
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAir-stable four-coordinate cobalt(II) single-ion magnets: experimental and ab initio ligand field analyses of correlations between dihedral angles and magnetic anisotropy†
Sandeep K.
Gupta
 a,
Shashank V.
Rao
b,
Serhiy
Demeshko
a,
Sebastian
Dechert
a,
Eckhard
Bill
c,
Mihail
Atanasov
*bd,
Frank
Neese
*b and
Franc
Meyer
*a
a,
Shashank V.
Rao
b,
Serhiy
Demeshko
a,
Sebastian
Dechert
a,
Eckhard
Bill
c,
Mihail
Atanasov
*bd,
Frank
Neese
*b and
Franc
Meyer
*a
aInstitut für Anorganische Chemie, Universität Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: franc.meyer@chemie.uni-goettingen.de
bMax Planck Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: mihail.atanasov@kofo.mpg.de; neese@kofo.mpg.de
cMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany
dInstitute of General and Inorganic Chemistry, Bulgarian Academy of Sciences, Akad. Georgi Bontchev Street 11, 1113 Sofia, Bulgaria
First published on 12th May 2023
Abstract
For single-ion magnets (SIMs), understanding the effects of the local coordination environment and ligand field on magnetic anisotropy is key to controlling their magnetic properties. Here we present a series of tetracoordinate cobalt(II) complexes of the general formula [FL2Co]X2 (where FL is a bidentate diamido ligand) whose electron-withdrawing –C6F5 substituents confer stability under ambient conditions. Depending on the cations X, these complexes adopt structures with greatly varying dihedral twist angle δ between the N–Co–N′ chelate planes in the solid state (48.0 to 89.2°). AC and DC field magnetic susceptibility measurements show this to translate into very different magnetic properties, the axial zero-field splitting (ZFS) parameter D ranging from −69 cm−1 to −143 cm−1 with substantial or negligible rhombic component E, respectively. A close to orthogonal arrangement of the two N,N′-chelating σ- and π-donor ligands at the Co(II) ion is found to raise the energy barrier for magnetic relaxation to above 400 K. Multireference ab initio methods were employed to describe the complexes' electronic structures, and the results were analyzed within the framework of ab initio ligand field theory to probe the nature of the metal–ligand bonding and spin–orbit coupling. A relationship between the energy gaps of the first few electronic transitions and the ZFS was established, and the ZFS was correlated with the dihedral angle δ as well as with the metal–ligand bonding variations, viz. the two angular overlap parameters eσ and eπs. These findings not only give rise to a Co(II) SIM showing open hysteresis up to 3.5 K at a sweep rate of 30 Oe s−1, but they also provide design guidelines for Co(II) complexes with favorable SIM signatures or even switchable magnetic relaxation properties.
Introduction
The observation of slow relaxation of the magnetization in a molecular complex, first discovered for the “Mn12 acetate” cluster,1 was an impactful landmark for interdisciplinary research in chemistry, physics, and materials science.1,2 Such single-molecule magnets (SMMs) promised great potential for applications in the fields of high-density data storage, quantum computing, and spintronics.2a,3 Furthermore, it was the first time quantum tunneling of the magnetization (QTM) was observed experimentally as steps in the magnetic hysteresis.4 This, in addition to fundamental interest, spurred the development of multinuclear exchange-coupled paramagnetic complexes with a very high ground spin-state (S), since the energy barrier for spin reversal (Ueff) is given by Ueff = |D|S2 (for integer spins) or Ueff = |D|(S2 − 1/4) (for half-integer spins).5 Here D is the axial zero-field splitting (ZFS) parameter, a measure of the magnetic anisotropy of the system. However, it was soon pointed out that maximization of S does not always lead to a higher energy barrier because D is inversely correlated with S.6 This directed the research efforts towards maximizing D in molecular complexes featuring large anisotropy, and ultimately towards single-ion magnets (SIMs).For SIMs based on transition metal ions, Co(II) complexes are particularly prominent, but relatively few of them exhibit slow relaxation dynamics in the absence of an applied magnetic field.7 The extremely air-sensitive mononuclear Co(II) complex [Co(SPh)4][PPh4]2 was the first reported zero-field 3d-SIM.8 Since then several Co(II) SIMs with a coordination number ranging from two to six have been discovered.8–14 However, some complexes with two,9 three,10a four8,11a–o and six13a–d coordinate metal ions feature the most favorable properties, and the largest energy barriers for any 3d SIMs have been observed for linear two-coordinate complexes where the ligand field is weak, thus maximizing the orbital contribution to the magnetic moment.9 Among the prominent examples of this family is the recently reported linear two-coordinate dialkyl Co(II) complex Co(C(SiMe2ONaph)3)2 that exhibits a non-Aufbau ground state L = 3 with a limiting magnetic anisotropy for a Co(II) ion.9b This is comparable with magnetic anisotropy exhibited by mono-coordinate Co atoms deposited on a MgO surface.15 However, these low coordinate complexes are extremely air and moisture sensitive.
Four-coordinate Co(II) complexes are usually more rugged than the low-coordinate complexes. In order to maximize the orbital contribution through second order spin–orbit coupling in distorted tetrahedral Co(II) complexes and increase the ZFS, which ultimately determines the magnetic anisotropy, great efforts are being made to understand the underlying factors that govern the magnitude of the ZFS and relaxation processes.11a,b,q,16 In this context, magneto-structural correlations have been developed for four-coordinate Co(II) SIMs, allowing to rationalize the effects of the structural distortion around the central paramagnetic ion on the sign and magnitude of the ZFS (Chart 1).11a–e,g,i,r,v,17 Most studied are four-coordinate [CoIIX4]2− complexes where X is an O-, S-, Se-, or Te-donor based monodentate anionic ligand A in Chart 1.11a–e It has been shown that the magnitude and sign of D are highly dependent on the nature of the donor atoms of the coordinating ligand X (viz., O, S, Se or Te). π-Anisotropic ligand systems with decreasing ligand field strength, i.e., with softer donor atoms, in tetragonally elongated D2d geometries lead to significantly enhanced negative D.8,11a Magneto-structural correlations for these [CoIIX4]2− systems have been established mainly for two types of structural distortions. The first one relates to the distortion 2γ of the first coordination sphere (where γ is the angle between the S4-rotation axis and the Co–X bond) and the second one (ω) describes torsion angles involving the second coordination sphere (R–S–Co–S4 axis; A in Chart 1).11a–e Their analysis has revealed that a large negative D value is associated with smaller 2γ values (tetragonal elongation) and the maximum negative D is achieved for ω = 0°. Similar magneto-structural correlations have been reported for CoL2X2 systems (B in Chart 1, L = neutral donor ligands and X = anions).18 Variations of the donor atom/coordinating anions in CoL2X2 systems have provided additional insights, indicating that the heavier and softer donor atoms significantly enhance the ZFS in Co(II) SIMs.11k,17e,19 Lately, there has been interest in understanding the effect of molecular vibrations concerning the relaxation mechanism operating in SIMs as well.16c,d
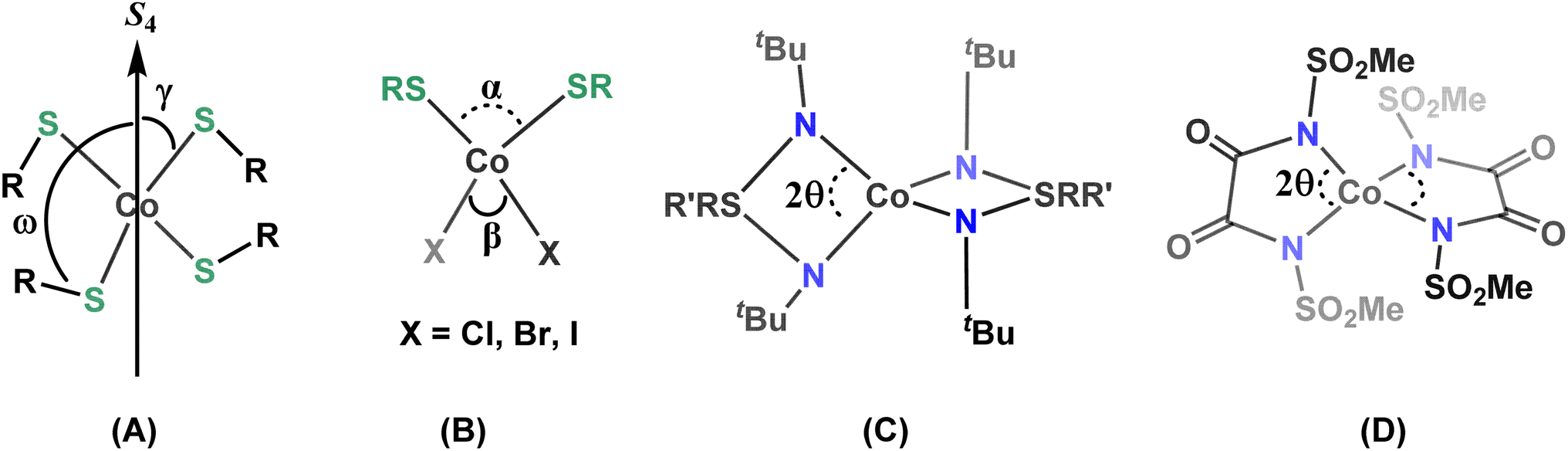 | ||
| Chart 1 Selected examples of magneto-structural correlations (A–D) studied for four-coordinate Co(II) SIMs. | ||
In addition to [CoX4]2− or CoL2X2 systems with monodentate ligands, enhanced magnetic bistability has recently been reported for four-coordinate Co(II) complexes derived from bidentate chelate ligands (C and D in Chart 1, and E and F in Chart 2).11f–j,11p,q In 2015, some of us found appreciable single-ion anisotropy (D = −58 cm−1) and slow relaxation of magnetization in zero-field for a Co(II) triimidosulfonate complex (C),11f and later the groups of Sarkar and van Slageren reported a Co(II) bis(sulfonamido)benzene complex that exhibits hysteresis at 1.8 K with coercivity at a sweep rate of 500 Oe s−1 (E).11h Recently the influence of the N–Co–N bite angle in four-coordinate Co(II) triimidosulfonate complexes11g (C in Chart 1) and Co(II) bis(methanesulfonyl)oxamidate complexes11i (D in Chart 1) has been elucidated, and bite angles around 76–78° and 81°, respectively, were found to be ideal for harnessing large ZFS. The Plass group has investigated different solvates of pseudo-tetrahedral bis-chelate {N2O2} Co(II) SIMs and concluded that the spin-reversal barrier increases with stronger distortion away from the tetrahedral toward a square-planar geometry; in the studied solvomorphs the dihedral twist angles between the N–Co–O chelate planes of the two Schiff-base ligands varied between 86° and 55.9°, with D values in the range from −25 to −40 cm−1.20
 | ||
| Chart 2 Examples of homoleptic dianionic four-coordinate Co(II) SIMs (E–G). | ||
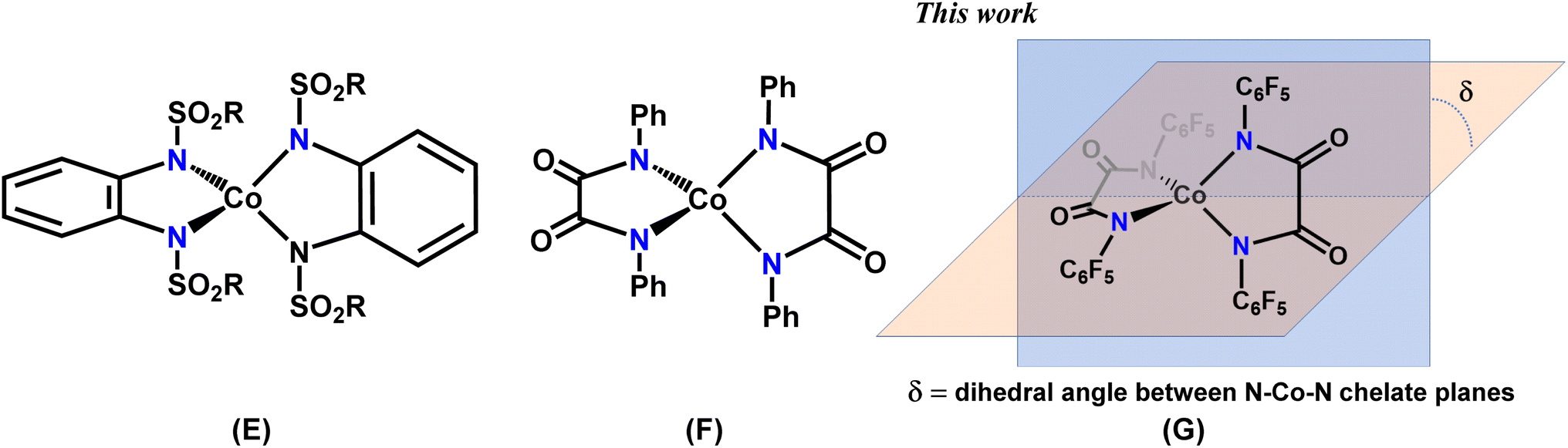
In the present work, we report a family of tetracoordinate dianionic cobalt(II) complexes of the general formula [FL2Co]X2 where FL is a bidentate N,N′-bis(perfluorophenyl)oxalamido ligand, and X are different cations. While metal amido bonds tend to be susceptible to hydrolysis, the strongly electron-withdrawing C6F5 substituents on the coordinating amido nitrogen atoms impart ambient air and moisture stability. Variation of the non-coordinating cations leads to very different dihedral twist angles (δ) of the N–Co–N coordination planes (G in Chart 2) in the solid state, which is shown to translate into significant alterations of the magnetic properties. We present a combined experimental and theoretical study on this set of complexes in order to unravel the magneto-structural correlations and to decipher and benchmark the factors that lead to the dihedral angle dependence of the magnetic properties.
Results and discussion
Synthesis and structural characterization
The diamido ligand, N,N′-bis(perfluorophenyl)oxalamide (FLH2) was synthesized using a reported literature procedure.21 The –C6F5 substituent was chosen to decrease the net charge density on the coordinating amido nitrogen atom, translating into a decreased ligand field strength. Additionally, this improves the robustness of the metal-amido bonds which are usually known to be susceptible to hydrolysis (vide infra). The mononuclear cobalt complexes were readily synthesized by the reaction of the dipotassium salt of the ligand in dry THF and anhydrous CoCl2 in a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under an inert atmosphere (Scheme 1). Cation exchange with tetrabutylammonium (TBA) bromide and tetraphenylphosphonium bromide introduced different non-coordinating counterions and allowed for the isolation of [FL2Co](TBA)2 (1) and [FL2Co](PPh4)2 (2), respectively. To further understand the effect of the charge-balancing cations on the magnetic properties, 18-crown-6 ether was directly added to the reaction mixture to isolate [FL2Co(K@18-crown-6)2]·2Et2O (3), where the K+ ions are hosted in the crown ether but additionally coordinated by the carbonyl oxygen atoms of the oxalamide ligands.
:1 under an inert atmosphere (Scheme 1). Cation exchange with tetrabutylammonium (TBA) bromide and tetraphenylphosphonium bromide introduced different non-coordinating counterions and allowed for the isolation of [FL2Co](TBA)2 (1) and [FL2Co](PPh4)2 (2), respectively. To further understand the effect of the charge-balancing cations on the magnetic properties, 18-crown-6 ether was directly added to the reaction mixture to isolate [FL2Co(K@18-crown-6)2]·2Et2O (3), where the K+ ions are hosted in the crown ether but additionally coordinated by the carbonyl oxygen atoms of the oxalamide ligands.
