
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific chirality-conferred structural compaction differentially mediates the cytotoxicity of Aβ42†
Gongyu
Li‡
 *ac,
Chae Kyung
Jeon‡
d,
Min
Ma‡
b,
Yifei
Jia
a,
Zhen
Zheng
f,
Daniel G.
Delafield
b,
Gaoyuan
Lu
b,
Elena V.
Romanova
e,
Jonathan V.
Sweedler
e,
Brandon T.
Ruotolo
*d and
Lingjun
Li
*b
*ac,
Chae Kyung
Jeon‡
d,
Min
Ma‡
b,
Yifei
Jia
a,
Zhen
Zheng
f,
Daniel G.
Delafield
b,
Gaoyuan
Lu
b,
Elena V.
Romanova
e,
Jonathan V.
Sweedler
e,
Brandon T.
Ruotolo
*d and
Lingjun
Li
*b
aState Key Laboratory of Pharmaceutical Chemical Biology, Research Center for Analytical Science and Tianjin Key Laboratory of Biosensing and Molecular Recognition, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: ligongyu@nankai.edu.cn
bSchool of Pharmacy and Department of Chemistry, University of Wisconsin–Madison, 777 Highland Ave., Madison, WI 53705, USA. E-mail: lingjun.li@wisc.edu
cHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China
dDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: bruotolo@umich.edu
eDepartment of Chemistry and The Beckman Institute for Advanced Science and Technology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA
fSchool of Pharmacy, Tianjin Medical University, Tianjin 300070, China
First published on 8th May 2023
Abstract
Growing evidence supports the confident association between distinct amyloid beta (Aβ) isoforms and Alzheimer's Disease (AD) pathogenesis. As such, critical investigations seeking to uncover the translational factors contributing to Aβ toxicity represent a venture of significant value. Herein, we comprehensively assess full-length Aβ42 stereochemistry, with a specific focus on models that consider naturally-occurring isomerization of Asp and Ser residues. We customize various forms of D-isomerized Aβ as natural mimics, ranging from fragments containing a single D residue to full length Aβ42 that includes multiple isomerized residues, systematically evaluating their cytotoxicity against a neuronal cell line. Combining multidimensional ion mobility-mass spectrometry experimental data with replica exchange molecular dynamics simulations, we confirm that co-D-epimerization at Asp and Ser residues within Aβ42 in both N-terminal and core regions effectively reduces its cytotoxicity. We provide evidence that this rescuing effect is associated with the differential and domain-specific compaction and remodeling of Aβ42 secondary structure.
Introduction
Naturally occurring D-amino acid substitution, also known as amino acid D-epimerization, has been observed in many disease-associated peptides and proteins, including amyloid beta (Aβ), one of the putative biomarkers and drug targets for Alzheimer's disease (AD).1–6 Aβ is of significant interest due to the prevalence of post-translational D-epimerization in AD brain samples.7 In brain samples extracted from AD patients, the two most common residues isomerized are aspartic acid and serine.5,8,9 Notably, previous studies have experimentally demonstrated that Aβ D-epimerization is age-dependent and that such isomerization is long lived.2,5,8 Although most prior reports aimed at AD biomarker and drug target discovery lack the ability to detect and evaluate the impact of D-isomerized Aβ due to technical limitations,5,10–12 an increasing number of recent reports have focused on the role of Aβ D-epimerization in AD through novel assays capable of D-isomerized Aβ-targeted identification and purification.7,13,14 Recently, directed D-epimerization has also been intensively explored in terms of its impact on the cytotoxicity and oligomerization of Aβ.3,4,15–17 Notably, point D-amino acid substitution can effectively reduce Aβ cytotoxicity, highlighting the critical role of Ser26 residue.16,17 Meanwhile, Makarov and coworkers have found that, isomerization at Asp7 residue alone increases the amyloid cytotoxicity in both mouse models and human neuronal cells.18,19 Although a systematic chirality survey of Asp and Ser residues within Aβ42 is still lacking, emerging data supports the prospects of a chiral regulation mechanism capable of rescuing Aβ in cytotoxicity,3,15–17 which may be further probed in the context of enzymatic regulation and phage display.20,21 Remaining challenges confronting a systematic survey concerning the chiral effects of Asp and Ser residues of Aβ42 consist not only of those associated with the analytical discrimination and preparation of Aβ42 stereoisomers, but also of the inadequate molecular understanding of the combinatorial structural consequences of D-epimerization at all Asp and Ser residues within different domains of Aβ42.While many prior reports have described technical advancements associated with the chiral discrimination and separation of D-amino acid containing peptides,22–30 there remains a dearth of tools capable of targeting Aβ42 stereochemistry. Traditional biophysical techniques frequently suffer from an inability to reconcile the mixture states created from the relatively low structural stability and high aggregation propensity of Aβ42. These challenges are amplified when one maps the subtle structural impacts induced by D-epimerization at few sites onto the mixture of oligomeric states created within most Aβ samples. Ion mobility-mass spectrometry (IM-MS) has increasingly become an important alternative for the chiral separation of Aβ stereoisomers.13,31–37 IM-MS offers high analytical speed, low sample consumption and the ability to resolve small structural differences in peptide analytes, driven by recent technological advancements in IM-MS.34–39 While molecular dynamics (MD) simulations have been combined with IM-MS to reveal the structural consequences of D-epimerization within small neuropeptides,40 Li et al. has recently reported a multi-dimensional IM-MS (md-IM-MS)-based structural analysis strategy, based on the metal-bound chiral amplification and oligomer-resolved data integration method, to facilitate the study of the chiral effects on monomer structure, oligomeric propensity and receptor binding for Aβ fragments.34,41 However, results from truncated Aβ (e.g. Aβ N-terminus and core region fragments) cannot be reliably extrapolated to predict those for the full length bioactive Aβ forms (e.g. Aβ42).
To fully probe the chiral effects in full length, bioactive Aβ42 including the differential roles of the N terminal and core regions, we herein provide an improved md-IM-MS34,41 approach for the systematic study of the structural consequences of domain-specific Asp/Ser D-epimerization within Aβ42 (Table 1 & Scheme S1†). Cytotoxicity of Aβ isoforms in the context of neuronal cell lines were examined using a series of custom-synthesized peptides, which were also subjected to structural analysis using an improved md-IM-MS platform based on composite IM measurements across multiple instrument platforms. Analytical challenges facing the MS characterization of full length Aβ42 have been successfully tackled through an improved sample preparation workflow. Moreover, IM-MS-guided replica exchange MD (REMD) simulations were employed to construct and refine the 3D structures of different Aβ42 stereoisomers. Our analysis reveals that co-D-epimerization at Asp and Ser residues of Aβ42 in both the N-terminal and core region effectively and synergistically reduces its cytotoxicity, seemingly related to the differential and domain-specific changes in both overall and local 3D shapes, among which structural compaction exhibits the most significant effect.
