
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formal synthesis of kibdelomycin and derivatisation of amycolose glycosides†
Manuel G.
Schriefer‡
,
Laura
Treiber‡
and
Rainer
Schobert
 *
*
Organic chemistry laboratory, University of Bayreuth, Universitaetsstr. 30, 95447 Bayreuth, Germany. E-mail: Rainer.Schobert@uni-bayreuth.de
First published on 3rd March 2023
Abstract
A convergent total synthesis of bacterial gyrase B/topoisomerase IV inhibitor kibdelomycin (a.k.a. amycolamicin) (1) was devised starting from inexpensive D-mannose and L-rhamnose, which were converted in new efficient ways to an N-acylated amycolose and an amykitanose derivative as late building blocks. For the former, we developed an expeditious, general method for the introduction of an α-aminoalkyl linkage into sugars via 3-Grignardation. The decalin core was built up in seven steps via an intramolecular Diels–Alder reaction. These building blocks could be assembled as published previously, making for a formal total synthesis of 1 in 2.8% overall yield. An alternative order of connecting the essential fragments was also made possible by the first protocol for the direct N-glycosylation of a 3-acyltetramic acid.
Introduction
Amycolamicin (1) (Scheme 1) was first mentioned in 2008/2009 in patents by Igarashi et al. who had isolated it from the bacterium Amycolatopsis sp. MK575-fF5.1 In 2010, proposals for its structure and for the biosynthesis of its N-acylated amycolose constituent 4, featuring an unusual α-aminoethyl branched sugar, were put forward.2 In 2011 Singh and coworkers isolated a compound from Kibdelsporangium sp. MA 7385 which they dubbed kibdelomycin and which they assumed to comprise a largely inverted amykitanose moiety when compared to the purported structure of amycolamicin.3 They recognised its extraordinary efficacy mainly against Gram-positive bacteria, including multidrug resistant pathogens from the ESKAPE panel. In 2012, a Japanese group disclosed a first crystal structure of the β-methyl anomer of amycolose and a revised structure of amycolamicin differing from the earlier one in the configuration of a stereogenic centre in the amykitanose.4 In 2014, Singh et al. settled the dispute over structure and stereochemistry with an X-ray diffraction analysis of crystals of kibdelomycin (1) bound to gyrase B/topoisomerase IV.5 They revised their original structure proposal and so proved that kibdelomycin and amycolamicin are one and the same.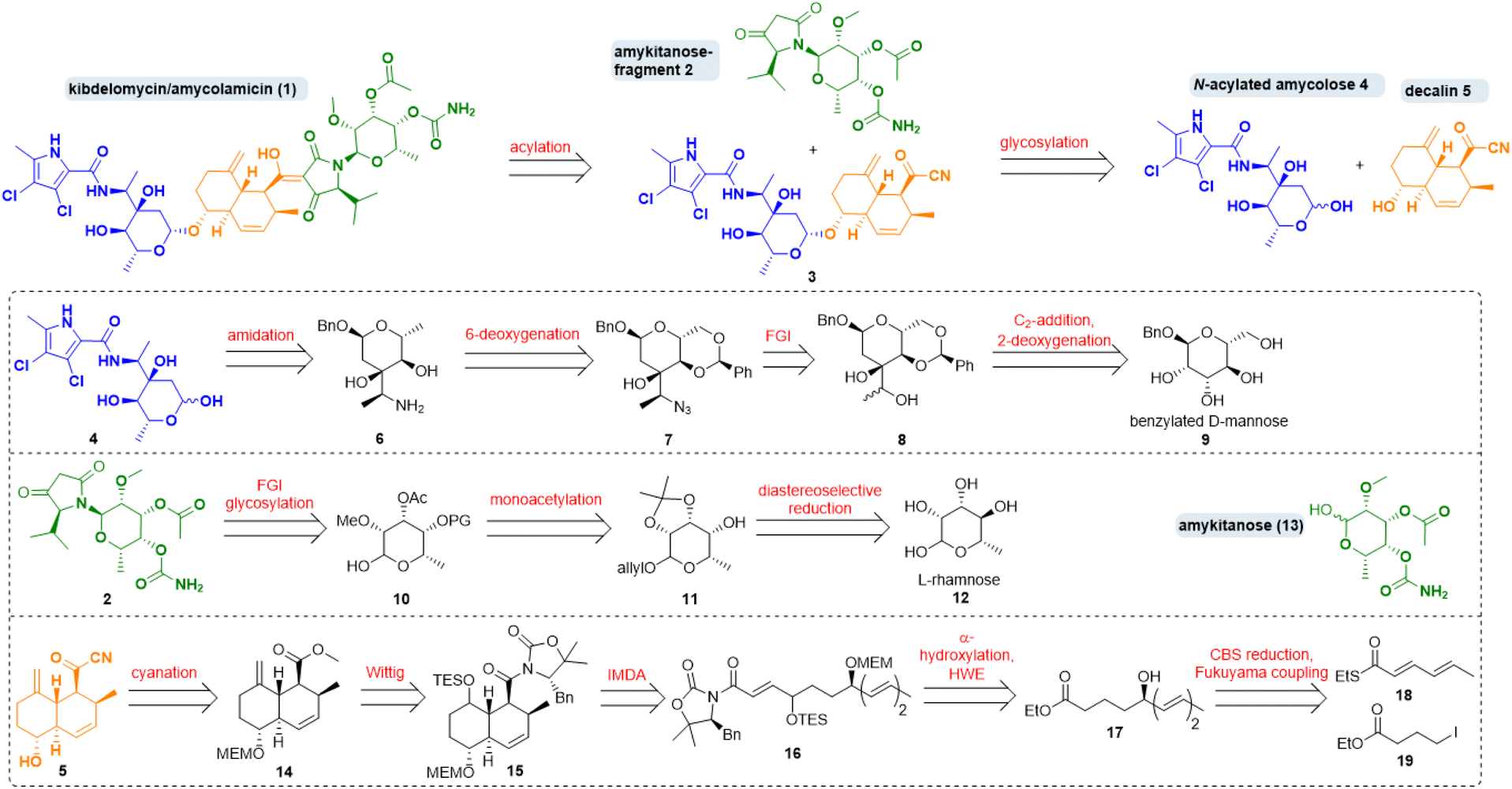 | ||
| Scheme 1 Retrosynthesis of kibdelomycin (1) and key fragments. FGI: functional group interconversion; IMDA: intramolecular Diels–Alder, HWE: Horner–Wadsworth–Emmons; CBS: Corey–Bakshi–Shibata. | ||
Singh et al. also undertook extensive studies of structure activity relationships.5 Their crystal structure revealed a horseshoe-like conformation in which the dichlorinated pyrrole of amycolose amide 4 penetrates the ATP-binding pocket of gyrase B/topoisomerase IV which is the usual target of known topoisomerase IV inhibitory antibiotics. In contrast to gyrase-inhibiting antibiotics like novobiocin, the decalin, the tetramic acid and the amykitanose fragments of kibdelomycin protrude from the usual binding pocket, a possible explanation for it not showing cross resistance with established gyrase inhibitors.
Regarding its synthesis, kibdelomycin (1) can be dissected in three main parts, which are interesting synthetic targets in their own right. There is a decalinoyltetramic acid, a compound class known for its diverse biological activities.6 The decalin is O-glycosidically bound to a 3-α-aminoethyl-3,6-dideoxyhexopyra-nose. A 6-deoxygenated talose, carrying a methyl ether, an acetate and a carbamic acid, is attached to the tetramic acid by an N-glycosidic bond. The first synthetic foray towards kibdelomycin was the preparation of N-acyl amycolose 4 by Kuwahara et al. in 2019.7 Then, in quick succession, the groups of Li, Kuwahara and Baran published total syntheses of kibdelomycin within less than one year from December 2021 until 2022.8,9
Results and discussion
Our retrosynthetic strategy for kibdelomycin (1) took advantage of a convergent route (Scheme 1). Disconnections were set (i) between N-acyl amycolose 4 and decalin fragment 5, requiring a challenging glycosylation of a 2-deoxy sugar in the forward direction, (ii) between decalin fragment 5 and N-amykitanosyltetramic acid 2, to be linked via a 3-acylation of the latter, and (iii) between amykitanose (13) and 5-isopropyltetramic acid as present in fragment 2. This strategy would harness our experience with decalinoyl- and N-glycosylated tetramic acids.10 While working on this project the three mentioned total syntheses were released, so that we decided not to frantically avoid a few of their obvious reaction steps but to concentrate on employing new and more efficient functional group interconversions for the sugar chemistry and to develop an expeditious formal total synthesis of 1.The first synthesis by Yang et al. resembles ours most because of its convergence and the similarity of some retrosynthetic fragments.9 However, we chose distinctly different routes to decalin 5, N-acylated amycolose 4 and amykitanose 13. For the latter two we used a glycal approach with the advantage of not having to build up every single stereogenic centre by means of expensive catalysts and starting materials. For amycolose derivative 4 we decided to start from inexpensive benzylated D-mannose 9, which first had to be deoxygenated at 2-position, and in which it was necessary to instal an oxidised ethyl group at 3-position. After a second deoxygenation at 6-position and formation of the 3-(α-aminoethyl)sugar 6 the amidation with a dichlorinated pyrrole carboxylic acid should afford 4. For the synthesis of amykitanose fragment 2 we wanted to start from affordable L-rhamnose (12) instead of expensive L-fucose or L-talose. Key steps were the inversion at 4-position, the regioselective monoacetylation at 3-position, the N-glycosylation of 5-isopropyltetramic acid, and carbamate formation at C-4. For the synthesis of decalin fragment 5 any reaction other than an intramolecular Diels–Alder (IMDA) cycloaddition was out of the question. In a few steps, starting from thioester 18 and iodide 19, triene 16 should be accessible via Fukuyama coupling, stereoselective reduction of the resulting δ-ketoester to give hydroxyester 17, α-hydroxylation of the latter, and a chain-lengthening HWE-olefination. The following IMDA should afford mainly the trans-decalin scaffold, which had to be olefinated once more and converted to acyl cyanide 5. Due to the complexity of kibdelomycin (1) we had to pursue different synthetic routes to these key fragments. Foundered and abandoned attempts are detailed in the ESI.†
For the synthesis of N-acylated amycolose 4, benzyl protected D-mannose 9 was reacted with benzaldehyde dimethyl acetal (BDMA) and camphorsulfonic acid (CSA) to give bisbenzylidene acetal 20. This was treated, without prior purification, with nBuLi at −78 °C to undergo a Klemer–Rodemeyer fragmentation upon warming to −35 °C, affording ketone 21 in 78% yield over two steps (Scheme 2).11 It is worthy of note that a p-methoxyphenyl (PMP) instead of a methyl, benzyl or propargyl protecting group at the anomeric position was cleaved under these conditions with release of PMPOH. The subsequent Grignard addition of vinyl magnesium bromide occurred exclusively from the side opposite to the neighbouring 4,6-benzylidene acetal. For the introduction of the amino group we intended an initial stereoselective formation of a secondary alcohol at the ethylene group, accessible via epoxidation and ensuing ring opening by a metal hydride, and its SN2-type substitution with sodium azide. The enantio- and diastereoselective Sharpless and VO(acac)2/TBHP epoxidations failed, whereas the Prilezhaev epoxidation gave the epoxides 24 and 23 in 88% yield as a separable 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers which could both be used for the synthesis of 4. Epoxide opening by LiAlH4 afforded diols 25 (from 23) and 27 (from 24) quantitatively. Applying the Mosher ester method, alcohol 27 was found to be (S)-configured (Fig. 1, top).12 For the retention of its terminal stereogenic centre, diol 25 was submitted to two consecutive SN2-like reactions. Epoxide formation between the secondary and tertiary alcohol with Tf2O/pyridine afforded compound 26 which was treated immediately with NaN3 to furnish azide 7 in 81% over two steps. For the inversion of the terminal stereogenic centre of diol 27, it was first converted to the sulfite 28. This was oxidised with RuCl3/NaIO4 to sulfate 29 which was reactive enough to render azide 7 (61% over 4 steps) upon treatment with NaN3 and subsequent acidic hydrolysis of the intermediate sodium sulfate ester (cf. ESI† for details). While on small scale this hydrolysis was possible using aqueous H2SO4 (70% yield), at a larger scale aqueous H2SO4 led to cleavage of the benzylidene acetal and had to be replaced by a pH 4 citric acid buffer. For the 6-deoxygenation of 7 we followed the protocol of Dang et al. and employed a system of DTBP/TIPST for its radical-chain redox rearrangement to give benzoate 30.13 After an extensive optimisation this step proceeded with at least 50% yield, which spared us the use of the alternative Hanessian–Hullar reaction with subsequent dehalogenation.14 Treatment of benzoate 30 with LiAlH4 led to concomitant azide and benzoate reduction with 79% yield. The resulting amine 6 was selectively acylated with carboxylic acid 31 and EDC·HCl/HOBt to give amide 32 in 83% yield. Other amidation reagents such as BOP or HATU were less effective. Because of the potential hydrogenative dechlorination of the pyrrole we used BCl3 rather than Pd/C and H2 for the final debenzylation step. We obtained a mixture of α- and β-anomers of 4, the ratio of which was strongly dependent on the solvent and purification. Next, we checked the applicability of this synthesis to other sugars (Scheme 3). We chose L-rhamnose to test the introduction of an α-aminoalkyl residue. Benzylated L-rhamnose 34 was regioselectively 3-acetylated using a molybdenum catalyst.15 The hydroxyl groups at 2- and 4-position were MEM-protected (→ 35, 80%), because the downstream Grignard reaction would not work with bulky (TBS, Bn) or no protecting groups. After removal of the acetyl group by DIBAL (82%) and DMP-oxidation, ketone 36 was obtained with good yield. Its reaction with vinyl magnesium bromide gave the tertiary allyl alcohol 37 in 79% yield and dr > 30:1. A 2D-NOESY-experiment proved that the Grignard reagent had attacked from the site opposite to the C4-OMEM group (Fig. 1, bottom). This finding also shows that the group at C4, directing diastereoselective additions, need not be a large 4,6-benzylidene acetal. Next, alkene 37 was converted to primary alcohol 38 by ozonolysis which was tosylated to give 39 that was converted to azide 40. After Staudinger reaction, the resulting amine 41 was acylated with pyrrole carboxylic acid 31 to give amide 42 in 81% yield. Finally, the benzyl group at the anomeric position as well as both MEM protecting groups of 42 were removed by BCl3 in a single step to give the rhamnose derivative 43 in excellent 17% yield over 11 steps. With the synthesis of amycolose and a rhamnose derivative, we demonstrated that this method may be used in general to introduce an α-aminoalkyl linkage in sugars. Moreover, the vinyl group is amenable to a good many other functionalisations (cf. ESI†). This aspect might facilitate diversity-oriented syntheses of highly functionalised sugars, including even amycolose, given its known cell growth suppression and possible application as an anticancer medication.16