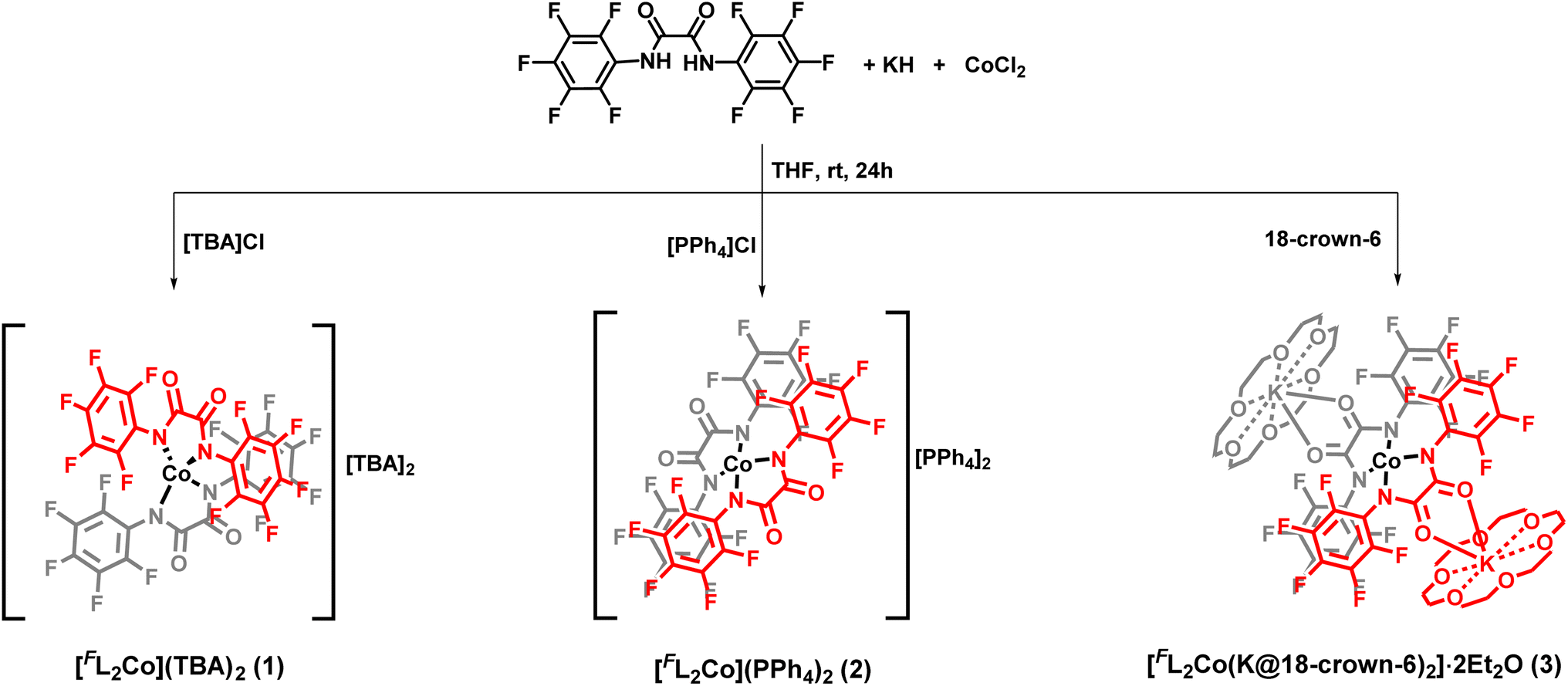 | ||
| Scheme 1 Syntheses of the tetracoordinate Co(II) complexes 1–3. | ||
The iso-structural zinc(II) complexes [FL2Zn](TBA)2 (4), [FL2Zn](PPh4)2 (5), and [FL2Zn(K@18-crown-6)2]·2Et2O (6) were prepared using a similar synthetic procedure. All the complexes were found to be stable under ambient aerobic conditions, both in the solid state and solution phase. All six complexes were characterized by several analytical and spectroscopic techniques as well as by elemental analysis (see below and ESI for details†). Cyclic voltammetry (CV) was measured for 1–3 in MeCN solution. The complexes exhibit an irreversible first oxidation at anodic peak potentials Epa = +0.09 V (1, 2) or +0.05 V (3; all at 100 mV s−1; vs. the Fc+/Fc couple; Fig. S23–S25†). These values are significantly higher by 0.31 or 0.27 V, respectively, compared to the irreversible first oxidation at Epa = −0.22 V (Fig. S26†) recorded under the same conditions for a related complex [ClL2Co](TBA)2 (H)11q that has less electron withdrawing 4-chlorophenyl substituents in the ligand [ClL]2− instead of the pentafluorophenyl substituent in [FL]2−. These findings are in line with the observed robustness of 1–3 under ambient conditions.
Solutions of these compounds appear orange in different solvents and solid compounds 2 and 3 are also orange, but complex 1 was isolated as a magenta solid. Attempts to grow crystals of complex 1 from different solvents, either by slow diffusion of diethyl ether into the orange solutions or by cooling, always resulted in the isolation of magenta crystals. Block-shaped orange single crystals of complex 2 as well as block-shaped orange-yellow crystals of 3 were obtained by slow diffusion of diethyl ether into DMF solutions. X-ray diffraction analyses revealed the molecular structures of the anions [FL2Co]2− as shown in Fig. 1 ([FL2Co(K@18C6)2] in case of 3).
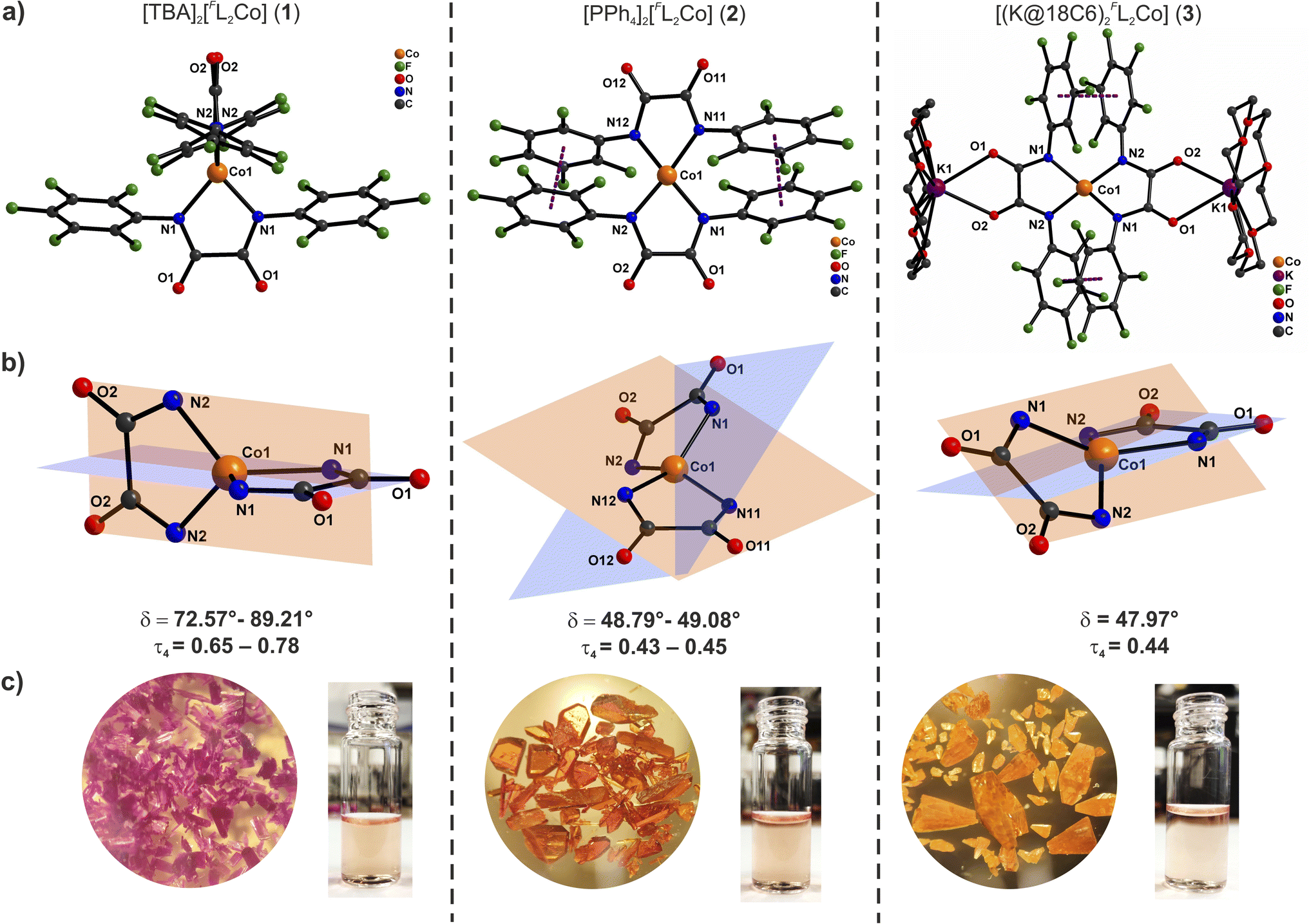 | ||
| Fig. 1 (a) Molecular structures of the anions of 1 (left) and 2 (middle), and 3 (right); cations have been omitted for clarity in case of 1 and 2. (b) Core structures of 1–3 showing the two intersecting N–Co–N chelate planes. (c) Complexes 1–3 in the solid state and DMF solution. | ||
Complex 1 crystallizes in the monoclinic space group P2/c with four crystallographically independent molecules in the asymmetric part of the unit cell (1a–1d). The Co(II) ions are coordinated by two orthogonally oriented dianionic ligands [FL]2− (Fig. 1, S27 and S28†) with average Co–N bond lengths of 1.992 Å, the charge being balanced by TBA cations in the crystal lattice. The N–Co–N bite angles from the bidentate oxanilido ligands (av. 81.2°) are considerably smaller than the other N–Co–N angles (av. 124.8°) leading to an approximate D2d symmetry or elongated tetrahedron (τ4 = 0.65–0.78).22 The relatively small ligand bite angle is quite close to the value of 76–78° recently proposed as an ideal bite angle for inducing large axial zero-field splitting in {N4}-ligated Co(II) complexes.11g The dihedral angle δ between the N–Co–N chelate planes ranges from 72.6 to 89.2° for the four molecules in the unit cell (average 83.08°). A continuous shape measure (CSM) analysis of the local coordination environment of the Co(II) ions reveals significant deviation from an ideal tetrahedral geometry (see Table S3†). The nearest Co(II) ions are separated by a minimum distance of 11.43 Å in the lattice due to the presence of the bulky charge-balancing TBA cations.
Complex 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) where the asymmetric part of the unit cell contains one and a half molecules of [FL2Co]2− and three tetraphenylphosphonium cations. The Co(II) ions are found in a distorted seesaw coordination environment (Fig. 1, S29 and S30†) with an average Co–N bond length of 1.985 Å. The entire ligand system is disordered over two positions, but overall the anions are structurally similar and roughly superimposable (Fig. S30d†); no disorder was observed for any of the tetraphenylphosphonium cations. The average N–Co–N bite angles from the bidentate oxanilido ligands (82.4°) are comparable to those of 1, but the other four N–Co–N angles show a wider range (104.4–154.6°), reflecting the distorted seesaw coordination sphere of the Co(II) ions (approximate D2 symmetry with τ4 = 0.43–0.45, Table S3†). The average dihedral twist angle δ between the N–Co–N chelate planes is 48.56°. The electron-deficient aryl rings appear to be involved in π–π interactions with distances between adjacent centroids in the range 3.41–3.73 Å. Nearest Co(II) ions within the lattice are separated by more than 10 Å.
where the asymmetric part of the unit cell contains one and a half molecules of [FL2Co]2− and three tetraphenylphosphonium cations. The Co(II) ions are found in a distorted seesaw coordination environment (Fig. 1, S29 and S30†) with an average Co–N bond length of 1.985 Å. The entire ligand system is disordered over two positions, but overall the anions are structurally similar and roughly superimposable (Fig. S30d†); no disorder was observed for any of the tetraphenylphosphonium cations. The average N–Co–N bite angles from the bidentate oxanilido ligands (82.4°) are comparable to those of 1, but the other four N–Co–N angles show a wider range (104.4–154.6°), reflecting the distorted seesaw coordination sphere of the Co(II) ions (approximate D2 symmetry with τ4 = 0.43–0.45, Table S3†). The average dihedral twist angle δ between the N–Co–N chelate planes is 48.56°. The electron-deficient aryl rings appear to be involved in π–π interactions with distances between adjacent centroids in the range 3.41–3.73 Å. Nearest Co(II) ions within the lattice are separated by more than 10 Å.
Complex 3 crystallizes in the monoclinic space group C2/c, and the structural features of its [FL2Co]2− core are very similar to those of 2. The asymmetric part of the unit cell contains half of a molecule with the Co(II) ion in a distorted seesaw coordination environment (Fig. 1, S31 and S32†). The K+ ions are hosted in the 18-crown-6 macrocycle and are additionally coordinated by the amide oxygen atoms of the [FL]2− ligands. In this case, and in contrast to 2, the ligands in 3 are not disordered, likely due to the steric constraints imposed by the 18-crown-6 moiety on the free rotation of the –C6F5 groups. The average Co–N bond length is 1.982 Å and the average N–Co–N bite angle is 82.8°. The average dihedral angle δ between the N–Co–N chelate planes is 48.0°, similar to the situation in 2 (approximate D2 symmetry with τ4 = 0.44, Table S3†). As observed for complex 2, the aryl rings are involved in π–π interactions with a distance of 3.531 Å between the nearest ring centroids. The nearest Co(II) neighbors are separated by a distance of more than 12 Å in the lattice.
Experimental absorption spectra
Solutions of all three complexes 1–3 in DMF or MeCN appear similar in color (Fig. S19†), and their UV-Vis-NIR spectra are almost identical (Fig. S19 and S20†); they exhibit three distinct absorption bands around 17600 cm−1, 9700 cm−1, and 8500 cm−1 with similar molar extinction coefficients (Fig. S19 and S20†). This observation indicates that although complexes 1–3 adopt different structures in solid-state, in solution the anions [FL2Co]2− are essentially identical and independent of the type of cation (NBu4+ in 1, PPh4+ in 2, [K@18-crown-6]+ in 3). In the range 7100–12500 cm−1 the solution spectra of 1–3 are similar to the solid-state spectra of 2 and 3 (vide infra) with a characteristic double-humped band peaking around 9600 cm−1 and 8500 cm−1. However, the relatively sharp and intense band at 17600 cm−1 with its high-energy shoulder (Fig. S19†) is more reminiscent of the absorption feature observed for solid 1 (vide infra). This suggests that in solution the complexes might be present in a conformation with a dihedral twist angle δ in between those found for 1vs.2 (or 3) in solid-state.