| D-Residues | N-terminus Aβ10/Aβ16a | Core region Aβ(17–36)b | Full length Aβ42c |
|---|---|---|---|
| a Aβ10/Aβ16: DAEFRHDSGY/DAEFRHDSGYEVHHQK. b Aβ(17–36): LVFFAEDVGSNKGAIIGLMV. c Aβ42: DAEFRHDSGY EVHHQKLVFF AEDVGSNKGA IIGLMVGGVV IA. | |||
| WT | — | — | — |
| dD | Asp1/7 | Asp23 | Asp1/7/23 |
| dS | Ser8 | Ser26 | Ser8/26 |
| dDdS | Asp1/7Ser8 | Asp23Ser26 | Asp1/7/23Ser8/26 |
Results and discussion
In vitro characterization of Aβ stereoisomer-receptor binding affinity
First, we customized a variety of Aβ stereoisomers with D-Asp and/or D-Ser residues to systematically construct site-specific, domain-constrained isomer models. As shown in Table 1, dD Aβ refers to Aβ isoforms with all Asp D-isomerized, dS Aβ refers to Aβ isoforms with all Ser D-isomerized, while dDdS Aβ refers to Aβ with all Asp and Ser co-D-isomerized. Scheme S1† shows the sequence and chemical structure for Aβ42, where N-terminal and core regions are highlighted in different colors. To be more specific, Aβ10/Aβ16 and Aβ(17–36) are designated as N-terminal and core peptides, respectively, all of which are synthesized in four isoforms, WT, dD, dS and dDdS (Table 1). In total, we studied 12 Aβ stereoisomers in three groups to inspect domain-constrained chiral effects.Immunoaffinity-based isolation is one of the most popular strategies in AD biomarker purification, which heavily relies on the binding specificity and affinity between the chosen antibody and the target (e.g. Aβ).7,10 Not surprisingly, we found that standard Aβ antibodies exhibit significantly varied performance in their ability to purify Aβ stereoisomers, as revealed by surface plasmon resonance (SPR) binding curves (Fig. 1). While in Fig. 1A the binding affinity for WT Aβ42 to its specific antibody, 6E10, is calculated to be around 5.55 μM, isomerization at both Asp and Ser residues significantly weakens Aβ42 binding. This series of experimental observations starkly illustrate the challenges involved in quantitatively evaluating the contribution from Aβ42 stereoisomers in AD pathology.
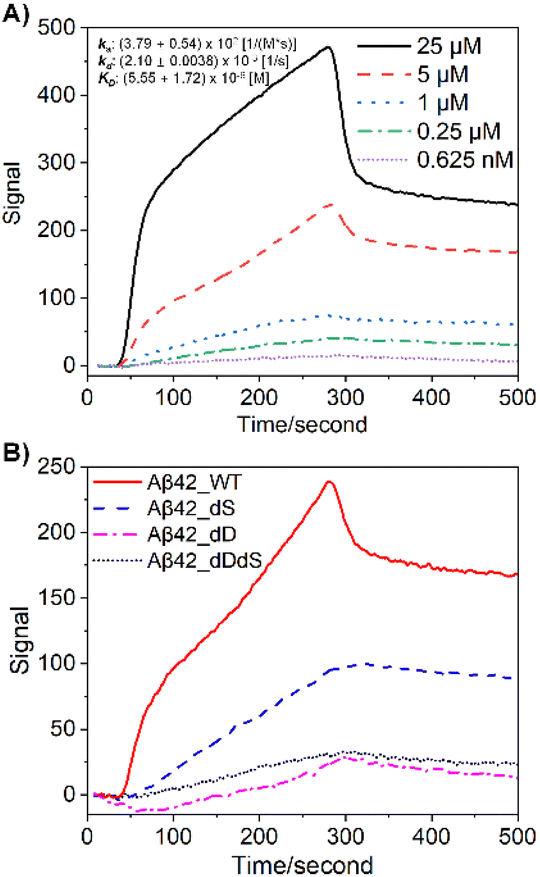 | ||
| Fig. 1 Differential antibody binding affinity mediated by site-specific chiral inversion at full-length Aβ42. (A) SPR response curves for various levels of WT Aβ42 with Aβ antibody, 6E10, immobilized on a COOH sensor chip. Calculated binding kinetics information: ka, (3.79 ± 0.54) × 102 1/(M*s); kd, (2.10 ± 0.0038) × 10−3 1/s; KD, (5.55 ± 1.72) × 10−6 M. (B) Response curves for WT and differentially D-isomerized Aβ42 at 5 μM each, showing the significantly decreased antibody binding affinity upon D-epimerization. | ||
Metal ions, particularly copper (Cu2+), have been considered as an important regulator for Aβ structural flexibility and self-assembly.34,42,43 As such, we performed a series of native IM-MS experiments on Aβ42-Cu2+ complexes in order to compare the copper binding affinities over differentially D-isomerized Aβ42 isoforms. Representative mass spectra in Fig. S1† support the differential copper binding capabilities (including affinity and stoichiometry) of Aβ42 stereoisomers. Further, triplicate binding affinity measurements enable quantitative comparison of their copper-binding affinities. As shown in Table 2, native MS reports strong copper binding to WT Aβ42 to form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex, with KD values of ∼40 nM, with significantly lower binding affinity measured for subsequent copper binding events (∼27 μM). While the formation of 1:2 Cu:Aβ complexes are not significantly affected upon site-specific isomerization (fold changes <2), 1:1 binding affinity diminishes significantly, resulting in a 7–34 fold change in Cu binding KD. Notably, the largest fold change was observed for dD Aβ42, which likely alters the known copper-binding domain located at N-terminus of Aβ42. Interestingly, the Cu affinity recorded for dDdS Aβ42 exhibits stronger affinity than the peptides carrying individual isomerized residues, which seemingly indicates the ability of Aβ42 to structurally compensate to create an altered binding pocket for Cu binding. In addition, these Cu-binding data may have implications for the biological activities of Aβ42 stereoisomers, as discussed above, Cu-regulated self-assembly and oligomerization is one of the putative molecular mechanisms for regulating Aβ neurotoxicity.42,44
:1 complex, with KD values of ∼40 nM, with significantly lower binding affinity measured for subsequent copper binding events (∼27 μM). While the formation of 1:2 Cu:Aβ complexes are not significantly affected upon site-specific isomerization (fold changes <2), 1:1 binding affinity diminishes significantly, resulting in a 7–34 fold change in Cu binding KD. Notably, the largest fold change was observed for dD Aβ42, which likely alters the known copper-binding domain located at N-terminus of Aβ42. Interestingly, the Cu affinity recorded for dDdS Aβ42 exhibits stronger affinity than the peptides carrying individual isomerized residues, which seemingly indicates the ability of Aβ42 to structurally compensate to create an altered binding pocket for Cu binding. In addition, these Cu-binding data may have implications for the biological activities of Aβ42 stereoisomers, as discussed above, Cu-regulated self-assembly and oligomerization is one of the putative molecular mechanisms for regulating Aβ neurotoxicity.42,44
| Aβ42 | K D1 [μM] (+1 Cu2+) | K D2 [μM] (+2 Cu2+) | Fold change | |
|---|---|---|---|---|
| K D1 | K D2 | |||
| a All data in this table represents triplicate measurements with SD values for error ranges. The calculation of KD fold changes is based on the normalization of WT group to be 1.0. | ||||
| WT | 0.04 ± 0.01 | 27.04 ± 1.88 | 1.00 | 1.00 |
| dD | 1.50 ± 0.02 | 33.31 ± 3.26 | 34.12 | 1.23 |
| dS | 0.70 ± 0.14 | 46.35 ± 3.85 | 15.84 | 1.71 |
| dDdS | 0.31 ± 0.06 | 19.77 ± 2.63 | 7.15 | 0.73 |
Cytotoxicity characterization of Aβ stereoisomers
Next, we aimed to interrogate and compare the biological impact of chiral inversion on Aβ42, targeting multiple sites of different functional domains. Cell viability experiments were then performed by using N2a cell lines. As shown in Fig. 2A, incubation of N2a cells with 20 μM WT Aβ42 reduced the cell viability by ∼82% and the half maximal inhibitory concentration (IC50) as calculated from the S curve was estimated to be ∼13 μM. While previous studies have reported effective reduction in cytotoxicity through chiral inversion of all amino acids within Aβ42,3 it was reported that isomerization at Asp7 residue alone increased the cytotoxicity burden.18,19 Our results demonstrate that D-epimerization of a few specific Asp and Ser residues may be sufficient to reduce its cytotoxicity. As WT Aβ42 showed high cytotoxicity at 20 μM and the trend lines for D-isomerized Aβ42 are well separated, we thus conducted experiments with concentrations no higher than 100 μM. The IC50 value for dD Aβ42 is ∼2.6 folds higher than WT Aβ42 while the cell viability only reduced by ∼18% with 20 μM incubation, suggesting a significantly reduced cytotoxicity within N2a cell line. However, the IC50 values for dS Aβ42 and dDdS Aβ42 cannot be derived from the concentration range studied herein. Noting their reduced viabilities are ∼0% and ∼12%, respectively, further expansion of our methodology may establish the possible cytotoxicity reduction. These dose–response experiments using N2a cell lines clearly reveal site-specific, domain-constrained structural inversion differentially reduces amyloid cytotoxicity of Aβ42. This agrees with previous studies on different cell lines that used similar Aβ42 stereoisomer samples.2,3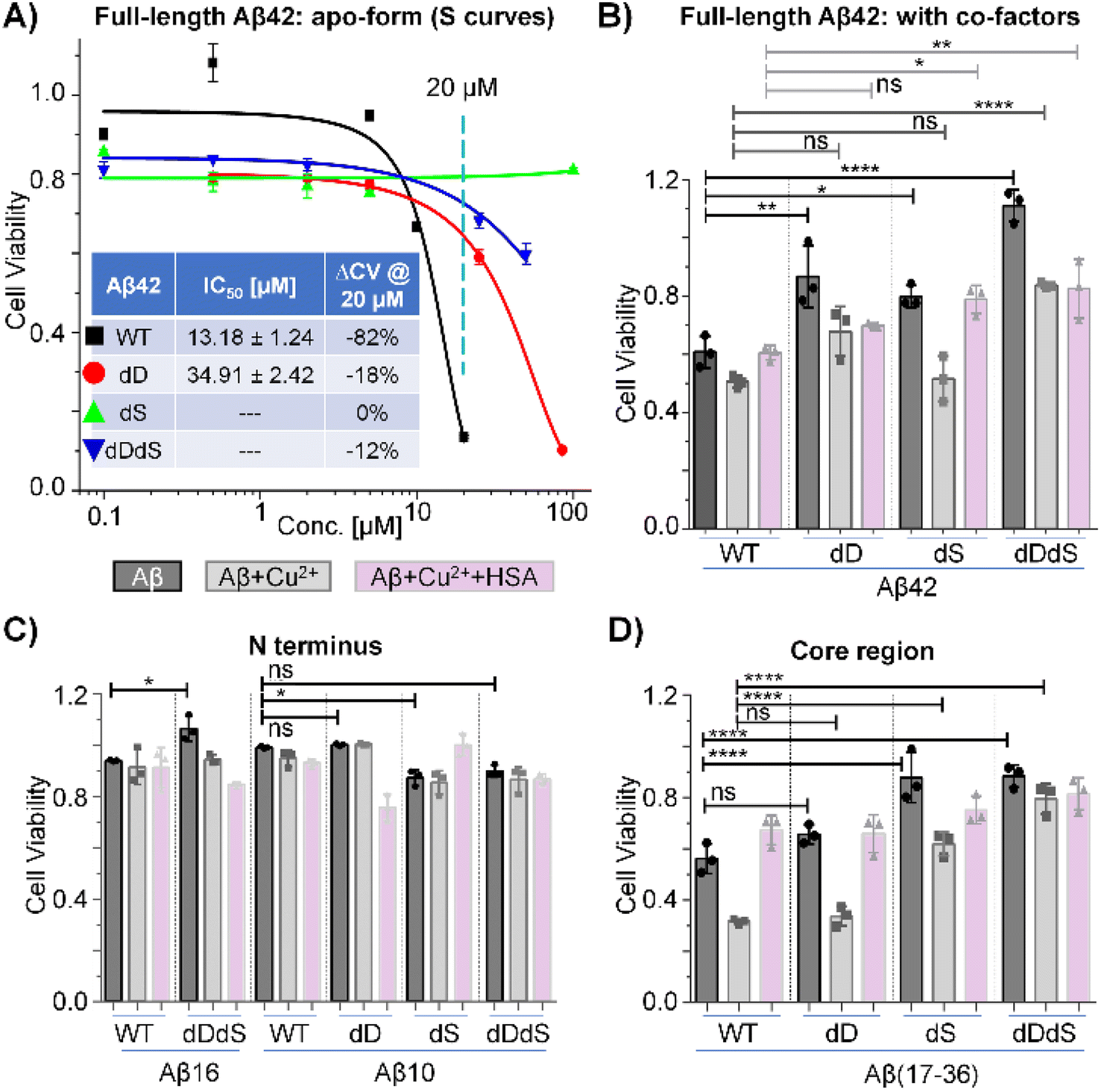 | ||
| Fig. 2 Amyloid cytotoxicity mediated by site-specific, domain-constrained structural flips. (A) Cellular viability of N2a cell line in response to varied dosing of four Aβ42 stereoisomers in the absence of co-factors. (B)–(D) Cell viability of N2a cells treated with Aβ42 (B), Aβ N-terminus (C), and Aβ core region fragment (D) samples generated upon 24 h preincubation of cofactors including Cu2+ and HSA. Error bars represent standard deviations from three independent experiments. Statistical comparisons are based on Student's t-tests: ns, no significance; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Conditions: [Aβ] = 10 μM; [Cu2+] = 10 μM; [HSA] = 10 μM. | ||
Notably, Aβ42 does not function alone. Instead, as previously reported, its biological activities rely on several cofactors including metal ions and cellular transporters (e.g. human serum albumin, HSA).42,43 Knowing this, it is of topical interest to inspect the cytotoxicity of Aβ42 in the presence of representative cofactors. Fig. 2B shows the viability results for four Aβ42 stereoisomers in the presence of Cu2+ and HSA. For WT Aβ42, the cell viability is approximately 60% at 10 μM in the absence of cofactors (dark bar), while the viability was reduced further by ∼10% with copper co-incubation (grey bar). Interestingly, the copper-induced cytotoxicity was successfully reduced with the addition of HSA (white bar), which is in accordance with previous reports.43 Agreeing with our previous dosing experiments (Fig. 2A), we also observed systematically reduced cytotoxicity of Aβ42 in the absence of cofactors. However, no significant viability changes were observed for dD Aβ42 with both copper and HSA co-incubation when compared to WT Aβ42. Contrasting this observation, we did detect significantly reduced cytotoxicity of dS Aβ42 and dDdS Aβ42 in the presence of both copper and HSA. To further interrogate the domain-specific cytotoxicity, we then measured cell viability in the presence of Aβ42 N-terminus (Fig. 2C) and core region fragments (Fig. 2D). While we observe the copper-enhanced and HSA-related cytotoxicity rescuing phenomenon in most groups, all N-terminus fragments of Aβ42 displayed only slight cytotoxicity whereas core regions present comparable cytotoxicity to that of full-length Aβ42. It should be noted that the observed reduction in cytotoxicity when screening the chiral Aβ42 core region correlates directly with our previous report that demonstrated reduced receptor binding affinity by ∼7–13-fold upon chiral inversion of this same truncated peptide.34 Taken together, we conclude that the site-specific, domain-constrained structural flip is beneficial for cytotoxicity reduction both in the presence and absence of cofactors. Our data demonstrate that Aβ42 cytotoxicity is largely mediated by the core region, with observed contribution from the N-terminus. Co-D-epimerization of Asp and Ser residues within Aβ42 likely imparts significant steric alteration of these two structural domains, resulting in cooperative reduction in cytotoxicity. The hypothesized synergistic relationship between N-terminus composition and Aβ toxicity is further reinforced by previous reports that D-isomerized Asp residues within this region.2 The results based on N2a cell line should represent a general trend for the cytotoxicity of Aβ stereoisomers from the perspective of a proof-of-concept demonstration, although it is also useful to extend future tests to more neuronal cell lines, including PC12 and SH-SY5Y.
Molecular mechanism for structure–activity relationship of Aβ42 stereoisomers
To understand the underlying molecular mechanisms behind the altered cytotoxicity induced by D-epimerization, we aimed to firstly characterize the structural impacts of chiral inversion of Aβ42. Our previously reported md-IM-MS strategy34,41 was the clear method-of-choice, given its ability to rapidly discriminating the subtle structural differences of Aβ42 stereoisomers. Expansion of this method for the analysis of full-length Aβ42 requires overcoming two pressing obstacles: (1) the propensity of Aβ42 to form aggregates in solution, crippling electrospray signal stability during IM-MS analysis; (2) the inadequate collision cross-section (CCS) resolution of IM-MS for Aβ42 stereoisomers when using a single IM regime. The first obstacle was resolved through our optimization of an Aβ42 sample preparation workflow. As shown (ESI†), the critical component that ensures analytical reproducibility and spray stability is the lyophilization and resuspension with desired native MS buffer (e.g. 10 mM ammonium acetate) and a tenfold dilution. Representative full MS spectra are shown in Fig. S2,† analyzing freshly prepared Aβ42 on a Synapt G2 traveling wave IMS (TWIMS) instrument. We observe similar charge state distribution across four Aβ42 stereoisomers (WT, dD, dS and dDdS), all of which predominantly carries a charge state of 4+. Though our measurement of the predominant 4+ charge state achieved a resolution near the maximum achievable on the TWIMS platform (R = 40),35 this resolving power was not capable of distinguishing highly similar stereoisomers or providing accurate collision cross-sectional measurement. This observation, which reinforces our stated second obstacle in Aβ42 analysis, necessitates the incorporation of novel analytical strategies.Addressing the challenge of limited structural resolution, we developed an improved md-IM-MS platform to assess the structural impacts of D-epimerization in full-length Aβ42. When employing TWIMS measurement, there were no noticeable CCS differences amongst the four Aβ42 stereoisomers (Fig. S3†). The inclusion of trapped ion mobility spectrometry (TIMS), which provides higher resolving power (R > 200) was able to reveal subtle CCS changes between Aβ42 isomers. Interestingly, D-epimerization of Asp and Ser residues induces a moderate reduction of CCS (Fig. 3A & S4†). The observation of multiple mobility peaks with similar distribution window for four stereoisomers suggests high conformational dynamics of Aβ42, with varied dominate conformers across stereoisomers that might contribute to their differences in activity. For WT Aβ42, at least three conformers were observed; cross sectional measurements were calculated as 844.6 Å2 (conf. #1), 874.6 Å2 (conf. #2) and 938.5 Å2 (conf. #3). While dD Aβ42 holds mainly conf. #2 and #3, both dS Aβ42 and dDdS Aβ42 adopt primarily conf. #1 and/or conf. #2. Stated briefly, D-epimerization at Asp and Ser residues generally makes Aβ42 more structurally compact. To further resolve and confirm the CCS differences amongst these Aβ42 stereoisomers, we incorporated analyses from a cyclic IM instrument (cIMS), representative of a new generation of high-resolution IMS. As shown in Fig. 3B, gradual separation was achieved at higher pass numbers, where the highest resolution measurements (10-passes) confirmed the negative trend in CCS that was observed in our TIMS measurements. These data allow us to definitively conclude that simultaneous chiral inversion of all Asp and Ser residues compacts the overall structure of full-length Aβ42 by ∼3.4% (CCS differences between conf. #1 and conf. #2).
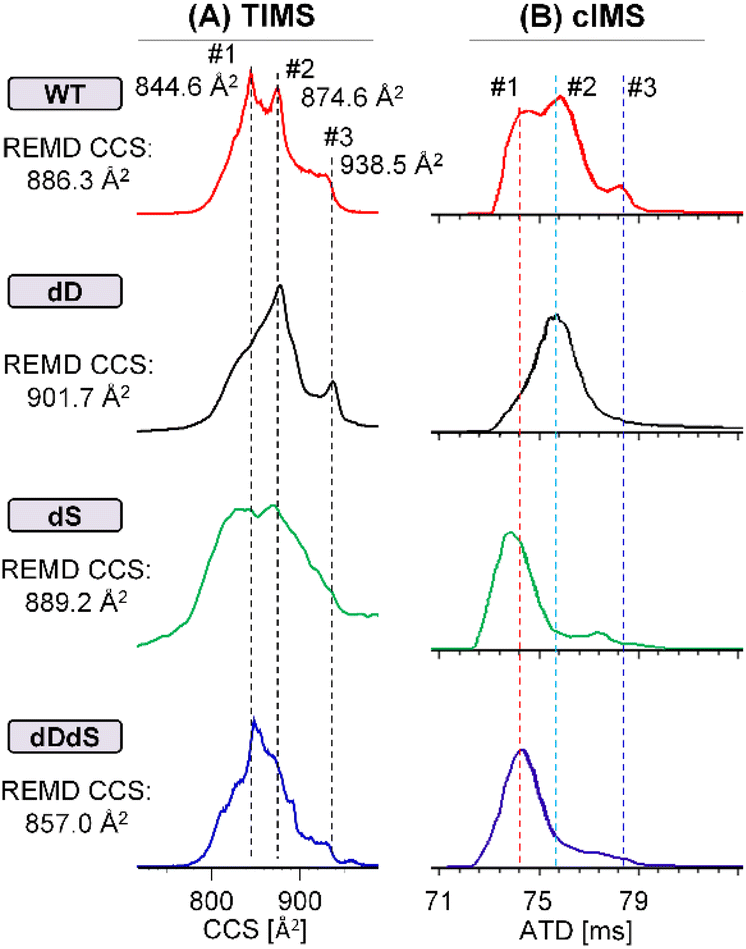 | ||
| Fig. 3 Chiral differentiation of full-length Aβ42 stereoisomers (WT/dD/dS/dDdS, 4+) by using high-resolution TIMS (A) and cIMS (10 passes) (B) instruments. | ||
We then comparatively summarized (Scheme 1) the effects of site-specific isomerization on Aβ42 cytotoxicity with the aim to understand potential correlations between toxicity and structural alteration (as indicated by CCS changes). As can be seen from Scheme 1, D-epimerization at Asp residues alone is sufficient to induce structural expansion and does not significantly affect Aβ42 cytotoxicity. On the contrary, D-epimerization at Ser residues alone promotes structural compaction and is beneficial for decreasing Aβ42 cytotoxicity. This latter trend of decreased cytotoxicity and compacted structure is consistent when evaluating co-D-epimerization of all Asp and Ser residues of full length Aβ42.
 | ||
| Scheme 1 Overview and comparative summary of the effects of site-specific structural flip of Aβ on (A) cytotoxicity and (B) CCS. ‡ Cytotoxicity data for Aβ42-D23S26 are derived from a previous literature (J. Org. Chem. 2020, 85, 1385). | ||
We also performed a series of further experiments to evaluate full-length Aβ42 D-isomerized at specific core region sites (Aβ42 D23S26). While it was previously reported that D-epimerization at D23S26 is not beneficial for decreasing Aβ42 cytotoxicity,17 our independent investigation provided clear supporting evidence that correlates D-epimerization at D23S26 with structural expansion (Fig. S5†). The striking difference in cytotoxicity and CCS in response to chiral inversion between Aβ42 dDdS group and Aβ42 D23S26 group suggests that the N-terminus plays an important regulatory role in conferring Aβ42 structure and function.
A further survey of Aβ42 domain-linked fragments enables more detailed observations of domain-constrained chiral effects. For N-terminus fragments, no significant Aβ cytotoxicity (Fig. 2) was observed alongside chiral inversion-induced structural expansion (Fig. S6–9, discussions in ESI†). For core region fragments, decreased Aβ cytotoxicity was observed in tandem chiral inversion-induced structural compaction (Fig. S6–9, discussions in ESI†).
In summary, co-D-epimerization at Asp and Ser residues of Aβ42 at both the N-terminus and within the core region effectively and synergistically reduces its cytotoxicity. This reduced toxicity coincides with the significant alteration of Aβ42 conformation, which are shown to display domain-specific structural features, including structural compaction.
Having overcome the two obstacles preventing reproducible, high-resolution analyses of full-length Aβ42, the data obtained reveal two additional questions of significant interest. First, is it possible to illuminate the relationship between specific chiral inversion and local conformation changes in Aβ42? Second, how do reciprocal CCS changes in domain specific fragments (+1.3% & −1.1%, Fig. S6–9, discussion in ESI†) contribute to the integrative CCS changes observed in full length Aβ42 (−3.4%, Fig. 3)?
To this end, we performed IM-MS-guided molecular dynamics (MD) simulations of Aβ42 stereoisomers. CCS values based on TIMS measurements were used to guide replica exchange MD (REMD), which means only structural models within experimental CCS error ranges were adopted to create final cluster models. REMD results in distinct cluster models that are energetically favorable for Aβ42 stereoisomers (Fig. S10–13†). Fig. 4 illustrates the lowest energy clusters for WT, dD, dS and dDdS Aβ42, ranging from 137.3 kJ mol−1 to 146.2 kJ mol−1. These chosen clusters of lowest energy are considered as representative conformers, as they possess significant percentages of total structure numbers, ranging from 107 (29% of all cluster models) to 1556 (61% of all cluster models). Aβ42 cluster models generated from 300 K REMD simulations displayed mean CCS values of 886.3 ± 3.2 Å2 (4+), 901.7 ± 2.0 Å2 (4+), 889.2 ± 2.1 Å2 (4+) and 857.0 ± 1.9 Å2 (4+), for WT, dD, dS and dDdS, respectively. The generated WT, dD and dS Aβ42 models match well with our experimental results (Fig. 3A), especially conf. #2, with CCS deviations spanning 1.3–3.0%. As well, the in silico dDdS Aβ42 model is in accordance with our experimentally observed conf. #1, deviating by only 1.5%. From these results, we can conclude not only a strong agreement between our experimental results and theoretical models, but also that our hypothesis regarding structural compaction post D-epimerization can be rigorously tested.
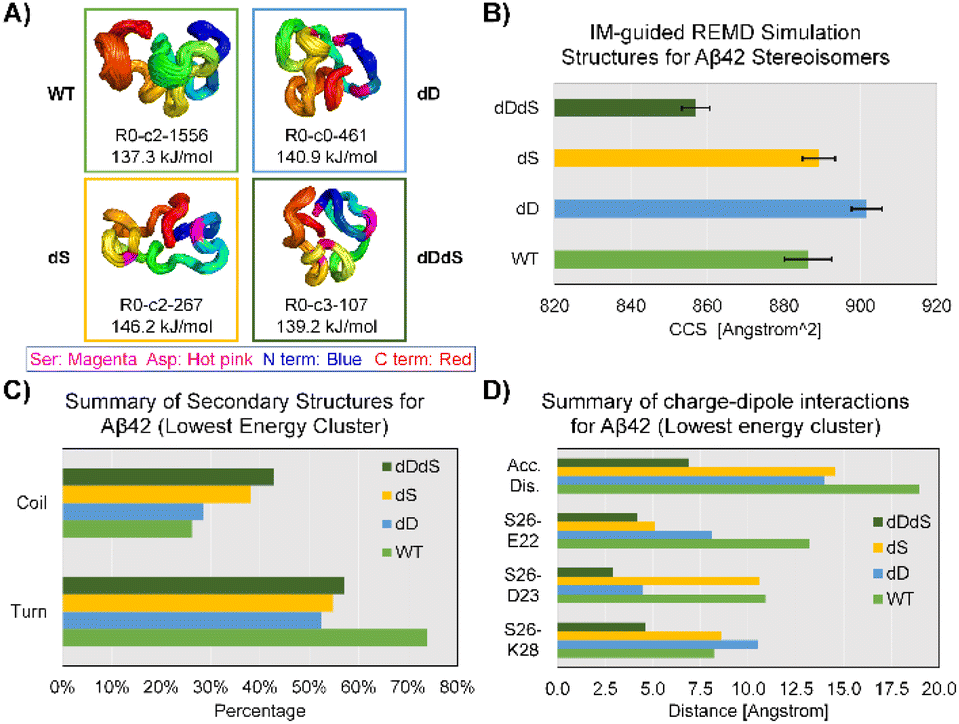 | ||
| Fig. 4 IM-guided REMD simulation results for full-length Aβ42 stereoisomers (WT/dD/dS/dDdS, [Aβ42 + 4H]4+) and corresponding structural analysis. (A) Lowest energy and most dominant/major REMD structural clusters for Aβ42 stereoisomers. Conformer name, R0-c3-107: R0 (rep #0), c3 (cluster #3), 107 (total structure numbers). N-terminus, blue; C-terminus, red; Ser residues, magenta; Asp residues, hot pink. Energy for each conformer is also listed right below the cartoon illustration. (B)–(D) Calculated CCS distributions, secondary structures and charge-dipole interactions for REMD-simulated Aβ42 stereoisomers. Charge-dipole interactions are characterized with the spatial distances between certain residues (e.g. S26–E22 (d1), S26–D23 (d2) and S26–K28 (d3)), which is termed Accumulated Distance (Acc. Dis.). Accumulated Distance is calculated from the Euclidian distance between all interacting residues: dacc = (d1^2 + d2^2 + d3^2)^0.5. | ||
In order to distill more structural detail, we further evaluated the REMD-generated Aβ42 stereoisomer models. Secondary structure analysis, as shown in Fig. 4C, suggests an increased coil fraction but decreased turn content in Aβ42 after chiral inversion. The D-isomerized Aβ42 clusters exhibit a coil fraction range of 29–43% while the turn fraction ranges between 52–57%. These observed changes of secondary structural elements appear to be linked to the structural compaction observed in Aβ42 isomers. Taking Ser26 as an example, we found that charge-dipole interactions17 are effectively altered at both long (WT: S26–E22 distance up to 13 Å, d1, Fig. 4D) and short ranges (dDdS: S26–E22 distance of 4.2 Å). This type of alteration in charge-dipole attraction is also prevalent across two other pairs of residues, S26–D23 (d2) and S26–K28 (d3). To characterize these overall charge-dipole interactions, we cumulatively evaluated the distances of these three pairs (dacc, Fig. 4D). In total, the dacc values for Ser26 were diminished from ∼19 Å (WT Aβ42) to 6.9 Å (dDdS Aβ42), indicative of the elevated charge-dipole attractions upon D-epimerization. Therefore, REMD simulations suggest that the major consequences of D-epimerization are the enhanced S26-related dipole-charge attractions and increased coil fraction at the C-terminus, which together result in the compaction of full-length Aβ42.
Conclusions
Collectively, we present integrated, comprehensive IM-MS datasets on the analyses of Aβ42 stereochemistry, with focus on two most frequently observed D-epimerization residues in nature, Asp and Ser. Site-specific, domain-constrained chirality inversion weakens both antibody and metal ion binding capacity of full-length Aβ42. Simultaneously, D-epimerization at all Asp and Ser residues of Aβ42 inhibits its cytotoxicity, likely by inhibiting the membrane disruption pathway that shown to be directed by the core or C-terminal regions.34,45 This is inferred from the structural compaction of full-length Aβ42 and core region fragments, observed both experimentally and in silico. The findings might be of substantial interest to the field and potentially generalizable to other intrinsically disordered proteins and peptides.Our further efforts in both IM-MS and REMD give rise to refined structural models of all stereoisomers in the gas phase, providing unprecedented information regarding local structural features that confirm our hypothesis surrounding Aβ42 structural compaction-based toxicity modulation. As shown, domain-constrained amino acid isomerization alters and generally compacts Aβ42 while promoting differential, cooperative and domain-specific structural changes. Our results suggest that enhanced interactions between Ser26 and neighbouring residues, as well as the increased amount of coil structure at the C-terminus are accommodated in Aβ42 chiral variants. This study provides a comprehensive insight into the molecular mechanisms behind the naturally occurring D-epimerization of Ser/Asp in Aβ42 and serves to inform on the molecular foundation that enables amino acid-level chiral control that can reduce Aβ42 cytotoxicity3,15–17 through enzymatic regulation and phage display.20,21 The analytical approaches described here may lay the groundwork for future AD drug development and clinical trials targeting the control of D-epimerization-promoting enzymes with specific residues in amyloid protein as substrates, such as racemase.20,46 It should be noted that, not all D-substitutions will lead to effective attenuation of cytotoxicity. In contrast, only specific site mutations, mostly Ser26-based, are beneficial for decreasing cytotoxicity. This may rationalize the fact that aged AD patients will accumulate all kinds of Aβ stereoisomers but they still suffer from the disease, and are not effectively reduced by the beneficial isoforms when combating with other harmful forms. Future efforts, from a clinical application perspective, may be directed to elevate the level of D-substitution towards Ser26 residue rather than other sites.
Data availability
Data can be available from the corresponding authors (G. L., L. L. and B. T. R.) upon reasonable request.Author contributions
G. L., L. L. and B. T. R. designed the project. G. L. collected MS data, C. K. J. performed REMD, M. M. carried out cell experiments. G. L. drafted the manuscript. Y. J., Z. Z., D. G. D., Ga. L. and E. R. helped the collection of MS data and involved in figure preparation. All authors involved in the data analysis, wrote the paper and approved the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. L. thanks the support by the National Key R&D Program of China (2022YFA1305200), the National Natural Science Foundation of China (22104064, 22204121, 22293030, 22293032), joint funding support from the Fundamental Research Funds for the Central Universities (Nankai University), and the Haihe Laboratory of Sustainable Chemical Transformations for financial support. We thank the useful discussions from Dr Jevgenij Raskatov (UC Santa Cruz) and his generous providing of some control samples for our study. The authors also gratefully appreciate Dr Yu Gao and Dr Xinyu Zhao (Waisman Center, UW-Madison) for their generous providing of cell lines and Waters Corporation and the three Chemists who helped with the cyclic IMS data collection (Brad J. Williams, Barbara J. Sullivan and Alexandre F. Gomes). This work was funded in part by NIH (R01DK071801, R56DK071801, U01CA231081, R01NS031609, P30DA018310 and RF1AG052324), and NSF (CHE-2108223). L. L. acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy. B. T. R. thanks the National Science Foundation Division of Chemistry under Grant 1808541 (with co-funding from the Division of Molecular and Cellular Biosciences) for their support of this work.Notes and references
- S. Ritztimme and M. Collins, Racemization of aspartic acid in human proteins, Ageing Res. Rev., 2002, 1, 43–59 CrossRef CAS PubMed.
- T. Sugiki and N. Utsunomiya-Tate, Site-specific aspartic acid isomerization regulates self-assembly and neurotoxicity of amyloid-beta, Biochem. Biophys. Res. Commun., 2013, 441, 493–498 CrossRef CAS PubMed.
- S. Dutta, A. R. Foley, C. J. A. Warner, X. Zhang, M. Rolandi, B. Abrams and J. A. Raskatov, Suppression of Oligomer Formation and Formation of Non-Toxic Fibrils upon Addition of Mirror-Image Abeta42 to the Natural l-Enantiomer, Angew. Chem., Int. Ed., 2017, 56, 11506–11510 CrossRef CAS PubMed.
- C. J. Warner, S. Dutta, A. R. Foley and J. A. Raskatov, Introduction of d-Glutamate at a Critical Residue of Abeta42 Stabilizes a Prefibrillary Aggregate with Enhanced Toxicity, Chem. – Eur. J., 2016, 22, 11967–11970 CrossRef CAS PubMed.
- T. Kubo, Y. Kumagae, C. A. Miller and I. Kaneko, β-Amyloid Racemized at the Ser26Residue in the Brains of Patients with Alzheimer Disease: Implications in the Pathogenesis of Alzheimer Disease, J. Neuropathol. Exp. Neurol., 2003, 62, 248–259 CrossRef CAS PubMed.
- A. Banreti, S. Bhattacharya, F. Wien, K. Matsuo, M. Refregiers, C. Meinert, U. Meierhenrich, B. Hudry, D. Thompson and S. Noselli, Biological effects of the loss of homochirality in a multicellular organism, Nat. Commun., 2022, 13, 7059 CrossRef CAS PubMed.
- S. Mukherjee, K. A. Perez, L. C. Lago, S. Klatt, C. A. McLean, I. E. Birchall, K. J. Barnham, C. L. Masters and B. R. Roberts, Quantification of N-terminal amyloid-beta isoforms reveals isomers are the most abundant form of the amyloid-beta peptide in sporadic Alzheimer's disease, Brain Commun., 2021, 3, fcab028 CrossRef PubMed.
- N. Fujii, Y. Kaji and N. Fujii, D-Amino acids in aged proteins: Analysis and biological relevance, J. Chromatogr. B, 2011, 879, 3141–3147 CrossRef CAS PubMed.
- Y. M. Kuo, M. R. Emmerling, A. S. Woods, R. J. Cotter and A. E. Roher, Isolation, chemical characterization, and quantitation of A beta 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits, Biochem. Biophys. Res. Commun., 1997, 237, 188–191 CrossRef CAS PubMed.
- A. Nakamura, N. Kaneko, V. L. Villemagne, T. Kato, J. Doecke, V. Dore, C. Fowler, Q. X. Li, R. Martins, C. Rowe, T. Tomita, K. Matsuzaki, K. Ishii, K. Ishii, Y. Arahata, S. Iwamoto, K. Ito, K. Tanaka, C. L. Masters and K. Yanagisawa, High performance plasma amyloid-beta biomarkers for Alzheimer's disease, Nature, 2018, 554, 249–254 CrossRef CAS PubMed.
- I. Kaneko, K. Morimoto and T. Kubo, Drastic neuronal loss in vivo by β-amyloid racemized at Ser26 residue: conversion of non-toxic [D-Ser26]β-amyloid 1–40 to toxic and proteinase-resistant fragments, Neuroscience, 2001, 104, 1003–1011 CrossRef CAS PubMed.
- W. J. Ray and V. Buggia-Prevot, Novel Targets for Alzheimer's Disease: A View Beyond Amyloid, Annu. Rev. Med., 2021, 72, 15–28 CrossRef CAS PubMed.
- T. R. Lambeth, D. L. Riggs, L. E. Talbert, J. Tang, E. Coburn, A. S. Kang, J. Noll, C. Augello, B. D. Ford and R. R. Julian, Spontaneous Isomerization of Long-Lived Proteins Provides a Molecular Mechanism for the Lysosomal Failure Observed in Alzheimer's Disease, ACS Cent. Sci., 2019, 5, 1387–1395 CrossRef CAS PubMed.
- S. Du, E. R. Readel, M. Wey and D. W. Armstrong, Complete identification of all 20 relevant epimeric peptides in beta-amyloid: a new HPLC-MS based analytical strategy for Alzheimer's research, Chem. Commun., 2020, 56, 1537–1540 RSC.
- S. Dutta, T. S. Finn, A. J. Kuhn, B. Abrams and J. A. Raskatov, Chirality Dependence of Amyloid beta Cellular Uptake and a New Mechanistic Perspective, Chembiochem, 2019, 20, 1023–1026 CrossRef CAS PubMed.
- A. R. Foley, T. S. Finn, T. Kung, A. Hatami, H. W. Lee, M. Jia, M. Rolandi and J. A. Raskatov, Trapping and Characterization of Nontoxic Abeta42 Aggregation Intermediates, ACS Chem. Neurosci., 2019, 10, 3880–3887 CrossRef CAS PubMed.
- A. R. Foley, H. W. Lee and J. A. Raskatov, A Focused Chiral Mutant Library of the Amyloid beta 42 Central Electrostatic Cluster as a Tool To Stabilize Aggregation Intermediates, J. Org. Chem., 2020, 85, 1385–1391 CrossRef CAS PubMed.
- A. A. Kulikova, I. B. Cheglakov, M. S. Kukharsky, R. K. Ovchinnikov, S. A. Kozin and A. A. Makarov, Intracerebral Injection of Metal-Binding Domain of Aβ Comprising the Isomerized Asp7 Increases the Amyloid Burden in Transgenic Mice, Neurotoxic. Res., 2016, 29, 551–557 CrossRef CAS PubMed.
- V. A. Mitkevich, I. Y. Petrushanko, Y. E. Yegorov, O. V. Simonenko, K. S. Vishnyakova, A. A. Kulikova, P. O. Tsvetkov, A. A. Makarov and S. A. Kozin, Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells, Cell Death Dis., 2013, 4, e939 CrossRef CAS PubMed.
- S. Takagi, D. T. Balu and J. T. Coyle, Factors regulating serine racemase and d-amino acid oxidase expression in the mouse striatum, Brain Res., 2021, 1751, 147202 CrossRef CAS PubMed.
- X. Zhou, C. Zuo, W. Li, W. Shi, X. Zhou, H. Wang, S. Chen, J. Du, G. Chen, W. Zhai, W. Zhao, Y. Wu, Y. Qi, L. Liu and Y. Gao, A Novel d-Peptide Identified by Mirror-Image Phage Display Blocks TIGIT/PVR for Cancer Immunotherapy, Angew. Chem., Int. Ed., 2020, 59, 15114–15118 CrossRef CAS PubMed.
- D. H. Mast, J. W. Checco and J. V. Sweedler, Differential Post-Translational Amino Acid Isomerization Found among Neuropeptides in Aplysia californica, ACS Chem. Biol., 2020, 15, 272–281 CrossRef CAS PubMed.
- D. H. Mast, J. W. Checco and J. V. Sweedler, Advancing d-amino acid-containing peptide discovery in the metazoan, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1869, 140553 CrossRef PubMed.
- A. V. Patel, T. Kawai, L. Wang, S. S. Rubakhin and J. V. Sweedler, Chiral Measurement of Aspartate and Glutamate in Single Neurons by Large-Volume Sample Stacking Capillary Electrophoresis, Anal. Chem., 2017, 89, 12375–12382 CrossRef CAS PubMed.
- I. Livnat, H. C. Tai, E. T. Jansson, L. Bai, E. V. Romanova, T. T. Chen, K. Yu, S. A. Chen, Y. Zhang, Z. Y. Wang, D. D. Liu, K. R. Weiss, J. Jing and J. V. Sweedler, A d-Amino Acid-Containing Neuropeptide Discovery Funnel, Anal. Chem., 2016, 88, 11868–11876 CrossRef CAS PubMed.
- L. Bai, E. V. Romanova and J. V. Sweedler, Distinguishing endogenous D-amino acid-containing neuropeptides in individual neurons using tandem mass spectrometry, Anal. Chem., 2011, 83, 2794–2800 CrossRef CAS PubMed.
- L. Bai, S. Sheeley and J. V. Sweedler, Analysis of Endogenous D-Amino Acid-Containing Peptides in Metazoa, Bioanal. Rev., 2009, 1, 7–24 CrossRef PubMed.
- M. A. Ewing, J. Wang, S. A. Sheeley and J. V. Sweedler, Detecting D-amino acid-containing neuropeptides using selective enzymatic digestion, Anal. Chem., 2008, 80, 2874–2880 CrossRef CAS PubMed.
- S. A. Sheeley, H. Miao, M. A. Ewing, S. S. Rubakhin and J. V. Sweedler, Measuring D-amino acid-containing neuropeptides with capillary electrophoresis, Analyst, 2005, 130, 1198–1203 RSC.
- D. H. Mast, H. W. Liao, E. V. Romanova and J. V. Sweedler, Analysis of Peptide Stereochemistry in Single Cells by Capillary Electrophoresis-Trapped Ion Mobility Spectrometry Mass Spectrometry, Anal. Chem., 2021, 93, 6205–6213 CrossRef CAS PubMed.
- X. Zheng, L. Deng, E. S. Baker, Y. M. Ibrahim, V. A. Petyuk and R. D. Smith, Distinguishing d- and l-aspartic and isoaspartic acids in amyloid beta peptides with ultrahigh resolution ion mobility spectrometry, Chem. Commun., 2017, 53, 7913–7916 RSC.
- G. Nagy, K. Kedia, I. K. Attah, S. V. B. Garimella, Y. M. Ibrahim, V. A. Petyuk and R. D. Smith, Separation of beta-Amyloid Tryptic Peptide Species with Isomerized and Racemized l-Aspartic Residues with Ion Mobility in Structures for Lossless Ion Manipulations, Anal. Chem., 2019, 91, 4374–4380 CrossRef CAS PubMed.
- F. Berthias, M. A. Baird and A. A. Shvartsburg, Differential Ion Mobility Separations of d/l Peptide Epimers, Anal. Chem., 2021, 93, 4015–4022 CrossRef CAS PubMed.
- G. Li, K. DeLaney and L. Li, Molecular basis for chirality-regulated Abeta self-assembly and receptor recognition revealed by ion mobility-mass spectrometry, Nat. Commun., 2019, 10, 5038 CrossRef PubMed.
- G. Li, D. G. Delafield and L. J. Li, Improved structural elucidation of peptide isomers and their receptors using advanced ion mobility-mass spectrometry, Trends Anal. Chem., 2020, 124, 115546 CrossRef CAS.
- K. Jeanne Dit Fouque, A. Garabedian, J. Porter, M. Baird, X. Pang, T. D. Williams, L. Li, A. Shvartsburg and F. Fernandez-Lima, Fast and Effective Ion Mobility-Mass Spectrometry Separation of d-Amino-Acid-Containing Peptides, Anal. Chem., 2017, 89, 11787–11794 CrossRef CAS PubMed.
- C. Jia, C. B. Lietz, Q. Yu and L. Li, Site-specific characterization of (D)-amino acid containing peptide epimers by ion mobility spectrometry, Anal. Chem., 2014, 86, 2972–2981 CrossRef CAS PubMed.
- A. V. Tolmachev, I. K. Webb, Y. M. Ibrahim, S. V. Garimella, X. Zhang, G. A. Anderson and R. D. Smith, Characterization of ion dynamics in structures for lossless ion manipulations, Anal. Chem., 2014, 86, 9162–9168 CrossRef CAS PubMed.
- S. M. Stow, T. Causon, X. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. E. Rennie, E. S. Baker, R. D. Smith, J. A. McLean, S. Hann and J. C. Fjeldsted, An Interlaboratory Evaluation of Drift Tube Ion Mobility - Mass Spectrometry Collision Cross Section Measurements, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS PubMed.
- T. D. Do, J. W. Checco, M. Tro, J. E. Shea, M. T. Bowers and J. V. Sweedler, Conformational investigation of the structure-activity relationship of GdFFD and its analogues on an achatin-like neuropeptide receptor of Aplysia californica involved in the feeding circuit, Phys. Chem. Chem. Phys., 2018, 20, 22047–22057 RSC.
- X. Xu, L. Han, Z. Zheng, R. Zhao, L. Li, X. Shao and G. Li, Composite Multidimensional Ion Mobility-Mass Spectrometry for Improved Differentiation of Stereochemical Modifications, Anal. Chem., 2023, 95, 2221–2228 CrossRef CAS PubMed.
- M. Rozga and W. Bal, The Cu(II)/Abeta/human serum albumin model of control mechanism for copper-related amyloid neurotoxicity, Chem. Res. Toxicol., 2010, 23, 298–308 Search PubMed.
- T. S. Choi, H. J. Lee, J. Y. Han, M. H. Lim and H. I. Kim, Molecular Insights into Human Serum Albumin as a Receptor of Amyloid-beta in the Extracellular Region, J. Am. Chem. Soc., 2017, 139, 15437–15445 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- S. De, D. C. Wirthensohn, P. Flagmeier, C. Hughes, F. A. Aprile, F. S. Ruggeri, D. R. Whiten, D. Emin, Z. Xia, J. A. Varela, P. Sormanni, F. Kundel, T. P. J. Knowles, C. M. Dobson, C. Bryant, M. Vendruscolo and D. Klenerman, Different soluble aggregates of Abeta42 can give rise to cellular toxicity through different mechanisms, Nat. Commun., 2019, 10, 1541 CrossRef PubMed.
- X. Dai, E. Zhou, W. Yang, X. Zhang, W. Zhang and Y. Rao, D-Serine made by serine racemase in Drosophila intestine plays a physiological role in sleep, Nat. Commun., 2019, 10, 1986 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, Fig. S1 to S13. See DOI: https://doi.org/10.1039/d3sc00678f |
| ‡ G. L., C. K. J. and M. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |