossRef CAS.
- K. P. Dockery, J. P. Dinnocenzo, S. Farid, J. L. Goodman, I. R. Gould and W. P. Todd, J. Am. Chem. Soc., 1997, 119, 1876–1883 CrossRef CAS.
- 3a′ was obtained instead probably due to the initial SET oxidation of 2a rather than SET reduction of 1a with exited state Acr-1. A benzyl radical cation was formed and it released a TMS+ group preferentially because no suitable base (e.g. radical anion II in Fig. 1) accepted the proton, leading to the generation of a benzyl radical, which underwent radical addition to 1a and SET/protonation to produce 3a′. See the ESI† for the possible catalytic cycle with Acr-1 as the catalyst.
- X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed.
- X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC.
- X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed.
- X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791–797 CrossRef CAS.
- M.-Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969–984 CrossRef CAS.
- S. X. Dong, X. H. Liu and X. M. Feng, Acc. Chem. Res., 2022, 55, 415–428 CrossRef CAS PubMed.
- Y. H. Wen, X. Huang and X. M. Feng, Synlett, 2005, 2445–2448 CAS.
- X. Zhang, D. H. Chen, X. H. Liu and X. M. Feng, J. Org. Chem., 2007, 72, 5227–5233 CrossRef CAS PubMed.
- L. Dai, W. Liu, Y. Q. Zhou, Z. Zeng, X. Y. Hu, W. D. Cao and X. M. Feng, Angew. Chem., Int. Ed., 2021, 60, 26599–26603 CrossRef CAS PubMed.
- Q. W. He, D. Zhang, F. C. Zhang, X. H. Liu and X. M. Feng, Org. Lett., 2021, 23, 6961–6966 CrossRef CAS PubMed.
- D. Zhang, M. P. Pu, Z. Z. Liu, Y. Q. Zhou, Z. D. Yang, X. H. Liu, Y.-D. Wu and X. M. Feng, J. Am. Chem. Soc., 2023, 145, 4808–4818 CrossRef CAS PubMed.
- CCDC 2117167 (3b).
- X. Fan, P. Xiao, Z. Jiao, T. Yang, X. Dai, W. Xu, J. D. Tan, G. Cui, H. Su, W. Fang and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 12580–12584 CrossRef CAS PubMed.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
- Z. Zhou, X. Kong and T. Liu, Chin. J. Org. Chem., 2021, 41, 3844–3879 CrossRef CAS.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- A. Singh, K. Teegardin, M. Kelly, K. S. Prasad, S. Krishnan and J. D. Weaver, J. Organomet. Chem., 2015, 776, 51–59 CrossRef CAS.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, and CD spectra (PDF). X-ray crystallographic data for 3b. CCDC 2117167. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00919j |
| This journal is © The Royal Society of Chemistry 2023 |