Static magnetic properties of solid compounds
Magnetic studies for complexes 1–3 were performed on polycrystalline powdered samples packed in a polycarbonate capsule. The powdered samples were covered with low-viscosity perfluoropolyether-based inert oil Fomblin Y45 to prevent any torquing. The χMT value of 3.34 cm3 K mol−1 for complex 1 at 210 K is larger than the expected spin-only value of one isolated Co(II) ion (1.875 cm3 K mol−1 for S = 3/2 and g = 2.0), suggesting a significant orbital contribution to the observed magnetic moment. The χMT value decreases gradually upon lowering the temperature and falls sharply below 8 K, reaching a value of 2.50 cm3 K mol−1 at 2 K (Fig. 2a). Since the nearest Co(II) centres are separated by a distance of 11.43 Å in the crystal lattice and significant intermolecular interactions are thus unlikely, the sharp fall of χMT at lower temperatures can be associated with the presence of ZFS and large magnetic anisotropy. The magnetization for 1 increases linearly until 1 T at 2 K and more gradually above 2 T. It reaches a value of 2.4 μB at 7 T, however, without any complete saturation (Fig. S37†) and well below the expected Msat value of 3.0 μB for an isolated isotropic Co(II) ion (S = 3/2). Simultaneous fitting of the magnetic susceptibility data along with the variable-temperature variable-field (VTVH) magnetization data with the program julX_2S23 (see details in ESI†) yields the best fit with D = −143 cm−1 and gx = gy = 2.25 and gz = 3.15 (Fig. 2b). This translates into a large energy separation of ca. 286 cm−1 (411 K, 2D) between the well-isolated ground MS = ±3/2 Kramers doublet (KD) and the excited state MS = ±1/2 KD. The χMT values for 2 and 3 are 3.09 and 3.00 cm3 K mol−1 at 210 K, and 2.10 and 1.99 cm3 K mol−1 at 2 K, respectively (Fig. S38 and S41†). The corresponding simultaneous fit of the magnetic susceptibility and VTVH magnetization data yields a high negative ZFS parameter D along with a significant rhombic component E. The best fit parameters obtained are D = −75 cm−1, E/D = 0.067 and gx = gy = 2.37 and gz = 2.85 for 2 (Fig. S38 and S39†) and D = −69 cm−1, E/D = 0.072 and gx = gy = 2.36 and gz = 2.79 for 3 (Fig. S41 and S42†), respectively. Table 1 summarizes the structural features of 1–3 and the various magnetic parameters determined experimentally by SQUID magnetometry.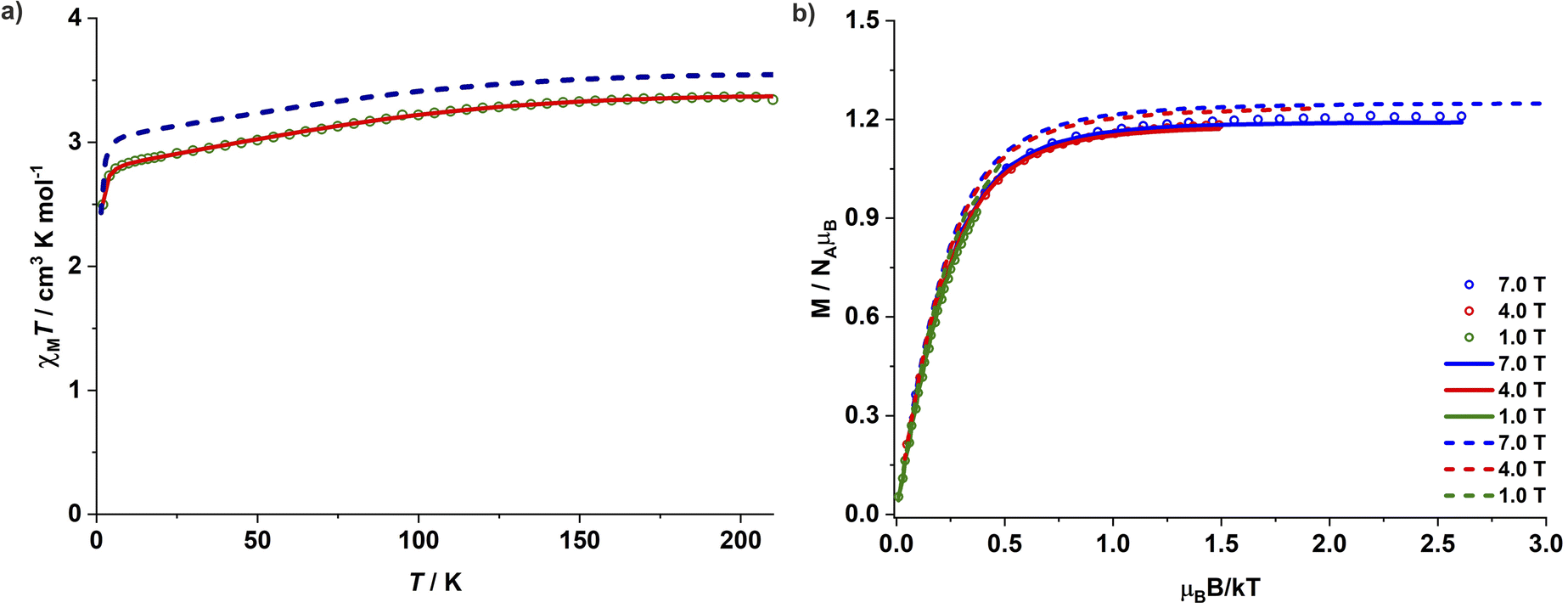 | ||
| Fig. 2 (a) Variable-temperature χMT product for complex 1 measured under an applied DC field of 0.5 T. (b) Variable-temperature variable-field magnetization for complex 1. The solid lines are the best fit with D = −143 cm−1, gx = gy = 2.25, and gz = 3.15 and the dashed lines are the theoretical predictions with CASSCF/NEVPT2. | ||
| Complex | δ (°) | Experiment | Theory (NEVPT2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g x | g y | g z | D (cm−1) | E/D | g x | g y | g z | D (cm−1) | E/D | E(4B2) (cm−1) | ||
| 1a | 88.2 | 2.25 | 2.25 | 3.15 | −143 | 0 | 1.50 | 1.51 | 3.53 | −149.7 | 0.001 | 94 |
| 1b | 89.2 | 1.52 | 1.53 | 3.52 | −149.2 | 0.001 | 100 | |||||
| 1c | 82.3 | 1.58 | 1.61 | 3.51 | −147.4 | 0.002 | 134 | |||||
| 1d | 72.6 | 1.65 | 1.74 | 3.48 | −142.7 | 0.007 | 186 | |||||
| 2a | 48.8 | 2.37 | 2.37 | 2.85 | −75 | 0.067 | 1.99 | 2.20 | 2.97 | −73.8 | 0.082 | 928 |
| 2b | 49.1 | 2.00 | 2.19 | 2.95 | −73.1 | 0.072 | 1013 | |||||
| 3 | 48.0 | 2.36 | 2.36 | 2.77 | −69 | 0.072 | 1.98 | 2.20 | 3.01 | −77.9 | 0.081 | 856 |
Dynamic magnetic properties of solid compounds
To gain insights into the magnetic relaxation dynamics, ac susceptibility measurements were carried out for complex 1 in the frequency range 0.1–1000 Hz at zero DC field. A clear frequency dependence of the maxima in the out-of-phase (χ′′M) components of the ac susceptibility is observed (Fig. 3 and S44†). The relaxation times were extracted by fitting the in-phase (χ′M) and out-of-phase (χ′′M) components of the ac susceptibility to a generalized Debye model using the CC-FIT2 program.24 The temperature dependence of the relaxation time was modelled by considering various potential relaxation pathways using eqn (1)| 1/τ = 1/τQTM + CTn + τ0−1exp(−Ueff/kBT) | (1) |
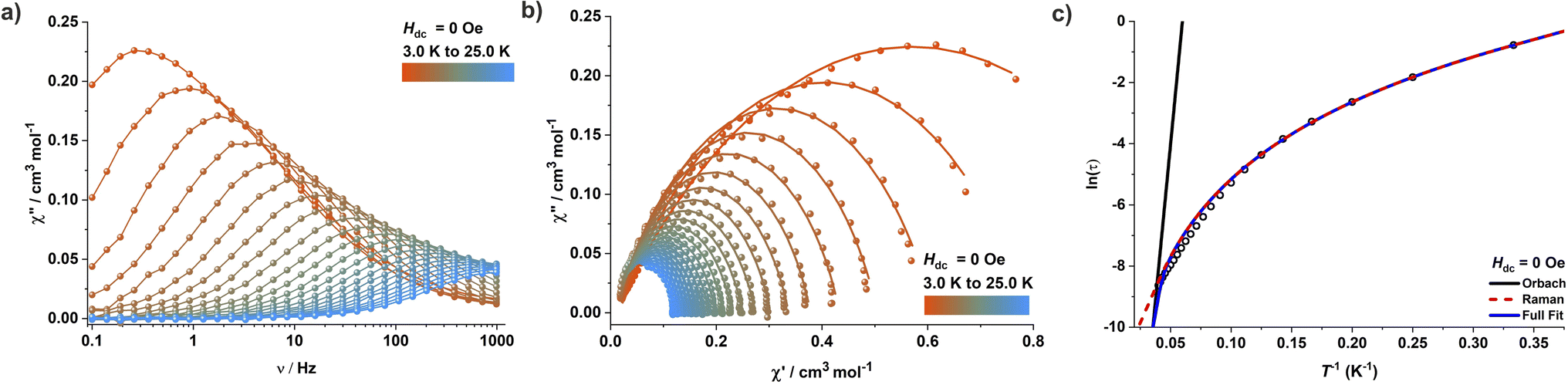 | ||
| Fig. 3 (a) Out-of-phase (χ′′M) component of the frequency-dependent (0.1–1000 Hz) AC susceptibility for 1 measured in an oscillating AC field of 3.0 Oe under zero DC field. (b) Cole–Cole plots for 1 under zero DC field. (c) Dependence of the natural logarithm of the relaxation time ln(τ) on inverse temperature (T−1); the solid blue line represents the best fit considering a combination of Orbach (Ueff = 410 K, τ0 = 2.25 × 10−11 s) and Raman (C = 0.0399 s−1 K−n, n = 3.64) relaxation pathways. | ||
The large energy separation between the ground state (MS = ±3/2) and excited state (MS = ±1/2) KD also prompted us to carry out magnetic hysteresis experiments. Variable-field magnetization data measured at a modest sweep rate of 30 Oe s−1 revealed a waist-restricted hysteresis loop up to 3.5 K with a non-negligible coercivity of ∼100 Oe at 1.8 K (Fig. 4a). The sharper magnetization decay towards zero field is most probably due to QTM between the low-lying KDs. These characteristic features are quite common among Co(II) SIMs and have also been observed in recently reported two and four-coordinate Co(II) based SIMs featuring very high energy barriers.9,11h,q The opening of the hysteresis loops becomes more apparent in the magnetically diluted material, with the coercivity increasing to ∼300 Oe at 1.8 K for a sample with ca. 2% 1 in 4 (1′′) (Fig. 4b). Magnetic blocking is also evident from the divergence of the zero-field cooled (ZFC) – field cooled (FC) variable temperature magnetization measurements. The maxima in the ZFC curve is at 3.5 K, whereas the divergence of the ZFC–FC variable temperature magnetization starts above 4.0 K (Fig. 4c). The combination of these properties with its ruggedness under ambient conditions makes this Co(II) complex 1 one of the best-performing 3d-SIMs.
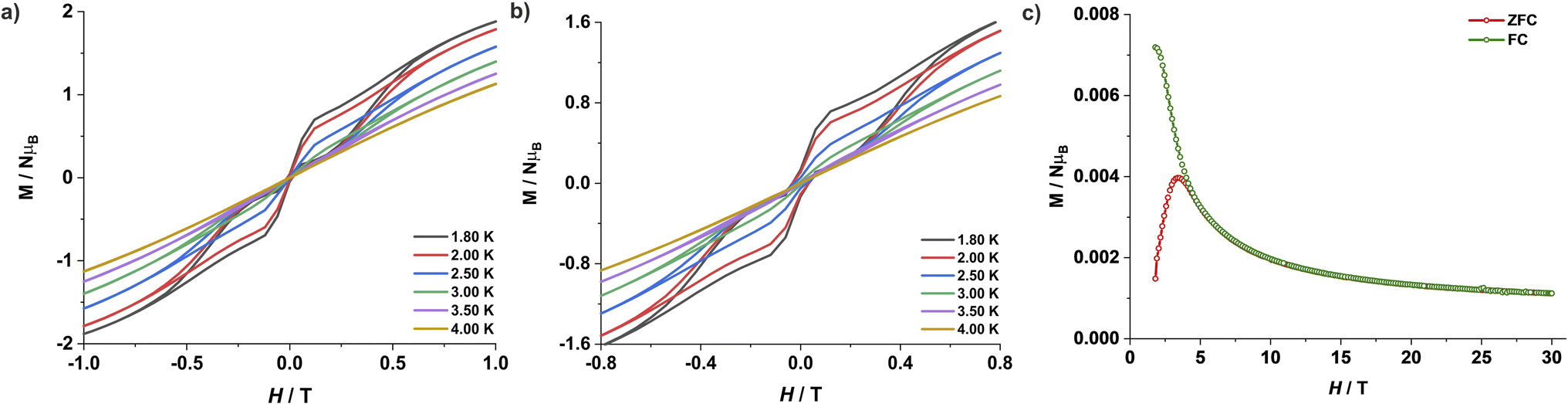 | ||
| Fig. 4 Variable field magnetization for (a) 1 and (b) 1′′ at a sweep rate of 30 Oe s−1 at indicated temperatures. (c) The plot of the zero field-cooled (red) and field-cooled (green) magnetization vs. temperature for 1′′ at an applied field of 50 Oe and a sweep rate of 2 K min−1. | ||
Interestingly, complexes 2 and 3 show dramatically different magnetic properties compared to 1, since both do not exhibit any maxima in the out-of-phase (χ′′M) AC susceptibility signals under zero applied DC field (Fig. S54 and S56†). While complex 3 features only very weak positive χ′′M signals at very low temperatures, χ′′M signals are completely absent in case of complex 2. These observations are not unexpected given the large rhombic ZFS parameter (E) derived from the DC susceptibility measurements, which is known to lead to undercutting of the over-barrier relaxation pathway via QTM. Application of an optimum DC field of 2000 Oe and 3000 Oe, respectively, led to the observation of well-resolved frequency-dependent χ′′M maxima in the AC susceptibility measurements of 2 (Fig. 5 and S55†) and 3 (Fig. S57†). Relaxation times were extracted by fitting the AC susceptibility data with a generalized Debye model; relaxation was found to be dominated by QTM and Raman pathways. The best-fit parameters obtained are C = 0.473 s−1 K−n and n = 5.02 (Raman) for 2 (Fig. 5c). Complex 3 exhibits similar relaxation dynamics (Fig. S57†), suggesting that the binding of charge-balancing cations to the ligand periphery (K@18C6 in 3) does not significantly influence the slow relaxation dynamics if the core coordination environment of the central paramagnetic ion, including the dihedral angle, is comparable.
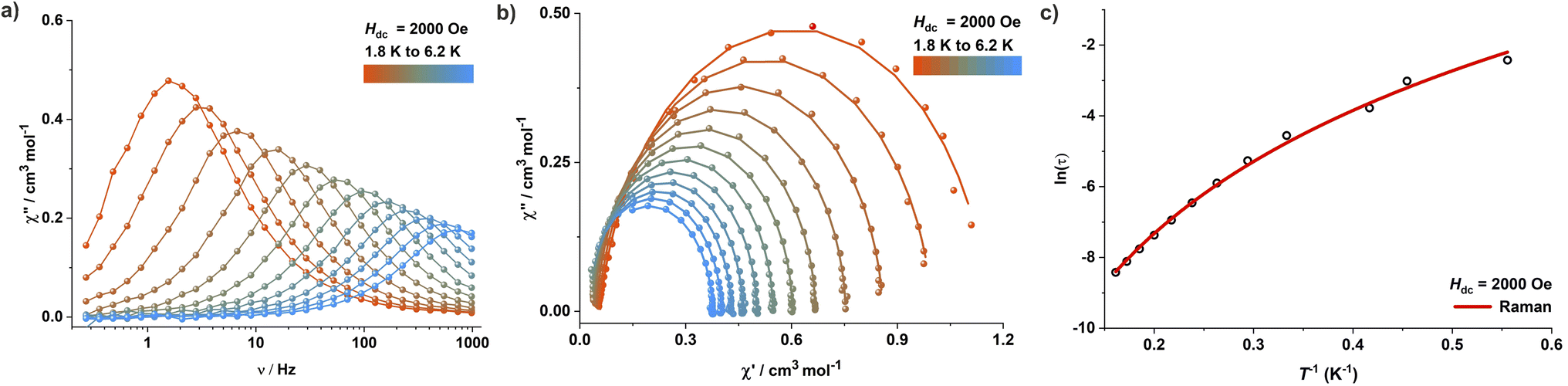 | ||
| Fig. 5 (a) Out-of-phase (χ′′M) component of the frequency-dependent (0.1–1000 Hz) AC susceptibility measured in an oscillating AC field of 3.0 Oe under an applied DC field of 2000 Oe for complex 2. (b) Cole–Cole plots for 2 under an applied DC field of 2000 Oe. (c) Dependence of the natural logarithm of the relaxation time ln(τ) on inverse temperature (T−1); the solid red line represents the best fit considering a Raman (C = 0.473 s−1 K−n, n = 5.02) relaxation pathway. | ||
In contrast to pristine undiluted material of 2 and 3, magnetically diluted samples containing ca. 10% 2 or 3 in the corresponding zinc(II) compounds 5 or 6 (called 2′ and 3′, respectively) exhibit slow relaxation dynamics even in zero DC field, although only below 6 K (Fig. S58†) or 8 K (Fig. S61†). This might be due to the suppression of dipolar exchange interactions in the diluted samples, which may result in additional quantum tunnelling pathways. These findings suggest that dipolar exchange coupling interactions can affect the slow relaxation dynamics even if the paramagnetic metal ions are more than 10 Å apart in the crystal lattice. QTM dominates the slow relaxation dynamics at lower temperatures for both 2′ and 3′, while the Raman process dominates at higher temperatures. The application of a DC field of 2000 Oe suppresses any QTM at lower temperatures in 2′ and 3′, which then show slow relaxation dynamics comparable with those observed for 2 and 3, respectively, under an applied DC field (Fig. S59 and S62†). Magnetic hysteresis experiments were performed on magnetically diluted samples containing ca. 2% 2 or 3 in the zinc(II) compound 5 or 6, respectively, viz.2′′ (Fig. S60†) and 3′′ (Fig. S63†), but magnetic hysteresis could not be observed in either case.
