
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMinimizing the off-target frequency of the CRISPR/Cas9 system via zwitterionic polymer conjugation and peptide fusion†
Yanjiao
Han
a,
Zhefan
Yuan
 b,
Sijin Luo
Zhong
b,
Haoxian
Xu
c and
Shaoyi
Jiang
*b
b,
Sijin Luo
Zhong
b,
Haoxian
Xu
c and
Shaoyi
Jiang
*b
aMolecular Engineering and Science Institute, University of Washington, WA 98195, USA
bMeinig School of Biomedical Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: sj19@cornell.edu
cDepartment of Material Science and Engineering, University of Washington, Seattle, WA 98195, USA
First published on 19th May 2023
Abstract
The clustered, regularly interspaced, short palindromic repeats (CRISPR)–associated protein 9 (Cas9) system is a powerful genome-editing tool that is widely used in many different applications. However, the high-frequency mutations induced by RNA-guided Cas9 at sites other than the intended on-target sites are a major concern that impedes therapeutic and clinical applications. A deeper analysis shows that most off-target events result from the non-specific mismatch between single guide RNA (sgRNA) and target DNA. Therefore, minimizing the non-specific RNA–DNA interaction can be an effective solution to this issue. Here we provide two novel methods at the protein and mRNA levels to minimize this mismatch issue by chemically conjugating Cas9 with zwitterionic pCB polymers or genetically fusing Cas9 with zwitterionic (EK)n peptides. The zwitterlated or EKylated CRISPR/Cas9 ribonucleoproteins (RNPs) show reduced off-target DNA editing while maintaining a similar level of on-target gene editing activity. Results show that the off-target efficiency of zwitterlated CRISPR/Cas9 is reduced on average by 70% and can be as high as 90% when compared with naive CRISPR/Cas9 editing. These approaches provide a simple and effective way to streamline the development of genome editing with the potential to accelerate a wide array of biological and therapeutic applications based on CRISPR/Cas9 technology.
Introduction
CRISPR/Cas9 system is now a widely used tool for genome editing in various organisms and cell types.1,2 Unfortunately, it can also cause unwanted mutations at off-target sites that resemble the on-target sequence.3 The off-target mutations are caused by the nonspecific recognition of DNA sequence by CRISPR/Cas9 RNPs.4 It has been demonstrated that besides the optimal PAM sequence 5-NGG-3, Cas9 can also cleave sites with a 5-NAG-3 or 5-NGA-3′PAM although less efficiently.5 In addition, a 20 nt single guide RNA (sgRNA) can recognize DNA sequences that harbor as many as 3–5 base pair mismatches with the sgRNA, suggesting there are up to thousands of possible binding sites for a given nuclease in the human genome.3 Furthermore, CRISPR/Cas9 can induce off-target cleavages with DNA sequences containing a few extra bases (‘DNA bulge’) or a few missing bases (‘RNA bulge’) compared to the RNA guide strand.6 Off-target DNA cleavages can give rise to mutations at unintended genomic loci and gross chromosomal rearrangements such as deletions,7,8 inversions,9 and translocations.10,11 These mutations at unwanted sites might disable a tumor-suppressor gene or activate a cancer-causing gene. Translocations have been known to be a possible reason for chronic myeloid leukemia.12 Preventing, avoiding, or at least reducing these off-target effects is crucial for the success of any downstream genome editing applications. Various strategies have been developed to reduce genome-wide off-target mutations of the commonly used Cas9 nuclease, including truncated sgRNAs bearing shortened regions of target complementarity,13,14 Cas9 mutants,15 paired Cas9 nickases,16,17 and dimeric fusions of catalytically inactive Cas9 to a non-specific FokI nuclease.18–20 However, these approaches are only partially effective and/or possess the potential to create more off-target sites. Furthermore, they may also require the expression of multiple sgRNAs and/or fusion of additional functional domains to Cas9, which can reduce the targeting range and create challenges for delivery using viral vectors which have a limited payload size of nucleic acids. Thus, a major challenge for the field remains the development of a simple robust strategy that can reduce the off-target effects of the CRISPR/Cas9 system.The “off-target” activity of the nucleases occurs fundamentally because the Cas9/sgRNA complex possesses more energy than what is needed for the effective recognition of its intended target DNA site.14,21 As a result, the complex lacks high specificity and can bind sequences that are similar to the on-target DNA strand. Therefore, it is hypothesized that the off-target effects of CRISPR/Cas9 might be minimized by reducing the non-specific interactions with its target DNA sites. Zwitterionic poly(carboxybetaine) (pCB) polymers and pseudo-zwitterionic alternating lysine (K) and glutamic acid (E) peptides are highly hydrated and effectively resistant to non-specific interactions.22 In our previous study, pCB polymers have been conjugated to chymotrypsin (CT),23 uricase,24 and interferon-α2a25 to preserve protein bioactivity. The super-hydrophilic nature of the polymer creates an environment to shift the equilibrium and favors the substrate and the binding site to interact. It has been demonstrated that a pCB conjugated protein exhibits reduced non-specific interactions with its surrounding environment. In our previous study, this reduction of nonspecific interactions was shown to significantly enhance protein circulation time and reduce protein-specific antibody production in vivo. Meanwhile, the zwitterionic (EK)n peptide, which is both biocompatible and biodegradable, would protect the protein in a similar way to the zwitterionic pCB polymer.26 It is believed that the distribution of equal amounts of oppositely charged E and K residues provides a zwitterionic layer to stabilize the protein and resist non-specific adsorptions.27–29 Both random and alternating EK sequences have been shown to confer nonfouling zwitterionic characteristics to surfaces and nanoparticles.
Since specific DNA–sgRNA matching is far stronger than non-specific interactions, we hypothesized that pCB conjugation or (EK)n peptide fusion was able to reduce non-specific interactions and still maintain specific interactions strong enough with on-target DNA strands. With this strategy, the off-target effects of CRISPR/Cas9 will be minimized. In this work, we chemically conjugated Cas9 with pCB polymers and examined its on-target and off-target efficiency. To assess the specificities of the pCB-conjugated CRISPR/Cas9 systems, we designed a series of mismatched sgRNAs containing single, double, or triple substitutions within multiple sgRNA target DNA interfaces. At the same time, a ‘one-step’ strategy called ‘EKylation’ was employed to genetically fuse zwitterionic (EK)n peptide to Cas9 protein at the mRNA level and to prove the same concept with another system. Different endogenous human genes were tested for both approaches. In this study, both pCB conjugates and (EK)n peptide fusions showed decreased off-target activity compared with the unmodified Cas9 while maintaining a similar level of on-target gene editing efficiency. We believe that pCB polymer conjugating or (EK)n peptide fusing may provide a simple, safe, and robust strategy for gene editing based on CRISPR/Cas9 system and other systems.
