
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulation of the CRISPR-Cas12a system by methylation and demethylation of guide RNA†
Zhian
Hu
ab,
Ao
Sun
c,
Jinlei
Yang
b,
Gul
Naz
 b,
Gongwei
Sun
b,
Zhengping
Li
a,
Jun-Jie
Gogo Liu
*cd,
Sichun
Zhang
*b and
Xinrong
Zhang
*b
b,
Gongwei
Sun
b,
Zhengping
Li
a,
Jun-Jie
Gogo Liu
*cd,
Sichun
Zhang
*b and
Xinrong
Zhang
*b
aBeijing Key Laboratory for Bioengineering and Sensing Technology, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, 30 Xueyuan Road, Haidian District, Beijing 100083, P. R. China
bDepartment of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: xrzhang@mail.tsinghua.edu.cn; sczhang@mail.tsinghua.edu.cn
cTsinghua-Peking Center for Life Sciences, School of Life Sciences, Tsinghua University, Beijing 100084, P. R. China. E-mail: junjiegogoliu@mail.tsinghua.edu.cn
dBeijing Advanced Innovation Center for Structural Biology & Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, P. R. China
First published on 9th May 2023
Abstract
Chemical modifications of CRISPR-Cas nucleases help decrease off-target editing and expand the biomedical applications of CRISPR-based gene manipulation tools. Here, we found that epigenetic modifications of guide RNA, such as m6A and m1A methylation, can effectively inhibit both the cis- and trans-DNA cleavage activities of CRISPR-Cas12a. The underlying mechanism is that methylations destabilize the secondary and tertiary structure of gRNA which prevents the assembly of the Cas12a-gRNA nuclease complex, leading to decreased DNA targeting ability. A minimum of three adenine methylated nucleotides are required to completely inhibit the nuclease activity. We also demonstrate that these effects are reversible through the demethylation of gRNA by demethylases. This strategy has been used in the regulation of gene expression, demethylase imaging in living cells and controllable gene editing. The results demonstrate that the methylation-deactivated and demethylase-activated strategy is a promising tool for regulation of the CRISPR-Cas12a system.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) – Cas technologies have revolutionized fields ranging from fundamental science to medical therapies.1,2 However, challenges of spatial and temporal control of CRISPR-Cas need to be addressed, which reduces potential off-target editing and broadens medical applications. Exogenously inducible CRISPR-Cas tools have been first developed,3 such as employing blue light4,5 and small drug molecules6–8 to precisely activate the CRISPR-Cas system to induce interest gene expression. To fully exert the potential of CRISPR in living cells, CRISPR tools need to be manipulated using crucial endogenous biomolecules for studying intracellular biomarkers and controlling the genome of specific cells such as cancer cells and stem cells.9,10 For example, a microRNA-inducible CRISPR platform has been proposed for serving as a stem cell genome-regulation tool and sensing the microRNA.9Chemical modifications on gRNA can also efficiently regulate the performance of CRISPR-Cas.8,11–13 Many chemical modifications on ribose sugar and the backbone have been investigated so far, including 2′-F,14,15 universal bases (inosine and 5′ nitroindole),16 locked nucleic acid (LNA),17 phosphorothioates (PS),18etc. These chemical modifications could significantly reduce off-target editing and strengthen CRISPR-Cas system bioavailability in vivo. Recently, conditional control of chemical modifications on gRNA, such as light caged modifications19–21 and small-ligand caged modifications,8,22 has sprung up. For example, Deiters' group has achieved light-controllable gene editing in living cells and zebrafish through 6-NPOM-caged modifications on gRNA of Cas9.13 These modifications can further spatiotemporally control CRSIPR in vivo for precise gene-editing and regulation. Despite the achievements, existing chemical modifications are commonly artificial rather than natural intracellular RNA modifications, which are scarcely related to cellular behaviours, such as epigenetic regulation.10 In general, endogenous chemical modifications, such as methylation, are often associated with vital cellular processes.23 By exploiting endogenous reversible chemical modifications to regulate the activity of CRISPR systems, we may be able to minimize their off-target effects while conferring them with cell-type or cell-state specificity, and even apply them for biosensing and therapeutic applications. Therefore, it is promising to investigate endogenous chemical modifications on gRNA to control CRISPR-Cas.12
The CRISPR-Cas effector is similar to the ribosome, working as ribonucleoprotein particles (RNPs).24,25 Emerging evidence demonstrates that epigenetic modifications on the RNA component can precisely regulate the function and structure of RNPs, playing an essential role in almost every aspect of cellular regulation and growth.26–28 Among these RNA epigenetic markers, N6-methyladenosine (m6A) and N1-methyladenosine (m1A) are two of the few RNA modifications whose reversible reaction pathway in cells has been fully elucidated.29 m6A could be erased from RNA by ALKBH5 and FTO demethylases while m1A could be removed by ALKBH3 demethylase.30–32 These demethylases maintain dynamic m6A and m1A expression in living cells and play vital roles in promoting cell proliferation and regulating RNA-protein interactions.33 Furthermore, regulation of CRISPR activity by naturally occurring mRNA modifications has scarcely been systematically studied before. Therefore, we expect that methylation and demethylation of CRISPR gRNA may be an effective way to control the activity of the CRISPR system when combined with demethylases.
In this work, we found that the m6A or m1A modification in the 5′ handle of gRNA could significantly inhibit the cis- and trans-cleavage activity of CRISPR-Cas12a. Referring to the structure of the Cas12a effector,34 methylation may hinder or disrupt the pseudoknot structure of gRNA, which prevents the formation of a stable complex between the Cas12a protein and gRNA, resulting in the loss of DNA cleavage capability. The gRNA structure could be restored by demethylases through erasing the methyl group. The m6A methylation-deactivated CRISPR-Cas12a nuclease can be robustly reactivated by ALKBH5 and FTO, while the m1A methylation-deactivated CRISPR-Cas12a can be reactivated by ALKBH3 demethylases. Using the trans-cleavage activity of Cas12a, we were able to successfully develop a reliable and sensitive fluorescence read-out strategy for monitoring the activity of demethylases. Moreover, we successfully used the demethylase to control gene editing. In addition, we constructed a specific dCas12a transcriptional circuit that permits inducible protein expression in living cells by demethylase-mediated gRNA activation. Overall, we proposed a novel strategy for manipulating CRISPR-Cas12a activity by controlling chemical modifications of gRNA (Scheme 1).
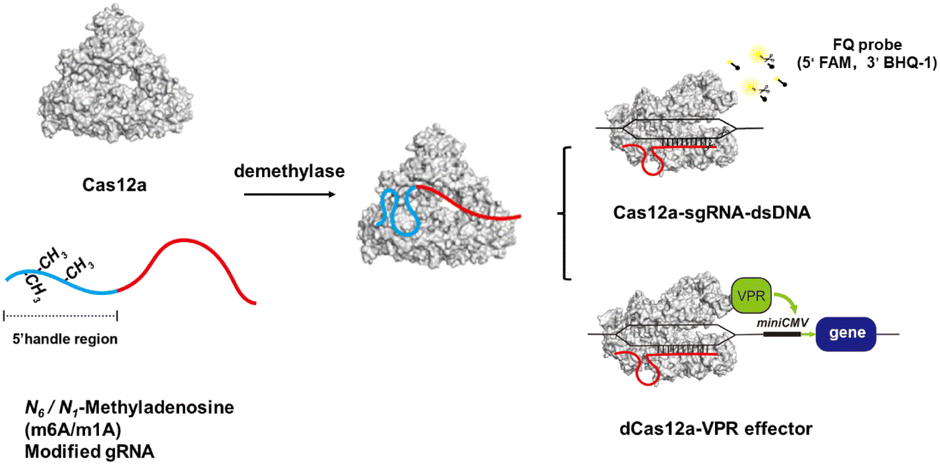 | ||
| Scheme 1 Illustration of a methylation-deactivated and demethylase-activated CRISPR-Cas12a system. m6A or m1A in the 5′ handle of gRNA disrupts the interaction between the Cas12a protein and gRNA. The deactivated CRISPR-Cas12a could be reactivated by demethylases. This strategy has enabled the in vitro and in vivo development of biosensors for demethylases and the precise regulation of gene expression. | ||
Results and discussion
The effects of m6A modifications on the cleavage activity of CRISPR-Cas12a
As a proof of concept, the effect of m6A modification on different regions of gRNA has been investigated in detail. As shown in Fig. 1A, the 5′ handle pseudoknot structure of gRNA (red box) is tightly folded and particularly sensitive to chemical modifications.12 The remaining 20 nts of the gRNA (blue box) serve as the spacer that recognizes and invades the target dsDNA. Both regions of the gRNA are important for efficient DNA cleavage. Generally, the methyl group in m6A causes a steric clash in Watson–Crick pairing of RNA (Fig. 1B).26,35 Therefore, we expect that this modification may destabilize the pseudoknot structure or the pairing between the spacer and target DNA. We constructed BC-gRNA (Fig. 1C) containing m6A at sites A2, A8, A16, and A18. As for the spacer, BR-gRNA (Fig. 1C) with m6A at sites A22, A27, A31, and A36 was designed. In addition, a gRNA with eight m6A methylations (8(m6A)-gRNA) was designed to effectively switch-off CRISPR-Cas12a (Fig. 1C).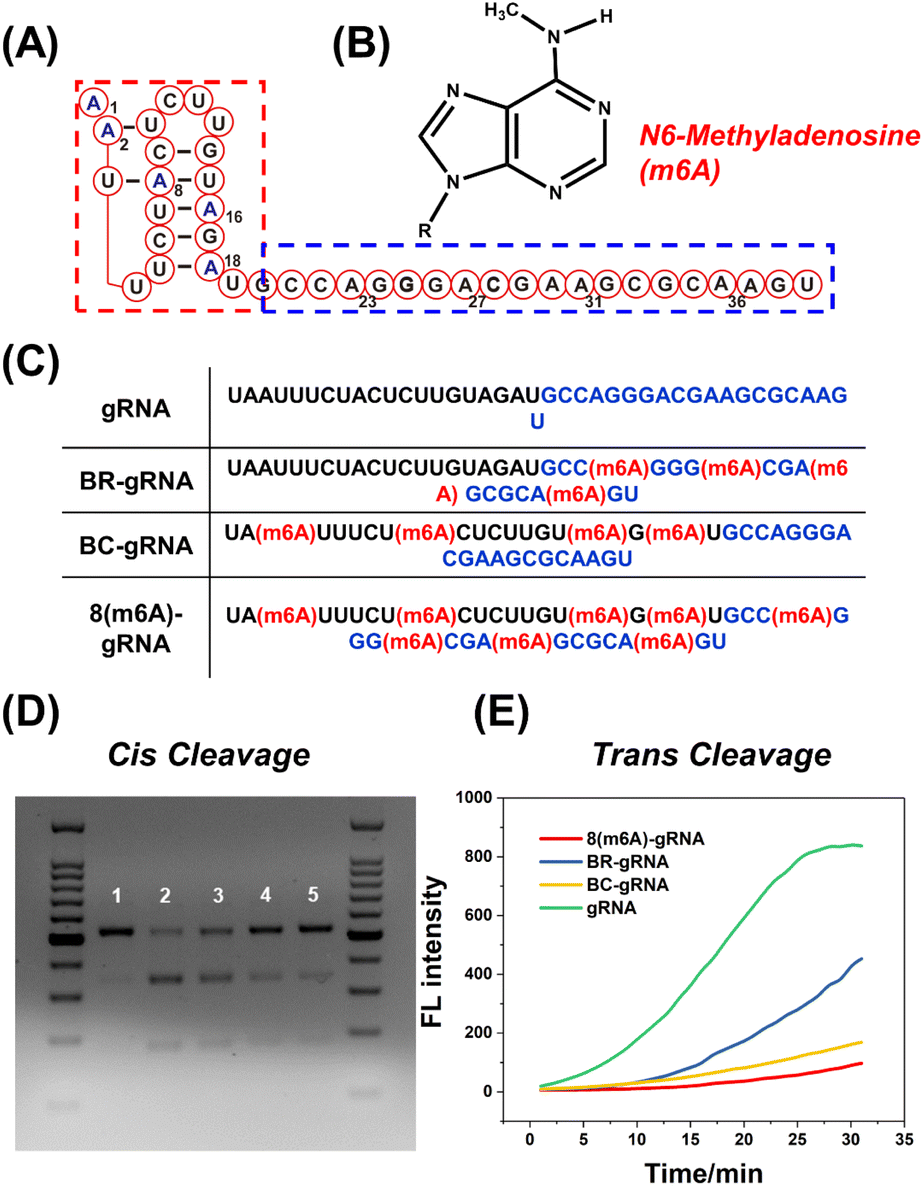 | ||
| Fig. 1 The effects of m6A modifications in gRNA on DNA cleavage activity of CRISPR-Cas12a. (A) The secondary structure of gRNA. The red box represents the 5′ handle of gRNA and the blue box denotes the spacer sequence of gRNA. (B) The chemical structure of m6A modification. (C) Locations of m6A modifications in different gRNAs are illustrated. (D) Agarose electrophoresis indicates the results of CRISPR-Cas12a cis-cleavage. (Lane 1) Only the dsDNA target; (Lane 2) non-modified gRNA + dsDNA + Cas12a; (Lane 3) BR-gRNA + dsDNA + Cas12a; (Lane 4) BC-gRNA + dsDNA + Cas12a; (Lane 5) 8(m6A)-gRNA + dsDNA + Cas12a. (E) Time course of Cas12a with different gRNA trans cleaving FQ probes. | ||
Generally, CRISPR-Cas12a holds two cleavage activities including target specific cleavage (cis-cleavage) activity and non-specific nucleic acid cleavage (trans-cleavage) activity. In this work, we conducted separate tests to assess the impact of methylated gRNAs on two discrete activities of CRISPR-Cas12a. First, the cis-cleavage activity of Cas12a to the dsDNA substrate was investigated. Gel electrophoresis revealed that dsDNA could be effectively cleaved by Cas12a in the presence of normal gRNA (Fig. 1D). BR-gRNA displayed a slight inhibitory effect on cis-cleavage. However, cis-cleavage activity decreased significantly when the gRNA was replaced by BC-gRNA, and few dsDNA substrates were cleaved. Actually, BC-gRNA has an inhibitory effect comparable to that of 8(m6A)-gRNA. These results demonstrate that m6A modification in the 5′ handle region of gRNA inhibited cis-cleavage of CRISPR-Cas12a significantly more than that in the 3′ spacer region of gRNA. We further investigated the inhibitory effect of m6A modification on the trans-cleavage activity of CRISPR-Cas12a. To monitor the trans-cleavage activity of Cas12a, a dually labelled substrate FQ-DNA probe was often used (5′ FAM and 3′ BHQ-1, Table S3†). The trans-cleavage would remove the quencher from the fluorophore on the FQ probe, resulting in fluorescence recovery. As shown in Fig. 1E, the m6A inhibition pattern during trans-cleavage was comparable to that of cis-cleavage. In comparison to natural gRNA, BR-gRNA could slightly suppress the trans-activity of Cas12a, with an inhibition rate of about 40%. Notably, BC-gRNA has a stronger inhibitory ability, with trans-cleavage activity decreasing by more than 70%. In addition, the trans-cleavage ability of Cas12a can be activated by a ssDNA activator (Fig. S2A†). As shown in Fig. S2B and C,† significant inhibition of trans-cleavage was also achieved by m6A in the 5′ handle of the gRNA using ssDNA as the activator.
Since m6A modification brings a significant inhibition effect to CRISPR-Cas12a activity, we further systematically investigated the m6A position in the 5′ handle region for the deactivation of Cas12a. As shown in Fig. 2A, the methyl group was modified at the second to fifth adenine N6 site in the 5′ handle of gRNA, respectively (m6A2, m6A3, m6A4, and m6A5). For cis-cleavage, there was no significant difference between natural gRNA and single-m6A modified gRNAs (Fig. 2B, Lane 2–6). Instead, m6Aall-gRNA showed minimal cleavage activity (Fig. 2B, Lane 7). Additionally, the inhibitory effect on trans-cleavage activity was investigated. The suppression patterns of dsDNA and ssDNA activators were consistent as shown in Fig. 2C and S3B.† It is difficult to deactivate CRISPR-Cas12a trans-cleavage by modifying gRNA with a single m6A. These findings show that the deactivation of CRISPR-Cas12a relies on the synergetic effect of multiple m6A modifications on the pseudoknot structure. Notably, m6A modifications at the sites A3, A4, and A5 have a stronger inhibitory effect than at the A2 site. The m6A modifications at the above three positions may play a vital role in deactivating CRISPR-Cas12a.
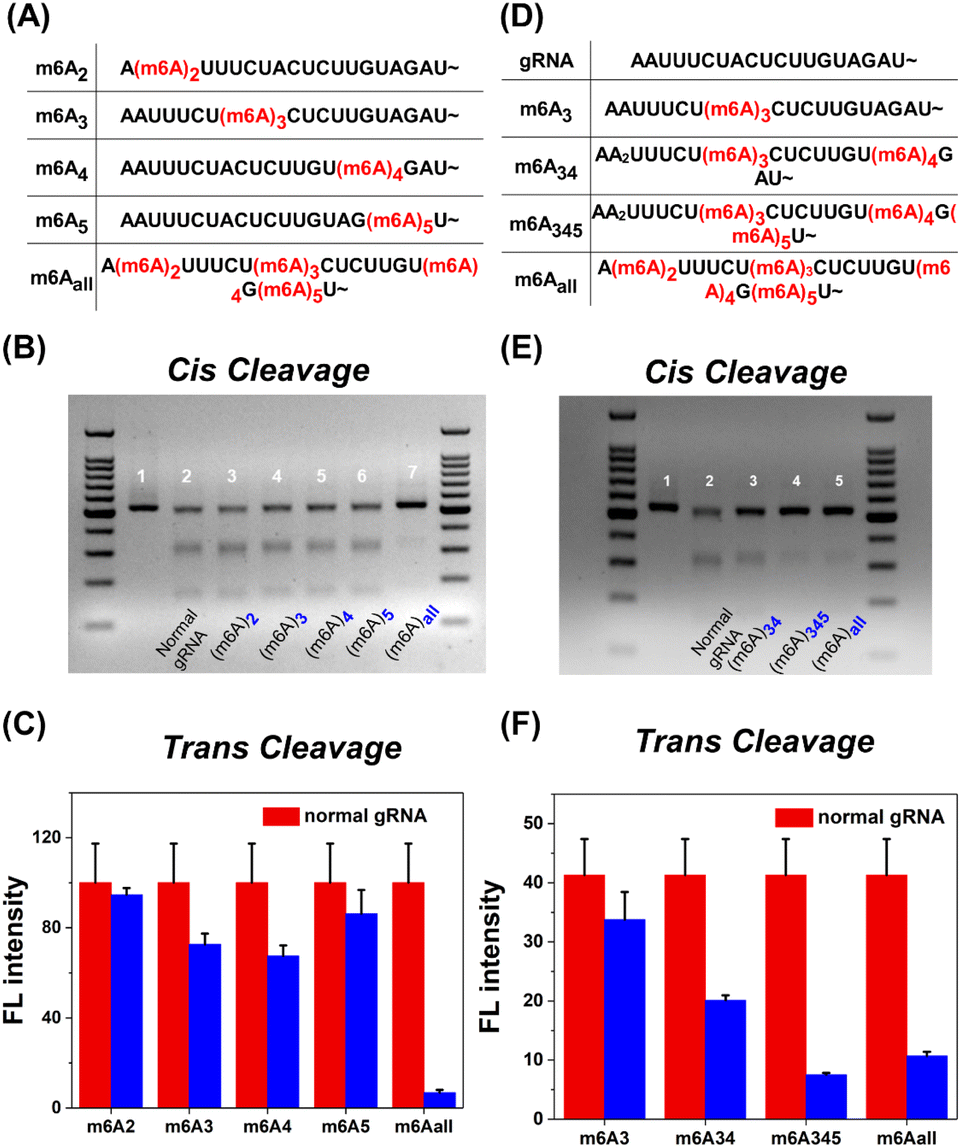 | ||
| Fig. 2 Systematic screening of the position and number of m6A in the 5′ handle of gRNA. (A) and (D) Locations of m6A modification in different gRNAs. (B) and (C) Screening of the position of m6A in the 5′ handle of gRNA. (B) Agarose gel electrophoresis (2%) characterized cis-cleavage. (Lane 1) Only the dsDNA target; (C) fluorescence results of trans cleaving the FQ-probe. (E) and (F) Screening the number of m6A in the 5′ handle. (E) Agarose gel electrophoresis (2%) characterized cis-cleavage. (Lane 1) Only the dsDNA target; (Lane 2) natural gRNA; (F) fluorescence results of trans cleaving the FQ-probe. The red column represents CRISPR-Cas12a with normal gRNA. The blue column represents CRISPR-Cas12a with m6A-modified gRNA. The error bar represents the standard deviation of three measurements. | ||
CRISPR-Cas12a deactivation is dependent on the number of m6A modifications on gRNA. Three gRNAs termed m6A34-gRNA, m6A345-gRNA and m6Aall-gRNA were designed as shown in Fig. 2D. The inhibitory effects on cis-cleavage were positively correlated with the number of m6A modifications (Fig. 2E). Among all designs, m6A345-gRNA was the most effective at inhibiting the cis-cleavage activity of CRISPR-Cas12a. Next, we investigated whether m6A345-gRNA could efficiently repress the trans-cleavage of Cas12a. The results of trans-cleavage are shown in Fig. 2F, S3D and S18.† The fluorescence results indicate that the trans-cleavage activity decreased with increasing m6A modifications on gRNA. Together, the m6A modification in the 5′ handle of gRNA has a similar inhibitory pattern on both the cis- and trans-cleavages. The cleavage activity of CRISPR-Cas12a can be efficiently deactivated by m6As at the three sites A3, A4, and A5 of the gRNA pseudoknot structure.
The deactivation mechanism of m6A modified gRNA was further tentatively studied using the previously reported atomic models of Cas12a effectors.34 As shown in Fig. 3A, Cas12a nuclease forms an open “thread cutter” architecture. The 5′ handle of gRNA is located at the hub of the “thread cutter”. The details of the 5′ handle are shown in Fig. 3B and C, and this pseudoknot structure is located in a pocket-like domain of Cas12a nuclease. Through multi-specific interactions with the Cas12a protein, the gRNA pseudoknot functions as a key structural element to recruit Cas12a, promote conformational–state transitions, and activate the nuclease activity of Cas12a.18,36 Previous research has indicated that the N6-methyl group could weaken the stability of the RNA secondary structure.37 We hypothesize that the m6A modifications in the 5′ handle may disrupt the interaction between gRNA and the Cas12a protein, thereby leaving Cas12a in a non-active state. To test this hypothesis, an electrophoretic mobility shift assay (EMSA) was used to determine the binding affinity between m6A modified gRNA and Cas12a. In this assay, the comigration ability of 3′ FAM labelled gRNA (Table S1†) at a 10 nM concentration was compared to the Cas12a concentration ranging from 0 to 80 nM (protein to RNA ratio from 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 8:1). We gradually increased Cas12a nuclease concentrations while maintaining constant gRNA levels. As shown in Fig. 3D, although normal gRNA began to shift at a protein to RNA ratio of 1:1, modified gRNA hardly shifted even at an 8:1 protein to RNA ratio. M6A modified gRNA requires more Cas12a protein for functional RNP assembly than natural gRNA. Furthermore, we systematically investigated the molecular interactions in the 5′ handle of gRNA to comprehend this inhibitory effect (Fig. S4 and S5†). Consistent with the aforementioned hypothesis, m6A in the 5′ handle suppressed gRNA's capacity to bind the Cas12a protein.
:1 to 8:1). We gradually increased Cas12a nuclease concentrations while maintaining constant gRNA levels. As shown in Fig. 3D, although normal gRNA began to shift at a protein to RNA ratio of 1:1, modified gRNA hardly shifted even at an 8:1 protein to RNA ratio. M6A modified gRNA requires more Cas12a protein for functional RNP assembly than natural gRNA. Furthermore, we systematically investigated the molecular interactions in the 5′ handle of gRNA to comprehend this inhibitory effect (Fig. S4 and S5†). Consistent with the aforementioned hypothesis, m6A in the 5′ handle suppressed gRNA's capacity to bind the Cas12a protein.
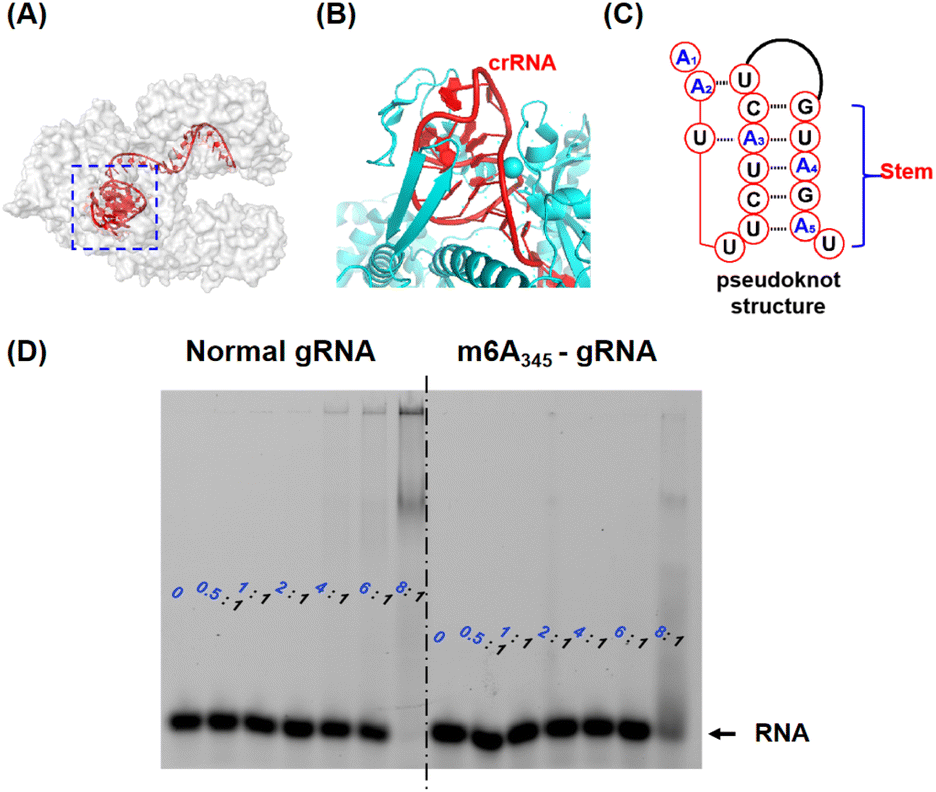 | ||
| Fig. 3 The analysis of the underlying inhibitory mechanism. (A) Crystal structure of the Cas12a-gRNA-dsDNA complex (Cas12a is shown as a grey surface model, and gRNA is marked in red). The 5′ handle of gRNA is denoted by a blue box. (PDB: 5XUZ) (B) The details of the 5′ handle in the Cas12a-gRNA-dsDNA complex. The Cas12a protein is marked in cyan and gRNA is marked in red. (C) The tertiary structure of the 5′ handle of gRNA. (D) The electrophoretic mobility shift assay (EMSA) picture (native 4% PAGE). The varying concentrations of Cas12a were incubated with 3′ labelled FAM gRNA (left graph, 20 nM) or 3′ labelled FAM m6A345-gRNA (right graph, 20 nM) respectively. The ratios listed represent the concentrations of Cas12a: the concentrations of gRNA. | ||
The inhibitory effect of m1A modification on CRISPR-Cas12a
In addition to m6A methylation, m1A is a well-characterized RNA modification containing a methyl group at the first nitrogen of adenosine and could entirely disrupt the Watson–Crick pairing between A and U (Fig. 4A). This chemical modification should therefore have a stronger inhibitory effect on CRISPR-Cas12a than m6A. The m1A-modified gRNAs were also evaluated (Fig. 4B). For cis-cleavage, m1A at the site A2 shows minimal inhibition which is consistent with m6A (Fig. 4C Lane 3). But the m1A at the site A3, A4, or A5 can significantly inhibit the activity of CRISPR-Cas12a (Fig. 4C Lane 4–6). When the sites A3, A4, and A5 were simultaneously modified with m1A, no products were observed, indicating that the cis-cleavage activity of CRISPR-Cas12a was nearly entirely suppressed (Fig. 4C Lane 8). In addition, m1A has a comparable inhibitory effect on trans-cleavage as it does on cis-cleavage (Fig. 4D and S6†). The most effective CRISPR-Cas12a inhibition was achieved by simultaneously locating m1A on sites A3, A4, and A5. The inhibitory effect of m1A exceeds 90% for both cis- and trans-cleavage, which is significantly higher than that of m6A modification. The mechanism of the inhibitory effect of m1A on CRISPR-Cas12a was subsequently investigated. We first used EMSA to examine the interaction between Cas12a and m1A modified gRNA. As shown in Fig. 4E, when the ratio of Cas12a to gRNA was 8:1, the normal gRNA was almost fully comigrated with Cas12a. However, for m1A-modified gRNA, there were few RNPs. The binding affinity of gRNA to Cas12a is significantly decreased by the m1A modification. In addition, the 3D-structural details were also evaluated. Fig. S7† demonstrates that the m1A modification at the sites A3, A4, and A5 prevented adenines and uracil from forming stable Watson–Crick base-pairing, hence directly destroying the pseudoknot structure of gRNA.
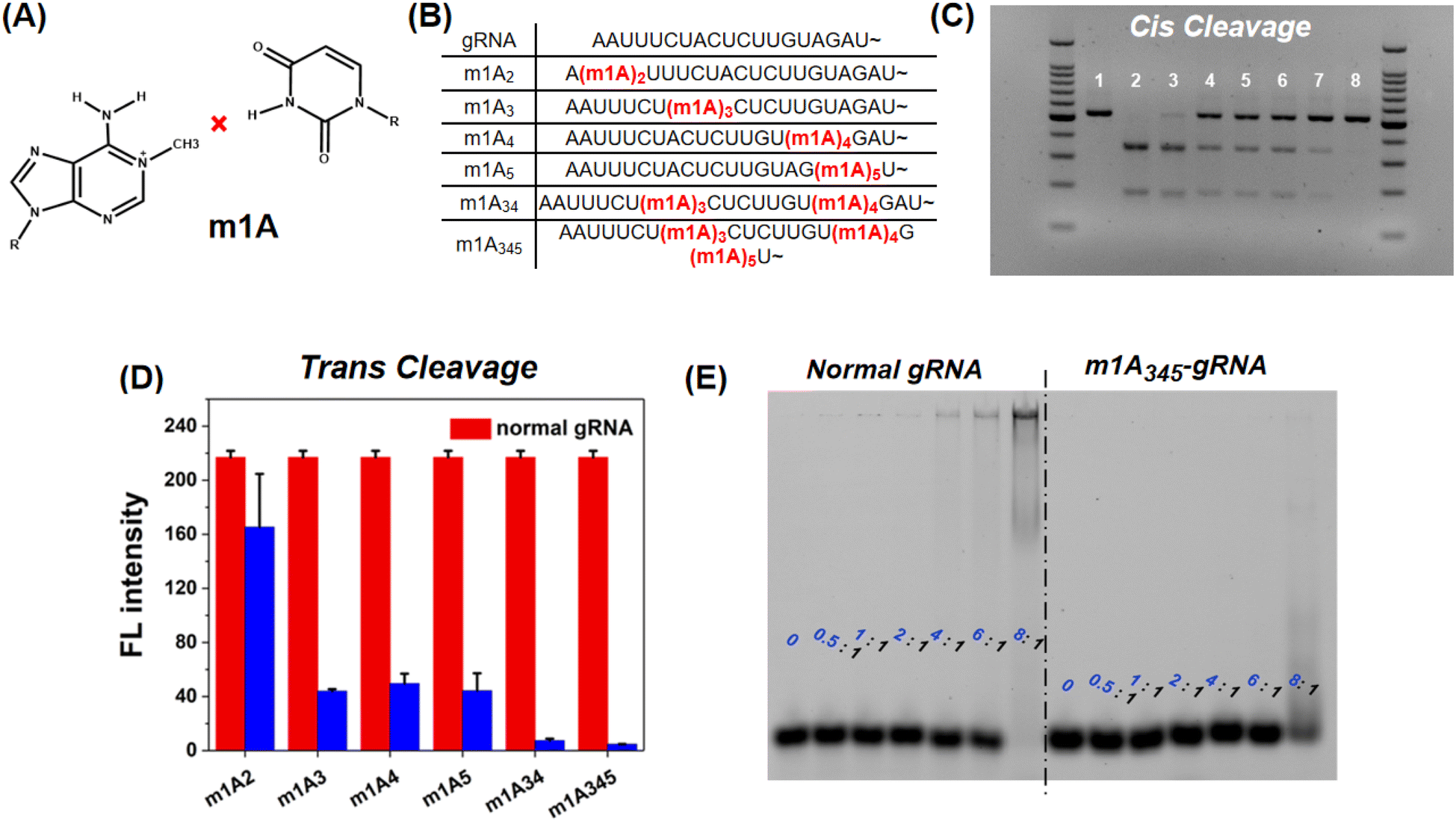 | ||
| Fig. 4 Systematic investigation of the m1A inhibition effect on CRISPR-Cas12a. (A) m1A destructing the Watson–Crick pairing between the adenine base (left) and uracil base (right). (B) The position and number of m1A in the 5′ handle of gRNA. (C) Gel electrophoresis (2%) result of cis cleavage. (Lane 1) Only the dsDNA target; (Lane 2) natural gRNA (50 nM); (Lane 3) m1A2-gRNA (50 nM); (Lane 4) m1A3-gRNA (50 nM); (Lane 5) m1A4-gRNA (50 nM); (Lane 6) m1A5-gRNA (50 nM); (Lane 7) m1A34-gRNA (50 nM); (Lane 8) m1A345-gRNA (50 nM); (D) fluorescence intensity at 525 nm after trans cleaving the FQ-probe (1 μM). The error bar represents the standard deviation of three measurements. (E) Fluorescence images of gel shift assays. The constant concentration of gRNA is 20 nM. And the ratios were calculated using the concentration of Cas12a: the concentration of gRNA. | ||
The m1A modified adenine lost its ability to form hydrogen bonds with nearby uracil, which is similar to a single-base mismatch at adenosine.38 Generally, mismatches between paired gRNA and the DNA target, particularly in the 5-bp PAM-proximal “seed region”, can significantly inhibit the cis- and trans-cleavage behavior of CRISPR-Cas12a.39 Thus, we further investigated the inhibitory effect of m1A on the spacer sequence in detail. As shown in Fig. 5A, we designed three gRNAs with the m1A modification evenly distributed across the spacer sequence (m1A1 is in the “seed region”; m1A3 is adjacent to the “seed region”; m1A6 is far away from the “seed region”). Surprisingly, for the dsDNA activator, all three m1A modified gRNAs effectively deactivated both cis- and trans-cleavage activity of CRISPR-Cas12a with nearly comparable inhibitory efficiency (Fig. 5B and C). For the ssDNA activator, m1A6-gRNA inhibits trans-cleavage significantly less than m1A1-and m1A3-gRNA (Fig. 5D). In general, the “seed region” is highly sensitive to mismatches, but the sequence near the 3′ end of the gRNA tolerates single-based mismatches.40,41 Consequently, m1A1- and m1A3-gRNA should have exhibited more inhibitory efficiency than m1A6-gRNA; rather, dsDNA activators behaved unusually. We hypothesized that m1A on the spacer sequence may inhibit the dsDNA unwinding and R-Loop conformational-state transitions16,42 in the cleavage process, except for preventing Watson–Crick pairing.
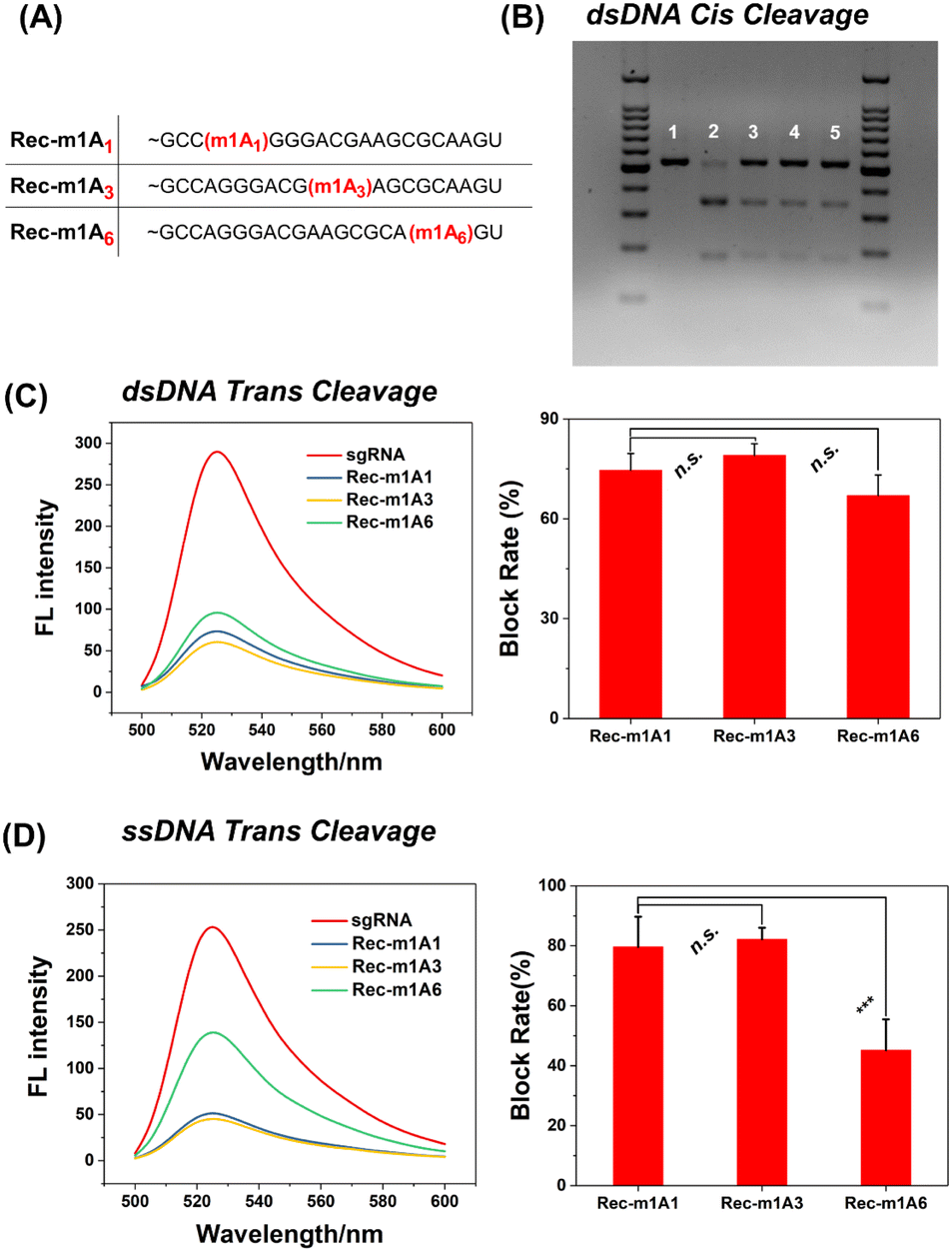 | ||
| Fig. 5 The deactivation of CRISPR-Cas12a cis- and trans-cleavage by m1A modifications in the gRNA 3′ spacer sequence. (A) The position of m1A in the gRNA spacer sequence. (B) The deactivation of CRISPR-Cas12a cis-cleavage. (Lane 1) Only the dsDNA target; (Lane 2) natural gRNA; (Lane 3) m1A1-gRNA; (Lane 4) m1A3-gRNA; (Lane 5) m1A6-gRNA. (C) For the dsDNA activator, the deactivation of CRISPR-Cas12a trans-cleavage by m1A modification. (D) For the ssDNA activator, the deactivation of CRISPR-Cas12a trans-cleavage by m1A modification. (Left graph) The fluorescence spectra result of CRISPR-Cas12a trans cleaving the FQ probe. (Right graph) Inhibitory efficiency of trans-cleavage activity. The error bars show the standard deviation of three parallel experiments. n.s. represents no significant differences and ***P < 0.001 was calculated by Student's t-test. | ||
Re-activating methylation-deactivated CRISPR-Cas12a via demethylases
Typically, the ALKBH5 demethylase can effectively remove m6A modifications on RNA. As shown in Fig. 6, the recovery of cis-cleavage was characterized by gel electrophoresis and the trans-cleavage was monitored by fluorescence assay. Fig. 6B shows that the cis-cleavage ability of CRISPR-Cas12a was successfully reactivated following ALKBH5 treatment. In addition, the fluorescence studies have shown that ALKBH5 activated trans-cleavage as well. The fluorescence intensity of ALKBH5-treated m6A345-gRNA (Fig. 6C line c) could recover to the same level as that of natural gRNA (Fig. 6C line a). ALKBH5, Cas12a protein, and dsDNA target were incubated without gRNA as a negative control. As expected, there were no dsDNA cleavage products and fluorescence signals (Fig. 6B lane d and Fig. 6C line d). To demonstrate the ALKBH5-mediated demethylation, the ALKBH5-treated m6A345-gRNA was hydrolyzed by P1 nuclease, and the hydrolysates were subsequently analyzed using HPLC-MS/MS. As shown in Fig. 6D and E, the amount of guanine (G) in m6A345-gRNA following ALKBH5 treatment remained unchanged. In contrast, the amount of m6A was significantly reduced compared to when ALKBH5 was absent. Overall, these results indicated that ALKBH5 reactivates deactivated CRISPR-Cas12a by demethylation of m6A-modified gRNA. Additionally, FTO is a well-characterized m6A demethylase. FTO demethylase has also been shown to restore m6A-deactivated CRISPR-Cas12a (Fig. S8†). We further attempted to activate the m1A-deactivated gRNA using ALKBH3 demethylase. As shown in Fig. S9,† m1A-deactivated CRISPR-Cas12a exhibited remarkable dsDNA cis-cleavage and ssDNA trans-cleavage abilities in the presence of ALKBH3 demethylase. Furthermore, the HPLC-MS/MS results revealed that m1A345-gRNA underwent a distinct demethylation process following ALKBH3 treatment (Fig. S9D†). Therefore, we successfully developed a demethylase-inducible CRISPR-Cas12a (DIC-Cas12a) strategy and found that both m6A- and m1A-deactivated CRISPR-Cas12a can be efficiently restored by demethylases.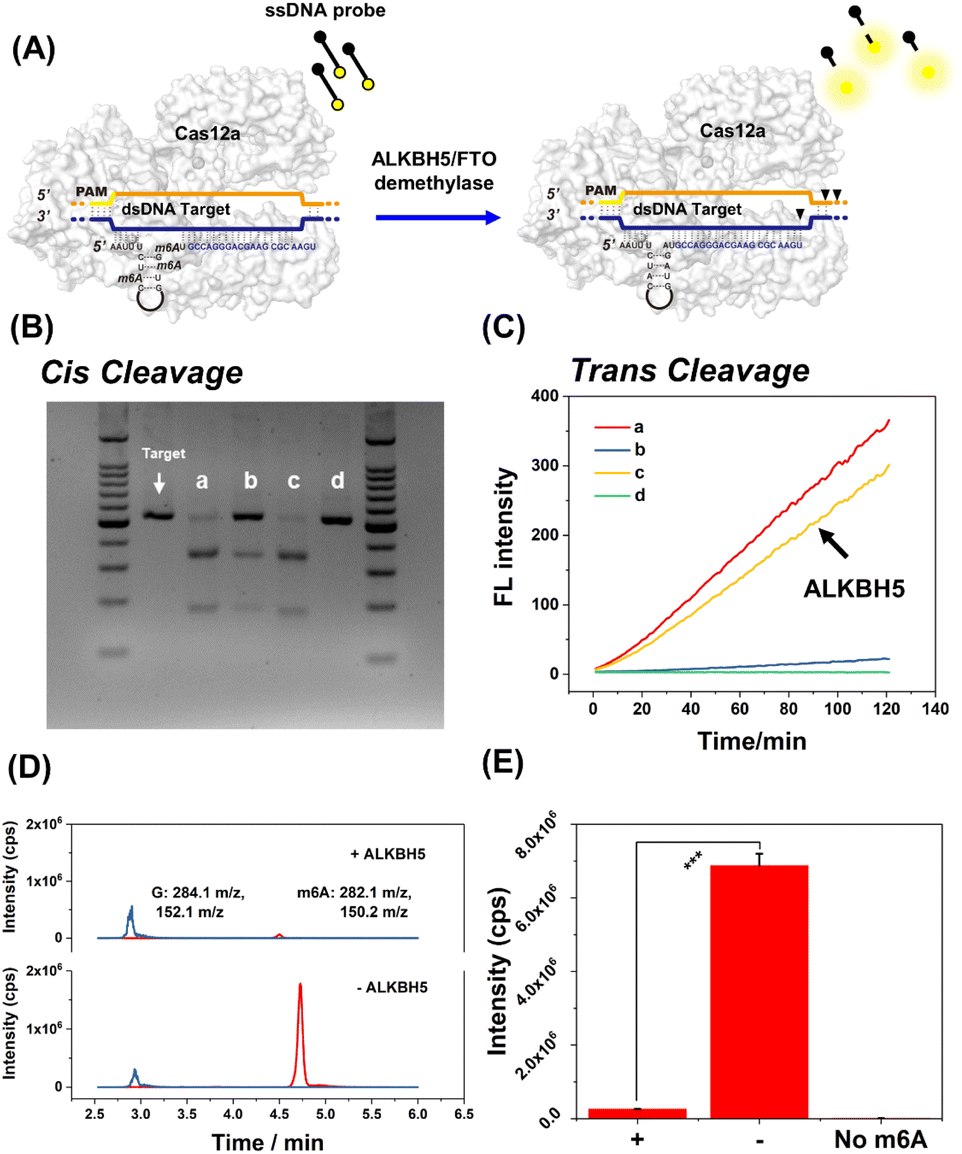 | ||
| Fig. 6 Activating m6A-deactivated CRISPR-Cas12a through demethylases. (A) Schematic illustration. The dsDNA was a substrate of cis-cleavage. And the trans-cleavage was monitored by fluorescence. (B) Activation of CRISPR-Cas12a cis-cleavage (2% agarose gel). (Lane 1) Only the dsDNA target; (Lane a) natural gRNA + Cas12a + dsDNA; (Lane b) m6A345-gRNA + Cas12a + dsDNA target; (Lane c) ALKBH5 demethylase + m6A345-gRNA + Cas12a + dsDNA target; (Lane d) negative control, ALKBH5 demethylase + Cas12a + dsDNA target. (C) Activation of CRISPR-Cas12a trans-cleavage. The experimental details of (a)–(d) were the same as those of (B) and the FQ probe was joined in these groups respectively. (D) and (E) The results of HPLC-MS/MS demonstrated that ALKBH5 demethylase removed m6A modification on gRNA. The mother ion of the G base is 284.1 m/z and the specific fragmental ion is 152.1 m/z in mass spectrometry. The mother ion of m6A is 282.1 m/z and the specific fragmental ion is 150.2 m/z. Error bars show the standard deviation of three parallel experimental groups. ***P < 0.001 was obtained by Student's t-test. | ||
Typical application of DIC-Cas12a based on trans-cleavage
The remarkable response of demethylases encouraged us to employ methylation-deactivated CRISPR-Cas12a for some potential applications. First, we intended to develop a fluorescence sensor for demethylase detection by trans-cleavage of CRISPR-Cas12a. Our demethylase biosensor consisted of methylated gRNA, the Cas12a protein, an ssDNA activator, and an ssDNA FQ probe. In the presence of demethylase, the methylated gRNA refolds structurally and binds to the ssDNA activator and Cas12a. In this case, CRISPR-Cas12a can be activated to cleave the FQ probes in a trans pattern, resulting in a continuous increase in fluorescence. The typical ALKBH5 demethylase was first measured. The concentrations of m6A345-gRNA were optimized for exceptional analytical performance (Fig. S10†). Then, fluorescence dynamic curves were established by detecting different concentrations of ALKBH5 (Fig. 7A). The fluorescence intensity increased dose-dependently from 0 nM to 300 nM for demethylases (Fig. 7B), with a good linear correlation and limit of detection (LOD). The LOD was about 0.417 nM (the concentrations corresponding to a signal 3 SD above the mean of 11 replicates of the zero calibrator). We also used this strategy to develop the FTO demethylase sensor as shown in Fig. S12.† Additionally, the m1A-deactivated CRISPR-Cas12a system was used to detect ALKBH3 demethylase (Fig. 7D and S11†). The calibration curve regression equation was derived from the range of 200–800 nM, and the LOD for ALKBH3 was 97.5 nM (Fig. 7E). The LODs of the above demethylases were comparable to or even superior to those of the reported method, and there were no laborious washing procedures or antibody incubation procedures, compared with the traditional ELISA strategy.43–45 Additionally, the selectivity of this strategy was assessed. The methylated gRNA was pretreated using endogenous proteins and small molecules respectively, including bovine serum albumin (BSA), polynucleotide kinase (PNK), and adenosine triphosphate (ATP). Fig. 7C and F show that the fluorescence response was highly selective for demethylases compared with other interferences. Collectively, these findings demonstrated that the DIC-Cas12a strategy offers a novel approach for expanding the bioanalytical applications of CRISPR-Cas systems.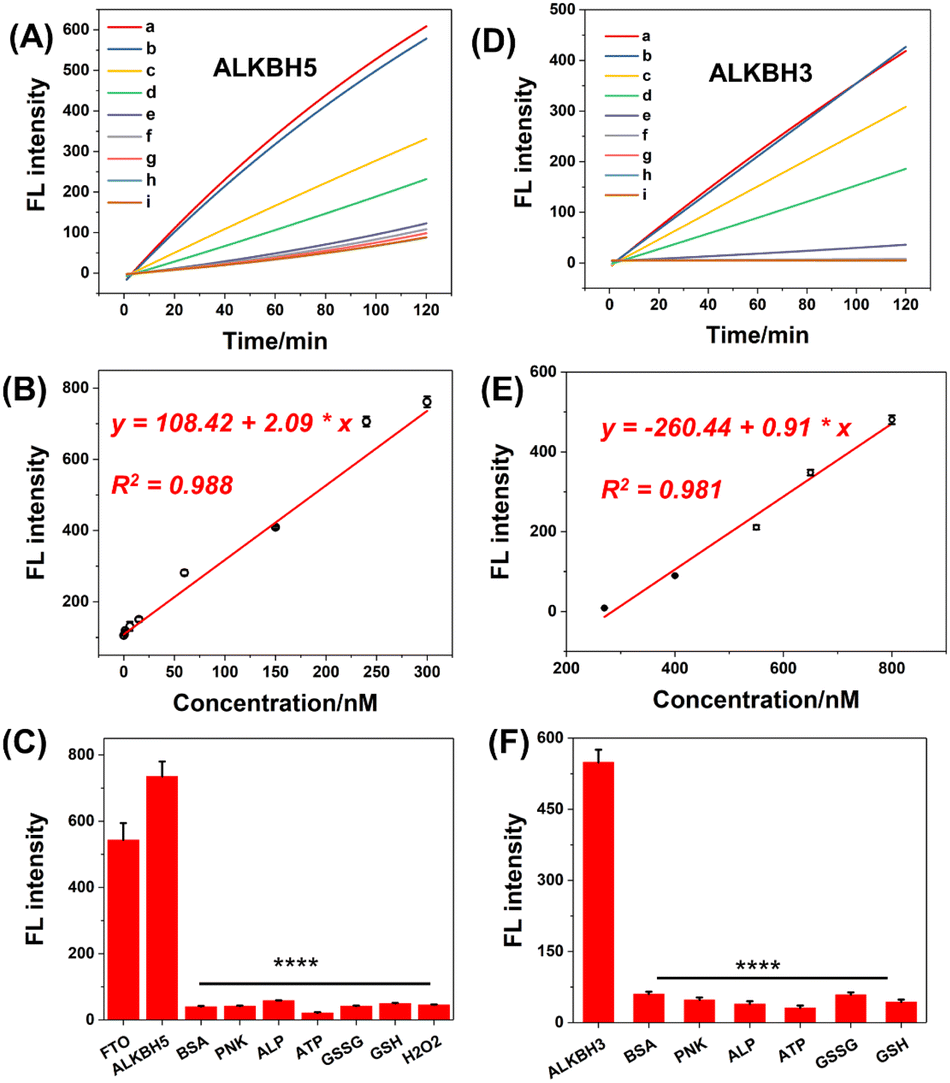 | ||
| Fig. 7 Demethylase sensor based on trans-cleavage ability of the DIC-Cas12a system. (A) Fluorescence signal increased as the concentrations of ALKBH5 increased ((a) 300 nM; (b) 240 nM; (c) 150 nM; (d) 60 nM; (e) 15 nM; (f) 6 nM; (g) 1.5 nM; (h) 0.6 nM; (i) 0; m6A345-gRNA: 25 nM). (B) Calibration curves of plotting the fluorescence intensity versus ALKBH5 concentrations. (C) The fluorescence intensity at 525 nm in the presence of m6A demethylases or other interferences. BSA (bovine serum albumin), PNK (T4 polynucleotide kinase), and ALP (alkaline phosphatase). **P< 0.01; ****P < 0.001 (Student's t-test and all results were compared with those of the ALKBH5 group). (D) Fluorescence signal increased as concentrations of ALKBH3 increased over time ((a) 1000 nM; (b) 800 nM; (c) 650 nM; (d) 550 nM; (e) 400 nM; (f) 270 nM; (g) 135 nM; (h) 62.5 nM; (i) 0 nM; m1A345-gRNA: 250 nM). (E) Calibration curves of ALKBH3. (F) Selectivity of the ALKBH3 biosensor. ****P < 0.001 (Compared with the ALKBH3 group, the P value was calculated by Student's t-test). | ||
Controllable gene editing based on the levels of cellular demethylases
In addition to in vitro sensors, we tentatively used the DIC-Cas12a system to knock down the endogenous gene. We chose the VEGAF loci in the HEK293 cell line as target for gene editing and the sequence of gRNA followed some literature studies.46 In general, if the CRISPR-Cas12a system works well, double-stranded breaks can be repaired by the non-homologous end-joining (NHEJ) pathway to induce random insertions and deletions (indels).We directly transfected the HEK293T cells with the m1A modified gRNAs. The gene editing results and quantitative indel frequency of targeted gene loci were monitored by the Sanger sequencing method. As shown in Fig. 8A and B, after transfecting the m1A modified gRNA, the sequencing map showed almost no disturbance caused by indels. The indel frequency caused by Cas12a with m1A modified gRNAs in HEK293 cells was about 2.63%. In the negative group, an indel frequency of 0.38% was detected in cells without any gRNA. In the positive group, normal gRNA caused an indel frequency of 24.93%. These results demonstrated that the m1A modifications on the gRNA conserved region effectively inhibited the CRISPR-Cas12a activity in living cells. Moreover, we pre-treated the HEK293 cell line with an ALKBH3 demethylase overexpressed plasmid, and the m1A-modified gRNA showed an indel frequency of 17.83%, which was comparable to that of normal gRNA and the editing efficiency increased about 8 fold. Obviously, the editing ability of CRISPR-Cas12a was activated by the overexpressed m1A demethylase in living cells. Since there are significant differences in the content and activity of demethylases in different cells, such as cancer cells and normal cells, this strategy has the potential to be used for cell-type-specific gene editing, which is valuable for gene therapy and diagnosis.
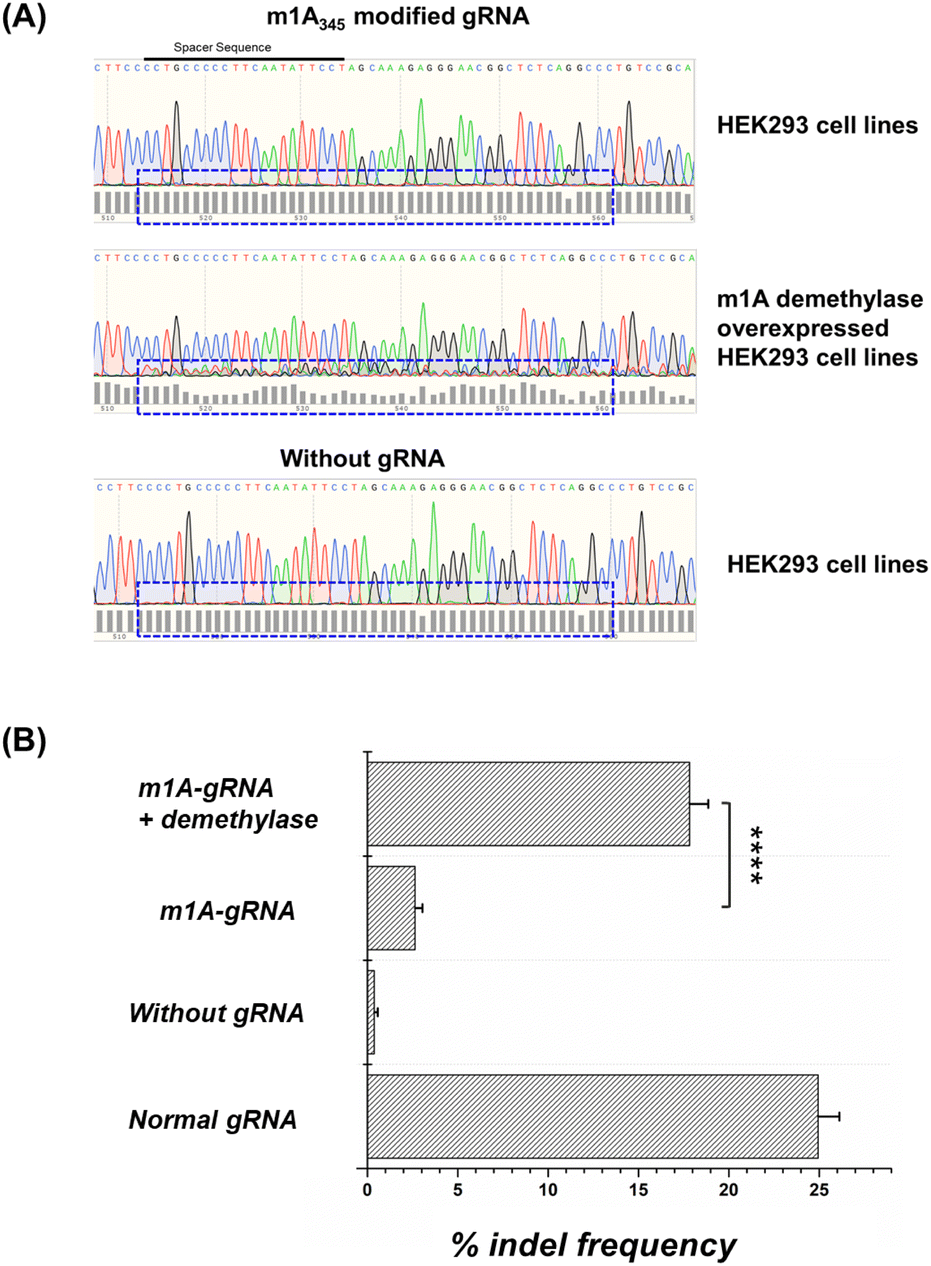 | ||
| Fig. 8 Controllable gene editing by m1A-gRNA and demethylases. (A) The results of Sanger sequencing under different conditions. The dashed lines suggest the level of disturbance as a visualization of indel formation. (B) Quantitative indel frequency of VEGAF gene loci under different conditions. The error bars show the standard deviation of three parallel experiments. ****P < 0.0001 was obtained by Student's t-test. | ||
Regulating the expression of genes based on the levels of cellular demethylases
CRISPR activation and interference have been widely used to modulate gene functions.47 In CRISPR activation technology, a non-cleavage Cas nuclease (dCas) is coupled with a transcriptional effector to regulate target gene expression.48,49 Therefore, we further designed a demethylase-inducible CRISPR activation strategy (DICa), in which a transcriptional circuit with two plasmids was designed to demonstrate our strategy (Fig. 9A).49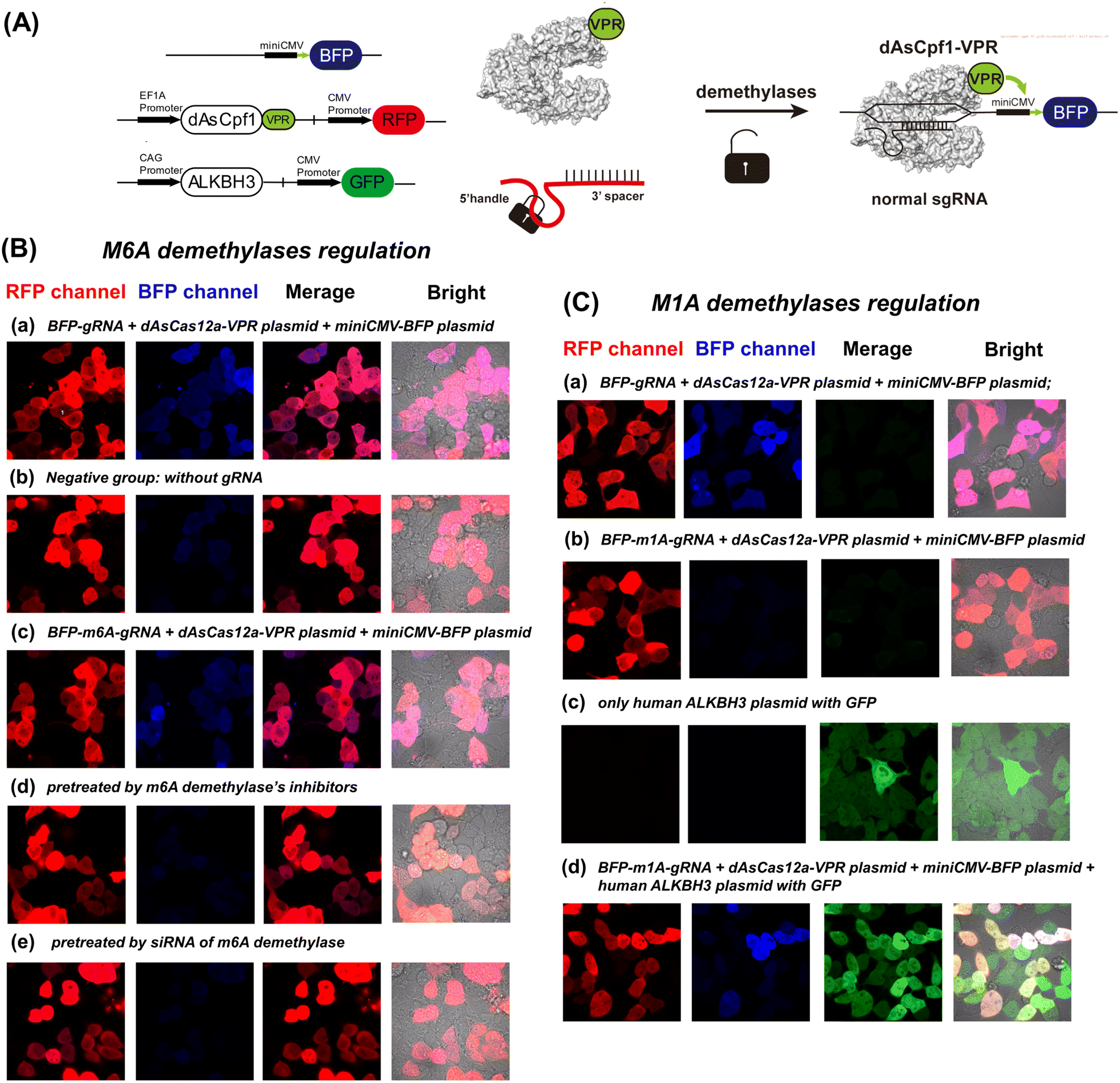 | ||
| Fig. 9 Results of m6A/m1A-demethylases inducible CRISPR-dCas12a transcriptional circuits. (A) Illustration of the transcriptional circuits activated by m6A- and m1A-demethylases. The first one is a TS-miniCMV-BFP plasmid. The second plasmid is dAsCas12a-VPR which exhibited red fluorescence in cells. The third plasmid is a commercially available human ALKBH3 plasmid which shows green fluorescence in cells. The dAsCas12a-VPR plasmid produced red fluorescence after being transfected into the cell. The expression of BFP can be activated by the BFP-gRNA and dCas12-VPR complex. (B) The results of confocal laser scanning microscopy (CLSM) under different conditions. (a) BFP-gRNA + dAsCas12a-VPR plasmid + miniCMV-BFP plasmid; (b) negative group: without gRNA; (c) BFP-m6A-gRNA + dAsCas12a-VPR plasmid + miniCMV-BFP plasmid; (d) pretreated with m6A demethylase inhibitors: entacapone (80 μM) and IOX-1 (60 μM). (e) Pretreated with siRNA before incubating with BFP-m6A-gRNA and two plasmids. (C) The results of CLSM under different conditions. (a) BFP-gRNA + dAsCas12a-VPR plasmid + miniCMV-BFP plasmid; (b) BFP-m1A-gRNA + dAsCas12a-VPR plasmid + miniCMV-BFP plasmid; (c) only human ALKBH3 plasmid with GFP; (d) BFP-m1A-gRNA + dAsCas12a-VPR plasmid + miniCMV-BFP plasmid + human ALKBH3 plasmid with GFP; the scale bar of CLSM is 20 μm. | ||
The HEK293 cells, which have been widely used in demethylases research as a model cell line, were used to establish the CRISPR activation system.31,50 As shown in Fig. 9B and C, all groups exhibited strong red fluorescence, indicating successful transfection of the dAsCas12a-VPR plasmid (Fig. S13†) into living cells. After transfection of no-modified gRNA, the illuminating BFP signal indicated that gRNA matched to the TS-miniCMV-BFP plasmid (Fig. S14†) successfully activates the transcriptional circuit (Fig. 9B(a) and 9C(a)). In the negative group, there is no BFP expression without any gRNA in cells. In general, the m6A demethylases are overexpressed in HEK cell lines, and as a result of m6A removal by endogenous demethylases, the m6A-deactivated gRNA could activate BFP expression similarly to the normal gRNA group (Fig. 9B(c)). We downregulated the FTO and ALKBH5 demethylases by using specific inhibitors (entacapone and IOX-1) and siRNAs, respectively. As expected, the m6A-modified gRNA cannot activate BFP expression in pre-treated HEK293 cells (Fig. 9B(d) and 9B(e)). Furthermore, an assay was performed to evaluate real-time imaging of the DICa strategy. As shown in Fig. S15,† the transcriptional circuit of DICa began about 6–8 h after m6A-deactivated gRNA transfection. The BFP fluorescence increased with time and reached a maximal level at 12 h.
To confirm the precise control of plasmid expression by m1A demethylase, we transfected m1A-modified gRNA into dCas12a-VPR overexpressed HEK293 cells. As shown in Fig. 9C(a) and (b), the m1A-modified gRNA did not induce blue fluorescence upon transfection. Western blot confirmed that the expression levels of ALKBH3 demethylase in the cytoplasm and nucleus of the HEK cell line are much lower than those of other demethylases (Fig. S16†). Then, we pre-treated the HEK293 cell line with a plasmid encoding m1A demethylase ALKBH3, and the results shown in Fig. 9C(c) and (d) demonstrate that m1A-modified gRNA with overexpressed ALKBH3 demethylase could re-activate BFP expression. A complementary technique to demonstrate the efficacy of this sensor in cells has been performed by qRT-PCR against the inducible gene. As shown in Fig. S17,† the mRNA level changes of BFP are consistent with the fluorescence results shown in Fig. 9. Overall, these results suggest that the methylation-deactivated CRISPR-Cas system could be specifically manipulated by the level of demethylases in cells. Naturally, an intracellular demethylase sensor based on the demethylase inducible CRISPR-Cas12a system could be established at the same time.
Conclusions
In summary, we found that both the m6A and m1A epigenetic modifications in the 5′ handle of gRNA displayed efficient inhibitory effects on DNA cleavage. In addition, this regulation is highly dependent on the position and number of methylation modifications. Especially for the m1A methylation, when the sites A3, A4, and A5 in the 5′ handle of gRNA are modified at the same time, the deactivation efficacy reached 98%. Besides, the inhibition mechanism was tentatively analysed in this work. The methylation in the 5′ handle of gRNA impedes the formation of RNPs, thereby preventing the cleavage behaviour of CRISPR-Cas12a. Moreover, a demethylase-inducible CRISPR-Cas12a tool was developed. Methylation-deactivated CRISPR-Cas12a displayed remarkable cleavage activity in the presence of demethylases. ALKBH5 and FTO demethylases can effectively activate the m6A-deactivated CRISPR-Cas12a. ALKBH3 demethylase can selectively activate m1A-deactivated CRISPR-Cas12a. The exceptional performance of DIC-Cas12a enables us to develop in vitro sensor strategies with a high degree of sensitivity. Due to these unique advantages, we further proposed a demethylase-inducible CRISPR activation strategy and demethylase-inducible gene editing strategy, which enabled plasmid gene expression in living cells to be activated and gene editing to be controlled. Specifically, m6A modified gRNA and m1A modified gRNA displayed different results in HEK293T cell lines. The m6A modified gRNA could directly activate the BFP gene expression in HEK293. In contrast, m1A-modified sgRNA requires a large amount of exogenous demethylase to activate the target gene in HEK293 cells. These results suggested that our demethylase-inducible CRISPR system could respond to specific cells according to the level of cellular demethylases. As for the limitations of this strategy, according to current experimental findings, m6A-modified gRNA exhibits lower stability in cells compared to m1A-modified gRNA, and its blocking efficiency is only 70% in vitro, which is significantly lower than the 98% blocking efficiency of m1A-modified gRNA. Besides, compared with unnatural nucleotide modifications, such as 2′-F and phosphorothioates, natural methylation modifications cannot effectively prevent the degradation of gRNA by RNase in cells. Despite these limitations, we believe that exploring the methylation effect on emerging Cas proteins, such as Cas13d and Cas12f, could expand the potential therapeutic applications of this strategy.Author contributions
Zhian Hu and Ao Sun planned and completed all experiments in this work. Zhian Hu and Ao Sun contributed equally. Jinlei Yang and Gongwei Sun assisted in some experiments, including cell experiments and CLSM detection. Gu Naz gave suggestions for some experiments and helped in article polishing. This manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Metabolomics Facility Center in the National Protein Science Technology Center of Tsinghua University for HPLC-MS/MS experiments. And we thank VectorBuilder Inc. (Guangzhou, China) for plasmid construction. We thank the Home for Researchers editorial team (https://www.home-for-researchers.com/) for language editing service. We thank Huasong Ai for assistance in EMSA experiments. This work was supported by the National Natural Science Foundation of China (Nos. 21974078, 21727813, and 22074077), National Science and Technology Major Project of the Ministry of Science and Technology of China (2022YFF0710200) and Fundamental Research Funds for the Central Universities (FRF-TP-22-022A1).Notes and references
- R. Torres-Ruiz and S. Rodriguez-Perales, CRISPR-Cas9 technology: applications and human disease modelling, Briefings Funct. Genomics, 2017, 16, 4–12 CrossRef CAS PubMed.
- J. A. Doudna and E. Charpentier, Genome editing. The new frontier of genome engineering with CRISPR-Cas9, Science, 2014, 346, 1258096 CrossRef PubMed.
- X. Dai, X. Chen, Q. Fang, J. Li and Z. Bai, Inducible CRISPR genome-editing tool: classifications and future trends, Crit. Rev. Biotechnol., 2018, 38, 573–586 CrossRef CAS PubMed.
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Development of Light-Activated CRISPR Using Guide RNAs with Photocleavable Protectors, Angew. Chem., Int. Ed. Engl., 2016, 55, 12440–12444 CrossRef CAS PubMed.
- Y. Nihongaki, F. Kawano, T. Nakajima and M. Sato, Photoactivatable CRISPR-Cas9 for optogenetic genome editing, Nat. Biotechnol., 2015, 33, 755–760 CrossRef CAS PubMed.
- L. E. Dow, J. Fisher, K. P. O'Rourke, A. Muley, E. R. Kastenhuber, G. Livshits, D. F. Tschaharganeh, N. D. Socci and S. W. Lowe, Inducible in vivo genome editing with CRISPR-Cas9, Nat. Biotechnol., 2015, 33, 390–394 CrossRef CAS PubMed.
- K. I. Liu, M. N. Ramli, C. W. Woo, Y. Wang, T. Zhao, X. Zhang, G. R. Yim, B. Y. Chong, A. Gowher, M. Z. Chua, J. Jung, J. H. Lee and M. H. Tan, A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing, Nat. Chem. Biol., 2016, 12, 980–987 CrossRef CAS PubMed.
- S. R. Wang, L. Y. Wu, H. Y. Huang, W. Xiong, J. Liu, L. Wei, P. Yin, T. Tian and X. Zhou, Conditional control of RNA-guided nucleic acid cleavage and gene editing, Nat. Comm., 2020, 11, 91 CrossRef PubMed.
- X. W. Wang, L. F. Hu, J. Hao, L. Q. Liao, Y. T. Chiu, M. Shi and Y. Wang, A microRNA-inducible CRISPR-Cas9 platform serves as a microRNA sensor and cell-type-specific genome regulation tool, Nat. Cell Biol., 2019, 21, 522–530 CrossRef PubMed.
- C. Gu, L. Xiao, J. Shang, X. Xu, L. He and Y. Xiang, Chemical synthesis of stimuli-responsive guide RNA for conditional control of CRISPR-Cas9 gene editing, Chem. Sci., 2021, 12, 9934–9945 RSC.
- Y. Fu, J. D. Sander, D. Reyon, V. M. Cascio and J. K. Joung, Improving CRISPR-Cas nuclease specificity using truncated guide RNAs, Nat. Biotechnol., 2014, 32, 279–284 CrossRef CAS PubMed.
- E. Rozners, Chemical Modifications of CRISPR RNAs to Improve Gene-Editing Activity and Specificity, J. Am. Chem. Soc., 2022, 144, 12584–12594 CrossRef CAS PubMed.
- W. Zhou, W. Brown, A. Bardhan, M. Delaney, A. S. Ilk, R. R. Rauen, S. I. Kahn, M. Tsang and A. Deiters, Spatiotemporal Control of CRISPR/Cas9 Function in Cells and Zebrafish using Light-Activated Guide RNA, Angew. Chem., Int. Ed. Engl., 2020, 59, 8998–9003 CrossRef CAS PubMed.
- B. Li, W. Zhao, X. Luo, X. Zhang, C. Li, C. Zeng and Y. Dong, Engineering CRISPR-Cpf1 crRNAs and mRNAs to maximize genome editing efficiency, Nat. Biomed. Eng., 2017, 1, 66 CrossRef CAS PubMed.
- M. A. McMahon, T. P. Prakash, D. W. Cleveland, C. F. Bennett and M. Rahdar, Chemically Modified Cpf1-CRISPR RNAs Mediate Efficient Genome Editing in Mammalian Cells, Mol. Ther., 2018, 26, 1228–1240 CrossRef CAS PubMed.
- A. R. Krysler, C. R. Cromwell, T. Tu, J. Jovel and B. P. Hubbard, Guide RNAs containing universal bases enable Cas9/Cas12a recognition of polymorphic sequences, Nat. Comm., 2022, 13, 1617 CrossRef CAS PubMed.
- C. R. Cromwell, K. Sung, J. Park, A. R. Krysler, J. Jovel, S. K. Kim and B. P. Hubbard, Incorporation of bridged nucleic acids into CRISPR RNAs improves Cas9 endonuclease specificity, Nat. Comm., 2018, 9, 1448 CrossRef PubMed.
- E. A. Ageely, R. Chilamkurthy, S. Jana, L. Abdullahu, D. O'Reilly, P. J. Jensik, M. J. Damha and K. T. Gagnon, Gene editing with CRISPR-Cas12a guides possessing ribose-modified pseudoknot handles, Nat. Comm., 2021, 12, 6591 CrossRef CAS PubMed.
- S. Wang, L. Wei, J. Q. Wang, H. Ji, W. Xiong, J. Liu, P. Yin, T. Tian and X. Zhou, Light-Driven Activation of RNA-Guided Nucleic Acid Cleavage, ACS Chem. Biol., 2020, 15, 1455–1463 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, F. Xie, J. Lin and L. Xu, Photocontrol of CRISPR/Cas9 function by site-specific chemical modification of guide RNA, Chem. Sci., 2020, 11, 11478–11484 RSC.
- D. Zhang, L. Liu, S. Jin, E. Tota, Z. Li, X. Piao, X. Zhang, X. D. Fu and N. K. Devaraj, Site-Specific and Enzymatic Cross-Linking of sgRNA Enables Wavelength-Selectable Photoactivated Control of CRISPR Gene Editing, J. Am. Chem. Soc., 2022, 144, 4487–4495 CrossRef CAS PubMed.
- M. Habibian, C. McKinlay, T. R. Blake, A. M. Kietrys, R. M. Waymouth, P. A. Wender and E. T. Kool, Reversible RNA acylation for control of CRISPR-Cas9 gene editing, Chem. Sci., 2019, 11, 1011–1016 RSC.
- P. Jones, Functions of DNA methylation: islands, start sites, gene bodies and beyond, Nat. Rev. Genet., 2012, 29(13), 484–492 CrossRef PubMed.
- J. S. Kim, Genome editing comes of age, Nat. Protoc., 2016, 11, 1573–1578 CrossRef CAS PubMed.
- J. J. Liu, N. Orlova, B. L. Oakes, E. Ma, H. B. Spinner, K. L. M. Baney, J. Chuck, D. Tan, G. J. Knott, L. B. Harrington, B. Al-Shayeb, A. Wagner, J. Brötzmann, B. T. Staahl, K. L. Taylor, J. Desmarais, E. Nogales and J. A. Doudna, CasX enzymes comprise a distinct family of RNA-guided genome editors, Nature, 2019, 566, 218–223 CrossRef CAS PubMed.
- J. Song and C. Yi, Chemical Modifications to RNA: A New Layer of Gene Expression Regulation, ACS Chem. Biol., 2017, 12, 316–325 CrossRef CAS PubMed.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Reading, writing and erasing mRNA methylation, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef CAS PubMed.
- D. V. Prokhorova, I. P. Vokhtantsev, P. O. Tolstova, E. S. Zhuravlev, L. M. Kulishova, D. O. Zharkov and G. A. Stepanov, Natural Nucleoside Modifications in Guide RNAs Can Modulate the Activity of the CRISPR-Cas9 System In Vitro, CRISPR J., 2022, 5, 799–812 CrossRef CAS PubMed.
- I. Barbieri and T. Kouzarides, Role of RNA modifications in cancer, Nat. Rev. Cancer, 2020, 20, 303–322 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C. M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W. L. Wang, S. H. Song, Z. Lu, R. P. Bosmans, Q. Dai, Y. J. Hao, X. Yang, W. M. Zhao, W. M. Tong, X. J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y. G. Yang and C. He, ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed.
- X. Li, X. Xiong, K. Wang, L. Wang, X. Shu, S. Ma and C. Yi, Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome, Nat. Chem. Biol., 2016, 12, 311–316 CrossRef CAS PubMed.
- I. A. Roundtree, M. E. Evans, T. Pan and C. He, Dynamic RNA Modifications in Gene Expression Regulation, Cell, 2017, 169, 1187–1200 CrossRef CAS PubMed.
- T. Yamano, B. Zetsche, R. Ishitani, F. Zhang, H. Nishimasu and O. Nureki, Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1, Mol. Cell, 2017, 67, 633–645 CrossRef CAS PubMed.
- C. Roost, S. R. Lynch, P. J. Batista, K. Qu, H. Y. Chang and E. T. Kool, Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification, J. Am. Chem. Soc., 2015, 137, 2107–2115 CrossRef CAS PubMed.
- S. Stella, P. Mesa, J. Thomsen, B. Paul, P. Alcón, S. B. Jensen, B. Saligram, M. E. Moses, N. S. Hatzakis and G. Montoya, Conformational Activation Promotes CRISPR-Cas12a Catalysis and Resetting of the Endonuclease Activity, Cell, 2018, 175, 1856–1871 CrossRef CAS PubMed.
- K. I. Zhou, M. Parisien, Q. Dai, N. Liu, L. Diatchenko, J. R. Sachleben and T. Pan, N(6)-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding, J. Mol. Biol., 2016, 428, 822–833 CrossRef CAS PubMed.
- Y. Chen, S. Yang, S. Peng, W. Li, F. Wu, Q. Yao, F. Wang, X. Weng and X. Zhou, N1-Methyladenosine detection with CRISPR-Cas13a/C2c2, Chem. Sci., 2019, 10, 2975–2979 RSC.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- E. M. Anderson, A. Haupt, J. A. Schiel, E. Chou, H. B. Machado, Ž. Strezoska, S. Lenger, S. McClelland, A. Birmingham, A. Vermeulen and A. Smith, Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity, J. Biotechnol., 2015, 211, 56–65 CrossRef CAS PubMed.
- Y. Tang and Y. Fu, Class 2 CRISPR/Cas: an expanding biotechnology toolbox for and beyond genome editing, Cell Biosci., 2018, 8, 59 CrossRef CAS PubMed.
- J. C. Cofsky, D. Karandur, C. J. Huang, I. P. Witte, J. Kuriyan and J. A. Doudna, CRISPR-Cas12a exploits R-loop asymmetry to form double-strand breaks, Elife, 2020, 9, 55143 CrossRef PubMed.
- A. Cheong, J. J. A. Low, A. Lim, P. M. Yen and E. C. Y. Woon, A fluorescent methylation-switchable probe for highly sensitive analysis of FTO N (6)-methyladenosine demethylase activity in cells, Chem. Sci., 2018, 9, 7174–7185 RSC.
- Q. Wang, K. Tan, H. Wang, J. Shang, Y. Wan, X. Liu, X. Weng and F. Wang, Orthogonal Demethylase-Activated Deoxyribozyme for Intracellular Imaging and Gene Regulation, J. Am. Chem. Soc., 2021, 143, 6895–6904 CrossRef CAS PubMed.
- X. Wang, X. Yi, Z. Huang, J. He, Z. Wu, X. Chu and J. H. Jiang, Repaired and Activated DNAzyme Enables the Monitoring of DNA Alkylation Repair in Live Cells, Angew. Chem., Int. Ed. Engl., 2021, 60, 19889–19896 CrossRef CAS PubMed.
- B. Zetsche, M. Heidenreich, P. Mohanraju, I. Fedorova, J. Kneppers, E. M. DeGennaro, N. Winblad, S. R. Choudhury, O. O. Abudayyeh, J. S. Gootenberg, W. Y. Wu, D. A. Scott, K. Severinov, J. van der Oost and F. Zhang, Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array, Nat. Biotechnol., 2017, 35, 31–34 CrossRef CAS PubMed.
- M. Kampmann, CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine, ACS Chem. Biol., 2018, 13, 406–416 CrossRef CAS PubMed.
- T. I. Jensen, N. S. Mikkelsen, Z. Gao, J. Foßelteder, G. Pabst, E. Axelgaard, A. Laustsen, S. König, A. Reinisch and R. O. Bak, Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi, Genome Res., 2021, 31, 2120–2130 CrossRef PubMed.
- Z. M. Ying, F. Wang, X. Chu, R. Q. Yu and J. H. Jiang, Activatable CRISPR Transcriptional Circuits Generate Functional RNA for mRNA Sensing and Silencing, Angew. Chem., Int. Ed. Engl., 2020, 59, 18599–18604 CrossRef CAS PubMed.
- J. Wei, F. Liu, Z. Lu, Q. Fei, Y. Ai, P. C. He, H. Shi, X. Cui, R. Su, A. Klungland, G. Jia, J. Chen and C. He, Differential m(6)A, m(6)A(m), and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm, Mol. Cell, 2018, 71, 973–985 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00629h |
| This journal is © The Royal Society of Chemistry 2023 |