Magnetic properties in frozen solution
The obvious differences in color and UV-Vis-NIR spectral properties between the solid samples and dissolved complexes prompted us to measure the static and dynamic susceptibilities of the complexes in frozen solutions. A reasonable signal-to-noise ratio could be obtained for 0.12 M DMF solutions of complexes 1 and 2 (solubility in MeCN is too limited); the experiment could not be performed for complex 3 due to the low solubility of the complex. The best fit of the magnetic susceptibility data along with the variable-field variable-temperature (VTVH) magnetization yields D = −78 cm−1, gx = gy = 2.16 and gz = 2.87 for 1 (Fig. 6, S64 and S65†) and D = −72 cm−1, gx = gy = 2.22 and gz = 2.72 for 2, respectively (Fig. S69 and S70†). The frozen DMF solutions of 1 and 2 display comparable slow relaxation dynamics under zero DC field with QTM dominating the slow relaxation process at lower temperatures (Fig. 6). The relaxation times extracted could be fitted assuming a combination of QTM and Raman relaxation mechanisms (Fig. 6c, S66d, and S71d†). Application of a DC field substantially suppresses the dominating QTM at low temperatures for the frozen DMF solutions of 1 and 2, which again display comparable slow relaxation dynamics (Fig. S67 and S72†); relaxation times over the entire temperature range could be fitted with a combination of Raman and QTM processes (Fig. S67d and S72d†). Parameters for the relaxation dynamics of 1 and 2 in frozen DMF solution are also similar to the ones observed for solids 2 and 3. These combined UV-Vis-NIR spectral and magnetic properties indicate that although complexes 1 and 2/3 exist in different structural forms in the solid state, their anions [FL2Co]2− adopt identical structures in solution, likely close to the ones found for crystalline 2 and 3. It is interesting to note that while no hysteresis was observed in the magnetically diluted solid samples 2′′ and 3′′, the frozen solutions of 1 and 2 display magnetic hysteresis at 1.8 K, although without coercivity (Fig. S68 and S73†).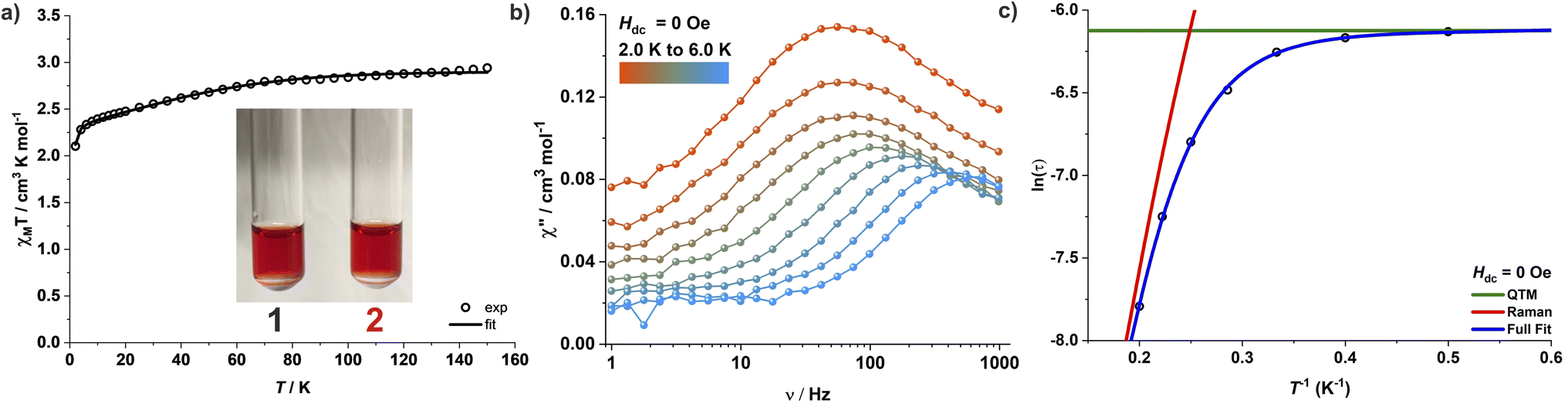 | ||
| Fig. 6 (a) Variable-temperature χMT product for a frozen DMF solution of 1 measured under an applied DC field of 0.5 T. Inset: DMF solution of complex 1 and 2 in a sealed NMR tube. (b) Out-of-phase (χ′′M) component of the frequency-dependent (0.1–1000 Hz) AC susceptibility in an oscillating AC field of 3.0 Oe for a frozen DMF solution of 1 under zero DC field. (c) Dependence of the natural logarithm of the relaxation time ln(τ) on inverse temperature (T−1); the solid blue line represents the best fit considering a combination of Raman (C = 0.047 s−1 K−n, n = 6.61), and QTM (τQTM = 2.19 × 10−3 s) relaxation pathways. | ||
Computed electronic structure
Since 1–3 show identical UV-Vis-NIR spectra in solution while packing in the crystal lattice seems to distort the complex from this ideal geometry, structures were not computationally optimized but calculations were based on coordinates obtained from the X-ray crystallographic structure determinations. In the presence of several disordered forms of the anion [FL2Co]2−, all the structures were analyzed (1a–d; 2a,b). Computations were carried out with the ORCA package,26 and the spectrum of the Hamiltonian was computed using the state averaged complete active space self-consistent field method (SA-CASSCF)27 with n-electron valence perturbation theory to second-order (NEVPT-2).28 Further details are provided below and in the ESI.†In Fig. 7, the molecular orbital energies of complex 3 are compared to the energies of an idealized tetrahedral complex. The lowest energy MOs are in order: dz2, dx2−y2, dxy, dyz, dxz. As expected, the e orbitals are lower in energy than the t2 orbitals. The electronic structure of the quartets in an ideal Td geometry is in the order 4A2(F) < 4T2(F) < 4T1(F) < 4T1(P). Upon reduction of the symmetry from Td to D2d, the 4A2 state reduces to 4B1 while 4T2 splits into a 4B2 and a 4E state, and the 4T1 splits into a 4A2 and a 4E state. Computations yield the ordering of energies in the D2d point group according to 4B1 < 4B2 < 4E < 4A2 < 4E < 4A2 < 4E. Further crystal distortions lift the degeneracy of the 4E state in the CASSCF/NEVPT2 calculations. A comparison of scalar relativistic energies obtained at the NEVPT2 level with the spin-allowed transitions deduced from optical absorption spectroscopy is made in the section below (Table 2). Analysis of the scalar relativistic wave functions (Fig. 8) shows that the ground state is 4B1 (D2d point group) dominated by the configuration dz22dx2−y22dxy1dyz1dxz1 while the excited state is 4B2 where one electron is excited from dx2−y2 to dxy resulting in a configuration given by dz22dx2−y21dxy2dyz1dxz1.
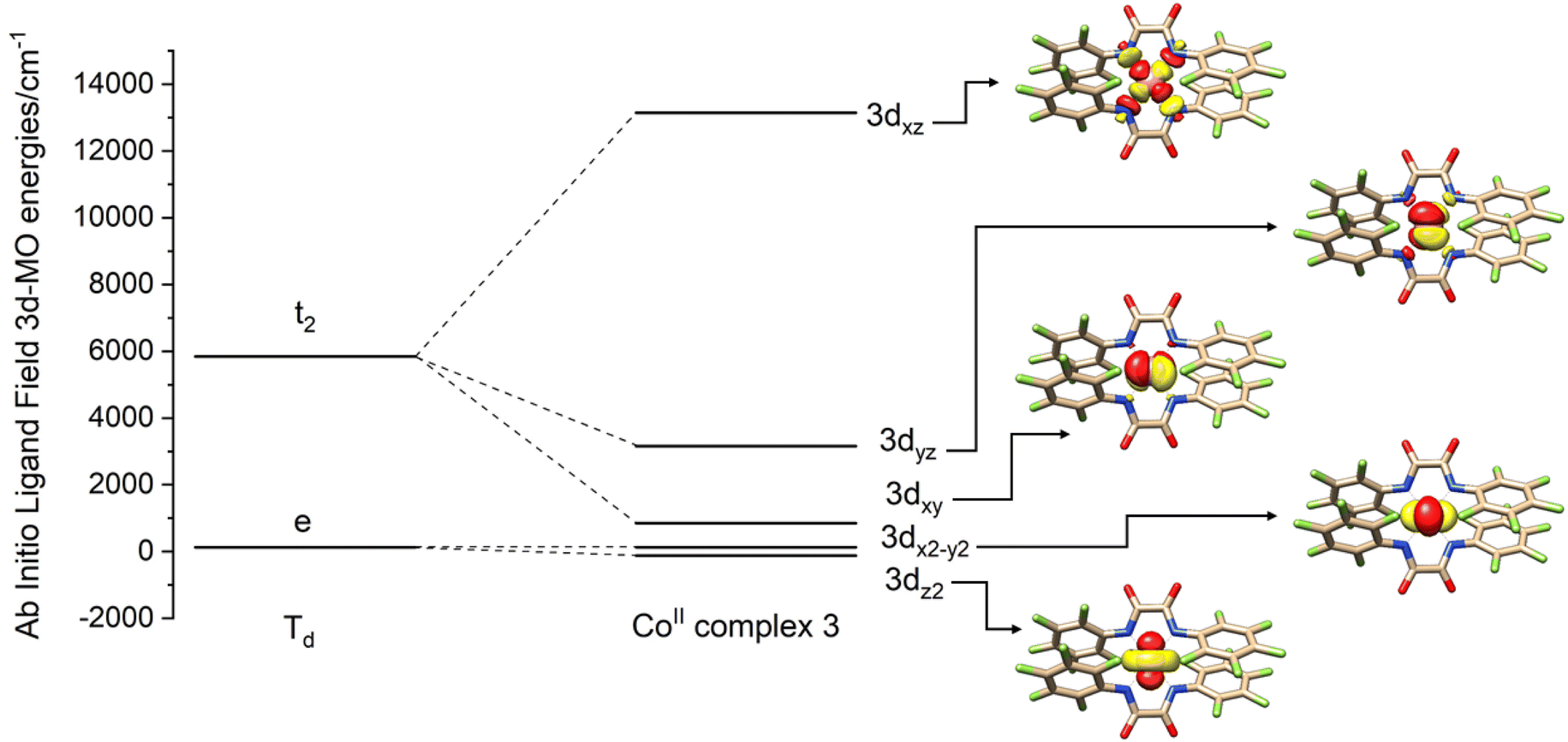 | ||
| Fig. 7 AILFT molecular orbital diagram for complex 3 at the NEVPT2 level. The geometry is determined crystallographically. Orbital images are shown on the right. While the orbital ordering is generally preserved across the complexes, the gap between 3dxy and 3dx2−y2 orbitals is reduced in complex 1. | ||
| Point group | Experimental energies (cm−1) | Point group | NEVPT2 energies (cm−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T d | 1 | 2 | 3 | D 2d | 1a | 1b | 1c | 1d | 2a | 2b | 3 |
| 4T2(F) | — | — | — | 4B2 | 76 | 80 | 115 | 172 | 928 | 1013 | 856 |
| 4E | 8733 | 8628 | 7789 | 6311 | 4191 | 4103 | 4008 | ||||
| 8882 | 8744 | 8384 | 7315 | 6432 | 6536 | 6148 | |||||
| 4T1(F) | 8470 | 8460 | 8580 | 4A2 | 9603 | 9315 | 9703 | 10142 |
10855 |
10047 |
10737 |
| 4E | 9521 | 9233 | 9974 | 10195 |
11662 |
12557 |
11379 |
||||
| 10575 |
10395 |
10483 |
11276 |
15348 |
15849 |
15190 |
|||||
| 4T1(P) | 17280 |
17720 |
17700 |
4A2 | 22727 |
22458 |
22511 |
22495 |
22809 |
21749 |
22638 |
| 18600 |
19190 |
19410 |
4E | 22494 |
22469 |
21895 |
21487 |
24214 |
24465 |
23961 |
|
| 22901 |
22660 |
23397 |
24582 |
28335 |
27937 |
28301 |
|||||
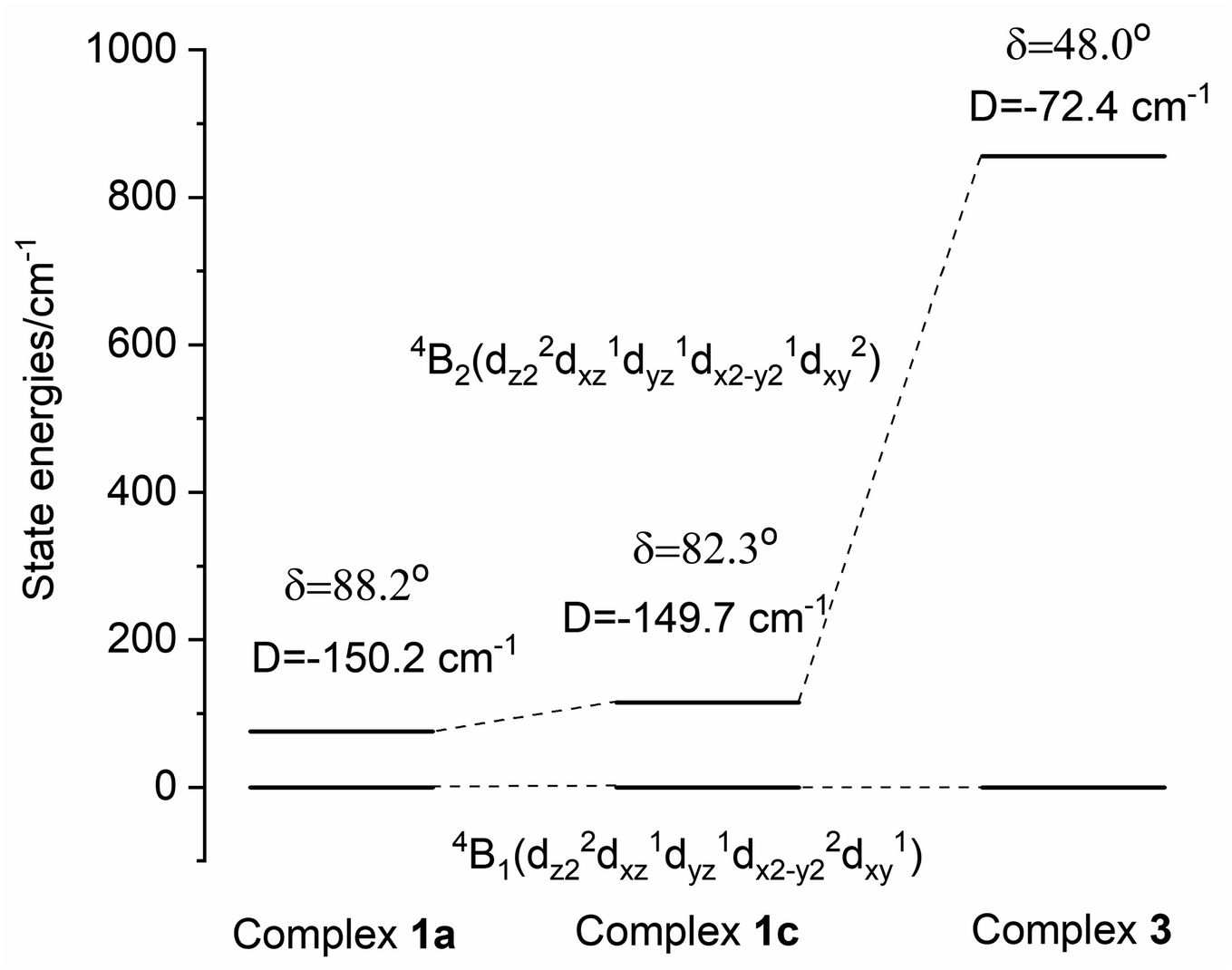 | ||
| Fig. 8 Non-relativistic energies, ZFS parameter and dihedral angles for selected structures. The 4B2 state has an energy smaller than the magnitude of spin–orbit coupling and therefore mixes strongly with the 4B1 state. | ||
Computed absorption spectra
The UV-Vis-NIR spectrum of solid 1 (Fig. 9) shows distinct features compared to the spectra of complexes 2 and 3 (Fig. 10). Complex 1 displays a sharp band at 17280 cm−1 with a shoulder peak around 18600 cm−1. The broad band extending from 12500 cm−1 to the NIR region (peak at 8470 cm−1) probably comprises several transitions. Complexes 2 and 3 exhibit similar bands in their absorption spectra with distinct bands at 19190 cm−1, 17720 cm−1, 8460 cm−1 (2) and at 19410 cm−1, 17700 cm−1, 8580 cm−1 (3), respectively.