Results and discussion
To examine the effect of zwitterionic pCB polymer conjugation on the off-target efficiency of the CRISPR/Cas9 system, we prepared a series of pCB–Cas9 conjugates with different numbers of polymer chains per protein. pCB–Cas9 conjugates were synthesized by reacting N-hydroxysuccinimide (NHS) ester groups of the polymer with available amine groups on the protein. The reaction scheme is illustrated in Fig. S1.† The polymer density was controlled by altering the molar ratio between Cas9 and NHS-pCB in the reaction. In this work, we synthesized pCB10–Cas9, pCB20–Cas9, and pCB50–Cas9 at the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 1:20, and 1:50, respectively. A native (unconjugated) Cas9 protein was used for comparison throughout the studies. The difference in size between native Cas9 and pCB–Cas9 conjugates was shown in Fig. S2,† which confirms the successful synthesis of the polymer–protein conjugates. Surface modification of a protein by covalent conjugation with polymers, such as PEGylation, lowers the in vitro biological activity of conjugated proteins.30,31 Therefore, we first tested whether the presence of zwitterionic polymers would compromise the on-target activity after conjugation. For these experiments, we used a well-established Cas9-induced GFP disruption assay that enabled the rapid quantification of targeted nuclease activities.3 In this assay, we targeted a genomic GFP reporter gene in human HEK293-GFP cells. The activities were quantified by measuring the loss of fluorescence signal in human HEK293-GFP cells, which is caused by the on-target CRISPR/Cas9 cleavage (Fig. 1a). The cells were treated with 50 ng sgRNA and 200 ng native Cas9/Cas9-equivalent pCB–Cas9 conjugates with CRISPRMAX (Fig. S3 and S4†) in DMEM containing 10% FBS for 48 hours to induce the disruption of GFP reporter gene. As shown in Fig. 1b, we found that pCB10–Cas9 and pCB20–Cas9 showed comparable editing efficiency to native Cas9, in which about 60% of cells lost their GFP expression after the treatment. The encouraging results demonstrated that the presence of pCB polymer did not compromise the “on-target” editing efficiency of the CRISPR/Cas9 system. However, only 40% GFP negative cells were found when treated with pCB50–Cas9 (Fig. 1b). This is due to the presence of longer pCB polymers, which may physically impede the binding between DNA and CRISPR/Cas9 RNP.
:10, 1:20, and 1:50, respectively. A native (unconjugated) Cas9 protein was used for comparison throughout the studies. The difference in size between native Cas9 and pCB–Cas9 conjugates was shown in Fig. S2,† which confirms the successful synthesis of the polymer–protein conjugates. Surface modification of a protein by covalent conjugation with polymers, such as PEGylation, lowers the in vitro biological activity of conjugated proteins.30,31 Therefore, we first tested whether the presence of zwitterionic polymers would compromise the on-target activity after conjugation. For these experiments, we used a well-established Cas9-induced GFP disruption assay that enabled the rapid quantification of targeted nuclease activities.3 In this assay, we targeted a genomic GFP reporter gene in human HEK293-GFP cells. The activities were quantified by measuring the loss of fluorescence signal in human HEK293-GFP cells, which is caused by the on-target CRISPR/Cas9 cleavage (Fig. 1a). The cells were treated with 50 ng sgRNA and 200 ng native Cas9/Cas9-equivalent pCB–Cas9 conjugates with CRISPRMAX (Fig. S3 and S4†) in DMEM containing 10% FBS for 48 hours to induce the disruption of GFP reporter gene. As shown in Fig. 1b, we found that pCB10–Cas9 and pCB20–Cas9 showed comparable editing efficiency to native Cas9, in which about 60% of cells lost their GFP expression after the treatment. The encouraging results demonstrated that the presence of pCB polymer did not compromise the “on-target” editing efficiency of the CRISPR/Cas9 system. However, only 40% GFP negative cells were found when treated with pCB50–Cas9 (Fig. 1b). This is due to the presence of longer pCB polymers, which may physically impede the binding between DNA and CRISPR/Cas9 RNP.
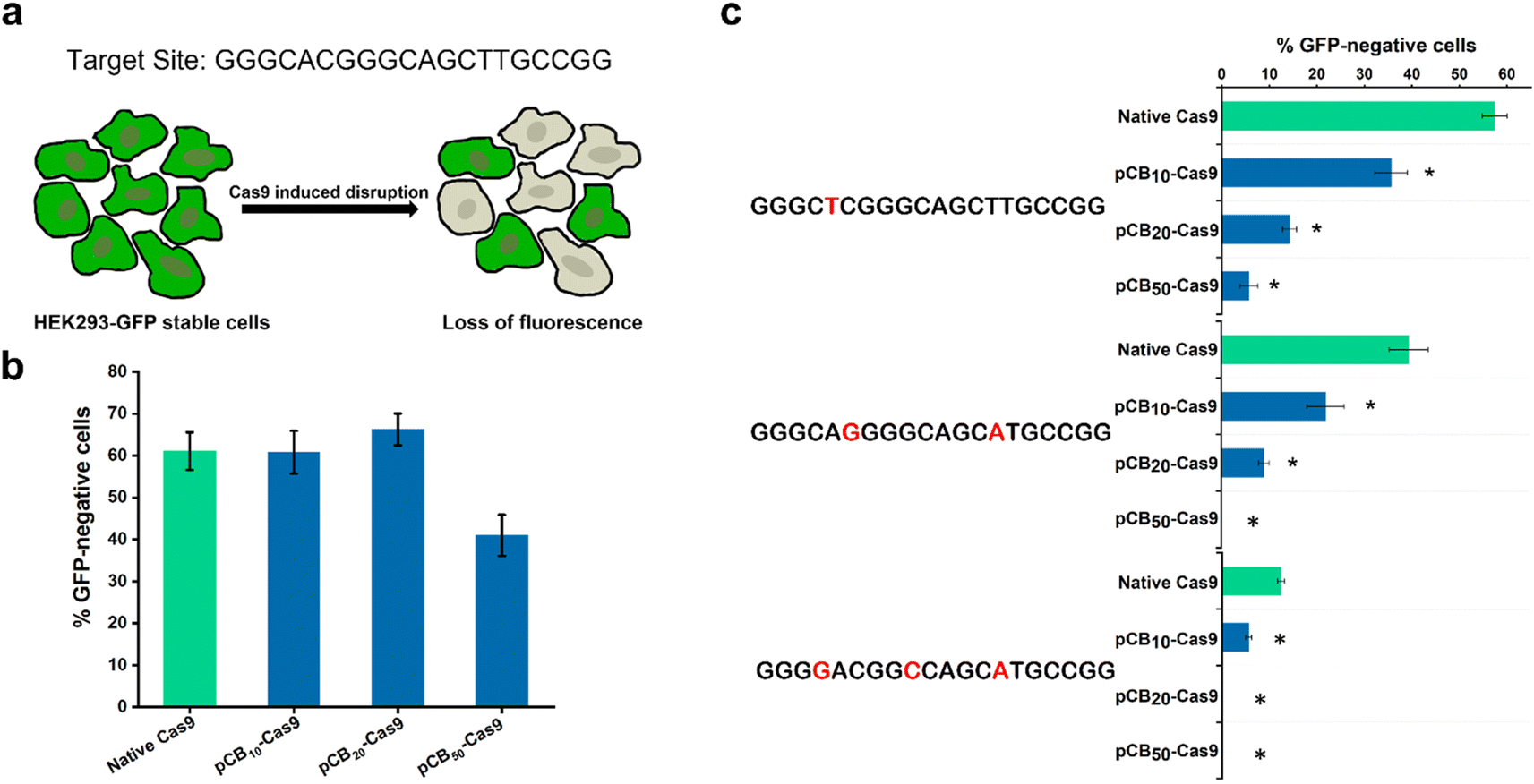 | ||
| Fig. 1 (a) Schematic overview of the GFP disruption assay and the target site used in the GFP gene; (b) efficiency of GFP disruption in HEK293-GFP cells mediated by native Cas9 and pCB–Cas9 conjugates. Error bars represent s.e.m. for n = 3. (c) Off-target editing efficiency of native Cas9 and pCB–Cas9 conjugates with mismatched sgRNA harboring one, two, or three nucleotide mutations in GFP disruption assay. Mutated nucleotides are colored red. *p < 0.05. Error bars represent s.e.m. for n = 3. | ||
To explore the potential of pCB conjugates in reducing the off-target activity, we randomly generated variant sgRNAs for the target site with one, two, or three mismatched nucleotides and tested whether these mismatched sgRNAs could drive off-target GFP disruption in human cells (Fig. 1c). If pCB conjugation could reduce off-targeting, then pCB–Cas9 conjugates would be less tolerant of mismatches than native Cas9. As presented in Fig. 1c, native Cas9 can still induce substantial GFP gene disruption in human cells when using mismatched sgRNA. In contrast, pCB–Cas9 conjugates showed a significant reduction of GFP disruption efficiency when mismatched sgRNAs were used (Fig. 1c). pCB10–Cas9 induced 35.6%, 21.9%, and 5.6% GFP disruption while pCB20–Cas9 led to 14.2%, 8.9%, and 0, respectively when one, two, or three nucleotide mismatches were present in the sgRNA. pCB50–Cas9 generated no detectable GFP disruption when 2 or 3 nucleotide mismatches were present. These data suggest that pCB conjugation can significantly reduce off-target gene editing when mismatched sgRNAs are used. Taking both the on-target and off-target efficiency into consideration, we selected pCB20–Cas9, which shows complete on-target efficiency and significantly reduces off-target efficiency. In the following study, pCB20–Cas9 will be denoted as pCB–Cas9 thereafter.
To further evaluate the effects of mismatches on pCB–Cas9, we created more sgRNA bearing one, two, or three nucleotide mutations. As shown in Table 1, native Cas9 exhibited significant off-target editing in a single-mismatch scenario (57.4%, 60.6%, and 49.3%). These off-target editing efficiencies are very close to their on-target editing efficiency in the perfect-match scenario (62.6%) as we discussed above. In contrast, using the same mismatched sgRNAs, pCB–Cas9 conjugate showed a significantly reduced off-target editing efficiency (14.2%, 12.6%, and 6.8%). This editing efficiency is more than 80% lower than its editing efficiency in the perfect-match scenario (67.4%). This observation was confirmed in another two scenarios when the sgRNAs have two or three mismatches. When 3 mismatches are presented in a sgRNA, the native Cas9 still exhibited positive editing efficiencies (12.4%, 7.9%, 9.3%). In contrast, no “off-target” efficiency is observed for pCB–Cas9 conjugate groups. This indicates that pCB-conjugated CRISPR/Cas9 has a high resolution in DNA editing and can distinguish the mismatch on a single-base level.
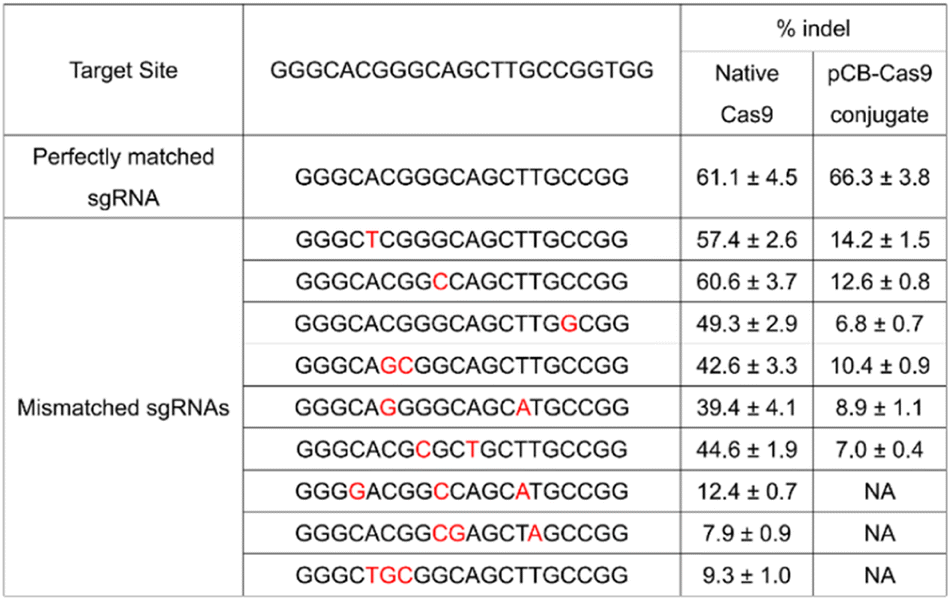
|
It is known that the “off-target” activity of the nucleases is fundamentally caused by the extra energy that the Cas9/sgRNA complex possesses, leading to the lack of perfect specificity.14,21 Such extra energy comes mainly from the nonspecific forces-hydrophobic and electrostatic in particular. Coating a protein with non-fouling polymeric materials can alter these nonspecific interactions and is the key to lowering energy and promoting specific binding.32,33 Zwitterionic materials based on naturally occurring betaines such as pCB have particularly high hydration. As a result, ultra-low nonspecific adsorption in complex biological media has been observed in different scenarios.22 Here we hypothesized that the conjugation of pCB to Cas9 could reduce the nonspecific binding force between the Cas9/sgRNA complex and the target DNA. As shown in Fig. 2, for native Cas9, the energy that the Cas9/sgRNA complex possesses is much higher than the minimum energy required for on-target binding between the sgRNA and the DNA. As a result, the RNP complex still possesses enough energy even if one or more mismatched nucleotides are present. With the pCB conjugates, the aforementioned nonspecific binding, especially the hydrophobic–hydrophobic interaction, is decreased significantly. The complex is unable to bind the double-strand DNA without sufficient energy when mismatched nucleotides are present in the sequence. As a result, off-target effects can be reduced significantly. In addition, benefiting from the super-hydrophilicity of the polymer, a tightly bound water layer is formed around and the nonspecific interactions between RNPs and zwitterionic polymers can be minimized as we observed before.25 As a result, the bioactivity of Cas9 can be retained after the polymer conjugation, which is very important to keep the on-target efficiency.