:1 mixture of diastereomers which could both be used for the synthesis of 4. Epoxide opening by LiAlH4 afforded diols 25 (from 23) and 27 (from 24) quantitatively. Applying the Mosher ester method, alcohol 27 was found to be (S)-configured (Fig. 1, top).12 For the retention of its terminal stereogenic centre, diol 25 was submitted to two consecutive SN2-like reactions. Epoxide formation between the secondary and tertiary alcohol with Tf2O/pyridine afforded compound 26 which was treated immediately with NaN3 to furnish azide 7 in 81% over two steps. For the inversion of the terminal stereogenic centre of diol 27, it was first converted to the sulfite 28. This was oxidised with RuCl3/NaIO4 to sulfate 29 which was reactive enough to render azide 7 (61% over 4 steps) upon treatment with NaN3 and subsequent acidic hydrolysis of the intermediate sodium sulfate ester (cf. ESI† for details). While on small scale this hydrolysis was possible using aqueous H2SO4 (70% yield), at a larger scale aqueous H2SO4 led to cleavage of the benzylidene acetal and had to be replaced by a pH 4 citric acid buffer. For the 6-deoxygenation of 7 we followed the protocol of Dang et al. and employed a system of DTBP/TIPST for its radical-chain redox rearrangement to give benzoate 30.13 After an extensive optimisation this step proceeded with at least 50% yield, which spared us the use of the alternative Hanessian–Hullar reaction with subsequent dehalogenation.14 Treatment of benzoate 30 with LiAlH4 led to concomitant azide and benzoate reduction with 79% yield. The resulting amine 6 was selectively acylated with carboxylic acid 31 and EDC·HCl/HOBt to give amide 32 in 83% yield. Other amidation reagents such as BOP or HATU were less effective. Because of the potential hydrogenative dechlorination of the pyrrole we used BCl3 rather than Pd/C and H2 for the final debenzylation step. We obtained a mixture of α- and β-anomers of 4, the ratio of which was strongly dependent on the solvent and purification. Next, we checked the applicability of this synthesis to other sugars (Scheme 3). We chose L-rhamnose to test the introduction of an α-aminoalkyl residue. Benzylated L-rhamnose 34 was regioselectively 3-acetylated using a molybdenum catalyst.15 The hydroxyl groups at 2- and 4-position were MEM-protected (→ 35, 80%), because the downstream Grignard reaction would not work with bulky (TBS, Bn) or no protecting groups. After removal of the acetyl group by DIBAL (82%) and DMP-oxidation, ketone 36 was obtained with good yield. Its reaction with vinyl magnesium bromide gave the tertiary allyl alcohol 37 in 79% yield and dr > 30:1. A 2D-NOESY-experiment proved that the Grignard reagent had attacked from the site opposite to the C4-OMEM group (Fig. 1, bottom). This finding also shows that the group at C4, directing diastereoselective additions, need not be a large 4,6-benzylidene acetal. Next, alkene 37 was converted to primary alcohol 38 by ozonolysis which was tosylated to give 39 that was converted to azide 40. After Staudinger reaction, the resulting amine 41 was acylated with pyrrole carboxylic acid 31 to give amide 42 in 81% yield. Finally, the benzyl group at the anomeric position as well as both MEM protecting groups of 42 were removed by BCl3 in a single step to give the rhamnose derivative 43 in excellent 17% yield over 11 steps. With the synthesis of amycolose and a rhamnose derivative, we demonstrated that this method may be used in general to introduce an α-aminoalkyl linkage in sugars. Moreover, the vinyl group is amenable to a good many other functionalisations (cf. ESI†). This aspect might facilitate diversity-oriented syntheses of highly functionalised sugars, including even amycolose, given its known cell growth suppression and possible application as an anticancer medication.16