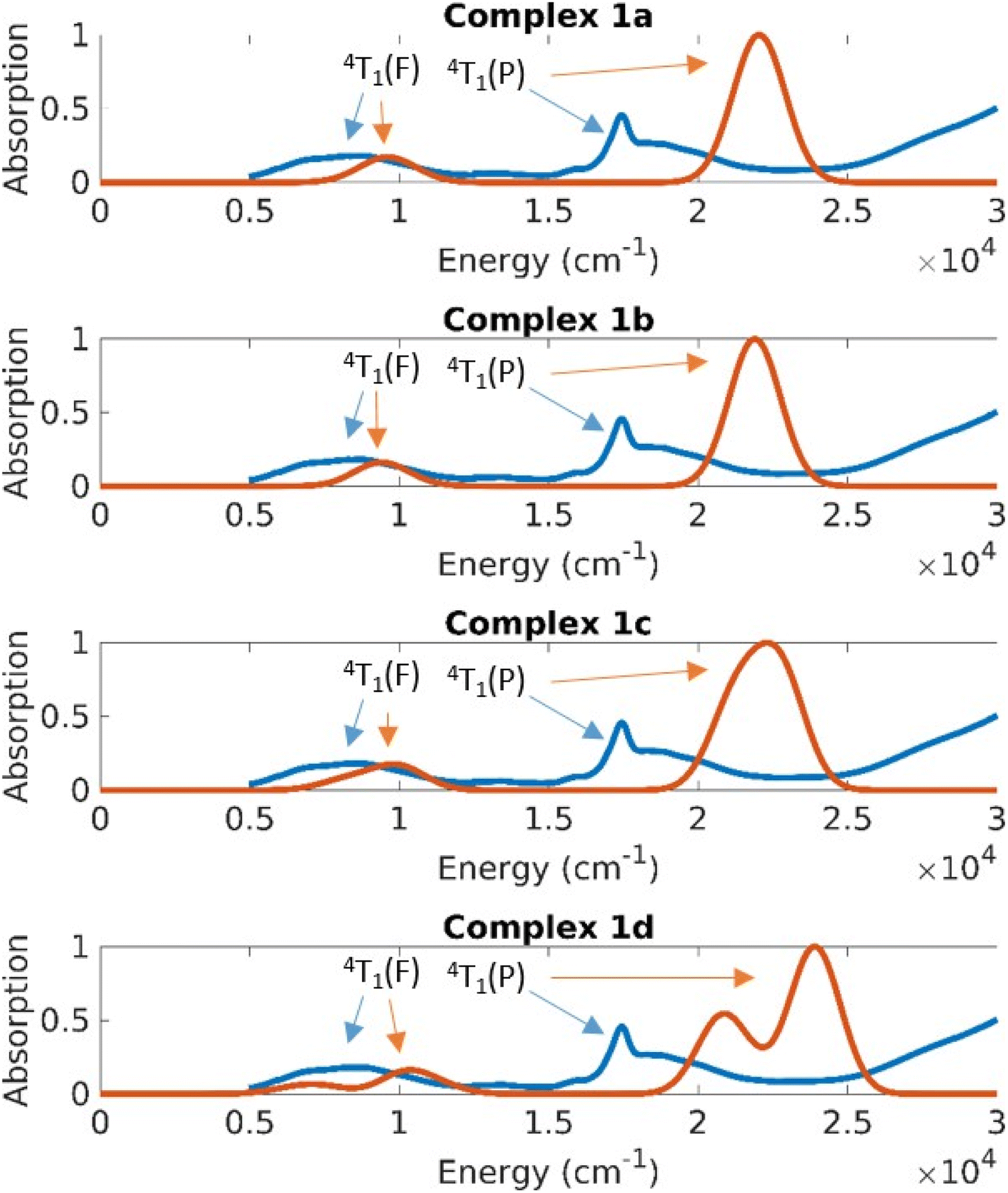 | ||
| Fig. 9 Absorption spectra obtained by experiment (blue spectrum for solid 1) and theory (red lines are calculated spectra for the different structures of 1). | ||
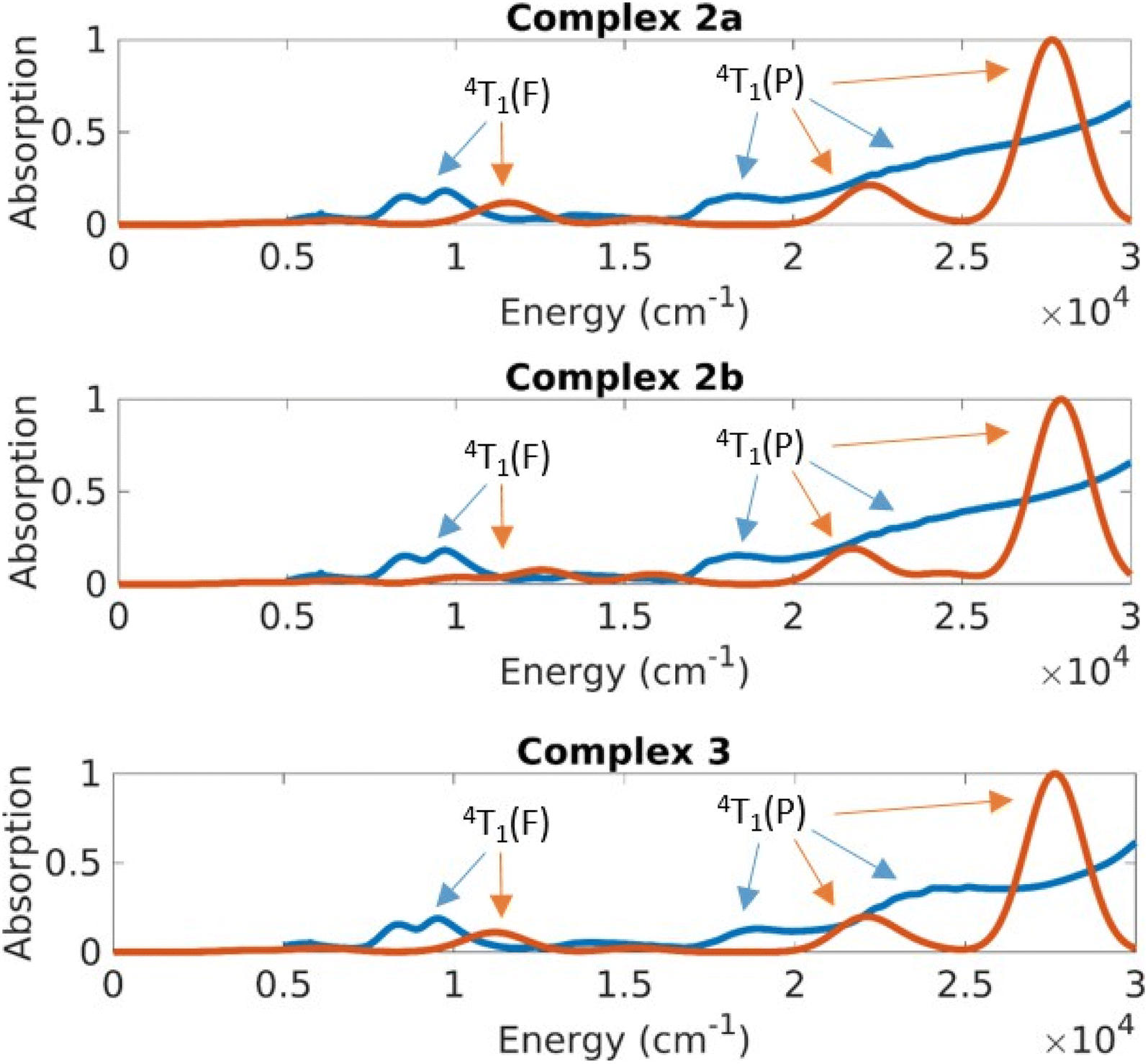 | ||
| Fig. 10 Absorption spectra obtained by theory and experiment for solid-state measurement for the structures of complexes 2 and 3. Red is the theoretical prediction for the different structures and blue is the experimental measurement. | ||
The CASSCF/NEVPT2 results predict excitations from the ground state with a character of 4A2(F) to the 4T2(F), 4T1(F) and 4T1(P) states (Td symmetry notations). The 4A2→4T2(F) transition is quite low in energy and is usually not seen in the spectrum of Co(II) complexes. Based on the CASSCF/NEVPT2 calculations it is possible to assign the bands observed in the ∼8000 cm−1 range as the excitation to the 4T1(F) states and the bands at ∼17000 cm−1 as excitations to the 4T1(P) states. The overlap due to the broad nature of these peaks prevents any further resolution of the spectral assignment. The error range between theory and experiment for the 4T1(F) states is about 2500 cm−1 and the error for the 4T1(P) states is about 5000 cm−1; the closer proximity of the 4T1(P) to the charge transfer state is suspected to cause the larger error in the latter case.
The differences in the absorption spectra of complex 1 compared to complexes 2 and 3 are also seen in their computed spectra. On plotting the energies in a linear scale of energy, one can see that the d–d excitations lie in the 5000–20000 cm−1 energy range (Fig. 9 and 10). Above this range, the absorption is dominated by charge transfer transitions which are beyond the scope of the CASSCF wave function in minimal active space.
On average the theoretical absorption spectra are blue-shifted by about 2000 cm−1 relative to the experimental spectra. This error is due to an underestimation of the dynamical correlation in the CASSCF wavefunction, which is not completely captured by the NEVPT2 correction. However, the intensities and splitting for excitations to both terms (4T1(F) and 4T1(P)) are predicted qualitatively correct. The unusual shape of the 4P band in the experimental spectrum of 1 (whose solid sample contains all four structures 1a–1d) results from a splitting of that band in structure 1d. This is understood in terms of the large deviation of the dihedral angle δ of 1d from the ideal 90° in D2d geometry.
Ligand field analysis
Ab initio ligand field analysis via the ab initio ligand field theory (AILFT)29 was employed to analyze the splitting of the d-orbitals. CASSCF energies and eigenvalues were used to reconstruct the ligand-field Hamiltonian. At the NEVPT2 level of theory, corrections to the energies of d–d transition have been taken into account. The ligand field matrix elements can be parametrized linearly in terms of ligand field parameters, the Racah parameters B and C and the 5 × 5 ligand field matrix, and hence these parameters can be obtained from a least-squares fit to their corresponding ab initio matrix counterparts. The Racah parameter A describes a mean repulsion common for all pairs of d-electrons. Therefore, the analyses of such systems focus on the parameters B and C, which account for the splitting of the free-ion states. Considering the fact that the spin–orbit coupling in these complexes is usually of the same form as the free ions, in the ligand-field analysis a single parameter ζ can be adjusted to fit spin–orbit coupling matrix elements.Ab initio ligand field analyses have been performed for all the systems reported in this study, viz. for the structures of the four crystallographically independent molecules of 1 (1a–1d) as well as for 2 and 3, in order to examine whether their different magnetic properties are associated with different Co–N bond character. As a next step in the analysis, the Racah parameters are extracted from fitting the AILFT matrix to the CASSCF/NEVPT2 eigenvectors and eigenvalues (as mentioned in the computational methods section; see ESI†). For parameters B and C the range obtained is 948.6 to 1006.8 cm−1 and 3640.7 to 3770.3 cm−1, respectively. Inspection of these parameters reveals no significant differences between the complexes. Furthermore, values for the effective nuclear charge parameter ζ are in the narrow range from 505.5 to 507.7 cm−1. This confirms that qualitatively there is no difference in the electronic structures due to these parameters.
Further insight into the interaction between the metal ion and the ligands can be obtained by using a ligand field parametrization scheme, which is additive. For this purpose, the angular overlap model for analysis with the AOMX program has been used.30 The nitrogen donor atoms are sp2 hybridized, which means that one p orbital is available for out-of-plane π-bonding and one sp2 orbital is available for σ-bonding. Thus the expression describing the ligand field Hamiltonian is given by eqn (2)31
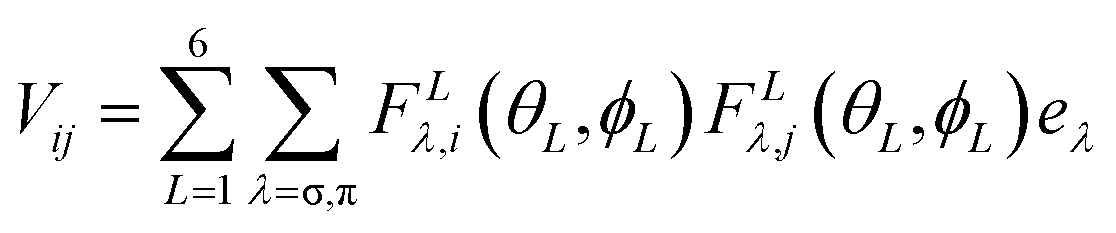 | (2) |
| B (cm−1) | C (cm−1) | e σ (cm−1)32 | e πs (cm−1)32 | ζ (cm−1) | D (cm−1) | E/D | Dihedral angle δ (°) | Energy of the 4B2 state (cm−1) | |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 949 | 3760 | 5389 | 1792 | 505 | −149.7 | 0.001 | 88.2 | 94 |
| 1b | 950 | 3771 | 5273 | 1741 | 506 | −149.2 | 0.001 | 89.2 | 100 |
| 1c | 951 | 3770 | 5260 | 1730 | 506 | −147.4 | 0.002 | 82.4 | 134 |
| 1d | 964 | 3763 | 5321 | 1612 | 507 | −142.7 | 0.007 | 72.6 | 186 |
| 2a | 1007 | 3666 | 5906 | 1756 | 507 | −73.8 | 0.082 | 48.8 | 928 |
| 2b | 1006 | 3641 | 5987 | 1721 | 506 | −73.1 | 0.072 | 49.1 | 1013 |
| 3 | 998 | 3687 | 5756 | 1546 | 508 | −77.9 | 0.081 | 48.0 | 856 |
Computational correlation of structures and magnetic properties
The magnetization and magnetic susceptibility were calculated using the exact magnetic sublevels computed by the CASSCF/NEVPT2 calculations (S2). The theoretically predicted curves closely correspond to the experimental measurements (Fig. 2 and S38–S42†). The gradual tapering upwards of the graph at higher values is due to temperature-independent paramagnetism (TIP). Table 1 (vide supra) shows that the computed gz of complex 1 in all four computed structures (corresponding to the four crystallographically independent molecules 1a–1d found in the unit cell of 1) is in the range 3.53 to 3.48 while the gx and gy are in the range of 1.50 to 1.74. This anisotropy is much higher than those of complexes 2 and 3 where gz is in the range of 2.97 to 3.01 and the gx and gy values are in the range of 1.98 to 2.20.Furthermore, starting from the D2d idealized geometry of complex 1 the dihedral angle δ was gradually swept in increments of 5°, and the resulting structures were then analyzed for the effect on D and E/D. The black solid lines in Fig. 11a and b shows the variation of D and E/D in an idealized geometry upon variation of the dihedral angle δ, while keeping the θ angle constant, in line with the rigidity of the N,N′-chelating ligand (the bite angle is 2θ = 82°); the black solid circles indicate the parameters D and E/D obtained from fitting the experimental SQUID data whereas those extracted from the CASSCF/NEVPT2 results of the different structures are indicated with open circles. It can be seen that the idealized geometry is in reasonable agreement with the calculations for the exact structures, which is, in turn, consistent with the parameters derived from the experimental measurements.
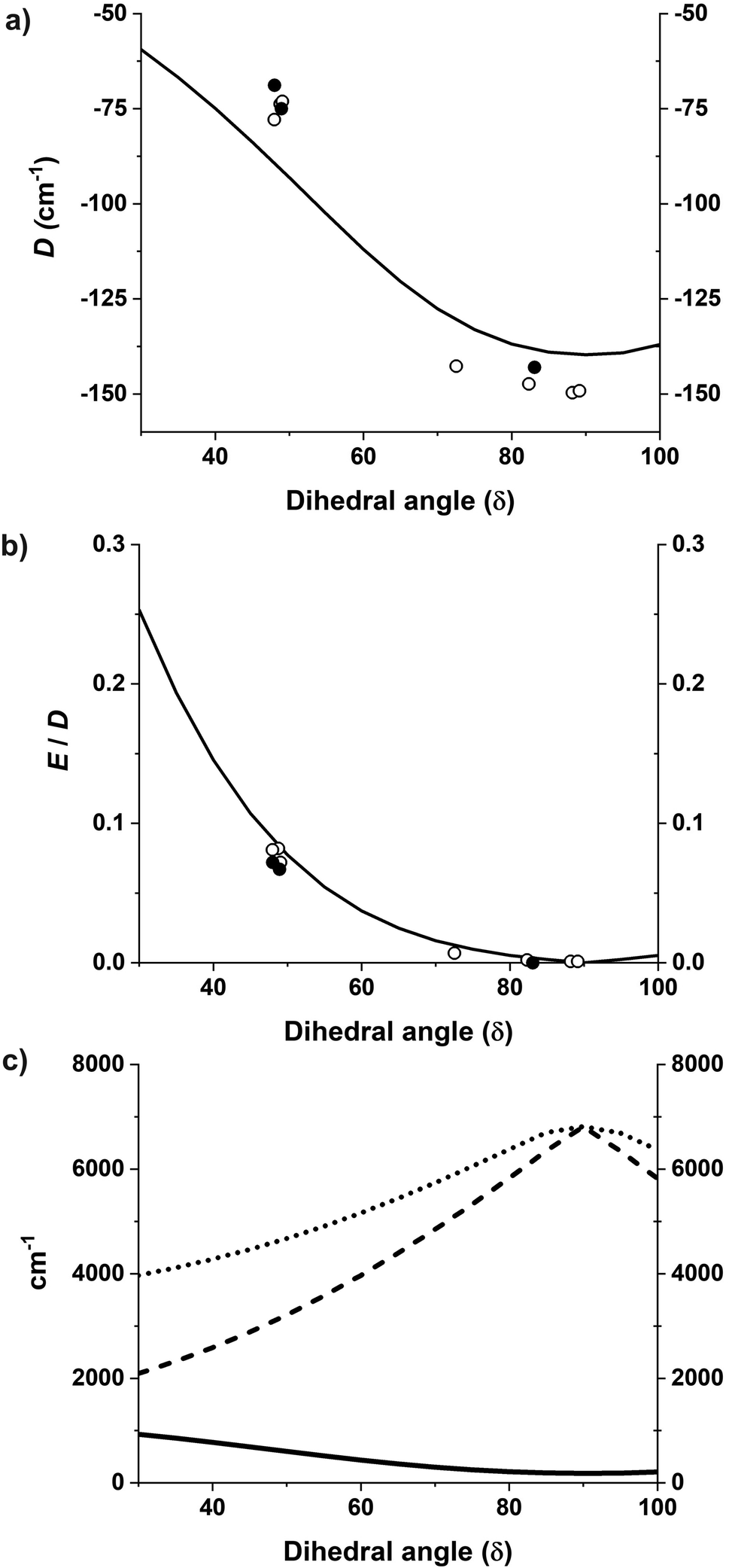 | ||
| Fig. 11 (a and b) D and E/D plotted as a function of the dihedral angle where the solid black line shows the CASSCF calculations33 with rotated geometries starting from an idealized D2d geometry. Open circles show the CASSCF/NEVPT2 results on the experimental structures and black solid circles show values obtained from fitting the experimental data. (c) Energies of the first, second, and third excited non-relativistic state energies as a function of the dihedral angle δ shown by solid black, dashed, and dotted lines respectively. | ||
In the absence of an external magnetic field and in axial symmetry, the spin–orbit coupling and ligand field induce a splitting of the S = 3/2 state into two distinct pairs of Kramers doublets: Ms = ±3/2 and Ms = ±1/2 with an energy difference that is equal to twice the zero-field splitting parameter (D). When the Ms = ±3/2 state is lower in energy the D value is said to be negative. In addition to the axial ligand field, an orthorhombic distortion can lead to the mixing of the MS = ±3/2 and MS = ±1/2 sublevels.
The magnetization and susceptibility curves also yield information regarding these D and E/D parameters using the spin Hamiltonian:
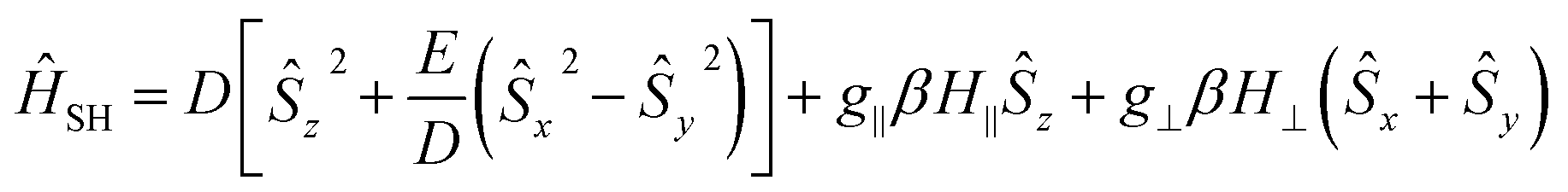 | (3) |
In these calculations, complex 1 gave D and E/D values in the range of −142.7 to −149.7 cm−1 and 0.001 to 0.008, respectively, while complexes 2 and 3 gave D and E/D values in the range of −73.1 to −74.0 cm−1 and 0.072 to 0.082, respectively, in line with the much better SMM properties of 1; the calculated parameters are compiled in Table 1. The values generated by the analysis of the CASSCF/NEVPT2 results are in line with the trends seen for these parameters derived from the experimental data.
The correlation between the dihedral angle δ and the first excitation is evident from the black curve in Fig. 11c. In all the studied complexes, the energy separation between the ground and the lowest excited state are of the same order of magnitude as the spin–orbit coupling constant (complexes 2a, 2b and 3), inducing unquenched orbital momenta responsible for the negative D for all complexes. With an energy separation much smaller in complexes 1a–1d this results in the largest negative values of D and is reflected in their SMM properties.
Using the angular overlap model, the one-electron ligand-field Hamiltonian was parametrized for a D2 point group symmetry as per equations given in ESI Section S4.† Average values of the Racah parameters from AILFT fits at the NEVPT2 level and the spin–orbit coupling obtained from CASSCF were used to calculate the many-particle, relativistic ligand field Hamiltonian matrix within the basis of the S = 3/2 ground state. Using this space of magnetic sublevels we extracted the parameters of the zero-field splitting spin-Hamiltonian (D and E) in dependence on the two structural parameters investigated here, viz. the dihedral angle (δ) and bite angle (2θ), as well as on the two angular overlap parameters eσ and eπs. Two-dimensional slices showing correlations between the zero-field splitting D and pairs of the parameters are presented in Fig. 12 and S86–S88;† they provide a basis for understanding the current system and predicting future systems of interest. The bite angle for the complexes in consideration in the current study is constant at about 82° due to the rigidity of the chelating ligand. Fig. 12 (left) shows the variation of D as a function of eπs and δ using a fixed bite angle of 82° and the average value of eσ of all the complexes studied in this work, i.e. 5556 cm−1. The zero-field splitting is rather sensitive to both parameters eπs and δ, but the sensitivity varies across the range. Close to a dihedral angle of 90° the value of D is more sensitive to the extent of the ligand π-bonding strength, whereas the sensitivity of D with respect to the out-of-plane π-bonding is greatly reduced when the dihedral angle tends toward 50°. Likewise, at higher values of eπs the sensitivity to the dihedral angle is lower.
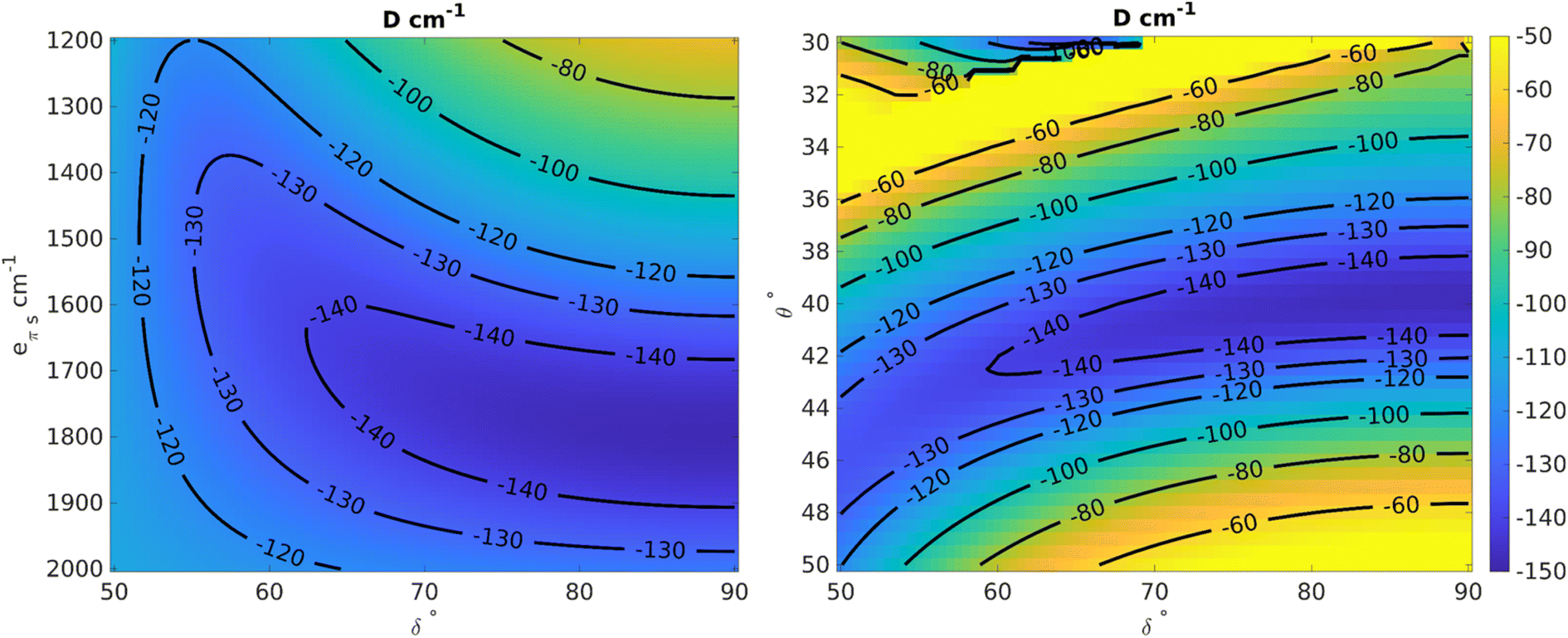 | ||
| Fig. 12 Left: Variation of D as a function of eσ and the dihedral angle δ at fixed values of bite angle 2θ = 82° and average value of eσ = 5556 cm−1. Right: Variation of D as a function of dihedral angle δ and bite angle at fixed average values of eπs = 1700 cm−1 and eσ = 5556 cm−1. | ||
In order to analyze the effect of the dihedral angle δ and the bite angle 2θ on D values, both eσ and eπs were fixed to their average values for the present set of complexes, viz. to 5556 and 1700 cm−1, respectively. Fig. 12 (right) shows that the D value is highly sensitive to the ligand bite angle, and its value of 82° enforced by the N,N′ chelate ligand in the present series of complexes is close to optimal for maximizing the zero-field splitting. With a given bite angle 2θ = 82°, increasing the dihedral angle δ increases the magnitude of the zero-field splitting systematically up to about 90°. This result from the AOM scheme is in qualitative correspondence with the ab initio NEVPT2 calculations (Fig. 12).
The zero-field splitting was also analyzed with respect to variations in eσ and eπs for a fixed bite angle 2θ of 82° and a dihedral angle δ of 90° (Fig. S86†). One can understand the effect of these parameters at a deeper level using the difference in the orbital energies of the 3dxy and 3dx2−y2 orbitals (Fig. S88 and Section S80†) which are responsible for the energy separation between the 4B1 ground state and the 4B2 state. The one-electron Hamiltonian is diagonal for this geometry and thus the orbital energy gap reduces to the equation E(3dxy) − E(3dx2−y2) = (0.55)eσ − (1.72)eπs. Thus, with increasing eσ the orbital energy separation becomes greater, leading to a larger separation between the 4B1 and 4B2 states, which results in a lower magnitude of D (as explained by perturbation theory, see Section 4.2 in ESI†). However, increasing eπs has a lowering effect on the orbital energy separation and therefore the magnitude of D becomes larger with increasing eπs. We also see from the equation that the effect of eπs is more pronounced than the effect of eσ, and in the opposite direction.
Finally, the dependence of D on eσ and the dihedral angle δ at a fixed eπs = 1700 cm−1 and a bite angle 2θ of 82° shows (Fig. S87†) that the sensitivity with respect to the sigma bonding is highest at a dihedral angle of 90° whereas closer to 50° there is hardly any dependence of the zero-field splitting on eσ. This further validates the choice of δ = 90° for the correlation shown in Fig. S86.†
To corroborate the viability of the proposed magneto-structural correlations, we examined a series of related Co(II) complexes previously reported, viz. Co(II) bis(sulfonamido)benzene complexes11h,p (E in Chart 2 and Table S9†) as well as Co(II) complexes with chelating oxamido backbone architecture and comparable bite angles (D and F in Charts 1 and 2, and Table S10†).11i,j In the former case, it was found that D increases (from −110 cm−1 to −130 cm−1) with increasing dihedral angle δ (from 83.28° to 86.62°; Table S9†). A similar trend is observed in the case of Co(II) complexes with chelating oxamido backbone (Table S10†) where D increases from −107.3 cm−1 to −128.2 cm−1 with the increase of dihedral angle from 87.49° to 89.32°. Deviations from the trend can likely be understood based on the effects of the different bite angles, cations, and electronic effects of the substituents at the ligand backbone.
Conclusions
In summary, the use of a highly electron-withdrawing group (–C6F5) as a substituent on a chelating diamido ligand [FL]2− led to the isolation of a family of rugged tetracoordinate Co(II) complexes [FL2Co]2−. Depending on the cations used (NBu4+ in 1, PPh4+ in 2, [K@18-crown-6]+ in 3), pronounced variations of the dihedral twist angle δ between the N–Co–N′ chelate planes are observed for the crystalline compounds, with δ ranging from 48.0 to 89.2°. These structural variations translate into drastically different magnetic properties of solid materials. A close to the orthogonal disposition of the two N,N′-chelating ligands at the Co(II) ion, as found for 1, proved advantageous for the SIM properties, increasing the magnetic anisotropy (D = −143 cm−1) and raising the energy barrier for relaxation to above 400 K, with magnetic blocking of 4.0 K. In contrast, a smaller dihedral twist angle δ gives rise to much smaller anisotropy and a substantial rhombic component (large E/D), which adversely affects the slow relaxation dynamics. CASSCF/NEVPT2 calculations successfully reproduced the absorption spectra as well as the magnetic properties of the [FL2Co]2− complexes with different dihedral angles, including their magnetic susceptibility, VTVH signatures, and g-factors. Computationally sweeping the dihedral twist angle between the two N–Co–N′ chelate planes allowed to analyze the effect of structural variation on the D and E/D values of the system and to derive correlations between the energy of the first excited scalar-relativistic state and the zero-field splitting. Indeed, for tetracoordinate Co(II) complexes with two N,N’-chelating σ- and π-donor ligands an orthogonal situation, viz. a dihedral twist angle of 90°, leads to a small energy gap between the ground and first excited state, translating into the largest D and a vanishing E/D. This differs from the conclusion reached for a series of bis-chelate {N2O2} Co(II) SIMs with π-acceptor Schiff-base N-donors, for which it was proposed that both the axial and rhombic ZFS parameters D and E increase with increasing distortion towards a square-planar coordination geometry, viz. with smaller dihedral twist angle δ.20 In the present study a chelate bite angle 2θ of around 80° is found to be most favorable, in line with the results of recent studies for related ligand types.11f,g,11i Furthermore, ab initio ligand field analyses with a fully analytical parametrization of the ligand field theory matrix now allowed to correlate the different magnetic properties not only with the structural but also with the bonding variations of the [FL2Co]2− molecules, viz. the two angular overlap parameters eσ and eπs.The findings of this work establish valuable design guidelines for the synthesis of Co(II) complexes with favorable SIM properties. In the present case, for magnetically diluted [FL2Co](TBA)2 (1) with favorable (average) dihedral twist angle (δ ≈ 83°) and N–Co–N bite angle (2θ ≈ 81°), magnetic blocking has been observed up to 4.0 K with an open hysteresis up to 3.5 K at a sweep rate of 30 Oe s−1, which is quite impressive for SIMs based on 3d metal ions. An additional benefit of these Co(II) complexes based on [FL]2− is their increased air and moisture stability both in solid-state and solution, which we attribute to the decreased net charge density on the coordinating nitrogen atoms imparted by the electron-withdrawing –C6F5 substituents. The magnetic studies of complexes 1–3 in frozen solution evidence that the different structures of their anions [FL2Co]2− in the crystalline materials are governed by solid-state effects, and the presence of several molecules [FL2Co]2− in the unit cell of 1 with dihedral angles varying between 73° and 90° suggests that twisting of the bis(N,N′-chelated) core is relatively facile. This raises interesting perspectives for the targeted tuning or even switching of magnetic relaxation in bis(chelated) four-coordinate Co(II) complexes via manipulating their dihedral angle δ by additives and/or second sphere interactions.
Experimental and computational methods
Materials and instrumentation
The solvents employed in the synthesis were dried according to standard procedures and distilled prior to use. The syntheses of the complexes were carried out under a dry and inert nitrogen atmosphere. Unless otherwise stated, all chemicals used were purchased from commercial sources. 1H NMR and 13C NMR spectra were recorded on Bruker Avance 300 MHz and 400 MHz spectrometers at room temperature. Chemical shifts are reported in parts per million relative to residual proton and carbon signals of the solvent. Infrared spectra were recorded on a Cary 630 FTIR spectrometer equipped with Dial Path Technology and analysed by FTIR MicroLab software. UV-Vis-NIR spectra in solution and solid-state were recorded with a Varian Cary 5000 spectrophotometer. ESI-MS were collected using a Bruker HCT ultra spectrometer. Elemental analyses were performed by the analytical laboratory of the Institute of Inorganic Chemistry at the University of Göttingen using an Elementar Vario EL III instrument. Cyclic voltammetry (CV) experiments were performed with an Interface 1000B potentiostat at room temperature. A three-electrode setup was used with a glassy carbon working electrode, a platinum wire as a counter electrode, and a silver wire in 0.1 M [TBA]PF6 solution in acetonitrile as a pseudo-reference electrode. [TBA]PF6 (0.1 M solution in MeCN) was used as the supporting electrolyte. Ferrocene was used as an internal standard, and the data were analyzed with Gamry Framework software.Synthesis protocols
Single crystal structure determinations
Crystal data and details of the data collections are given in Tables S1 & S2.† X-ray data were collected on a STOE IPDS II or a BRUKER D8-QUEST diffractometer (monochromated Mo-Kα radiation, λ = 0.71073 Å) by use of ω or ω and ϕ scans at low temperature. The structures were solved with SHELXT and refined on F2 using all reflections with SHELXL.34 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and assigned to an isotropic displacement parameter of 1.5 or 1.2 Ueq(C).Crystals of 1 could only be obtained in rather poor quality, which is especially evident by the high Rint value. Furthermore, two ethyl groups of NEt4+ cations were found to be disordered (occupancy factors: 0.59(2)/0.41(2) & 0.53(2)/0.47(2)). SADI restraints were applied for two (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )C–C(O) bonds. Likewise, in case of the isostructural 4 (a = 23.3421(13), b = 25.4937(10), c = 22.5825(11) Å, β = 106.256(4)°, P2/c) the arrangement of the atoms could be established, but the overall quality of the structure determinations prevents a detailed discussion of the bonding parameters. In case of 2 a pair of ligands is disordered about two positions in both crystallographically independent molecules (occupancy factors: 0.8554(14)/0.1446(14) & 0.539(2)/0.461(2)). Fig. S36† emphasizes the disorder. In the second crystallographically independent molecule the cobalt atom resides on a center of inversion, so that ligand pairs are symmetry related. SAME and SIMU restraints have been applied to model the disorder. Crystals of 3 were found to be twinned (twin law: 0.899 −0.002 −0.199, 0.002 −1.001 0.004, −0.969 −0.010 −0.895). The BASF was refined to: 0.303(1). Each ligand in 5 was found to be disordered about two positions (occupancy factors: 0.648(2)/0.352(2) & 0.650(2)/0.350(2)). SAME and RIGU restraints and EADP constraints were applied to model the disorder. The disorder is comparable to the disorder observed in 2. As in 3 crystals of 6 were found to be twinned (twin law: 0.906 0 −0.188, 0 −1 0, −0.953 0 −0.906). The BASF was refined to: 0.115(1). Face-indexed absorption corrections were performed numerically with the program X-RED35 or in case of 1 by the multi-scan method with SADABS.36
)C–C(O) bonds. Likewise, in case of the isostructural 4 (a = 23.3421(13), b = 25.4937(10), c = 22.5825(11) Å, β = 106.256(4)°, P2/c) the arrangement of the atoms could be established, but the overall quality of the structure determinations prevents a detailed discussion of the bonding parameters. In case of 2 a pair of ligands is disordered about two positions in both crystallographically independent molecules (occupancy factors: 0.8554(14)/0.1446(14) & 0.539(2)/0.461(2)). Fig. S36† emphasizes the disorder. In the second crystallographically independent molecule the cobalt atom resides on a center of inversion, so that ligand pairs are symmetry related. SAME and SIMU restraints have been applied to model the disorder. Crystals of 3 were found to be twinned (twin law: 0.899 −0.002 −0.199, 0.002 −1.001 0.004, −0.969 −0.010 −0.895). The BASF was refined to: 0.303(1). Each ligand in 5 was found to be disordered about two positions (occupancy factors: 0.648(2)/0.352(2) & 0.650(2)/0.350(2)). SAME and RIGU restraints and EADP constraints were applied to model the disorder. The disorder is comparable to the disorder observed in 2. As in 3 crystals of 6 were found to be twinned (twin law: 0.906 0 −0.188, 0 −1 0, −0.953 0 −0.906). The BASF was refined to: 0.115(1). Face-indexed absorption corrections were performed numerically with the program X-RED35 or in case of 1 by the multi-scan method with SADABS.36
Magnetic measurements
Magnetic measurements were carried out with a Quantum-Design MPMS3 SQUID magnetometer equipped with a 7.0 T magnet. Direct current (dc) magnetic susceptibility measurements were performed under an applied DC field with powdered polycrystalline samples in the range from 210 K to 2 K. The powdered samples were packed in a polycarbonate or gelatine capsule and covered with low-viscosity perfluoropolyether-based inert oil Fomblin Y45 in a non-magnetic sample holder. Each raw data for the measured magnetic moment was corrected for the diamagnetic contribution of the capsules including the inert oil if used according to Mdia(capsule) = χgmH, with an experimentally obtained gram susceptibility of the capsules including the inert oil. The diamagnetic contribution of the compounds was corrected using Pascal's constants. Experimental data were modelled with the julX_2S program23 using a fitting procedure to the spin Hamiltonians in eqn (3). Alternating current (ac) susceptibility measurements were carried out in an oscillating AC field of 3.0 Oe and frequencies ranging from 0.1 to 1000 Hz.Theoretical calculations
Computations were carried out with the ORCA package26 using coordinates obtained from the X-ray crystallographic structure determinations. In the presence of several disordered forms, all structures were analyzed. The spectrum of the Hamiltonian was computed using the state-averaged complete active space self-consistent field method (SA-CASSCF)27 with n-electron valence perturbation theory to second-order (NEVPT-2).28 Scalar relativistic effects were taken into account via the Douglas–Kroll–Hess (DKH) method of second order.37 Ahlrichs polarized def2-TZVPP basis sets38 optimized for DKH method39 were used for all calculations. Resolution of identity along with corresponding auxiliary basis set40 were used to speed up calculations. An active space of seven electrons on five active was 3d molecular orbitals CAS(7,5) was employed. The S = 3/2 (S = 1/2) states of the free ion (4F, 4P and 2P, 2D,2F, 2G and 2H, respectively) give rise to ten quartets (fourty doublets) of the complex. Absorption spectra of the complex are plotted with the orca_mapspc utility. The zero-field splitting tensor D of the spin Hamiltonian41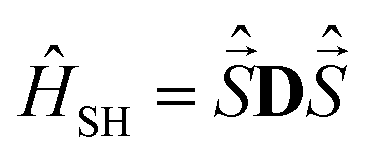 was extracted using effective Hamiltonian theory from a mapping onto the full spin–orbit coupling of many particle energy eigenvalues and wavefunctions (more details are provided in the ESI†).
was extracted using effective Hamiltonian theory from a mapping onto the full spin–orbit coupling of many particle energy eigenvalues and wavefunctions (more details are provided in the ESI†).
Data availability
CCDC 2240414 (1), 2240415 (2), 2240416 (3), 2240417 (5) and 2240418 (6) contain the supplementary crystallographic data for this paper. All other data for this paper, including data from experimental methods and computational studies, are available at the Göttingen Research Online server at https://doi.org/10.25625/BV9SCD.Author contributions
SKG carried out the synthesis, spectroscopic characterization, magnetic measurements, and analyses. SVR carried out the theoretical modeling and calculations. SD and EB helped with magnetic data collection and analysis. SD helped with the in-house single-crystal X-ray data collection and analysis. MA, FN, and FM conceived the study and supervised the experiments and analyses. The manuscript was written through the contributions of all authors and all the authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. K. G. is associated with the DFG-funded Research Training Group BENCh (RTG 2455) funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project 389479699) and thanks the Alexander von Humboldt Foundation and the International Centre, University of Göttingen, for postdoctoral fellowships. F.M. acknowledges the University of Göttingen for basic support. Purchase of the SQUID magnetometer and the X-ray diffractometer was enabled by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project numbers INST 186/1329-1 FUGG and INST 186/1327-1 FUGG, respectively) and the Niedersächsische Ministerium für Wissenschaft und Kultur (MWK). S. V. R. and F. N. acknowledge support from the MPI für Kohlenforschung and the Max Planck Society.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- (a) E. Coronado, P. Delhaès, D. Gatteschi and J. S. Miller, Molecular magnetism: from molecular assemblies to the devices, Springer, 1996 Search PubMed; (b) R. A. Layfield, Organometallics, 2014, 33, 1084–1099 CrossRef CAS; (c) A. Caneschi, D. Gatteschi, R. Sessoli, A. L. Barra, L. C. Brunel and M. Guillot, J. Am. Chem. Soc., 1991, 113, 5873–5874 CrossRef CAS.
- (a) S. V. Rao, J. M. Ashtree and A. Soncini, Phys. B, 2020, 592, 412237 CrossRef CAS; (b) D. Gatteschi, R. Sessoli and J. Villain, Molecular nanomagnets, Oxford University Press, New York, 2011 Search PubMed; (c) C. Cervetti, A. Rettori, M. G. Pini, A. Cornia, A. Repollés, F. Luis, M. Dressel, S. Rauschenbach, K. Kern, M. Burghard and L. Bogani, Nat. Mater., 2016, 15, 164–168 CrossRef CAS PubMed; (d) M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Nature, 2016, 531, 348–351 CrossRef CAS PubMed; (e) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed; (f) M. Urdampilleta, S. Klyatskaya, J. P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef CAS PubMed; (g) D. Aravena and E. Ruiz, Dalton Trans., 2020, 49, 9916–9928 RSC.
- L. Thomas, F. Lionti, R. Ballou, D. Gatteschi, R. Sessoli and B. Barbara, Nature, 1996, 383, 145–147 CrossRef CAS.
- (a) A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926–4929 CrossRef CAS PubMed; (b) A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed; (c) M. Murugesu, M. Habrych, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2004, 126, 4766–4767 CrossRef CAS PubMed.
- (a) F. Neese and E. I. Solomon, Inorg. Chem., 1998, 37, 6568–6582 CrossRef CAS PubMed; (b) J. Cirera, E. Ruiz, S. Alvarez, F. Neese and J. Kortus, Chem.–Eur. J., 2009, 15, 4078–4087 CrossRef CAS PubMed; (c) F. Neese and D. A. Pantazis, Faraday Discuss., 2011, 148, 229–238 RSC; (d) O. Waldmann, Inorg. Chem., 2007, 46, 10035–10037 CrossRef CAS PubMed.
- (a) A. Sarkar, S. Dey and G. Rajaraman, Chem.–Eur. J., 2020, 26, 14036–14058 CrossRef CAS PubMed; (b) S. Tripathi, A. Dey, M. Shanmugam, R. S. Narayanan and V. Chandrasekhar, in Organometallic Magnets, ed. V. Chandrasekhar and F. Pointillart, Springer International Publishing, 2019, pp. 35–75 Search PubMed; (c) S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 379–392 CrossRef; (d) M. Murrie, Chem. Soc. Rev., 2010, 39, 1986–1995 RSC.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed.
- (a) X.-N. Yao, J.-Z. Du, Y.-Q. Zhang, X.-B. Leng, M.-W. Yang, S.-D. Jiang, Z.-X. Wang, Z.-W. Ouyang, L. Deng, B.-W. Wang and S. Gao, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef CAS PubMed; (b) P. C. Bunting, M. Atanasov, E. Damgaard-Møller, M. Perfetti, I. Crassee, M. Orlita, J. Overgaard, J. v. Slageren, F. Neese and J. R. Long, Science, 2018, 362, eaat7319 CrossRef CAS PubMed.
- (a) Y.-F. Deng, Z. Wang, Z.-W. Ouyang, B. Yin, Z. Zheng and Y.-Z. Zheng, Chem.–Eur. J., 2016, 22, 14821–14825 CrossRef CAS PubMed; (b) Y.-F. Deng, T. Han, B. Yin and Y.-Z. Zheng, Inorg. Chem. Front., 2017, 4, 1141–1148 RSC.
- (a) E. A. Suturina, D. Maganas, E. Bill, M. Atanasov and F. Neese, Inorg. Chem., 2015, 54, 9948–9961 CrossRef CAS PubMed; (b) E. A. Suturina, J. Nehrkorn, J. M. Zadrozny, J. Liu, M. Atanasov, T. Weyhermüller, D. Maganas, S. Hill, A. Schnegg, E. Bill, J. R. Long and F. Neese, Inorg. Chem., 2017, 56, 3102–3118 CrossRef CAS PubMed; (c) D. Maganas, S. Sottini, P. Kyritsis, E. J. J. Groenen and F. Neese, Inorg. Chem., 2011, 50, 8741–8754 CrossRef CAS PubMed; (d) S. Vaidya, S. Tewary, S. K. Singh, S. K. Langley, K. S. Murray, Y. Lan, W. Wernsdorfer, G. Rajaraman and M. Shanmugam, Inorg. Chem., 2016, 55, 9564–9578 CrossRef CAS PubMed; (e) S. Tripathi, S. Vaidya, K. U. Ansari, N. Ahmed, E. Rivière, L. Spillecke, C. Koo, R. Klingeler, T. Mallah, G. Rajaraman and M. Shanmugam, Inorg. Chem., 2019, 58, 9085–9100 CrossRef CAS PubMed; (f) E. Carl, S. Demeshko, F. Meyer and D. Stalke, Chem.–Eur. J., 2015, 21, 10109–10115 CrossRef CAS PubMed; (g) C. M. Legendre, E. Damgaard-Møller, J. Overgaard and D. Stalke, Eur. J. Inorg. Chem., 2021, 2021, 3108–3114 CrossRef CAS; (h) Y. Rechkemmer, F. D. Breitgoff, M. van der Meer, M. Atanasov, M. Hakl, M. Orlita, P. Neugebauer, F. Neese, B. Sarkar and J. van Slageren, Nat. Commun., 2016, 7, 10467 CrossRef CAS PubMed; (i) M. Wang, H. J. Xu, T. M. Sun, H. H. Cui, Y.-Q. Zhang, L. Chen and Y. F. Tang, J. Solid State Chem., 2021, 299, 122209 CrossRef CAS; (j) H.-H. Cui, F. Lu, X.-T. Chen, Y.-Q. Zhang, W. Tong and Z.-L. Xue, Inorg. Chem., 2019, 58, 12555–12564 CrossRef CAS PubMed; (k) M. R. Saber and K. R. Dunbar, Chem. Commun., 2014, 50, 12266–12269 RSC; (l) X.-N. Yao, M.-W. Yang, J. Xiong, J.-J. Liu, C. Gao, Y.-S. Meng, S.-D. Jiang, B.-W. Wang and S. Gao, Inorg. Chem. Front., 2017, 4, 701–705 RSC; (m) J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209–217 CrossRef CAS; (n) J. Vallejo, E. Pardo, M. Viciano-Chumillas, I. Castro, P. Amorós, M. Déniz, C. Ruiz-Pérez, C. Yuste-Vivas, J. Krzystek, M. Julve, F. Lloret and J. Cano, Chem. Sci., 2017, 8, 3694–3702 RSC; (o) R. Boča, J. Miklovič and J. Titiš, Inorg. Chem., 2014, 53, 2367–2369 CrossRef PubMed; (p) H. Bamberger, U. Albold, J. Dubnická Midlíková, C.-Y. Su, N. Deibel, D. Hunger, P. P. Hallmen, P. Neugebauer, J. Beerhues, S. Demeshko, F. Meyer, B. Sarkar and J. van Slageren, Inorg. Chem., 2021, 60, 2953–2963 CrossRef CAS PubMed; (q) S. K. Gupta, H. H. Nielsen, A. M. Thiel, E. A. Klahn, E. Feng, H. B. Cao, T. C. Hansen, E. Lelièvre-Berna, A. Gukasov, I. Kibalin, S. Dechert, S. Demeshko, J. Overgaard and F. Meyer, JACS Au, 2023, 3, 429–440 CrossRef CAS PubMed; (r) T. Wu, Y.-Q. Zhai, Y.-F. Deng, W.-P. Chen, T. Zhang and Y.-Z. Zheng, Dalton Trans., 2019, 48, 15419–15426 RSC; (s) M. S. Fataftah, J. M. Zadrozny, D. M. Rogers and D. E. Freedman, Inorg. Chem., 2014, 53, 10716–10721 CrossRef CAS PubMed; (t) D. Tu, D. Shao, H. Yan and C. Lu, Chem. Commun., 2016, 52, 14326–14329 RSC; (u) P. Kumar, D. J. SantaLucia, K. Kaniewska-Laskowska, S. V. Lindeman, A. Ozarowski, J. Krzystek, M. Ozerov, J. Telser, J. F. Berry and A. T. Fiedler, Inorg. Chem., 2020, 59, 16178–16193 CrossRef CAS PubMed; (v) M. S. Fataftah, S. C. Coste, B. Vlaisavljevich, J. M. Zadrozny and D. E. Freedman, Chem. Sci., 2016, 7, 6160–6166 RSC.
- C. Rajnák, J. Titiš, O. Fuhr, M. Ruben and R. Boča, Inorg. Chem., 2014, 53, 8200–8202 CrossRef PubMed.
- (a) V. V. Novikov, A. A. Pavlov, Y. V. Nelyubina, M.-E. Boulon, O. A. Varzatskii, Y. Z. Voloshin and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137, 9792–9795 CrossRef CAS PubMed; (b) B. Yao, Y.-F. Deng, T. Li, J. Xiong, B.-W. Wang, Z. Zheng and Y.-Z. Zhang, Inorg. Chem., 2018, 57, 14047–14051 CrossRef CAS PubMed; (c) J. Vallejo, I. Castro, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, G. De Munno, W. Wernsdorfer and E. Pardo, J. Am. Chem. Soc., 2012, 134, 15704–15707 CrossRef CAS PubMed; (d) M. R. Saber, M. K. Singh and K. R. Dunbar, Chem. Commun., 2020, 56, 8492–8495 RSC; (e) A. A. Pavlov, D. Y. Aleshin, S. A. Savkina, A. S. Belov, N. N. Efimov, J. Nehrkorn, M. Ozerov, Y. Z. Voloshin, Y. V. Nelyubina and V. V. Novikov, ChemPhysChem, 2019, 20, 1001–1005 CrossRef CAS PubMed; (f) Y.-Y. Zhu, C. Cui, Y.-Q. Zhang, J.-H. Jia, X. Guo, C. Gao, K. Qian, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. Sci., 2013, 4, 1802–1806 RSC; (g) Y.-Z. Zhang, S. Gómez-Coca, A. J. Brown, M. R. Saber, X. Zhang and K. R. Dunbar, Chem. Sci., 2016, 7, 6519–6527 RSC; (h) P. K. Sahu, A. Mondal and S. Konar, Dalton Trans., 2021, 50, 3825–3831 RSC; (i) A. Landart-Gereka, M. M. Quesada-Moreno, I. F. Díaz-Ortega, H. Nojiri, M. Ozerov, J. Krzystek, M. A. Palacios and E. Colacio, Inorg. Chem. Front., 2022, 9, 2810–2831 RSC.
- (a) L. Chen, J. Wang, J.-M. Wei, W. Wernsdorfer, X.-T. Chen, Y.-Q. Zhang, Y. Song and Z.-L. Xue, J. Am. Chem. Soc., 2014, 136, 12213–12216 CrossRef CAS PubMed; (b) L. Rigamonti, N. Bridonneau, G. Poneti, L. Tesi, L. Sorace, D. Pinkowicz, J. Jover, E. Ruiz, R. Sessoli and A. Cornia, Chem.–Eur. J., 2018, 24, 8857–8868 CrossRef CAS PubMed.
- I. G. Rau, S. Baumann, S. Rusponi, F. Donati, S. Stepanow, L. Gragnaniello, J. Dreiser, C. Piamonteze, F. Nolting, S. Gangopadhyay, O. R. Albertini, R. M. Macfarlane, C. P. Lutz, B. A. Jones, P. Gambardella, A. J. Heinrich and H. Brune, Science, 2014, 344, 988–992 CrossRef CAS PubMed.
- (a) S. Tripathi, S. Vaidya, N. Ahmed, E. Andreasen Klahn, H. Cao, L. Spillecke, C. Koo, S. Spachmann, R. Klingeler, G. Rajaraman, J. Overgaard and M. Shanmugam, Cell Rep. Phys. Sci., 2021, 2, 100404 CrossRef CAS; (b) M. Craven, M. H. Nygaard, J. M. Zadrozny, J. R. Long and J. Overgaard, Inorg. Chem., 2018, 57, 6913–6920 CrossRef CAS PubMed; (c) M. Atanasov and F. Neese, J. Phys.: Conf. Ser., 2018, 1148, 012006 CrossRef; (d) A. Lunghi and S. Sanvito, J. Chem. Phys., 2020, 153, 174113 CrossRef CAS PubMed.
- (a) T. J. Woods, M. F. Ballesteros-Rivas, S. Gómez-Coca, E. Ruiz and K. R. Dunbar, J. Am. Chem. Soc., 2016, 138, 16407–16416 CrossRef CAS PubMed; (b) A. Sarkar, S. Tewary, S. Sinkar and G. Rajaraman, Chem.–Asian J., 2019, 14, 4696–4704 CrossRef CAS PubMed; (c) L. Devkota, D. J. SantaLucia, A. M. Wheaton, A. J. Pienkos, S. V. Lindeman, J. Krzystek, M. Ozerov, J. F. Berry, J. Telser and A. T. Fiedler, Inorg. Chem., 2023, 62, 5984–6002 CrossRef CAS PubMed; (d) Y. Chen, Q. Yang, G. Peng, Y.-Q. Zhang and X.-M. Ren, Dalton Trans., 2021, 50, 13830–13840 RSC; (e) S. Vaidya, S. K. Singh, P. Shukla, K. Ansari, G. Rajaraman and M. Shanmugam, Chem.–Eur. J., 2017, 23, 9546–9559 CrossRef CAS PubMed; (f) A. K. Mondal, M. Sundararajan and S. Konar, Dalton Trans., 2018, 47, 3745–3754 RSC; (g) S. Gomez-Coca, E. Cremades, N. Aliaga-Alcalde and E. Ruiz, J. Am. Chem. Soc., 2013, 135, 7010–7018 CrossRef CAS PubMed; (h) R. Bruno, J. Vallejo, N. Marino, G. De Munno, J. Krzystek, J. Cano, E. Pardo and D. Armentano, Inorg. Chem., 2017, 56, 1857–1864 CrossRef CAS PubMed; (i) F. Yang, Q. Zhou, Y. Zhang, G. Zeng, G. Li, Z. Shi, B. Wang and S. Feng, Chem. Commun., 2013, 49, 5289–5291 RSC; (j) M. Idešicová, J. Titiš, J. Krzystek and R. Boča, Inorg. Chem., 2013, 52, 9409–9417 CrossRef PubMed.
- S. Vaidya, P. Shukla, S. Tripathi, E. Rivière, T. Mallah, G. Rajaraman and M. Shanmugam, Inorg. Chem., 2018, 57, 3371–3386 CrossRef CAS PubMed.
- S. Vaidya, A. Upadhyay, S. K. Singh, T. Gupta, S. Tewary, S. K. Langley, J. P. S. Walsh, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. Commun., 2015, 51, 3739–3742 RSC.
- (a) M. H. Pohle, M. Böhme, T. Lohmiller, S. Ziegenbalg, L. Blechschmidt, H. Görls, A. Schnegg and W. Plass, Chem.–Eur. J., 2023, 29, e202202966 CrossRef CAS PubMed; (b) S. Ziegenbalg, D. Hornig, H. Görls and W. Plass, Inorg. Chem., 2016, 55, 4047–4058 CrossRef CAS PubMed; (c) M. Böhme, S. Ziegenbalg, A. Aliabadi, A. Schnegg, H. Görls and W. Plass, Dalton Trans., 2018, 47, 10861–10873 RSC.
- K. Yamaguchi, G. Matsumura, N. Haga and K. Shudo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 558–559 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- E. Bill, julX_2S, Program for Simulation of Molecular Magnetic Data, Max-Planck Institute for Chemical Energy Conversion, Mülheim/Ruhr, Germany, 2014 Search PubMed.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- K. Chakarawet, P. C. Bunting and J. R. Long, J. Am. Chem. Soc., 2018, 140, 2058–2061 CrossRef CAS PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed; (c) F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- (a) P. Siegbahn, A. Heiberg, B. Roos and B. Levy, Phys. Scr., 1980, 21, 323–327 CrossRef CAS; (b) P. E. M. Siegbahn, J. Almlöf, A. Heiberg and B. O. Roos, J. Chem. Phys., 1981, 74, 2384–2396 CrossRef; (c) B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- (a) C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS; (b) C. Angeli and R. Cimiraglia, Theor. Chem. Acc., 2002, 107, 313–317 Search PubMed; (c) C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- (a) M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC; (b) M. Atanasov, D. Ganyushin, K. Sivalingam and F. Neese, Molecular electronic structures of transition metal complexes II, 2011, pp. 149–220 Search PubMed.
- H. Adamsky, AOMX: A FORTRAN Program for the Calculation of dn terms within the Angular Overlap Model with Interelectronic Repulsion and Spin-Orbit Coupling, Institute of Theoretical Chemistry, Heinrich-Heine-Univ., 1995 Search PubMed.
- C. E. Schäffer, A perturbation representation of weak covalent bonding, Springer, Berlin, 1968 Search PubMed.
- AOM parameters extracted from a fitting of the respective CASSCF/NEVPT2 one-electron AILFT matrices using the geometry of each complex determined by X-ray diffraction.
- CASSCF values do not differ much from NEVPT2 and is therefore used to generate the curves for the several rotated versions of the idealized geometry reduce computational cost.
- (a) G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (b) G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- X-RED, STOE & CIE GmbH, Darmstadt, Germany, 2002 Search PubMed.
- SADABS, BRUKER AXS GmbH, Karlsruhe, Germany, 2016 Search PubMed.
- (a) A. Wolf, M. Reiher and B. A. Hess, J. Chem. Phys., 2002, 117, 9215–9226 CrossRef CAS; (b) B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742–3748 CrossRef CAS PubMed; (c) M. Reiher and A. Wolf, J. Chem. Phys., 2004, 121, 10945–10956 CrossRef CAS PubMed; (d) A. Wolf and M. Reiher, J. Chem. Phys., 2006, 124, 064102 CrossRef PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef; (c) A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- F. Neese, J. Chem. Phys., 2003, 119, 9428–9443 CrossRef CAS.
- (a) F. Neese, L. Lang and V. G. Chilkuri, Topology, Entanglement, and Strong Correlations, 2020 Search PubMed; (b) R. Maurice, R. Bastardis, C. d. Graaf, N. Suaud, T. Mallah and N. Guihéry, J. Chem. Theory Comput., 2009, 5, 2977–2984 CrossRef CAS PubMed; (c) V. Vallet, L. Maron, C. Teichteil and J.-P. Flament, J. Chem. Phys., 2000, 113, 1391–1402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR, UV-Vis and ESI mass spectra, cyclic voltammograms, crystallographic information, magnetic characterization, and details of the theoretical calculations. CCDC 2240414 (1), 2240415 (2), 2240416 (3), 2240417 (5) and 2240418 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00813d |
| This journal is © The Royal Society of Chemistry 2023 |