
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisualization of multiple localizations of GLUT4 by fluorescent probes of PYP-tag with designed unnatural warhead†
Miyako
Nishiura
a,
Yuichiro
Hori
*b,
Maho
Umeno
a and
Kazuya
Kikuchi
 *acd
*acd
aDepartment of Applied Chemistry, Graduate School of Engineering, and Division of Applied Chemistry, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kkikuchi@mls.eng.osaka-u.ac.jp
bDepartment of Chemistry, Faculty of Science, Kyushu University, Fukuoka, Fukuoka 819-0395, Japan. E-mail: hori@chem.kyushu-univ.jp
cImmunology Frontier Research Center, Osaka University, Suita, Osaka 565-0871, Japan
dQuantum Information and Quantum Biology Division, Osaka University, Suita, Osaka 565-0871, Japan
First published on 15th May 2023
Abstract
Within a cell, multiple copies of the same protein coexist in different pathways and behave differently. Being able to individually analyze the constant actions of proteins in a cell is crucial to know the pathways through which they pass and which physiological functions they are deeply involved in. However, until now, it has been difficult to distinguish protein copies with distinct translocation properties by fluorescence labeling with different colors in living cells. In this study, we have created an unnatural ligand with an unprecedented protein-tag labeling property in living cells and overcome the above-mentioned problem. Of special interest is that some fluorescent probes with the ligand can selectively and efficiently label intracellular proteins without binding to cell-surface proteins, even if the proteins are present on the cell membrane. We also developed a cell-membrane impermeable fluorescent probe that selectively labels cell-surface proteins without labeling of intracellular proteins. These localization-selective properties enabled us to visually discriminate two kinetically distinct glucose transporter 4 (GLUT4) molecules that show different multiple subcellular localization and translocation dynamics in live cells. Taking advantage of the probes, we revealed that N-glycosylation of GLUT4 influences intracellular localization. Furthermore, we were able to visually distinguish active GLUT4 molecules that underwent membrane translocation at least twice within an hour from those that remained intracellularly, discovering previously unrecognized dynamic behaviors of GLUT4. This technology provides not only a valuable tool for study on multiple localization and dynamics of proteins but also important information on diseases caused by protein translocation dysfunction.
Introduction
After translation and folding, each protein that comes to exist within a cell undergoes various processes throughout its life including various modifications, regeneration, preservation and degradation.1–8 As such, even the same protein can coexist in multiple locations and undergo intracellular translocation to regulate its function. Analysis of the multiple subcellular distributions and the dynamics of the protein molecules is essential for gaining deeper understanding of the true purposes and functions of the proteins, and such studies are attracting much attention lately.9–11 Conventional methods employing western blotting and immunostaining of dead cells have shown that different copies of the same protein can differ in their pattern of glycosylation and localization. However, the required samples do not represent live cells, and, in addition, protein dynamics and translocation pathway cannot be examined by those methods. Visualization of a protein of interest (POI) in a live cell can be achieved using a method that labels a POI fused with a tag protein such as SNAP-tag, Halo-tag, PYP-tag, TMP-tag, CLIP-tag and BL-tag9,12–22 with a fluorescent probe that interacts with the tag protein. Using this method, it has been possible to distinguish POIs that are present within the cell membrane from those that exist in the cytoplasm.19 However, such a method only allows labeling of different POIs with probes having different fluorescence wavelengths. Hence different molecules of the same POI become tagged with the same fluorescent label, resulting in their detection as the same signal regardless of whether or not their locations and dynamics differ. Herein, we successfully solved this problem by developing fluorescent probes that selectively label proteins fused to the same protein tag in different subcellular locations with different colors, enabling differentiation of proteins that show distinct translocation dynamics.Our group has been developing fluorescent probes23–27 that can specifically label a protein tag based on photoactive yellow protein (PYP), a 14 kDa blue-light photoreceptor protein from a purple sulfur bacterium28 that binds 4-hydroxycinnamic acid as its cofactor. In the current study, we focused our attention on the characteristics required of a high-performance probe, namely fast labeling rate, high labeling efficiency necessary for intracellular imaging of live cells and unique attributes of the probe itself, and designed a novel probe by selecting chemical motifs that would confer those characteristics to the probe. For the ligand of the probe, we shortlisted 12 candidates compounds that employed a new labeling mechanism and were structurally similar to but sufficiently different from the natural PYP-tag ligand for testing. The new probes were developed first by establishing the most suitable probe framework after examining the reactivity, speed of reaction and other properties of the candidate ligands, then tethering a dye to the framework. As a result, the new probe was able to label its target rapidly and with high efficiency intracellularly. Most importantly, the probe had a unique ability, which allows selective labeling of intracellular PYP-tagged proteins without binding to cell surface proteins with the same tag, even if the same protein molecules were present on the plasma membrane as well as in the cytoplasm. Thus, by using the probe in conjunction with a membrane-impermeable probe, we were able to separately label the POI from inside and outside of the cell. This property was extremely useful in individually analyzing POIs with differential dynamics. In addition, using the newly developed probe framework, we also prepared a four-color palette by synthesizing probes that carried dyes with different wavelengths. We applied these probes for multicolor imaging of glucose transporter 4 (GLUT4), which shows dynamic translocation in a live cell.29–31 We successfully labeled GLUT4 molecules with different translocation dynamics using two different fluorescent probes. The result indicated that the unique selective labeling property of the newly developed probes made it possible to uncover previously unknown details of the multiple intracellular localization of GLUT4. Specifically, using this labeling system, we revealed that N-glycan of GLUT4 changed the fate of its intracellular trafficking. It was previously suggested that the N-glycan increased the intracellular protein stability.36 We imaged intracellular localization of glycan-deficient GLUT4 and revealed that some of the GLUT4 molecules are translocated to the lysosome due to the lack of the glycan and some others undergo transient but dynamic translocation to the plasma membrane. Moreover, we were able to identify mobile and immobile GLUT4 molecules in a single cell, verifying that the former type of GLUT4 are translocated to the membrane at least twice within an hour, while the latter immobile molecules remained within the cell without reaching the plasma membrane at all. These results show that, for the first time, these novel probes can be used to visualize and track the multiple subcellular localization and multiple dynamics of the same protein in the same cell with different fluorescent colors.
Results
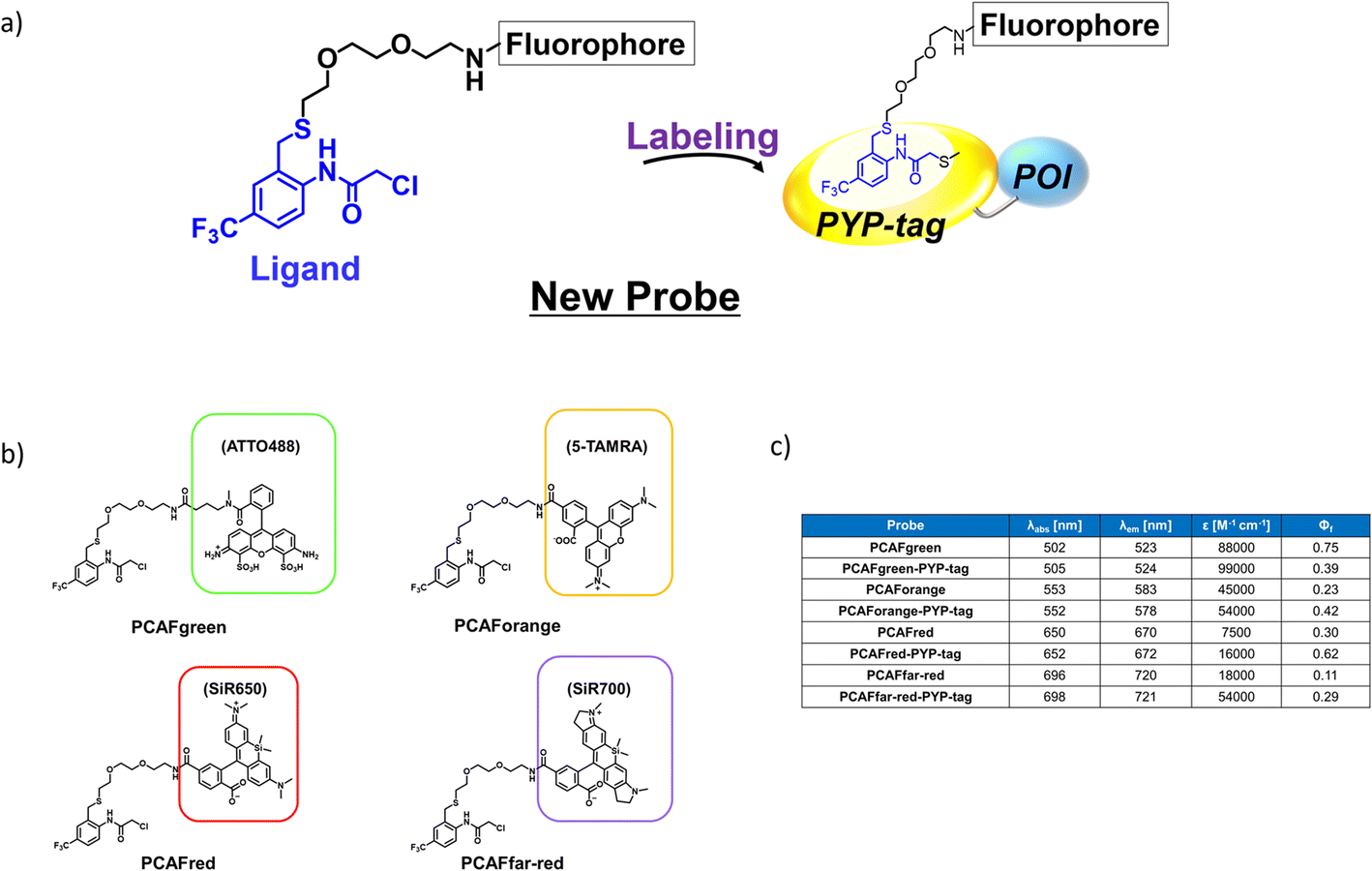
Design and synthesis of the probe framework with novel ligands
The existing probes with cinnamic acid-based ligands we developed earlier24,27,29 had a thioester functional group tethered to a thiophenol derivative as a leaving group.32 The probe labels PYP-tag fluorescently when the ligand of the probe enters the pocket within the PYP-tag and reacts with the Cys69 residue (Fig. 1a). A dinitrobenzene (DNB) moiety is incorporated into the leaving group and is required to accelerate the speed of the labeling reaction.24 However, DNB-containing leaving groups are highly hydrophobic and give the probe a higher tendency to aggregate in an aqueous environment, potentially harming the rate of reaction. Since it was imperative to resolve the existing problem before being able to design a high-performing probe, we set out to seek for a new labeling reaction that differed mechanistically from the currently existing system. As a start, we set out to determine the design of the ligand including the reactive site that would become covalently bound to the reactive cysteine residue of PYP-tag. To make the reaction mechanism non-reliant on the use of the conventional large leaving groups, we selected the leaving group to be either a bromo or chloro group to help improve the water solubility of the probe. Of the two leaving group candidates, the compounds containing a bromo group (12, Fig. 1b) was unstable (ESI Fig. 1†). Therefore, we chose to employ a chloro group as a leaving group for further development. Then, we combined various reactive groups with motifs that resembled the natural PYP-tag ligand to come up with 11 analogs as our prospective ligands (Figure 1b, 1–11). For the selection of the motifs, we considered accessibility of chemical synthesis, and commercial availability of starting materials. Thus, some of the candidates are selected to contain a synthetically accessible amide group, which is convertible with the double bond of the natural ligand, because these functional groups are related as a bioisostere. Those compounds were synthesized according to the methods described in the ESI and ESI Scheme 1† except for commercially available 6 and 7. | ||
| Fig. 1 Design and evaluation of the ligands to be incorporated into the new fluorogenic probes for fast and efficient labeling of PYP-tag. (a) The chemical structure of the previous probe with cinnamic acid (red) as the ligand, and the basic mechanism of fluorogenic labeling of PYP-tag. (b) The chemical structures of the newly designed 12 analogs of the natural PYP-tag ligand. (c) Reactivity of compounds 1–11 with PYP-tag analyzed in the competition assay in the presence of TMBDMA (8 μM), shown in ESI Fig. 2.† Compounds with “High” reactivity, 5 and 6, were completely bound to PYP-tag even with only slightly higher concentration (10 μM) than that of TMBDMA. Compounds classified as “Middle”, 7–10, and “Low”, 4, require relatively higher and much higher concentrations (20 and 100 μM), respectively, for complete binding. Binding of compounds, 1–3, to PYP-tag was not detected (“ND”) under this experimental condition. (d) Binding of the preferentially reactive compounds 5 (red circle), 6 (blue diamond) and 7 (green square) at 20 μM with 10 μM PYP-tag analyzed in a competition assay using TMBDMA previously reported.25 See Methods for details. | ||
Next, we examined the reactivity of the compounds 1–11 with PYP-tag by running competitive reactions with the previously developed PYP-tag probe, TMBDMA.25 Based on the SDS-PAGE analyses of the reactions, 5, 6 and 7 were determined to react more preferentially with PYP-tag than other candidate ligands (Fig. 1c and ESI Fig. 2†). Further characterizations of the three ligands by time-course analysis indicated that 5 exhibited the fastest reaction rate, followed by 6 and 7 (Fig. 1d and ESI Fig. 3†). In addition, it is known that glutathione, which is present in a live cell at a relatively high concentration of 1 to 10 mM,33 carries a strongly nucleophilic thiol group that may interfere with the reaction between the probe and PYP-tag. Therefore, we also investigated which of the compounds 5, 6 and 7 was least affected by glutathione for further development of the probe. Upon analyzing the reaction between glutathione and 5, 6 and 7 over time by HPLC, it was determined that 5 was least reactive with glutathione (ESI Fig. 4†). Based on these results, we concluded that the most suitable ligand structure for the new probe would be 5, which employed a chloroacetyl group at the reactive site and a trifluoromethyl group within the ligand portion. The reason 5 exhibited high reactivity against PYP-tag was thought to be due to the electron-withdrawing trifluoromethyl functionality elevating the reactivity of the chloroacetyl group34 and noncovalent interaction between PYP-tag and 5 that is absent in glutathione. Thus, incorporation of those structural features into the novel probe design was expected to accelerate the labeling reaction rate of the probe. This observation suggested that the probe could exhibit high stability during live cell imaging, an essential characteristic for the type of probe we were aiming to develop. Therefore, we decided the overall framework of the new probe for the PYP-tag-labeling system to have a structure where a fluorophore is conjugated to the new ligand which we named PCAF (PYP ligand with chloroacetamide and trifluoromethyl) via a linker (Fig. 2a). Another advantage this probe framework offers is that the fluorophore can be replaced easily with other functional elements such as substrates and inhibitors, allowing a quick expansion of the range of applications of this probe. As a first step in testing our concept, we synthesized a probe named PCAForange by linking the fluorophore 5-carboxytetramethylrhodamine (5-TAMRA) to the new basic framework (Fig. 2b and ESI Scheme 2†).
 | ||
| Fig. 2 PCAF probes and protein labelling. (a) Labeling of PYP-tag using a PCAF ligand (blue) employed in fluorescent probes. (b) The chemical structures of newly designed probes with a PCAF ligand, PCAFgreen, PCAForange, PCAFred and PCAFfar-red. (c) Spectroscopic properties of the probes and their complexes with PYP-tag. | ||
Protein labeling with the new probe PCAForange
As an initial characterization of PCAForange, we conducted in vitro experiments to test if the probe would bind to PYP-tag. Purified PYP-tag recombinantly produced in Escherichia coli was reacted with PCAForange in either buffer alone or HeLa cell lysate, and the reaction was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The results indicated that fluorescent bands with the molecular weight of PYP-tag were observed under both conditions only in the samples with PYP-tag and PCAForange present (Fig. 3a). Since the samples were heated and denatured before being subjected to electrophoresis, the probe and PYP-tag were considered to be covalently linked. For the labeling reaction performed in the lysate, the PYP-tag band yielded a strong probe-derived fluorescence signal. On the other hand, fluorescence signals from bands of other proteins were much weaker than that from the PYP-tag band, suggesting that non-specific labeling reactions were suppressed with the probe (Fig. 3b and ESI Fig. 5†). This result indicated that PCAForange would be able to label PYP-tag effectively in the presence of contaminating proteins. We also examined whether glutathione might influence the labeling reaction between PCAForange and PYP-tag. For this, we allowed PCAForange to react with PYP-tag both in the presence and absence of glutathione and analyzed the reactions by SDS-PAGE (ESI Fig. 6†). As a result, we were able to confirm that PCAForange was capable of labeling PYP-tag without being affected by glutathione that would be present in a live cell.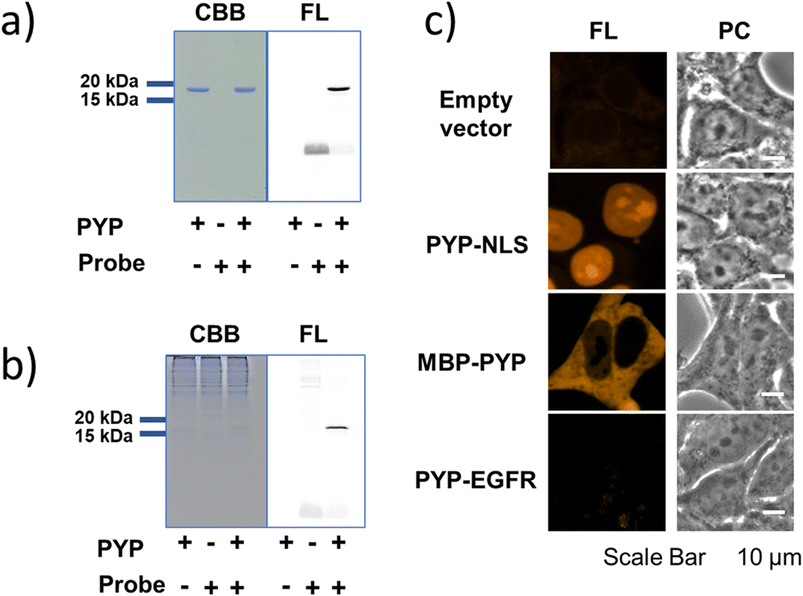 | ||
| Fig. 3 Protein labeling experiments with PCAForange. (a) In vitro protein labeling experiments, analyzed by SDS-PAGE. CBB: Coomassie brilliant blue image, FL: Fluorescence image. (b) PYP labeling reactions in HeLa cell lysate, analyzed by SDS-PAGE. PYP (1 μM) was reacted with PCAForange (2 μM) in HeLa cell lysate. (c) Live-cell imaging of PYP-NLS, MBP-PYP and PYP-EGFR fusion proteins expressed in HEK293T cells labeled with PCAForange. The cells were incubated with 500 nM PCAForange for 60 min at 37 °C, washed and imaged with a confocal fluorescence microscope. Fluorescence (FL, λex = 559 nm) and phase contrast (PC) images are shown. See Methods for details. | ||
The optical characteristics of PCAForange was determined by observing the spectra change of the reaction between PCAForange and PYP-tag (ESI Fig. 7†). PCAForange was found to increase its fluorescence intensity in terms of the fluorescence quantum yield ΦF by approximately two folds upon completion of the PYP-tag labeling reaction (Fig. 2c and ESI Fig. 7†). Lavis and Lionnet et al. reported that TAMRA exhibited a twisted intramolecular charge transfer (TICT) in the excited state, causing nonradiative decay, which is responsible for decrease in its fluorescence quantum yield.35 It is possible that TICT could be suppressed upon protein labeling likely due to a fluorophore–protein interaction, leading to increase in the fluorescence quantum yield. We also calculated the second-order rate constant for the labeling reaction by following the reaction using SDS-PAGE. Under the reaction condition examined, the PYP-tag labeling rate of PCAForange was faster than any of the conventional probes carrying a cinnamic acid-based ligand (ESI Fig. 8 and ESI Table 1†). Lastly, we performed fluorescence imaging to confirm the labeling capability of the new probe within a live cell. HEK293T cells were transformed to express PYP-tag fused with either a nuclear localization signal (NLS) that transfers protein molecules into the nucleus, maltose-binding protein (MBP) that exists in the cytoplasm or epidermal growth factor receptor (EGFR) that is a transmembrane protein for labeling with PCAForange. The results indicated that PCAForange was able to label PYP-NLS and MBP-PYP that were both expressed intracellularly, demonstrating that PCAForange could permeate through the cell membrane (Fig. 3c). We conducted imaging with and without cell washing after addition of PCAForange and compared the images, showing that the specific signals indicative of PYP-tagged proteins were obtained from both washed and unwashed cells expressing PYP-NLS or MBP-PYP (ESI Fig. 9†). In contrast, PYP-EGFR present on the plasma membrane was not labeled with PCAForange (Fig. 3c). The lack of labeling of PYP-EGFR was thought to arise from PCAForange being smaller than the conventional probes. Because of its small size, PCAForange permeated through the membrane and accumulated inside the cell faster than it could label PYP-tag on the membrane surface. This attribute of PCAForange to be able to selectively label intracellular PYP-tag gives a unique handle in tracking POIs that do not translocate to the membrane and remain only inside the cell, a trait particularly useful in studying the dynamics of POIs.
Comparison of live-cell imaging performance by the conventional probe TAMRA-DNB and the new probe PCAForange
Since PCAForange was shown to be able to label PYP-tag inside a live cell, we performed a comparative study using the conventional probe TAMRA-DNB29 (Fig. 4d), which has the same fluorophore TAMRA as PCAForange but is based on a conventional probe framework. First, we examined the time it takes for each of the probes to label nuclear-localized PYP-NLS expressed in live HEK293T cells. Under this experimental condition, it took TAMRA-DNB 80 minutes for the fluorescence intensity to reach saturation, while PCAForange reached saturation in only 30 minutes. Thus, the rate of intracellular labeling by PCAForange was more than twice as high as TAMRA-DNB (Fig. 4a and b). Similarly, comparison of the brightness of the labeled nuclei between TAMRA-DNB and PCAForange showed that PCAForange was able to label the nuclei 12 times brighter than TAMRA-DNB (Fig. 4c). To decipher why these two probes, which differed in their framework structure but shared the exact same fluorophore, exhibited such a substantial difference in their brightness, we examined the efficiencies of the two probes in labeling PYP-NLS expressed in a live cell. Live HEK293T cells expressing hemagglutinin tag-PYP tag fusion protein (HA-PYP) were labeled with PCAForange and TAMRA-DNB separately. Subsequently, the HA-tagged fusion proteins were isolated from the cell lysate by HA-tag immunoprecipitation. SDS-PAGE analyses of the isolated proteins indicated, while the fluorescence intensity of the TAMRA-DNB-labeled bands was weak, strongly fluorescent bands were clearly observed in PCAForange-labeled samples. Also, almost all of the HA-PYP band shifted its mobility as the PCAForange-labeling reaction proceeded (Fig. 4e). This result implied that PCAForange was able to label nearly all of HA-PYP present in the cell, suggesting that the labeling efficiency of PCAForange was substantially improved from the conventional probes. The observed enhancement in the labeling efficiency of PCAForange was thought to be achieved by the compounding effect of multiple factors, such as improved membrane permeability, reduced steric hindrance and increased water solubility, that were brought about by the structural modification of the probe. Based on these findings, we believe that, unlike the conventional probes, PCAForange is a probe that can unleash the full potential of PYP-tag as a tag protein.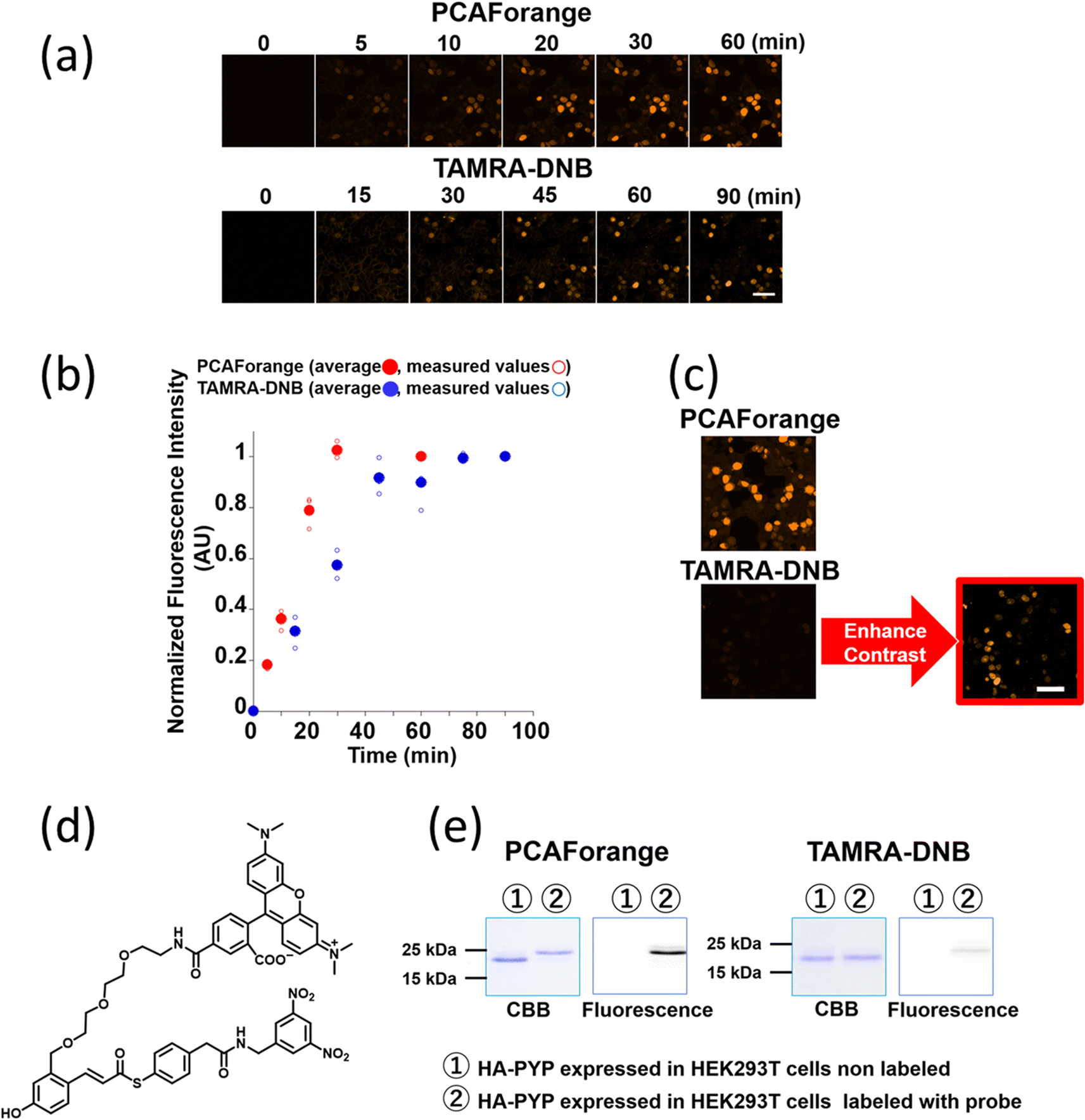 | ||
| Fig. 4 Comparison of the labeling performance of PCAForange against TAMRA-DNB. (a) Time-lapse imaging of PYP-NLS expressed in HEK293T cells labeled with 1 μM PCAForange or 1 μM TAMRA-DNB. Scale bar: 50 μm. (b) Quantification of the fluorescence intensity of PYP-NLS labeled with PCAForange (red solid circles) and TAMRA-DNB (blue solid circles). Data points plotted are means of the mean of triplicate measurements (open circles) with error bars representing the standard deviations. (c) Comparison of the brightness of PCAForange- and TAMRA-DNB-labeled PYP-NLS in HEK293T cells imaged at the same laser intensity. Due to low signal intensity, TAMRA-DNB-labeled cells could only be visualized with contrast enhancement. Scale bar: 50 μm. (d) Chemical structure of TAMRA-DNB. (e) SDS-PAGE analysis of labeling efficiency of PCAForange and TAMRA-DNB in live HEK293T cells. | ||
Development of multicolor PCAF probes
Our investigations described above denoted that the structural modifications incorporated into the new probe framework contributed significantly to the improvements in the labeling efficiency and reaction rate of the probe. Therefore, we decided to synthesize multicolor probes PCAFgreen, PCAFred and PCAFfar-red by coupling the PCAF ligand with fluorophores of different wavelengths ATTO 488, SiR650 and SiR700, respectively, to be used in combination with PCAForange (Fig. 2b, ESI Scheme 2†). Similar to PCAForange, PCAFgreen, PCAFred and PCAFfar-red were confirmed to be able to label PYP-tag in vitro, and glutathione had no effect on their labeling ability (Fig. 2c, ESI Fig. 6–8, 10 and ESI Table 1†). Lastly, we performed live-cell fluorescent imaging to examine the labeling capabilities of the new probes within live cells. For this study, we expressed PYP-NLS, MBP-PYP and PYP-EGFR in HEK293T cells and labeled them with PCAFgreen, PCAFred and PCAFfar-red in vivo. PCAFgreen was able to label PYP-tag on the cell surface (Fig. 5a), because it was the only probe carrying the membrane-impermeable dye ATTO 488. Experiments were also conducted to confirm that PCAFgreen is membrane-impermeable (ESI Fig. 11†). Thus, we were able to establish that PCAFgreen is a probe that can selectively label only the extracellular PYP-tag. On the other hand, similar to PCAForange, PCAFred and PCAFfar-red have the characteristics of selectively labeling intracellular POIs such as PYP-NLS and MBP-PYP but not those on the cell surface like PYP-EGFR (Fig. 5b and c). Thus, by combining these three probes with PCAFgreen, which is capable of selectively labeling POIs on the cell surface, it becomes possible to separately trace different copies of the same POI that exist at different locations. We surmised that such a probe set would be a powerful tool for analyzing the dynamics of a single POI that is known to travel across the cell membrane.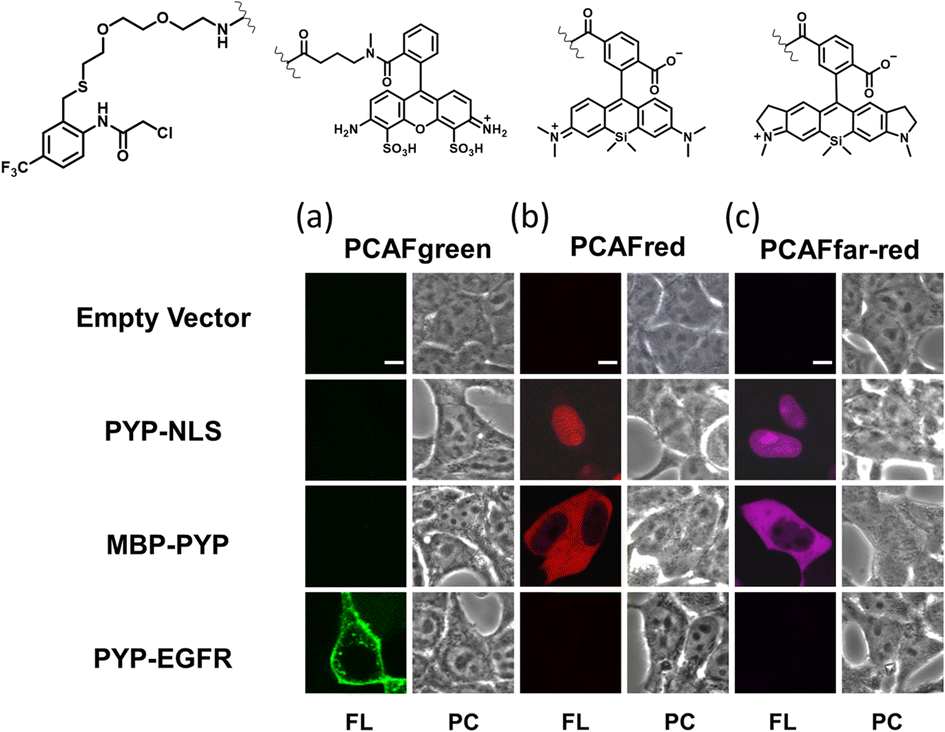 | ||
| Fig. 5 Imaging of PYP-NLS, MBP-PYP and PYP-EGFR with the PCAF probes in live HEK293T cells. HEK293T cells transfected with a plasmid carrying the gene of either the PYP-NLS, MBP-PYP or PYP-EGFR fusion protein were incubated with 500 nM of (a) PCAFgreen, (b) PCAFred or (c) PCAFfar-red for 60 min at 37 °C, washed and imaged under a confocal fluorescence microscope (designated by FL). Excitation wavelengths of λex = 473 nm was used for PCAFgreen, while λex = 635 nm was used for PCAFred and PCAFfar-red. | ||
Imaging of multiple localizations of PYP-GLUT4
With the set of new probes in hand, we decided to perform imaging of localization and dynamics of glucose transporter 4 (GLUT4). GLUT4 is a protein that contributes to the maintenance of homeostasis of the body by transiting from intracellular storage vesicles to the cell membrane to take up glucose in the presence of insulin stimulation and becoming intracellularly internalized in the absence of insulin stimulation.29,36–38 Since translocation and recycling of GLUT4 have been implicated in insulin resistance,30,31 understanding those phenomena and the underlying mechanisms would be vital to developing treatments and drug discovery for diseases associated with insulin resistance.38,39 Previously, it was possible to visualize the shift in GLUT4 localization with the use of fluorescent proteins or fluorescent probe-based labeling techniques. However, those methods detected all of the intracellular GLUT4 molecules expressed in the same timeline as a single identical signal, and it was not possible to recognize the behavioral changes of those molecules arising from their differential localization and dynamics. Hence, we postulated that previously unobservable signals and dynamics of GLUT4 could be visualized by performing highly sensitive multicolor imaging enabled by the advanced functionality and characteristics of our newly developed probes. Previously we reported that N-glycosylation at residue Asn57 of GLUT4 played an important role in the retention of GLUT4 on the membrane during insulin stimulation and showed that GLUT4 behaved differently with and without the N-glycan.29 For this study, a PYP-tag fusion protein of the GLUT4 mutant GLUT4N57Q (PYP-GLUT4N57Q) was used. GLUT4N57Q lacked N-glycosylation, because the asparagine-to-glutamine mutation at position 57 prevented the glycosylation of the molecule. To investigate whether the glycosylation affects not only the retention of GLUT4 on the cell membrane but also its intracellular localization, we constructed two stable cell lines based on HeLa cells, one expressing PYP-GLUT4N57Q and the other the wild-type counterpart, PYP-GLUT4WT. As to the PCAF probes to be used for the imaging experiments, we chose to use PCAFgreen to label PYP-GLUT4 that translocates to the cell surface. On the other hand, PCAForange was used to label PYP-GLUT4 that is located intracellularly. First, the stable cell lines expressing PYP-GLUT4N57Q and PYP-GLUT4WT were incubated for three hours in low-glucose Krebs–Ringer bicarbonate buffer (KRB) buffer.39 Subsequently, insulin and PCAFgreen were added to both cultures simultaneously to label majority of PYP-GLUT4 that translocated to the cell surface. Next, PCAForange was added to the cultures to selectively label the GLUT4 molecules that were present intracellularly. Upon insulin stimulation, the PCAFgreen signal was observed on the plasma membrane of cells expressing PYP-GLUT4WT as well as in the cytoplasm (Fig. 6a and ESI Fig. 12†). No fluorescence signal was found on the plasma membrane in the PYP-GLUT4N57Q cell line (Fig. 6a and ESI Fig. 12†). These results were in agreement with our previous finding.29 To further identify the intracellular PCAFgreen signal, we performed immunostaining using antibody (ESI Fig. 13†). First, stable strains of PYP-GLUT4WT and PYP-GLUT4N57Q HeLa cells were cultured and labelled with PCAFgreen, a cell-membrane impermeant probe, in the absence of insulin. The cells were then fixed and imaged using an anti-FLAG antibody and its secondary antibody conjugated to Cy3. The FLAG tag is included in the construct of PYP-GLUT4 proteins. PCAFgreen fluorescence was confirmed in the areas where Cy3 fluorescence was observed. No fluorescence signal was observed from cells not expressing PYP-GLUT4 proteins. These results demonstrate that the PCAFgreen signal observed in cells is derived not from nonspecific off-target proteins but from PYP-GLUT4WT or PYP-GLUT4N57Q, strongly indicating that PCAFgreen specifically labels transiently exocytosed PYP-GLUT4 proteins on the plasma membrane, which are then internalized, because PCAFgreen is a cell impermeant probe. Next, we looked at the distribution of localization of the PYP-GLUT4 molecules. Signals from both PCAForange and PCAFgreen were observed inside the cell. However, unlike in the previous study, signal from the membrane-impermeable probe was also observed in the PYP-GLUT4N57Q cells even when there was no insulin stimulation (Fig. 6a and ESI Fig. 12†). The reason for the different outcome was thought to be because PCAFgreen was able to efficiently label low level of PYP-GLUT4 that the previous probes could not label sufficiently. The live imaging results demonstrated that the use of distinct dyes afforded a new insight that, among PYP-GLUT4N57Q which lacks N-glycosylation, those that went through transient exocytosis to the cell surface and those that remained inside the cell localized differently within the cell.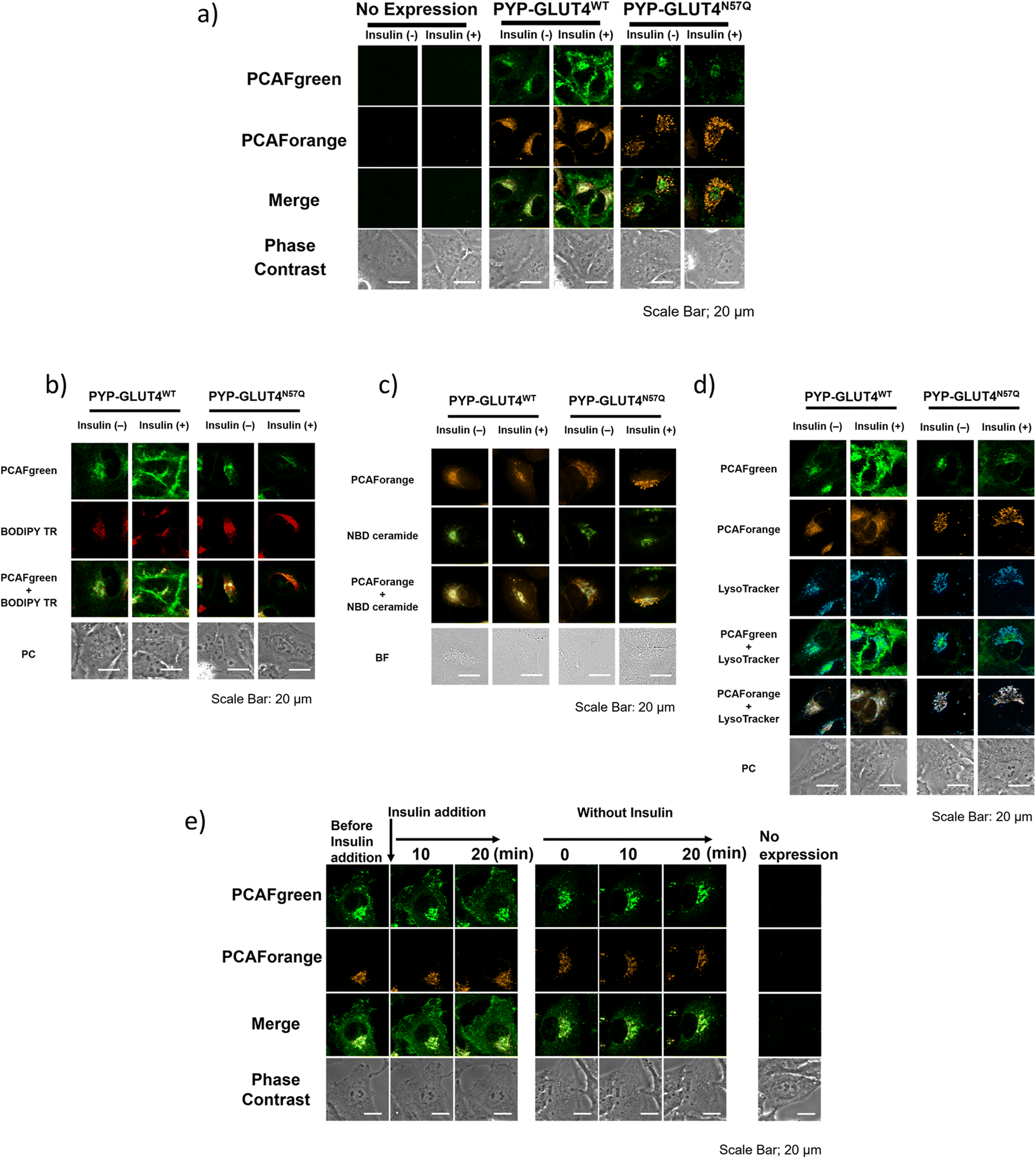 | ||
| Fig. 6 Multicolor live-cell imaging with the PCAF color palette to examine the localization of PYP-GLUT4WT and PYP-GLUT4N57Q with or without insulin stimulation. (a) HeLa cells stably expressing PYP-GLUT4WT and PYP-GLUT4N57Q along with un transfected HeLa cells (no expression) were labeled with 2 μM PCAFgreen or 1 μM PCAForange with or without insulin stimulation and visualized under a confocal fluorescence microscope. (b) Fluorescence and phase-contrast (PC) images of HeLa cells expressing PYP-GLUT4WT (left column) or PYP-GLUT4N57Q (right column) co-stained with PCAFgreen and BODIPYTM TR ceramide (BODIPY TR) in the presence (+) or absence (−) of insulin. For BODIPY TR labeling, BODIPYTM TR ceramide complexed to BSA (Invitrogen) was used. PCAFgreen: λex = 473 nm; BODIPY TR: λex = 559 nm. (c) Fluorescence and bright-field (BF) images of HeLa cells expressing PYP-GLUT4WT (left column) or PYP-GLUT4N57Q (right column) co-stained with PCAForange and NBD C6-ceramide (NBD ceramide) in the presence (+) or absence (−) of insulin. For NBD ceramide labeling, NBD C6-ceramide (6-((N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)hexanoyl)sphingosine) (Invitrogen) was used. PCAForange: λex = 470 nm; NBD ceramide: λex = 405 nm. (d) Fluorescence and phase-contrast (PC) images of HeLa cells expressing PYP-GLUT4WT (left column) or PYP-GLUT4N57Q (right column) co-stained with PCAFgreen, PCAForange and LysoTracker® Deep Red (LysoTracker, Invitrogen) in the presence (+) or absence (−) of insulin. PCAFgreen: λex = 473 nm; PCAForange: λex = 559 nm. (e) Time-lapse imaging of the dual dynamics of PYP-GLUT4WT in a single cell. | ||
We also focused on the difference in the intracellular signal distribution between the cells expressing PYP-GLUT4N57Q and PYP-GLUT4WT (Fig. 6a and ESI Fig. 12†). In the PYP-GLUT4WT cells, signals from PYP-GLUT4 that underwent a transient membrane transfer and those that did not were equally distributed inside the cells. In contrast, localization of those two signals was clearly segregated in the PYP-GLUT4N57Q cells. As quantitative analyses of localization of intracellular PCAFgreen- and PCAForange-labeled proteins, we determined Pearson's correlation coefficient using 10 cells and provided box-and-whisker plots to show the degree of their variation, indicating that both fractions of PYP-GLUT4WT proteins labeled with PCAFgreen or PCAForange are highly colocalized, whereas two different fractions of PYP-GLUT4N57Q proteins labeled with either of the probes are hardly colocalized (ESI Fig. 14†). This finding indicated that the intracellular distribution of PYP-GLUT4 differed depending on the presence of the N-glycan, suggesting that the N-glycan is controlling the intracellular localization of PYP-GLUT4. In addition, the cells were co-stained with fluorescence dyes for labeling the Golgi apparatus (BODIPY TR and NBD ceramide) and lysosomes (LysoTracker) to examine the organelle localization of PYP-GLUT4WT and PYP-GLUT4N57Q. The PCAFgreen signals in the cell of PYP-GLUT4WT, mostly colocalized with the Golgi-Tracker signals, indicating that PYP- PYP-GLUT4WT that went through a transient membrane transfer partially localized to the Golgi apparatus after internalization (Fig. 6b and c). On the other hand, in the PYP-GLUT4N57Q cells where localization of the PCAFgreen and PCAForange signals were clearly segregated, the majority of the PCAForange signals were found to localize with the lysosomes (Fig. 6d). This localization of the PCAForange signals to the lysosomes suggests that the cell recognized PYP-GLUT4N57Q to be different from the wild-type PYP-GLUT4, labeled it as an abnormal protein and sent it down the degradation pathway. In comparison, localization of PYP-GLUT4WT was shown to be globally distributed within the cell (Fig. 6a and ESI Fig. 12†), presumably because it resides at various intracellular locations and function as a normal protein. These results demonstrated that the N-glycan of PYP-GLUT4 is involved in quality control of PYP-GLUT4 and dictates the pathway PYP-GLUT4 takes within the cell. While these observations are in agreement with previous reports that indicated that the N-glycan plays an important role in quality control of PYP-GLUT4WT,34 this is the first report that actually visualized the distribution of different types of PYP-GLUT4 molecules within a live cell using fluorescent probes of different wavelengths.
Next, we compared quantitatively the intracellular mean pixel intensity to determine whether the N-glycan exerted control over PYP-GLUT4 that would undergo transient exocytosis. The quantitative comparison showed that the intracellular mean pixel intensity of the cells expressing N-glycanless PYP-GLUT4N57Q did not change regardless of whether the cells were given insulin stimulation or not (p = 0.1436) (ESI Fig. 15†). The results suggest that transient exocytosis of PYP-GLUT4N57Q occurs regardless of insulin stimulus, indicating that PYP-GLUT4N57Q is not immediately transported to the lysosomes after expression. For the PYP-GLUT4WT-expressing cells, the intracellular mean pixel intensity was clearly lower when insulin stimulation was given (p < 0.0001). Thus, retention of PYP-GLUT4WT on the cell membrane appeared to be correlated with the observed intracellular mean pixel intensity, we proceed to examine how insulin stimulation affects PYP-GLUT4WT present in the cell. Initially, both PCAFgreen and PCAForange probes were added to the cells without insulin stimulation and allowed accumulation of the labelled proteins within the cells. PCAFgreen-labeled PYP-GLUT4WT clearly became accumulated on the cell membrane within approximately 20 minutes after the insulin stimulation (Fig. 6e). We quantified the ratio of plasma membrane fluorescence intensity to that of a whole cell. The results also showed that only PCAFgreen-labeled PYP-GLUT4WT increased the ratio after addition of insulin (ESI Fig. 16†). Since labeling by PCAFgreen serves as a log of prior transient exocytosis, the PCAFgreen-labeled PYP-GLUT4WT on the membrane must have undergone membrane translocation for the second time and remained on the membrane. On the contrary, the signals from PCAForange that remained inside the cell did not translocate to the membrane even when insulin stimulation was given. These results show that we successfully visualized PYP-GLUT4WT that assumed at least two different dynamic modes with different colors using the newly developed PCAF probes. Also, the quantitative measurements of the intracellular mean pixel intensity revealed that the loss of intracellular mean pixel intensity occurred only when PYP-GLUT4WT was given insulin stimulation. This observed loss of intracellular mean pixel intensity was speculated to be caused by PYP-GLUT4WT that was labeled with PCAFgreen during the initial transient exocytosis becoming retained on the membrane after going through membrane translocation at least twice within one hour (Fig. 6e).
Discussion
In the current study, we carried out chemistry-based strategic designing of probe structure for developing high-performing fluorogenic probe capable of superior PYP-tag-labeling performance. The outcome was the creation of a PCAF ligand that was capable of fast and efficient labeling of the target molecule. A chemically interesting finding is that the PCAF ligand binds to PYP-tag with much higher reactivity than glutathione, enabling efficient protein labeling in living cells. More importantly, some of the probes with the PCAF ligand showed unique selectivity toward intracellular proteins. When used together with a PCAF probe carrying a cell-impermeable dye, we can separately label targets on the outer cell membrane and those inside the cells. These intracellular- and cell surface-selective probes enabled us to visually identify multiple localizations of PYP-GLUT4 molecules. We uncovered that intracellular PYP-GLUT4 molecules that were thus far detected as a same single signal were actually comprised of those that translocated from the membrane and those that remained within the cell, which were undistinguishable previously. Also, the probes allowed us to newly discover that the regular PYP-GLUT4WT and glycosylation-deficient PYP-GLUT4N57Q mutant localized differently within a cell, and the observed localization patterns of those molecules gave an insight into the role N-glycan plays in the biological quality control process of the cell.A previous report suggests that GLUT4N57Q undergoes ER-associated protein degradation (ERAD), while the protein escaped from the quality control system could reach the cell surface.37 Our results also show that some fractions of PYP-GLUT4N57Q proteins are transiently translocated to the plasma membrane, while others are present in the lysosomes, indicating that the former fraction is escaped from protein degradation, and the latter is subjected to the quality control system, consistent with the previous study. On the other hand, ERAD system generally utilizes proteasome. Although we do not exclude a possibility that PYP-GLUT4N57Q undergoes proteasome-mediated degradation, our results suggest that the protein undergoes lysosomal degradation. In fact, GLUT4 is reported to be decomposed in the lysosomes rather than the proteasome, according to the other study.40 Furthermore, a recent study reported the presence of ER-to-lysosome-associated degradation (ERLAD) system, in which proteins in the ER are sorted to the lysosome for degradation.41 The same group indicated that N-glycan is involved in the determination of the degradation pathway, ERAD or ERLAD.42 Thus, it is suggested that PCAForange could detect the translocation of PYP-GLUT4N57Q, which undergoes ERLAD, while PCAFgreen could image the proteins that could be escaped from ERLAD. In addition, since most fractions of PYP-GLUT4WT were not shown to be translocated to the lysosomes in this experimental time scale, it is also suggested that N-glycan influences the intracellular fate of GLUT4.
We further identified that PYP-GLUT4WT engaged in at least two different types of behaviors existed by our probes in different colors. One type of PYP-GLUT4WT molecules migrated to the membrane in the absence of insulin stimulation and became internalized after going through a transient exocytosis. In response to a subsequent insulin stimulation, those molecules underwent membrane translocation at least twice within one hour, and then remained on the membrane, whereas another type did not undergo membrane translocation and remained within the cell. The rapid and efficient labeling with PCAFgreen captured the transient exocytosis and enabled detection of the multiple membrane translocation. As demonstrated in this study, we designed and developed a new set of probes based on chemical principles and established a powerful tag protein labeling technology. The in-depth application of the labeling technology to a single protein is invaluable as a key for discovering what actually happens to the protein during its life cycle, such as post-translation modification process and recycling pathway. The technology we developed here would become a valuable tool in obtaining deeper understanding and making new discoveries of the complex biological phenomena that take place within a living organism.
Data availability
The data supporting the findings of this study can be found in both the main text and the ESI.†Author contributions
M. N. and M. U. performed experiments. M. N. and M. U. and Y. H. analyzed the data. M. N. and Y. H. co-wrote the initial draft. M. N., Y. H. and K. K. revised the final manuscript. The project was supervised by Y. H. and K. K.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the JSPS KAKENHI (Grant No. JP17H06409 “Frontier Research on Chemical Communications”, JP21H04706 and JP23H048810 to K. K.); JSPS KAKENHI (Grant No. JP20H02879, JP21K19048, JP21H05073 and JP21H05075 to Y. H.), Toray Science Foundation (19-6008 to Y. H.) and the Uehara Memorial Foundation. This work was inspired by the international and interdisciplinary environments of JSPS CORE-to-CORE Program “Asian Chemical Biology Initiative”. JSPS-ISF Joint Research Program (JPJSBP120218404 to K. K.).References
- F. Ardito, M. Giuliani, D. Perrone, G. Troiano and L. Lo Muzio, Int. J. Mol. Med., 2017, 40, 271–280 CrossRef CAS.
- W. Ren, Y. Sun and K. Du, Biochem. Biophys. Res. Commun., 2015, 460, 709–714 CrossRef CAS PubMed.
- D. Shental-Bechor and Y. Levy, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 8256–8261 CrossRef CAS PubMed.
- M. A. Kukuruzinska and K. Lennon, Crit. Rev. Oral Biol. Med., 1998, 9, 415–448 CrossRef CAS PubMed.
- D. Y. Lee, C. Teyssier, B. D. Strahl and M. R. Stallcup, Endocr. Rev., 2005, 26, 147–170 CrossRef CAS PubMed.
- A. Drazic, L. M. Myklebust, R. Ree and T. Arnesen, Biochim. Biophys. Acta, 2016, 1864, 1372–1401 CrossRef CAS PubMed.
- Y. Haga, K. Ishii, K. Hibino, Y. Sako, Y. Ito, N. Taniguchi and T. Suzuki, Nat. Commun., 2012, 3, 1–7 Search PubMed.
- T. Tai, Methods Enzymol., 2006, 415, 20–30 CAS.
- O. M. Crook, T. Smith, M. Elzek and K. S. Lilley, Proteomics, 2020, 20, e1900392 CrossRef PubMed.
- T. Endo and H. Sakaue, Biochem. Soc. Trans., 2019, 47, 1269–1277 CrossRef CAS PubMed.
- K. Sasaki and H. Yoshida, Cell Struct. Funct., 2019, 44, 85–94 CrossRef CAS PubMed.
- K. K. Sadhu, S. Mizukami, S. Watanab and K. Kikuchi, Chem. Commun., 2010, 46, 7403–7405 RSC.
- C. Jing and V. W. Cornish, ACS Chem. Biol., 2013, 8, 1704–1712 CrossRef CAS PubMed.
- M. Kumauchi, M. T. Hara, P. Stalcup, A. Xie and W. D. Hoff, Photochem. Photobiol., 2008, 84, 956–969 CrossRef CAS PubMed.
- C. J. Zhang, L. Li, G. Y. Chen, Q. H. Xu and S. Q. Yao, Org. Lett., 2011, 13, 4160–4163 CrossRef CAS PubMed.
- X. Sun, A. Zhang, B. Baker, L. Sun, A. Howard, J. Buswell, D. Maurel, A. Masharina, K. Johnsson, C. J. Noren, M. Xu and I. R. Corrêa Jr, Chembiochem, 2011, 12, 2217–2226 CrossRef CAS PubMed.
- T. Liu, P. Hsieh, Y. Zhuang, C. Hsia, C. Huang, H. Lai, H. Lin, I. Chen, H. Hsu and K. Tan, ACS Chem. Biol., 2014, 9, 2359–2365 CrossRef CAS PubMed.
- H. N. Duc and X. Ren, Bio-protoc., 2017, 7, e2526 Search PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa Jr, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed.
- Gr. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed.
- M. Yoshida, H. Chida, F. Kimura, S. Yamamura and K. Tawa, Micromachines, 2020, 11, 604 CrossRef PubMed.
- Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610–16611 CrossRef CAS PubMed.
- Y. Hori, K. Nakaki, M. Sato, S. Mizukami and K. Kikuchi, Angew. Chem., Int. Ed., 2021, 51, 5611–5614 CrossRef PubMed.
- Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360–12365 CrossRef CAS PubMed.
- Y. Hori, S. Hirayama, M. Sato and K. Kikuchi, Angew. Chem., Int. Ed., 2015, 54, 14368–14371 CrossRef CAS PubMed.
- Y. Hori, N. Otomura, A. Nishida, M. Nishiura, M. Umeno, I. Suetake and K. Kikuchi, J. Am. Chem. Soc., 2018, 140, 1686–1690 CrossRef CAS PubMed.
- T. E. Meyer, Biochim. Biophys. Acta, 1985, 806, 175–183 CrossRef CAS PubMed.
- S. Hirayama, Y. Hori, Z. Benedek, T. Suzuki and K. Kikuchi, Nat. Chem. Biol., 2016, 12, 853–859 CrossRef CAS PubMed.
- W. T. Garvey, L. Maianu, J. H. Zhu, G. Brechtel-Hook, P. Wallace and A. D. Baron, J. Clin. Invest., 1998, 101, 2377–2386 CrossRef CAS PubMed.
- F. S. Thong, C. B. Dugani and A. Klip, Physiology, 2005, 20, 271–284 CrossRef CAS PubMed.
- S. M. Al-Kindy and J. N. Miller, Luminescence, 2011, 26, 148–152 CrossRef CAS PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS PubMed.
- K. Chiba, Y. Hashimoto and T. Yamaguchi, Chem. Pharm. Bull., 2016, 64, 1647–1653 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- D. Leto and A. R. Saltiel, Nat. Rev. Mol. Cell Biol., 2012, 13, 383–396 CrossRef CAS PubMed.
- Y. Haga, K. Ishii and T. Suzuki, J. Biol. Chem., 2011, 286, 31320–31327 CrossRef CAS PubMed.
- J. B. Sadler, N. J. Bryant, G. W. Gould and C. R. Welburn, Int. J. Mol. Sci., 2013, 14, 9963–9978 CrossRef PubMed.
- K. G. Stenkula, V. A. Lizunov, S. W. Cushman and J. Zimmerberg, Cell Metab., 2010, 12, 250–259 CrossRef CAS PubMed.
- J. Shi and K. V. Kandror, Dev. Cell, 2005, 9, 99–108 CrossRef CAS PubMed.
- I. Fregno, E. Fasana, T. J. Bergmann, A. Raimondi, M. Loi, T. Soldà, C. Galli, R. D'Antuono, D. Morone, A. Danieli, P. Paganetti, E. van Anken and M. Molinari, EMBO J., 2018, 37, e99259 CrossRef PubMed.
- I. Fregno, E. Fasana, T. Soldà, C. Galli and M. Molinari, EMBO J., 2021, 40, e107240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00724c |
| This journal is © The Royal Society of Chemistry 2023 |