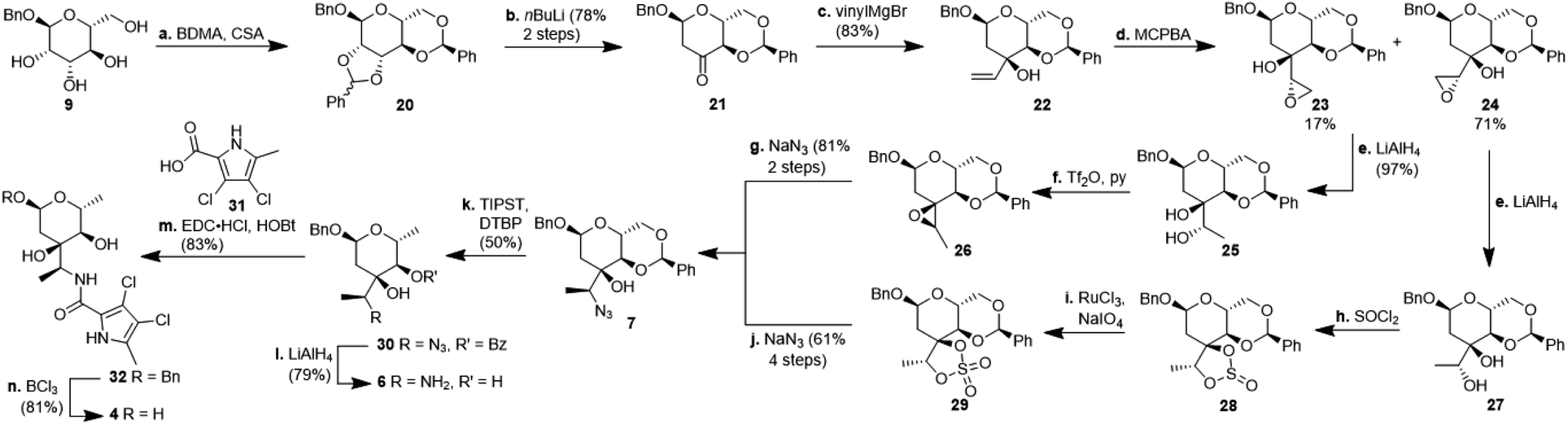 | ||
| Scheme 2 Synthesis of amycolose derivative 4. BDMA: benzaldehyde dimethyl acetal; CSA: camphorsulfonic acid; MCPBA: 3-chloroperbenzoic acid; Tf2O: triflic anhydride; TIPST: triisopropylsilanethiol; DTBP: di-tert-butylperoxide; EDC: N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide; HOBt: 1-hydroxybenzotriazole. | ||
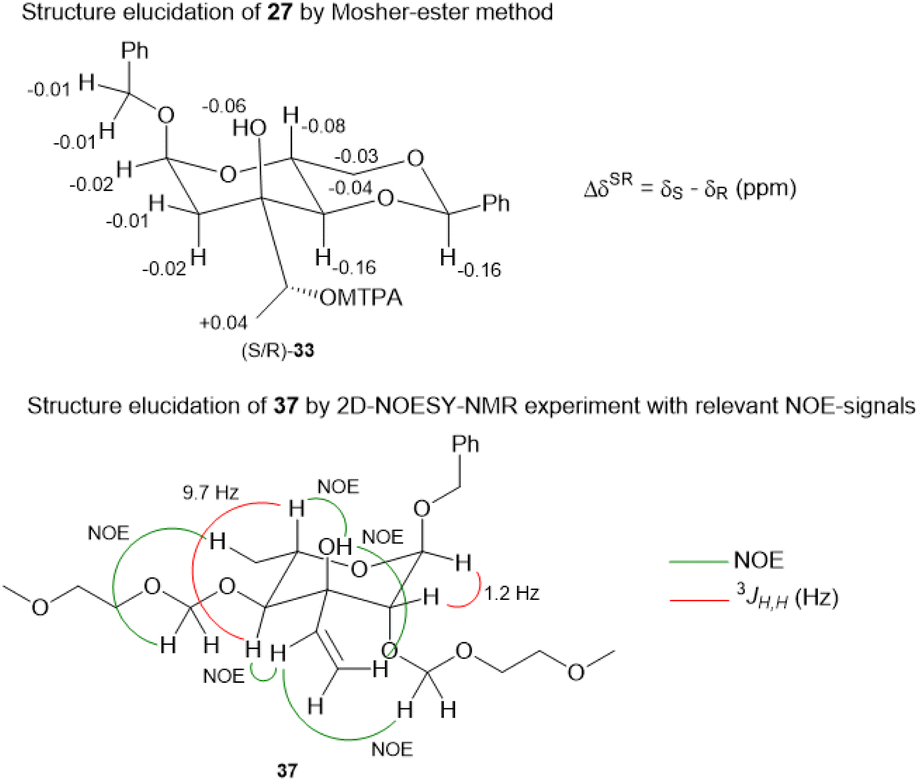 | ||
| Fig. 1 Structure elucidation of 27via Mosher ester method (top) and significant NOE-signals for the elucidation of the stereoconfiguration of 37 (bottom). | ||
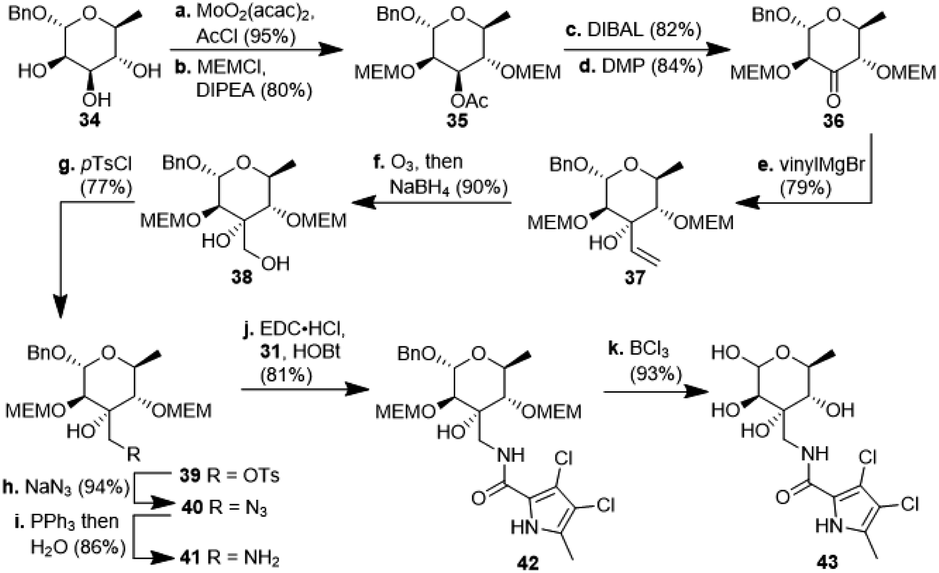 | ||
| Scheme 3 Synthesis of 3-aminomethyl-6-deoxyhexopyranose derivative 43 starting from benzylated L-rhamnose 34. MEM: methoxyethoxymethyl; DIPEA: diisopropylethylamine; DMP: Dess–Martin periodinane; Ts: tosyl. | ||
The synthesis of decalin fragment 5 started with a Fukuyama coupling between ethyl 4-iodobutyrate 19 and ethyl (2E,4E)-hexa-2,4-dienethioate 18 to give δ-ketoester 44 in 91% yield (Scheme 4).17 The ketone was reduced with BH3 in the presence of (S)-CBS-catalyst affording alcohol 17 with 90% yield and an ee of 91%. This protocol is easier to use on a laboratory scale than a recently published asymmetric Noyori-type hydrogenation of α,β,γ,δ-unsaturated ketones.18 Unlike other groups who applied a more than quantitative amount of CBS-catalyst, we realised that the reduction proceeded with higher ee when using a merely catalytic amount of CBS-catalyst. After MEM-protection of the alcohol to give ether 45 with 79% yield, a non-trivial α-hydroxylation had to be done at this post-Fukuyama stage, since α-hydroxylated esters from the chiral pool failed to undergo the Fukuyama coupling due to not forming the respective zinc organyl (cf. ESI†). After quite a few failed attempts with sulfonyloxaziridines, we identified MoOPH/KHMDS as a viable α-hydroxylating agent affording α-hydroxyester 46 with 89% yield and 1.9:1 dr. The TES-protected ester 47 was reduced with DIBAL to aldehyde 48 and the latter was submitted to a HWE-olefination with phosphonate 49 to give the triene 16 comprising the SuperQuat auxiliary (70%, two steps). Because HWE-reactions with Evans/Davies auxiliary bearing phosphonates only worked with α-hydroxylated aldehydes but not so with α-methylene substituted aldehydes (cf. ESI†) we had to postpone the introduction of the methylene group until after the decalin formation. We opted for Davies' SuperQuat auxiliary for the following Diels–Alder reaction, after many attempts to remove an Evans auxiliary had failed after successful Diels–Alder reaction and in accordance with the results of Frossard et al.19 Unlike most who use AlMeCl2 as a catalyst for the IMDA, we had better results when heating triene 16 in toluene at 80 °C over 3 d which afforded octalin 15 with 43% yield besides some separable undesired cis-octalin. Quantitative removal of the TES-protecting group with HF pyridine complex left the alcohol 50 which had its auxiliary cleaved off with sodium methoxide to give hydroxyester 51 with 90% yield. The introduction of the methylene unit was achieved by oxidising alcohol 51 with DMP (96%) and treating the resulting ketone 52 with methylenetriphenylphosphorane. The resulting ester 14 (90%) was reduced to aldehyde 53 in two steps, i.e. reduction to the corresponding alcohol with DIBAL and subsequent oxidation with DMP, because of overreduction issues. Reaction of aldehyde 53 with TMSCN led to a cyanohydrin, which was right away oxidised with DMP to acyl cyanide 54. Cleavage of the MEM-group, liberating decalin 5, proceeded best using LiBF4 compared to TiCl4 or TFA. This synthesis of the central decalin building block has an edge over those of the previous kibdelomycin syntheses due to its high yielding, simple steps and inexpensive starting materials. Most reactions were performed on a gram scale without yields decreasing.
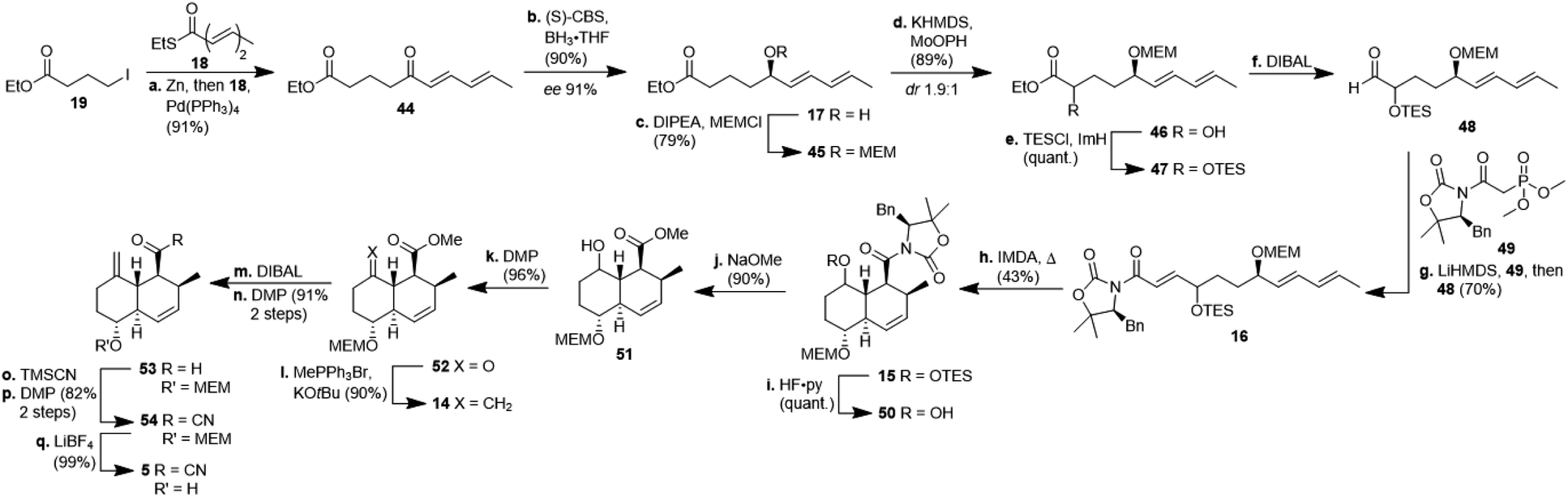 | ||