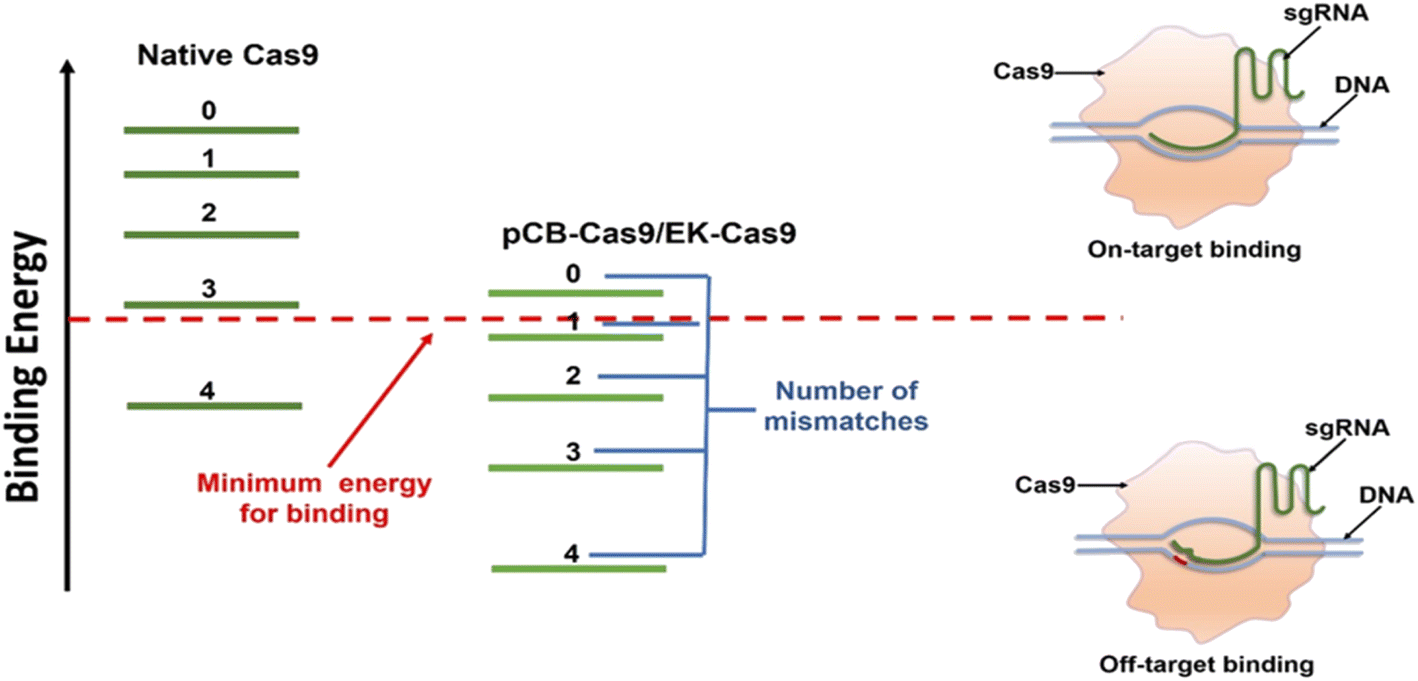 | ||
| Fig. 2 Mechanism of pCB conjugation or (EK)n peptide fusion in reducing the off-target efficiency of the CRISPR/Cas9 system. The Cas9/sgRNA complex possesses more energy than what is needed for optimal recognition of its target DNA site, leading to the cleavage of mismatched off-target sites. pCB polymer conjugation (EK)n peptide fusion eliminates the non-specific binding between Cas9/sgRNA complex and double-strand DNA, thereby decreasing the binding energy. The remained energy is strong enough for on-target binding, but not enough for mismatched binding. | ||
To examine whether pCB-conjugated CRISPR/Cas9 RNPs can reduce off-target effects on other DNA domains in human cells, we selected three new genomic loci in the VEGFA, EMX, and CLTA genes due to their potential biomedical relevance and widely use in Cas9 off-target studies.3 As presented in Fig. 3, for all three targets, CRISPR/pCB–Cas9 mediated indels at their endogenous loci were detected using the T7 endonuclease I (T7EI) assay. For each of these three target sequences, we examined the editing efficiencies of several potential off-target sites which have been observed in other studies. In this study, we observed similar trends as we showed in the previous examples. The rates of mutation at the selected off-target sites were very high, ranging from 9.4% to 93.6% when the cells were edited using native CRISPR/Cas9. In contrast, for the cells edited using pCB-conjugated CRISPR/Cas9, the off-target mutation rates were observed at a much lower level, ranging from 2.4% to 10.5%. It is noticeable that the editing efficiency of the pCB–Cas9 conjugate is slightly higher than that of native Cas9. This confirms our hypothesis that the bioactivity of Cas9 is reserved after conjugation.
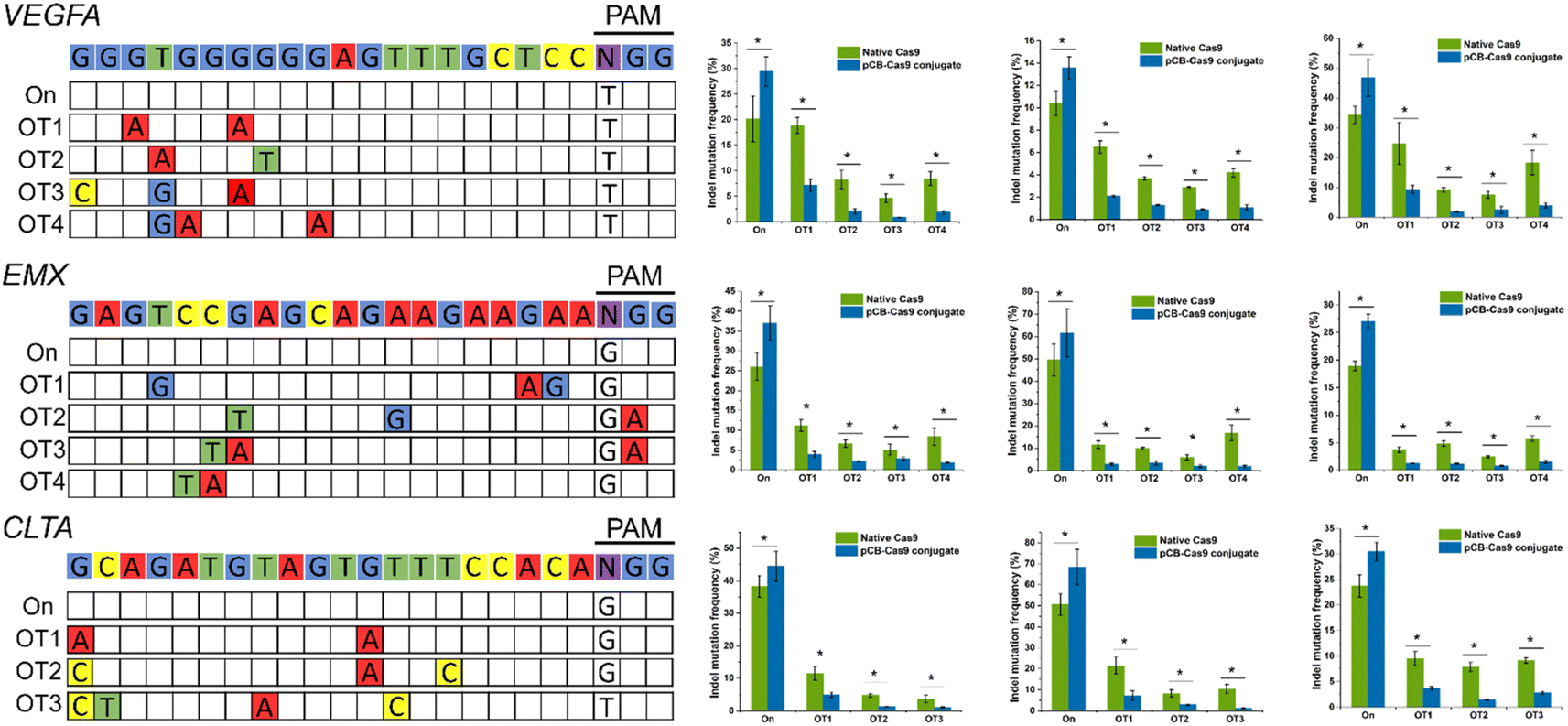 | ||
| Fig. 3 On-target and off-target DNA editing efficiencies on other DNA domains in human cells resulting from native Cas9 and pCB–Cas9. Three new genomic loci in the VEGFA, EMX, and CLTA genes in three different cell lines (HEK 293, U2OS, and K562) were selected. *p < 0.05. Error bars represent s.e.m. for n = 3. | ||
After pCB conjugation is demonstrated to reduce the off-target mutations of CRISPR/CAS9 in HEK293-GFP cells, we proceed to evaluate this zwitterlated Cas9 in other types of human cells. Here, we employed U2OS and K562 cell lines as they were also widely used to test the on-/off-target activity of CRISPR/Cas9. We also explored the editing efficiencies on three targets using either native Cas9 or pCB–Cas9 in U2OS and K562 cell editing. As expected, pCB–Cas9 conjugates generate similar or slightly higher editing efficiency when compared with native Cas9 at the target genomic locus (Fig. 3). For the off-target examinations using variant mismatches, the pCB–Cas9 conjugate groups produced less than 4% indels rates at 20 out of 22 off-target sites. In contrast, native Cas9 generated 2.9–24.7% off-target indels, of which six were higher than 10%. These results further demonstrated that pCB-conjugated CRISPR/Cas9 showed reduced off-target effects in different cell lines.
In the previous section, a reduction in the off-targeting efficacy of the CRISPR/Cas9 system was demonstrated using pCB–Cas9 conjugates at the protein level. Here, proof-of-concept experiments will be performed again, but at the mRNA level. Here zwitterionic (EK)n peptides were genetically fused to Cas9. In this work, human codon-optimized DNA encoding Cas9 nuclease from Streptococcus pyogenes with N and C terminal nuclear localization signal (NLS) was cloned into a pcDNA3.1 vector. DNA encoding poly(EK) with 10 kDa or 30 kDa length were commercially synthesized and appended to the C-terminal or both C- and N-terminals of the Cas9 gene to generate EK–Cas9 constructs (Fig. 4a). We constructed three EK–Cas9 plasmids, Cas9–(EK)10, (EK)10–Cas9–(EK)10, and Cas9–(EK)30, based on the length and fusion position of poly(EK). The Cas9 sequence without poly(EK) sequence was used as the control sequence. Since the constitutive presence of the plasmids and transcripts could result in high levels of undesired off-target gene editing, we turned to DNA-free CRISPR gene-editing systems by transfecting both in vitro transcribed sgRNA and Cas9 mRNA to achieve their desired gene editing effects. This system relies on the translation of Cas9 mRNA in cells, so polyadenylation (poly(A)) of Cas9 and required to prevent Cas9 mRNA from degradation before in vivo translation occurs. The polyadenylation reaction was started with the addition of the E-PAP enzyme and incubated for 30 min at 37 °C. The band shift after polyadenylation in the electrophoresis image (Fig. S5†) confirmed the presence of poly(A) tails.
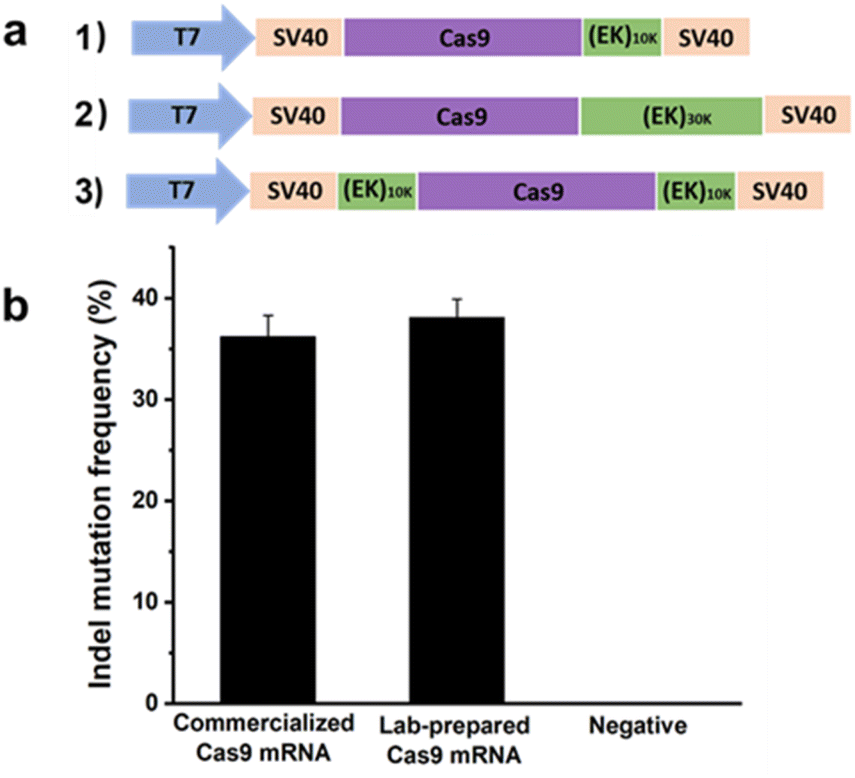 | ||
| Fig. 4 (a) Construction of expression plasmid encoding Cas9–(EK)n; (b) gene editing efficacy of Cas9 using commercialized Cas9 mRNA and lab-prepared Cas9 mRNA. Error bars represent s.e.m. for n = 3. | ||
After getting the mRNA, we first confirmed the activity of lab-prepared Cas9 mRNA in mammalian cells by comparing it with the commercialized Cas9 mRNA. The gRNA that targets GFP (GGGCACGGGCAGCTTGCCGG) was selected for this analysis. Two days after co-transfecting HEK293-GFP cells with mRNA expressing either commercialized Cas9 or lab-prepared Cas9 together with the GFP gRNA, the percentage of indel mutations was quantified by the T7EI assay. As shown in Fig. 4b, the lab-prepared Cas9 mRNA possesses similar on-target editing efficiency to the commercial Cas9 mRNA, which indicates the successful synthesis of the in vitro transcribed Cas9 mRNA. The purity of the Cas9 mRNA and EK–Cas9 mRNAs were confirmed from the traditional UV spectroscopy with A260/A280 ratio (Table S3†).
To investigate whether the EK–Cas9 mRNAs could be programmed by gRNAs to cleave chromosomal DNA in mammalian cells, we used the same assay to test the gene-editing efficiency of Cas9–(EK)10, (EK)10–Cas9–(EK)10, and Cas9–(EK)30 at the GFP site in HEK293-GFP cell. As shown in Fig. 5a, all three EK–Cas9 mRNAs show a similar editing level to the Cas9 mRNA. The presence of poly(EK) did not compromise the on-target gene editing efficiency on the selected on-target site. To approve that this effect is not targeted site-specific, the similar editing frequency was further verified with genomic loci VEGFA (Fig. 5b) and EMX (Fig. 5c). The quantified data are summarized and presented in Fig. 5d. To examine whether the presence of poly(EK) can reduce the off-target effects in human cells, we selected several potential off-target sites for VEGFA and EMA target loci. Similar to the study of pCB–Cas9, we employed 3 different cell lines to avoid the cell specificity. The results are shown in Fig. 6. For Cas9–(EK)10, the off-target activity was similar or slightly reduced for some off-target sites compared to the native Cas9. This is likely due to the insufficient length of poly(EK) to effectively reduce the nonspecific interactions between gRNA and double-strand DNA. However, both (EK)10–Cas9–(EK)10, and Cas9–(EK)30 showed a significantly reduced off-target editing efficiency, ranging from 1.2% to 3.5%. These results demonstrated that EK–Cas9 showed reduced off-target effects on different genomic loci.
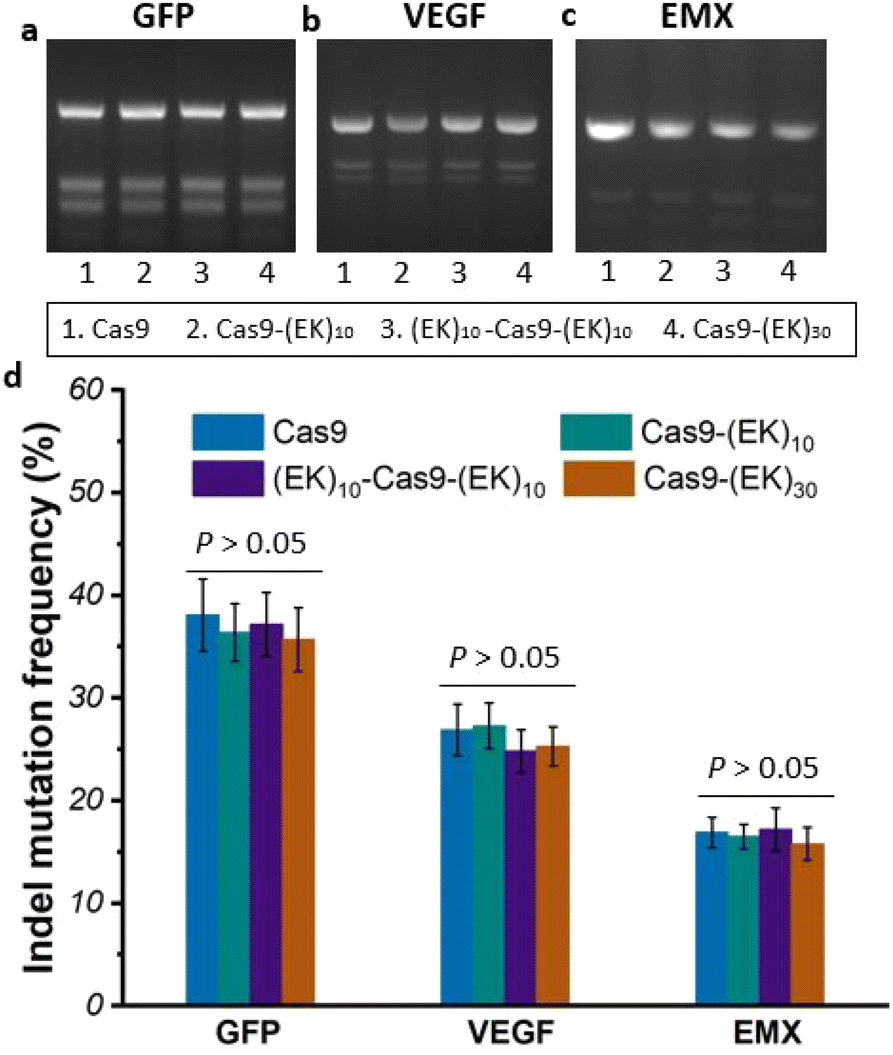 | ||
| Fig. 5 (a)–(c) Electrophoresis of on-target DNA editing resulting from native Cas9, Cas9–(EK)10, (EK)10–Cas9–(EK)10, and Cas9–(EK)30 for target sites GFP (a), VEGFA (b), and EMX (c) in HEK293-GFP cells; (d) quantified data of (a)–(c). Error bars represent s.e.m. for n = 3. | ||
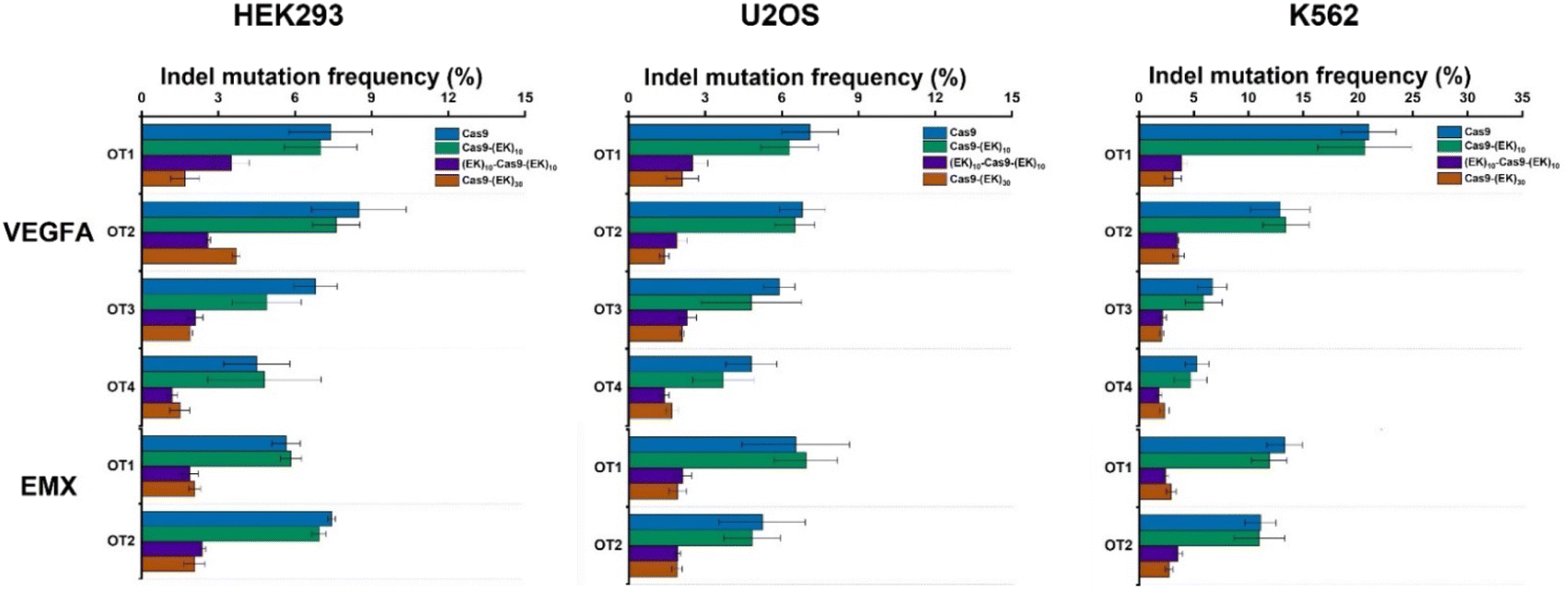 | ||
| Fig. 6 Off-target DNA editing efficiencies resulting from native Cas9, Cas9–(EK)10, (EK)10–Cas9–(EK)10, and Cas9–(EK)30 for target sites (VEGFA and EMX) in three different cell lines (HEK293, U2OS, and K562). Error bars represent s.e.m. for n = 3. P values are listed in Table S2.† | ||
Conclusions
In summary, a protein–polymer conjugation technology and a protein-peptide fusion strategy have been demonstrated in this study to address the “off-target” concern faced by the CRISPR/Cas9 system at both protein and mRNA levels. After being modified with a super-hydrophilic zwitterionic polymer or a biodegradable zwitterionic peptide, the CRISPR/Cas9 system shows significantly reduced “off-target” efficiency. More importantly, no reduced “on-target” frequency, which is usually noticed in many other strategies, was observed. These technologies will potentially provide a simple and robust strategy to improve the efficiency and safety of a wide range of CRISPR/Cas9-based biological research and the treatment of various genetic diseases.34–37Materials and methods
Synthesis of N-hydroxysuccinimide poly(carboxybetaine acrylamide) (NHS-PCBAA)
pCB-NHS was synthesized based on our previous procedure. Briefly, we firstly synthesized 3-acrylamido-N-(2-(tert-butoxy)-2-oxoethyl)-N,N-dimethylpropan-1-aminium (CBAAM-tBu). Then, a typical reversible addition–fragmentation chain transfer (RAFT) polymerization reaction was performed to yield the 10 kDa SH-pCB polymer. The final colorless NHS-activated polymer was formed by reaction with AMAS at 1:10 molar ratio in DI water (pH 6) for 30 min, followed by removal of unreacted AMAS via Amicon spin dialysis tubes and freeze-drying for 48 h.
Preparation and characterization of pCB–Cas9 conjugates
Conjugate of pCB–Cas9 was synthesized by reacting NHS ester groups of the polymer with available amine groups on the protein. In a typical conjugation reaction, Cas9 nuclease and NHS-pCB at 1:10, 1:20, or 1:50 molar ratio were dissolved in 50 mM sodium borate buffer, pH 9.0. The final protein concentration was ∼5 mg mL−1. The reaction mixture was stirred for 2 hours at 4 °C and stopped by adjusting the pH of the mixture to 4.5 with glacial acetic acid. The polymer–protein conjugate was isolated via molecular weight cut-off (MWCO) spin dialysis membrane. High-performance liquid chromatography (HPLC) was used to measure the hydrodynamic size of the protein conjugates.
In vitro synthesis of sgRNA
The in vitro synthesis of sgRNA was carried out using EnGen® sgRNA Synthesis Kit using the manufacturer's recommended conditions. The sgRNA product was purified using GeneJET RNA Cleanup and Concentration Micro Kit as described in the manual. The concentration of RNA was determined by measuring the absorbance at 260 nm on a microplate reader.Mammalian cell culture
HEK293-GFP cells were maintained in DMEM, medium supplemented with 10% FBS. U2OS cells were maintained in McCoy's 5A modified medium supplemented with 25 mM HEPES and 10% FBS. K562 cells were propagated in RPMI 1640 medium containing 10% FBS. After thawing, cells were passaged 4–5 times before using for transfection. When setting up the experiments for transfections, cultured cells were plated in 24-well format (500 μL volume) in a complete growth medium at a cell density necessary to reach ∼70% confluence the next day. Full serum media was replaced with the same media but containing no antibiotics at least 1 h before delivery. All cultures were maintained in 5% CO2 at 37 °C in a humidified incubator.In vitro co-delivery of Cas9 protein and sgRNA
For Cas9 protein transfection, 200 ng of purified Cas9 protein was added to 5 μL of Opti-MEM medium, followed by the addition of 50 ng gRNA. The molar ratio of gRNA to Cas9 protein was kept at approximately 1 to 1.2:1. The sample was mixed by gently tapping the tubes a few times and then incubated at room temperature for 10 min. In a separate test tube, 0.8 μL of Lipofectamine CRISPRMAX transfection reagent was diluted to 5 μL with Opti-MEM medium. The diluted transfection reagent was transferred to the tube containing Cas9 protein/gRNA complexes, followed by incubation at room temperature for 10 min, and then the entire solution was added to the cells in a 24-well plate and mixed by gently swirling the plate. The plate was incubated at 37 °C for 48 h in a 5% CO2 incubator.
Construction of EK–Cas9 plasmids and sgRNA
Human codon-optimized DNA encoding Cas9 nuclease from Streptococcus pyogenes with an N and C terminal nuclear localization signal (NLS) was cloned into a pcDNA3.1 vector (GenScript). DNA encoding a poly(EK) with various lengths was commercially synthesized and appended to the C-terminal or both C- and N-terminal of the Cas9 gene to generate a EK–Cas9 constructs. The Cas9 sequence without poly(EK) sequence was used as the control sequence. The sgRNAs that target GFP (GGGCACGGGCAGCTTGCCGG), VEGFA (GGGTGGGGGGAGTTTGCTCC), and EMX (GAGTCCGAGCAGAAGAAGAA) were purchased from Synthego. The potential off-target sites listed in Table 1 were used to measure the off-target efficiency in human cells.In vitro transcription of Cas9/EK–Cas9 mRNA
The Cas9 and EK–Cas9 plasmids were linearized using BbsI (New England Biolabs) according to the manufacturer's instructions. Following purification, the Cas9 and EK–Cas9 mRNAs were transcribed using mMESSAGE mMACHINE T7 Ultra Transcription Kit (ThermoFisher) according to manufacturer's instructions with a 2 hours incubation time at 37 °C. TURBO DNase was added to stop transcription. Before polyadenylation, a 2 μL aliquot was removed and diluted into 10 μL of nuclease-free water. This aliquot was used as an untailed control in the RNA gel. The polyadenylation reaction was started with the addition of the E-PAP enzyme and incubated for 30 min at 37 °C.In vitro delivery of Cas9/EK–Cas9 mRNA
One day before transfection, the cells were seeded in a 24-well plate at a cell density of 1–2 × 105 cells per well. 0.5 μg Cas9 or EK–Cas9 mRNA was added to 25 μL of Opti-MEM, followed by the addition of 50–100 ng gRNA. Meanwhile, 2 μL of Lipofectamine MessengerMax (ThermoFisher) was diluted into 25 μL of Opti-MEM and then mixed with the mRNA/gRNA sample. The mixture was incubated for 15 min before addition to the cells. Then the entire solution was added to the cells and mixed by gently swirling the plate. The plate was incubated at 37 °C for 48 h in a 5% CO2 incubator. Genomic DNA was extracted from cells using the Quick-DNA Miniprep Plus Kit (Zymo Research). The cut efficiency is assessed by T7E1 assays and site-specific Sanger sequencing of on-target and putative off-target sites.Determination of on- and off-target mutation frequencies in human cells
Genomic DNA was harvested 2 days after Cas9/pCB-Cas9 conjugates transfection or 3 days after Cas9/EK–Cas9 mRNA transfection from U2OS, HEK293, or K562 cells using the Quick-DNA Miniprep (Zymo Research), according to the manufacturer's instructions. 100 ng of isolated genomic DNA was used as a template to PCR amplify the targeted genomic sites with primer pairs. PCR products were purified with a PureLink™ PCR Purification Kit (Thermo Fisher) and quantified on a microplate reader. 250 ng of purified PCR DNA was combined with 2 μL of NEBuffer 2 (NEB) in a total volume of 19 μL and denatured then re-annealed with thermocycling at 95 °C for 5 min, 95–85 °C at 2 °C s−1; 85–20 °C at 0.2 °C s−1. The re-annealed DNA was incubated with 1 μL of T7 endonuclease I (10 U μL−1, NEB) at 37 °C for 30 min. Cas9-induced cleavage bands and the uncleaved band were visualized under UV light and quantified using ImageJ software30. The peak intensities of the cleaved bands were divided by the total intensity of all bands (uncleaved + cleaved bands) to determine the fraction cleaved, which was used to estimate gene modification levels. For each sample, transfections and subsequent modification measurements were performed in triplicate on different days. The off-target analysis was performed using a bioinformatics-based search tool to select potential off-target sites, which was also evaluated using the T7E1 mutation detection assay.Sanger sequencing
To better determine the mutation rate, the same purified PCR products used for the T7EI assay were sequenced to observe the individual mutations and determine the mutational spectra. Sanger sequencing was used to confirm the gene modification frequencies for the modified and unmodified CRISPR/Cas9 systems. The results were analyzed by ICE Analysis (Synthego).Author contributions
Y. H. and S. J. formulated the initial idea. Y. H., Z. Y., S. L. Z., and H. X. performed the experiments. Y. H. and S. J. prepared the manuscript with inputs from all authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
S. J. is a cofounder of ZWI Therapeutics, Inc. Y. H.; Z. Y.; S. Z.; S. J. are inventors of a pending patent (PCT/US2020/055095 filed 10/9/2020) filed by the University of Washington.Acknowledgements
We acknowledge financial support from the National Institute of Health (5R21GM128004-02) and start-up support from Cornell University, including Robert Langer 70 Family and Friends Professorship and Cornell NEXT Nano Initiative.References
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang and L. A. Marraffini, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- Y. Fu, J. A. Foden, C. Khayter, M. L. Maeder, D. Reyon, J. K. Joung and J. D. Sander, Nat. Biotechnol., 2013, 31, 822–826 CrossRef CAS PubMed.
- T. Koo, J. Lee and J. S. Kim, Mol. Cells, 2015, 38, 475 CrossRef CAS PubMed.
- S. H. Sternberg and J. A. J. Doudna, Mol. Cells, 2015, 58, 568–574 CAS.
- Y. Lin, T. J. Cradick, M. T. Brown, H. Deshmukh, P. Ranjan, N. Sarode, B. M. Wile, P. M. Vertino, F. J. Stewart and G. Bao, Nucleic Acids Res., 2014, 42, 7473–7485 CrossRef CAS PubMed.
- Y. Kim, J. Kweon, A. Kim, J. K. Chon, J. Y. Yoo, H. J. Kim, S. Kim, C. Lee, E. Jeong, E. Chung, D. Kim, M. S. Lee, E. M. Go, H. J. Song, H. Kim, N. Cho, D. Bang, S. Kim and J. S. Kim, Nat. Biotechnol., 2013, 31, 251–258 CrossRef CAS PubMed.
- H. J. Lee, E. Kim and J. S. Kim, Genome Res., 2010, 20, 81–89 CrossRef CAS PubMed.
- H. J. Lee, J. Kweon, E. Kim, S. Kim and J. S. Kim, Genome Res., 2012, 22, 539–548 CrossRef CAS PubMed.
- E. Brunet, D. Simsek, M. Tomishima, R. DeKelver, V. M. Choi, P. Gregory, F. Urnov, D. M. Weinstock and M. Jasin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10620–10625 CrossRef CAS PubMed.
- S. W. Cho, S. Kim, Y. Kim, J. Kweon, H. S. Kim, S. Bae and J. S. Kim, Genome Res., 2014, 24, 132–141 CrossRef CAS PubMed.
- P. Hasty and C. Montagna, Mol. Cell. Oncol., 2014, 1, e29904 CrossRef PubMed.
- S. Q. Tsai, Z. Zheng, N. T. Nguyen, M. Liebers, V. V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A. J. Iafrate, L. P. Le, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2015, 33, 187–197 CrossRef CAS PubMed.
- Y. Fu, J. D. Sander, D. Reyon, V. M. Cascio and J. K. Joung, Nat. Biotechnol., 2014, 32, 279–284 CrossRef CAS PubMed.
- B. P. Kleinstiver, M. S. Prew, S. Q. Tsai, V. V. Topkar, N. T. Nguyen, Z. Zheng, A. P. Gonzales, Z. Li, R. T. Peterson, J. R. J. Yeh, M. J. Aryee and J. K. Joung, Nature, 2015, 523, 481–485 CrossRef PubMed.
- P. Mali, J. Aach, P. B. Stranges, K. M. Esvelt, M. Moosburner, S. Kosuri, L. Yang and G. M. Church, Nat. Biotechnol., 2013, 31, 833–838 CrossRef CAS PubMed.
- F. A. Ran, P. D. Hsu, C.-Y. Lin, J. S. Gootenberg, S. Konermann, A. E. Trevino, D. A. Scott, A. Inoue, S. Matoba, Y. Zhang and F. Zhang, Cell, 2013, 154, 1380–1389 CrossRef CAS PubMed.
- S. Q. Tsai, N. Wyvekens, C. Khayter, J. A. Foden, V. Thapar, D. Reyon, M. J. Goodwin, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2014, 32, 569–576 CrossRef CAS PubMed.
- J. P. Guilinger, D. B. Thompson and D. R. Liu, Nat. Biotechnol., 2014, 32, 577–582 CrossRef CAS PubMed.
- N. Wyvekens, V. V. Topkar, C. Khayter, J. K. Joung and S. Q. Tsai, Hum. Gene Ther., 2015, 26, 425–431 CrossRef CAS PubMed.
- B. P. Kleinstiver, V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng and J. K. Joung, Nature, 2016, 529, 490–495 CrossRef CAS PubMed.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- A. J. Keefe and S. Jiang, Nat. Chem., 2012, 4, 59–63 CrossRef CAS PubMed.
- S. Liu and S. Jiang, Nano Today, 2016, 11, 285–291 CrossRef CAS.
- Y. Han, Z. Yuan, P. Zhang and S. Jiang, Chem. Sci., 2018, 9, 8561–8566 RSC.
- E. J. Liu, A. Sinclair, A. J. Keefe, B. L. Nannenga, B. L. Coyle, F. Baneyx and S. Jiang, Biomacromolecules, 2015, 16, 3357–3361 CrossRef CAS PubMed.
- A. J. Keefe, K. B. Caldwell, A. K. Nowinski, A. D. White, A. Thakkar and S. Jiang, Biomaterials, 2013, 34, 1871–1877 CrossRef CAS PubMed.
- A. K. Nowinski, F. Sun, A. D. White, A. J. Keefe and S. Jiang, J. Am. Chem. Soc., 2012, 134, 6000–6005 CrossRef CAS PubMed.
- S. Chen, Z. Cao and S. Jiang, Biomaterials, 2009, 30, 5892–5896 CrossRef CAS PubMed.
- C. S. Fishburn, J. Pharm. Sci., 2008, 97, 4167–4183 CrossRef CAS PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 CrossRef CAS PubMed.
- S. Chen, L. Li, C. Zhao and P. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
- Q. Shao, A. D. White and S. Jiang, J. Phys. Chem. B, 2014, 118, 189–194 CrossRef CAS PubMed.
- L. Zhang, L. Wang, Y. Xie, P. Wang, S. Deng, A. Qin, J. Zhang, X. Yu, W. Zheng and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12404–12408 CrossRef CAS PubMed.
- K. Huang, X. Liu, Y. Li, Q. Wang, J. Zhou, Y. Wang, F. Dong, C. Yang, Z. Sun and C. Fang, Adv. Sci., 2019, 6, 1900782 CrossRef PubMed.
- P. Wang, L. Zhang, W. Zheng, L. Cong, Z. Guo, Y. Xie, L. Wang, R. Tang, Q. Feng and Y. Hamada, Angew. Chem., Int. Ed., 2018, 57, 1491–1496 CrossRef CAS PubMed.
- P. Wang, L. Zhang, Y. Xie, N. Wang, R. Tang, W. Zheng and X. Jiang, Adv. Sci., 2017, 4, 1700175 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc07067g |
| This journal is © The Royal Society of Chemistry 2023 |