| Scheme 4 Synthesis of decalin fragment 5 starting from ethyl 4-iodobutyrate 19. CBS: Corey–Bakshi–Shibata catalyst; KHMDS: potassium hexamethyldisilazide; MoOPH: oxodiperoxymolybdenum(pyridine)(hexamethylphosphoric triamide); TES: triethylsilyl; ImH: imidazole; LiHMDS: lithium hexamethyldisilazide; TMSCN: trimethylsilyl cyanide. | ||
The second, amykitanose-related sugar fragment was synthesised starting from L-rhamnose (12) (Scheme 5). It was allylated at the anomeric position in 93% yield and its syn-diol was protected as an isopropylidene acetal using anhydrous CuSO4 (95%). The allyl protecting group was chosen since the cleavage of the comparable methyl acetal later on in the synthesis had failed in the presence of other necessary functional groups, e.g. because of the instability of the acetyl group. The configuration at the 4-position of the resulting compound 55 was inverted by a sequence of Swern oxidation and ensuing reduction with NaBH4 to give a single diastereomer of 11 in 88% over two steps. Benzylation of the hydroxyl group led to fully protected sugar 56a. After deprotection of the syn-diol, the hydroxyl group at 3-position was acetylated selectively under optimised conditions to afford sugar 57a in 74% yield over two steps.20 Methylation at 2-positon was difficult due to the acetyl group getting easily removed under basic conditions, but was eventually achieved using TMSCHN2 and HBF4. Deprotection of the anomeric position in acidic milieu under Pd-catalysis gave sugar 58a. All attempts at coupling it with any kind of tetramic acid via different customary methods in order to establish analogues of amykitanose fragment 2, as well as Dieckmann cyclisation based sequences failed (cf. ESI†).21 As a last resort and based on the first total synthesis of kibdelomycin by Li et al.,9 sugar 58a was esterified with carboxylic acid 59 and the resulting ester 60a was coupled with 3-cyclohexanoyl-tetramic acid 61via Au-catalysis affording N-glycoside 62a in a decent 58% yield.22 The cyclohexyl residue was to mimic the octalin moiety. As far as we know, this is the first example of a direct N-glycosylation of a 3-acyltetramic acid. The anomeric ratio of 10:1 was inferior to the 20:1 ratio reported by Li et al.9 for the N-glycosylation of 3H-5-isopropylpyrrolidin-2,4-dione. The divergent results could only be attributed to the different protecting groups at 4-position of the sugar (Bn vs. TES). To verify this assumption, we introduced a silyl protection group as in compound 58b. The following steps were identical to those for the 4-OBn analogues, albeit with slightly different reaction conditions because of the instability of the TBS-group in an acidic milieu. Even the esterification of 58b with carboxylic acid 59 showed the influence of the protecting group, since the anomeric ratio of the resulting sugar 60b increased to 10:1 α:β. After coupling with the 3-acyl tetramic acid 61, the N-glycoside 62b was isolated with an α:β-ratio of >30:1. For a strict formal total synthesis, the TES-protected sugar 65 was required (Scheme 6). So, we removed the benzyl group of compound 63, obtained from methylation of glycoside 57a, with in situ generated HI, and replaced it with a triethylsilyl group to afford compound 64. De-allylation of the latter and glycosylation with acid 59 gave ester 65 in excellent 15% yield over 12 steps, comparable with the corresponding sequence of the first total synthesis by Li et al.9 Glycoside 65 can then be coupled with 4-O-benzyl 5-isopropyltetramate 68 as shown in the first total synthesis of kibdelomycin.9 Tetramate 68 is readily accessible in one step and 63% yield from reaction of ketenylidenetriphenylphosphorane (66) with L-valine benzyl ester (67).23 Removal of the benzyl group in glycoside 63 also opened the door for the synthesis of amykitanose (13) in three more steps (cf. ESI† for a not yet optimised protocol). The formal synthesis of kibdelomycin (1) can be completed by esterification of amycolose derivative 4 with acid 59 to give 70 and subsequent use of the latter for glycosylation of decalin fragment 5 to give compound 71. Glycoside 65 can be converted to tetramic acid fragment 2 in four steps. Acylation of tetramic acid 2 with ketonitrile 71 using 1-hydroxy-7-azabenzotriazole (HOAt) and triethylamine finally affords kibdelomycin (1). For the completion of an alternative total synthesis exploiting the novel N-glycosylation of 3-acyltetramic acids cf. the ESI.†
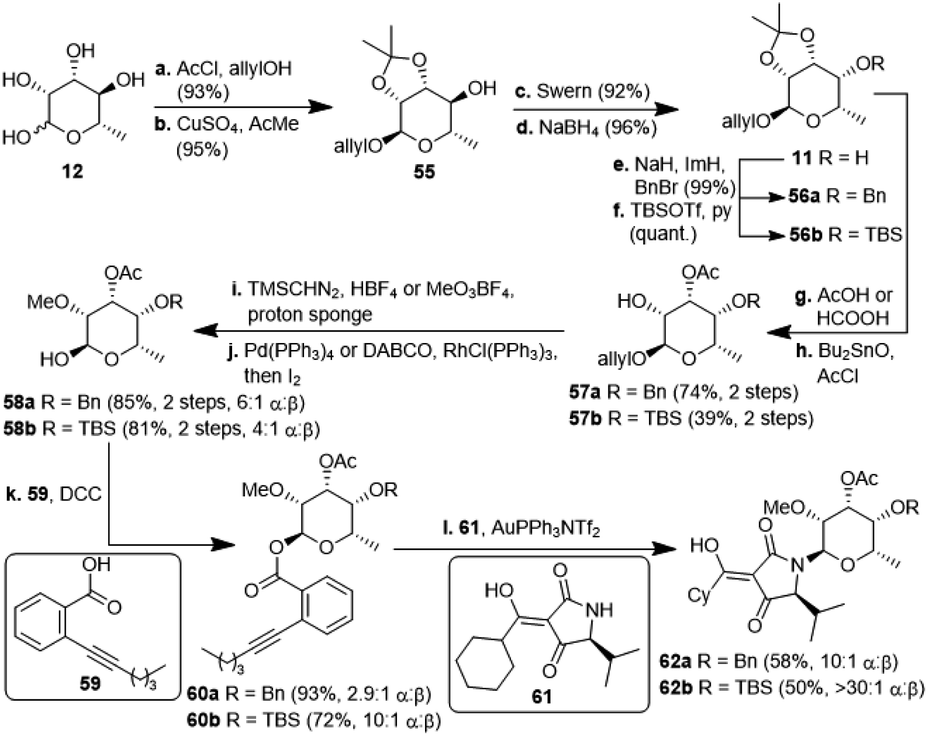 | ||
| Scheme 5 Synthesis of N-glycosylated 3-cyclohexanoylltetramic acids 62a/b. TBS: tertbutyldimethylsilyl; Tf: triflyl; DABCO: 1,4-diazabicyclo[2.2.2]octane; DCC: dicyclohexylcarbodiimide. | ||
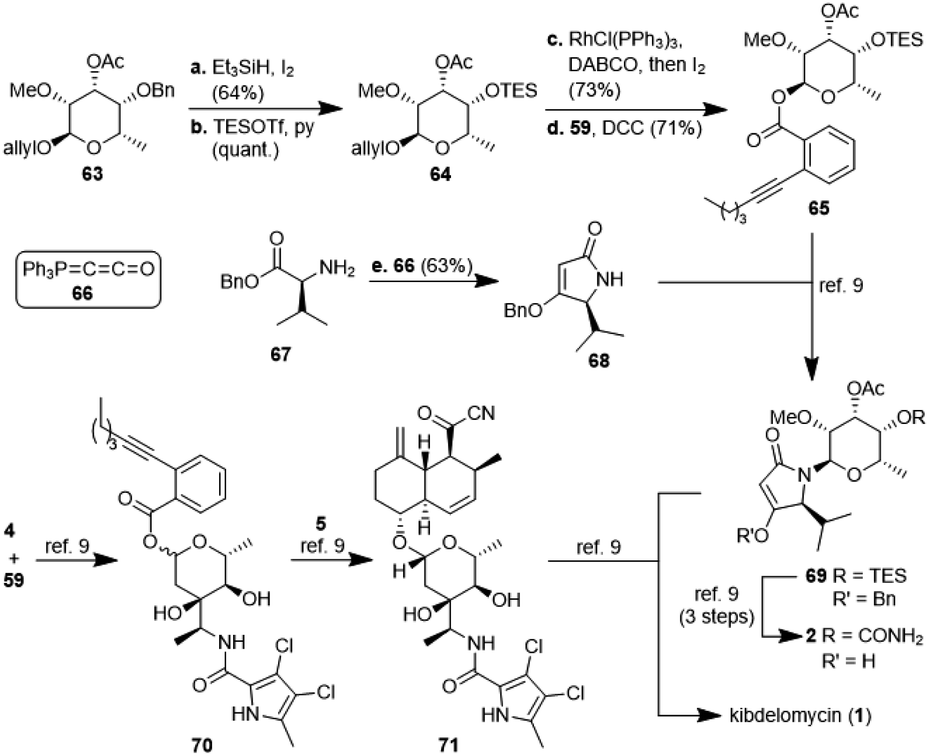 | ||
| Scheme 6 Synthesis of glycoside 65 and tetramate 68 as well as a formal synthesis of kibdelomycin (1) according to ref. 9. | ||
Conclusion
In summary we developed an expeditious formal synthesis of kibdelomycin (1) starting from inexpensive compounds and employing simple and high-yielding standard protocols, even on a large scale. The stereochemical information stems from the chiral pool or from highly diastereoselective reactions. The longest linear sequence of the factual synthesis of the fragments amounts to a competitive 19 steps. With all fragments in hand, a formal synthesis following the protocol of Yang et al. leads to kibdelomycin (1, 2.8% overall yield).9 During our research, we developed a method for introduction of an α-aminoalkyl linkage into sugars via Grignard addition to C3 which also opens access to a range of other functionalities. It could be used to synthesise different derivatives of kibdelomycin (1) for structure–activity relationship studies or for an optimisation of its applicability and efficacy. As a side benefit, we also report the first N-glycosylation of a 3-acyltetramic acid.Data availability
The datasets and spectra supporting this article have been uploaded as part of the ESI† material.Author contributions
M. G. S. planned and carried out all reactions concerning amycolose, planned the synthesis of derivatives of amycolose, and wrote parts of the manuscript. L. T. planned and carried out all syntheses concerning amykitanose and rhamnose derivatives and wrote parts of the manuscript. L. T. and M. G. S. planned and realised the synthesis of decalin fragment 5. R. S. supervised the syntheses and assisted with manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Alessandro Burger, Lina-Marie Beck, Gopal Gupta and Ines Bauer for their practical, synthetic contributions as part of their BSc projects.References
- M. Igarashi, R. Sawa and T. Honma, New compound amycolamicin, method for producing the same, and use of the same, JP Pat., JP2009203195A, 2009 Search PubMed.
- S. Tohyama, Y. Takahashi and Y. Akamatsu, Biosynthesis of amycolamicin: the biosynthetic origin of a branched alpha-aminoethyl moiety in the unusual sugar amycolose, J. Antibiot., 2010, 63, 147–149 CrossRef CAS.
- (a) D. L. Zink, M. Goetz, O. Genniloud, F. Vicente, S. Singh and J. D. Polishook, Antibacterial agents, Pat., ES 2368236A1, 2011 Search PubMed; (b) J. W. Phillips, M. A. Goetz, S. K. Smith, D. L. Zink, J. Polishook, R. Onishi, S. Salowe, J. Wiltsie, J. Allocco, J. Sigmund, K. Dorso, S. Lee, S. Skwish, M. de La Cruz, J. Martín, F. Vicente, O. Genilloud, J. Lu, R. E. Painter, K. Young, K. Overbye, R. G. K. Donald and S. B. Singh, Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus, Chem. Biol., 2011, 18, 955–965 CrossRef CAS.
- R. Sawa, Y. Takahashi, H. Hashizume, K. Sasaki, Y. Ishizaki, M. Umekita, M. Hatano, H. Abe, T. Watanabe, N. Kinoshita, Y. Homma, C. Hayashi, K. Inoue, S. Ohba, T. Masuda, M. Arakawa, Y. Kobayashi, M. Hamada, M. Igarashi, H. Adachi, Y. Nishimura and Y. Akamatsu, Amycolamicin: a novel broad-spectrum antibiotic inhibiting bacterial topoisomerase, Chem.–Eur. J., 2012, 18, 15772–15781 CrossRef CAS.
- J. Lu, S. Patel, N. Sharma, S. M. Soisson, R. Kishii, M. Takei, Y. Fukuda, K. J. Lumb and S. B. Singh, Structures of kibdelomycin bound to Staphylococcus aureus GyrB and ParE showed a novel U-shaped binding mode, ACS Chem. Biol., 2014, 9, 2023–2031 CrossRef CAS PubMed.
- (a) R. Schobert and A. Schlenk, Tetramic and tetronic acids: an update on new derivatives and biological aspects, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS PubMed; (b) G. Li, S. Kusari and M. Spiteller, Natural products containing ‘decalin’ motif in microorganisms, Nat. Prod. Rep., 2014, 31, 1175–1201 RSC.
- Y. Meguro, Y. Ogura, M. Enomoto and S. Kuwahara, Synthesis of the N-acyl amycolose moiety of amycolamicin and Its methyl glycosides, J. Org. Chem., 2019, 84, 7474–7479 CrossRef CAS.
- (a) C. He, Y. Wang, C. Bi, D. S. Peters, T. J. Gallagher, J. Teske, J. S. Chen, R. Corsetti, A. D'Onofrio, K. Lewis and P. S. Baran, Total synthesis of kibdelomycin, Angew. Chem., Int. Ed., 2022, 61, e202206183 CAS; (b) Y. Meguro, J. Ito, K. Nakagawa and S. Kuwahara, Total synthesis of the broad-spectrum antibiotic amycolamicin, J. Am. Chem. Soc., 2022, 144, 5253–5257 CrossRef CAS PubMed.
- S. Yang, C. Chen, J. Chen and C. Li, Total synthesis of the potent and broad-spectrum antibiotics amycolamicin and kibdelomycin, J. Am. Chem. Soc., 2021, 143, 21258–21263 CrossRef CAS PubMed.
- (a) M. Winterer, K. Kempf and R. Schobert, Synthesis of an isomer of the decalinoyltetramic acid methiosetin by a stereocontrolled IMDA reaction of a metal-chelated 3-trienoyltetramate, J. Org. Chem., 2016, 81, 7336–7341 CrossRef CAS; (b) M. Petermichl, S. Loscher and R. Schobert, Total synthesis of aurantoside G, an N-β-glycosylated 3-oligoenoyltetramic acid from Theonella swinhoei, Angew. Chem., Int. Ed., 2016, 55, 10122–10125 CrossRef CAS PubMed.
- A. Klemer, G. Rodemeyer and F.-J. Linnenbaum, Reaktionen von O-Isopropylidenzuckern mit lithiumorganischen Verbindungen zu ungesättigten Zuckern. Synthese von 4-Desoxy-4-eno-β-D-threo-pentose- und 5-Desoxy-5-eno-β-D-threo-hexulose-Derivaten, Chem. Ber., 1976, 109, 2849–2861 CrossRef CAS.
- J. A. Dale and H. S. Mosher, Nuclear magnetic resonance enantiomer reagents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS.
- H.-S. Dang, B. P. Roberts, J. Sekhon and T. M. Smits, Deoxygenation of carbohydrates by thiol-catalysed radical-chain redox rearrangement of the derived benzylidene acetals, Org. Biomol. Chem., 2003, 1, 1330–1341 RSC.
- (a) D. L. Failla, T. L. Hullar and S. B. Siskin, Selective transformation of O-benzylidene acetals into ω-bromo-substituted benzoate esters, Chem. Commun., 1966, 716–717 RSC; (b) S. Hanessian, The reaction of O-benzylidene sugars with N-bromosuccinimide, Carbohydr. Res., 1966, 2, 86–88 CrossRef CAS.
- E. V. Evtushenko, Regioselective benzoylation of glycopyranosides by benzoyl chloride in the presence of MoO2(acac)2, J. Carbohydr. Chem., 2010, 29, 369–378 CrossRef CAS.
- S. Tohyama, Amycolose derivative, and production process and use of same, EP Pat., EP2471788A1, 2012 Search PubMed.
- H. Tokuyama, S. Yokoshima, T. Yamashita and T. Fukuyama, A novel ketone synthesis by a palladium-catalyzed reaction of thiol esters and organozinc reagents, Tetrahedron Lett., 1998, 39, 3189–3192 CrossRef CAS.
- C. Li, W. Lu, B. Lu, W. Li, X. Xie and Z. Zhang, Ru-catalyzed chemo- and enantioselective hydrogenation of 2,4-pentadien-1-ones: synthesis of chiral 2,4-pentadien-1-ols, J. Org. Chem., 2019, 84, 16086–16094 CrossRef CAS PubMed.
- (a) S. G. Davies, I. A. Hunter, R. L. Nicholson, P. Roberts, E. D. Savory and A. D. Smith, N-α-Benzyloxyacetyl derivatives of (S)-4-benzyl-5,5-dimethyloxazolidin-2-one for the asymmetric synthesis of differentially protected α,β-dihydroxyaldehydes, Tetrahedron, 2004, 60, 7553–7577 CrossRef CAS; (b) T. M. Frossard, N. Trapp and K.-H. Altmann, Studies towards the total synthesis of amycolamicin: a chiral auxiliary-based Diels-Alder approach towards the decalin core, Eur. J. Org. Chem., 2022, 2022, e202200761 CrossRef CAS.
- M. A. Nashed and L. Anderson, Organotin derivatives and the selective acylation and alkylation of the equatorial hydroxy group in a vicinal, equatorial-axial pair, Tetrahedron Lett., 1976, 17, 3503–3506 CrossRef.
- R. N. Lacey, Derivatives of acetoacetic acid. Part VII. α-Acetyltetramic acids, J. Chem. Soc., 1954, 850–854 RSC.
- For the first use of an ortho-alkynylbenzoate/Au-catalytic system see (a) Y. Li, Y. Yang and B. Yu, An efficient glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors under the catalysis of Ph3PAuOTf, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS , for the use in N-glycosylations see; (b) Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Gold(I)-catalyzed glycosylation with glycosyl ortho-alkynylbenzoates as donors: general scope and application in the synthesis of a cyclic triterpene saponin, Chem.–Eur. J., 2010, 16, 1871–1882 CrossRef CAS PubMed; (c) Q. Zhang, J. Sun, Y. Zhu, F. Zhang and B. Yu, An efficient approach to the synthesis of nucleosides: gold(I)-catalyzed N-glycosylation of pyrimidines and purines with glycosyl ortho-alkynyl benzoates, Angew. Chem., Int. Ed., 2011, 50, 4933–4936 CrossRef CAS PubMed.
- J. Löffler and R. Schobert, Domino syntheses of five-, six- and seven-membered O-, N- and S-heterocycles from α-, β- and γ-substituted carboxylic esters, J. Chem. Soc., Perkin Trans. 1, 1996, 2799–2802 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses, characterization and NMR spectra of all new compounds. See DOI: https://doi.org/10.1039/d3sc00595j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |