
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile modification of phosphole-based aggregation-induced emission luminogens with sulfonyl isocyanates†
Nils
König
 a,
Yokari
Godínez-Loyola
b,
Fangshun
Yang
c,
Christian
Laube
c,
Michael
Laue
d,
Peter
Lönnecke
a,
Cristian A.
Strassert
*b and
Evamarie
Hey-Hawkins
*a
a,
Yokari
Godínez-Loyola
b,
Fangshun
Yang
c,
Christian
Laube
c,
Michael
Laue
d,
Peter
Lönnecke
a,
Cristian A.
Strassert
*b and
Evamarie
Hey-Hawkins
*a
aLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bInstitut für Anorganische und Analytische Chemie, CiMiC, SoN and CeNTech, Westfälische Wilhelms-Universität Münster, Heisenbergstraße 11, 48149 Münster, Germany. E-mail: cstra_01@uni-muenster.de
cLeibniz-Institut für Oberflächenmodifizierung e.V., Permoserstrasse 15, 04318 Leipzig, Germany
dLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Organic Chemistry, Johannisallee 29, 04103 Leipzig, Germany
First published on 3rd February 2023
Abstract
Phosphole oxides undergo a highly chemoselective reaction with sulfonyl isocyanates forming sulfonylimino phospholes in high yields. This facile modification proved to be a powerful tool for obtaining new phosphole-based aggregation-induced emission (AIE) luminogens with high fluorescence quantum yields in the solid state. Changing the chemical environment of the phosphorus atom of the phosphole framework results in a significant shift of the fluorescence maximum to longer wavelengths.
Introduction
Since Tang et al. first described the principle of aggregation-induced emission (AIE) for methylpentaphenylsilole about 20 years ago,1 the interest and importance of this research area have steadily increased, as indicated by the number of publications per year.2,3 The potential applications of AIE range from bioanalytics4–6 and materials science7 to optoelectronics.8 AIE describes the phenomenon of molecules that show minimal or no emission in solutions but significantly increased emission in the solid or aggregated state.9These molecular properties, especially in rotovibration mode-rich compounds, can be well explained by the structural characteristics of a molecule. Thus, it was noted that in AIE luminogens (AIEgens) in the excited state, the non-radiative decay is accelerated by intramolecular motions.13 These types of intramolecular rotations are strongly restricted in the aggregated state, mainly by intermolecular Caryl–H⋯π interactions, which leads to an enhancement of luminescence.14 This effect is clearly demonstrated in phenyl-rich systems shown in Fig. 1. In diluted solutions, low photoluminescence quantum yields were observed (ΦF = 0.1–0.3%),12,15 due to numerous possibilities for the rotation of the phenyl rings. However, in the solid state, these compounds become strongly luminescent (ΦF(solid) = 36–78%),12,15 because of the restriction of intramolecular rotations (RIRs). In addition to the well-known AIEgens of tetraphenylethenes (TPEs),10 phenylsiloles11 and phenylpyrazines,16 the incorporation of the phosphole motif into π-conjugated molecular frameworks and aryl-rich systems received considerable attention.17–23
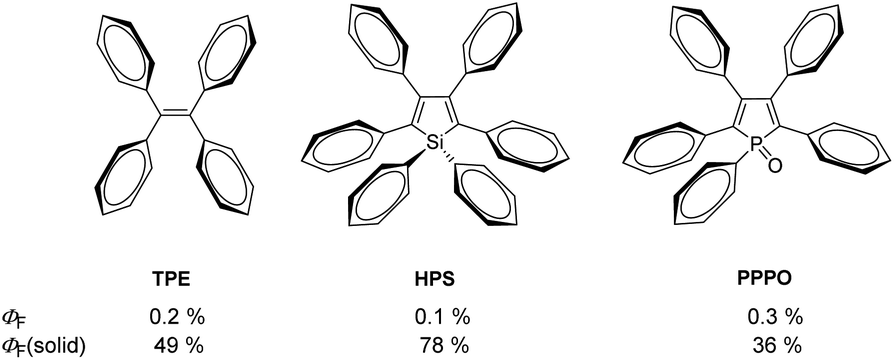 | ||
| Fig. 1 Molecular structures of AIEgens tetraphenylethene (TPE),10 hexaphenylsilole (HPS),11 and pentaphenylphosphole oxide (PPPO)12 and their fluorescence quantum yields in solution (ΦF) and the solid state (ΦF(solid)). | ||
Phospholes are five-membered organophosphorus heterocycles and the phosphorus analogues of pyrroles. Compared to pyrroles and other conjugated five-membered heterocycles, phospholes have much lower aromaticity due to the pyramidal environment at the phosphorus atom.25 Thus, the orientation of the lone pair of electrons at the P-atom does not favor interaction with the neighboring orbitals of the π system,26,27 allowing facile modifications.19 The change in the chemical environment of the phosphorus atom has a major influence on the photophysical properties of the molecule, as demonstrated for dithienyl-phosphole (DTP, Scheme 1). The advantages of incorporating phospholes in such π systems are based on their electronic structure. In general, a modification of the environment around the phosphorus atom leads to a reduction of the gap energies of the highest energy occupied molecular orbital (HOMO) and the lowest energy unoccupied molecular orbital (LUMO) resulting in lower energies for the emitted photons.24 Several options for the chemical modification of phospholes have already been described in the literature. Besides alkylation28 or oxidation with chalcogens (O, S, and Se),29–31 the electronic structure of the phosphole framework can be changed by complex formation with various metals.32–35
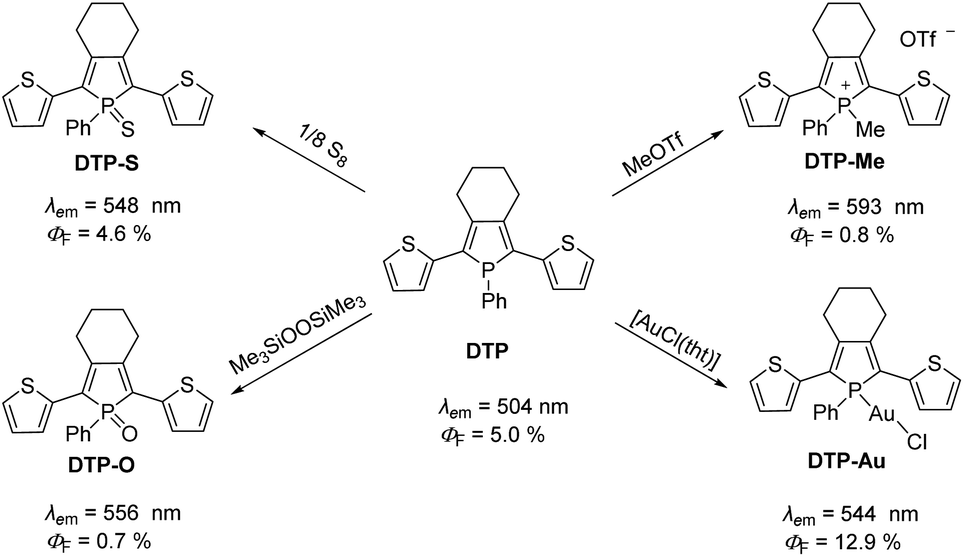 | ||
| Scheme 1 Chemical modification of dithienyl-phosphole (DTP) and changes in photophysical properties.21,24 | ||
In this work, we present a powerful synthetic strategy for selectively converting phosphole oxides by reaction with sulfonyl isocyanates into the corresponding sulfonylimino phospholes, which showed tunable phosphole-based AIE with high ΦF in the solid state.
Results and discussion
Only a few reactions in which cyclic phosphane oxides are selectively converted into their phosphane imines have already been reported in the literature.36,37 Herein, we investigated the reaction of four structurally different phosphole oxides 1–4 (Scheme 2) with chlorosulfonyl isocyanate (5a, CSI) and p-toluenesulfonyl isocyanate (5b, TSI). CSI 5a is the most reactive isocyanate known.38 In addition to the well-known cycloadditions, a wide variety of nucleophilic addition and substitution reactions are possible, making CSI a powerful reagent.39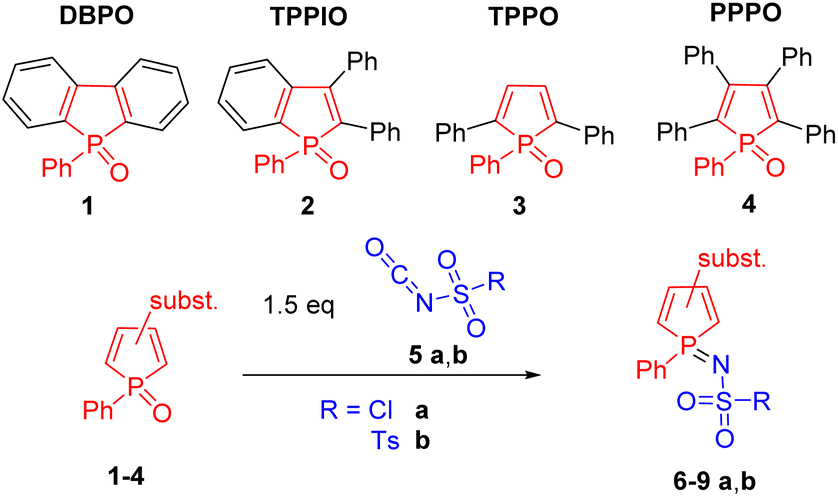 | ||
| Scheme 2 Synthesis of sulfonylimino phospholes 6–9 a,b. | ||
When phosphole oxides 1–4 were treated with a solution of 5a, we could observe a specific reaction behavior. 5a undergoes a highly chemoselective reaction with phosphole oxides 1–4 resulting in chlorosulfonylimino phospholes 6a–9a, where the reaction conditions strongly depend on the structure of the phosphole oxide used (Table 1). DBPO 1 exhibits the highest reactivity due to its planar structure and the good accessibility of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond. Complete conversion is achieved after 2 h at −30 °C. In contrast, TPPO 3 and PPPO 4 show the lowest reactivity as a result of the phenyl rings in the α position to the phosphorus atom and the stronger steric hindrance. In these cases, complete conversion was observed up to 24 h at RT. All the products were isolated via recrystallization giving 6a–9a in 90–98% yields. The 31P NMR signals of the compounds 6a–9a are shifted to a higher field by about 12–14 ppm compared to the corresponding phosphole oxides 1–4, allowing the reaction progress to be easily monitored by 31P NMR spectroscopy. Phospholes 6a–9a are moisture-insensitive in the crystalline form but show slow hydrolysis and substitution reactions in the presence of nucleophiles in solution. p-Tolylsulfonylimino phospholes 6b–9b can be obtained when TSI 5b is employed; nonetheless, higher reaction temperatures are required due to the lower reactivity of TSI 5b compared to CSI 5a. Phospholes 6b–9b were formed highly chemoselectively in good yields (80–97%). Here, the phosphole oxides TPPO 3 and PPPO 4 required temperatures up to 85 °C, which are significantly higher than in the case of DBPO 1. Derivatives 6b–9b are moisture and air stable and can be purified by flash chromatography in addition to recrystallization. The exchange of the chlorine group by a tosyl moiety leads to a higher electron density at the phosphorus atom, which is shown by a low field shift of 4 to 5 ppm in the 31P NMR spectrum. Even though a few examples of sulfonylimino phospholes have already been reported in the literature, they were obtained by the reaction of phospholes with sulfonyl azides40 or chloramine T.41,42 The advantage of our approach is the selective and facile synthesis of iminophospholes starting from air-stable and easily isolable phosphole oxides 1–4.
O double bond. Complete conversion is achieved after 2 h at −30 °C. In contrast, TPPO 3 and PPPO 4 show the lowest reactivity as a result of the phenyl rings in the α position to the phosphorus atom and the stronger steric hindrance. In these cases, complete conversion was observed up to 24 h at RT. All the products were isolated via recrystallization giving 6a–9a in 90–98% yields. The 31P NMR signals of the compounds 6a–9a are shifted to a higher field by about 12–14 ppm compared to the corresponding phosphole oxides 1–4, allowing the reaction progress to be easily monitored by 31P NMR spectroscopy. Phospholes 6a–9a are moisture-insensitive in the crystalline form but show slow hydrolysis and substitution reactions in the presence of nucleophiles in solution. p-Tolylsulfonylimino phospholes 6b–9b can be obtained when TSI 5b is employed; nonetheless, higher reaction temperatures are required due to the lower reactivity of TSI 5b compared to CSI 5a. Phospholes 6b–9b were formed highly chemoselectively in good yields (80–97%). Here, the phosphole oxides TPPO 3 and PPPO 4 required temperatures up to 85 °C, which are significantly higher than in the case of DBPO 1. Derivatives 6b–9b are moisture and air stable and can be purified by flash chromatography in addition to recrystallization. The exchange of the chlorine group by a tosyl moiety leads to a higher electron density at the phosphorus atom, which is shown by a low field shift of 4 to 5 ppm in the 31P NMR spectrum. Even though a few examples of sulfonylimino phospholes have already been reported in the literature, they were obtained by the reaction of phospholes with sulfonyl azides40 or chloramine T.41,42 The advantage of our approach is the selective and facile synthesis of iminophospholes starting from air-stable and easily isolable phosphole oxides 1–4.
To obtain a deeper insight into the possible reaction mechanism of the formation of sulfonylimino phospholes, computational studies using DBPO 1 and chlorosulfonyl isocyanate 5a were performed at the B3LYP-D3(BJ)/TZP level of theory43 using ADF.2018.107.44,45 Transition states were located by employing the Climbing Image Nudged Elastic Band (CI-NEB) method46 starting from the relaxed ground state structures of the reactants (Chapter 5, ESI†).
Fig. 2 shows the relative Gibbs free energy profile of the reaction regarding the starting materials (SM). Theoretical calculations indicated that the phosphole sulfonylimination reaction proceeded via an addition–elimination mechanism and could be considered a formal [2 + 2] cycloaddition. First, the oxygen atom of the PO double bond attacks the carbon atom of the isocyanate. In the resulting ionic intermediate (Int), the negatively charged nitrogen atom attacks the phosphorus atom of the phosphole framework, yielding the corresponding sulfonylimino phosphole after the elimination of CO2. In the exergonic reaction, an energy of 67.3 kJ mol−1 is released and the driving forces are the generation of a CO double bond, the elimination of CO2, and the formation of a resonance-stabilized P–N–S moiety. Interestingly, acyclic phosphane oxides, such as triphenylphosphane oxide, cannot be converted to the corresponding sulfonylimines by CSI 5a. The reason is the more stable and sterically protected PO double bond compared to phosphole oxides. However, TSI 5b exhibits higher thermal stability, which allowed the conversion of triphenylphosphane oxide to tosylimino phosphane at high temperatures and long reaction times (Fig. S1.2, ESI†).
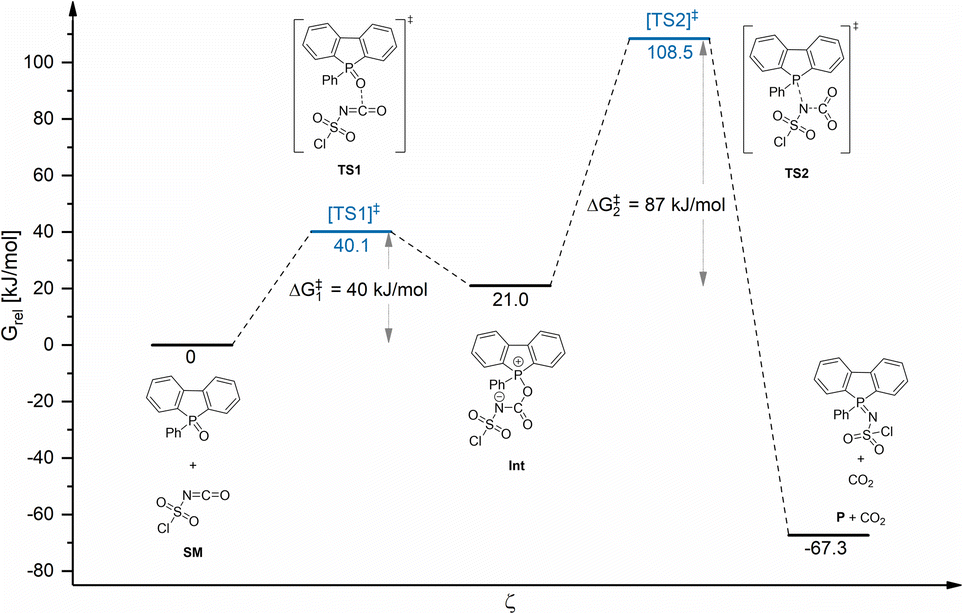 | ||
| Fig. 2 Relative Gibbs free energy profile of the reaction with respect to the starting materials. | ||
Sulfonylimino phospholes are characterized by excellent crystallization properties, readily furnishing single crystals suitable for X-ray diffractometric structure analysis by common crystallization methods (Table 2). The PN double bonds (1.603–1.613 Å) in phospholes 6a–9a (Fig. 3) agree with the calculated (1.61 Å) and reported values.47 Interestingly, the S1–N1 bond lengths are in a range of 1.550 to 1.561 Å and indicate double bond character. This observation suggests delocalization in the P–N–S moiety, which is stabilized by the neighboring electron-withdrawing groups. A comparable delocalized P–N–S unit has already been reported by Dehnicke et al. for sulfonylimino phosphoranes.47 The bond length of the characteristic S–Cl bond is in a range of 2.078 to 2.080 Å and slightly longer than in comparable compounds.47 The tolyl derivatives 6b–9b (Fig. 4) show structural differences compared to their chlorine derivatives 6a–9a. The PN bonds are shorter by 0.01 to 0.04 Å and the S–N bonds are longer by 0.02 to 0.04 Å, resulting in almost equal S–N and P–N bond lengths.
| 6a | 7a | 8a | 9a | |
|---|---|---|---|---|
| P1–C1 | 1.790(2) | 1.791(2) | 1.816(5) | 1.798(2) |
| P1–N1 | 1.603(1) | 1.610(2) | 1.609(4) | 1.613(1) |
| N1–S1 | 1.550(1) | 1.548(2) | 1.557(4) | 1.561(1) |
| S1–Cl1 | 2.0778(5) | 2.0803(9) | 2.079(2) | 2.077(1) |
| P1–N1–S1 | 127.13(8) | 123.2(1) | 122.3(3) | 125.61(8) |
| C1–P1–C4 | 93.61(7) | 94.0(1) | 94.8(2) | 94.69(7) |
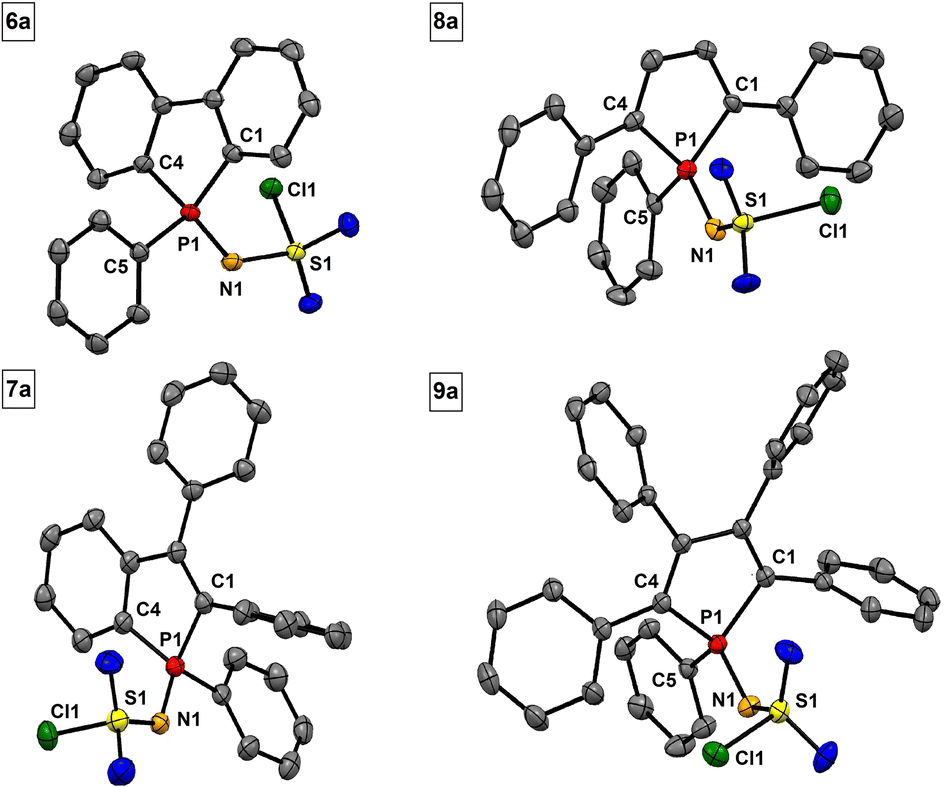 | ||
| Fig. 3 Molecular structures of 6a, 7a, 8a, and 9a. Thermal displacement ellipsoids shown at 50% probability. Hydrogen atoms have been omitted for clarity. | ||
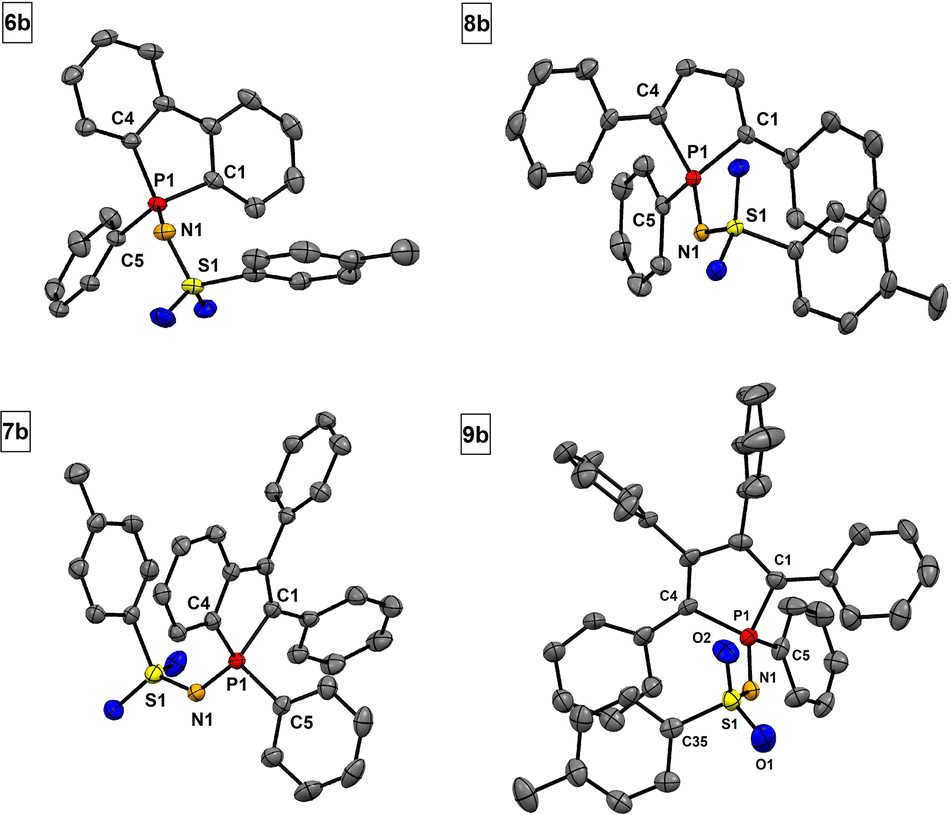 | ||
| Fig. 4 Molecular structures of 6b, 7b, 8b and 9b. Thermal displacement ellipsoids shown at 50% probability. Hydrogen atoms have been omitted for clarity. | ||
The interesting P–N–S bonding mode of sulfonylimino phospholes is possibly the cause of the high kinetic stability of this class of compounds.48 Compared to the chlorine substituent, the tolyl group does not have any electron-withdrawing properties, which leads to a stronger S–N single bond character and reduced delocalization (Table 3).49 For the annelated derivatives 6b and 7b, a significant increase of the P–N–S bond angle is observed compared to the corresponding chlorine derivatives, whereas a decrease is observed for the phospholes with exocyclic substituents 8b and 9b. This is due to intramolecular π interactions between the tosyl group and the fused rings, which leads to an increased tension of the P–N–S linker and increase of the bond angle. In 8b and 9b, the intramolecular π interaction takes place between the tosyl group and the exocyclic phenyl rings in the α position of the phospholes. In addition, the derivatives 6b–8b show a decrease in the C1–P1–C4 bond angle by 0.5 to 1.0°. Significant intermolecular π–π stacking could only be observed in compounds 6b, 8a, and 8b (see Chapter 2, ESI†).
Phosphole-based conjugated π systems are characterized by remarkable photophysical properties. Therefore, in-depth photophysical studies of phospholes 6–9 a,b were carried out to understand the influence of the new modification method on the optical properties. Relating to this, photoluminescence spectra and excited state lifetimes (τav_amp), as well as ΦF in fluid solution at RT, in a frozen glassy matrix at 77 K and in the solid state were measured (Table 4). In general, we observed that all time-resolved photoluminescence measurements yield biexponential decays, due to the coexistence of different conformers; thus, amplitude-weighted average lifetimes were used to estimate deactivation rate constants.50 It became apparent that the chemical modification of the phosphorus atom strongly influences the optical properties. All derivatives show that the transformation from oxides 1–4 into sulfonylimines 6–9 a,b leads to a bathochromic shift of the absorption (λex) and emission maxima (λem) (Fig. 5). Similar observations were already reported by Matano et al.40
| Compound | λ ex [nm] | ε [103 mol−1 dm3 cm−1] | λ em [nm] | τ av_amp (sol, RT) , [ns] | τ av_amp(77 K) [ns] | Φ F(IS) , [±2%] | Φ F(SR) , [±2%] | Φ F(77 K) , [±2%] |
|---|---|---|---|---|---|---|---|---|
| a Measured in MeCN solutions at RT. b Amplitude-weighted average lifetimes (τav_amp).50 c Frozen glassy matrices of butyronitrile at 77 K. d Absolute ΦF(IS) was obtained using a calibrated integrating sphere. e Φ F(SR) was determined by a comparative approach using 1,6-diphenyl-1,3,5-hexatriene (DPHT) in cyclohexane as the standard reference (ΦF(RT) = 0.78). f Not detectable due to weak fluorescence. | ||||||||
| 6a | 335 | 1.59 | 385 | 2.33 ± 0.01 | 3.82 ± 0.06 | 16 | 15 | 98 |
| 6b | 330 | 1.69 | 378 | 12.57 ± 0.04 | 13.7 ± 0.1 | 10 | 12 | 77 |
| 7a | 357 | 5.80 | 480 | 0.84 ± 0.01 | 12.21 ± 0.08 | 5 | 4 | 85 |
| 7b | 353 | 4.30 | 472 | 0.74 ± 0.01 | 12.16 ± 0.04 | 5 | 10 | 98 |
| 8a | 410 | 12.60 | 555 | 0.104 ± 0.002 | 6.46 ± 0.04 | <2 | <2 | 78 |
| 8b | 405 | 16.30 | 537 | 0.407 ± 0.001 | 7.63 ± 0.02 | 3 | 4 | 62 |
| 9a | 405 | 5.20 | 558 | n. d.f | 11.1 ± 0.1 | <2 | <2 | 83 |
| 9b | 398 | 5.00 | 547 | 3.54 ± 0.08 | 14.9 ± 0.1 | <2 | <2 | 89 |
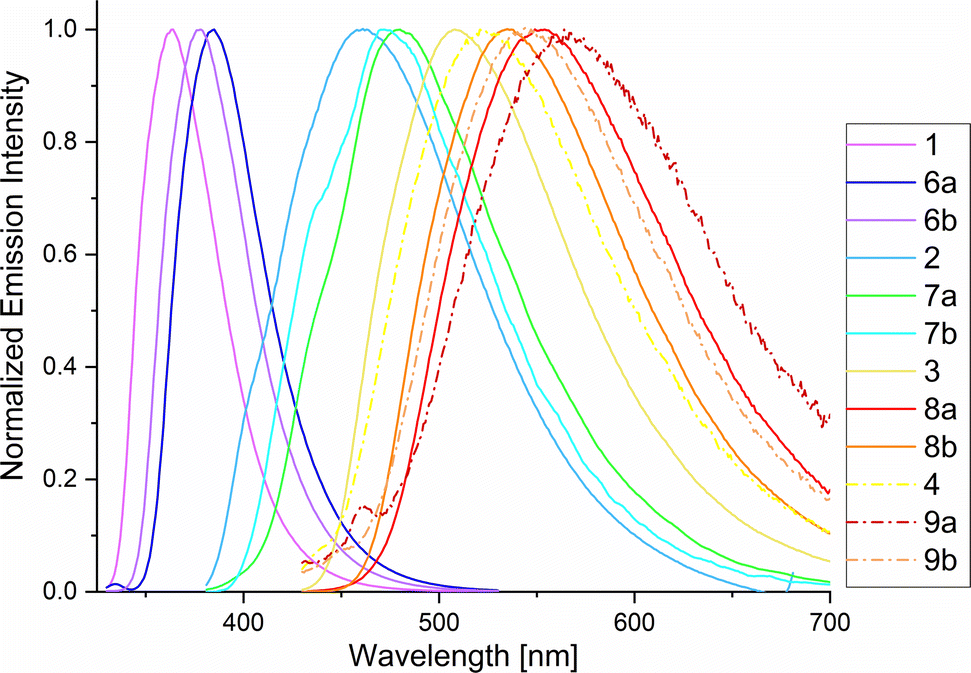 | ||
| Fig. 5 Photoluminescence spectra of 1–4 and 6–9 a,b in MeCN at RT (for λex, see Chapter 4, ESI†). | ||
The shifts in the UV/vis absorption spectra are in the range of 8–18 nm for the chlorine derivatives 6a–9a and 2–13 nm for the tosyl derivatives 6b–9b. However, this effect is even more evident in the steady-state fluorescence spectra, wherein the emission maxima show changes in a range of 21–48 nm for 6a–9a and 14–29 nm for 6b–9b, if compared with their oxide precursors. The largest bathochromic shift is observed in 8a, which shows a difference of 47 nm compared to the oxide 3 (λem = 508 nm). The bathochromic shift can be explained by the fact that sulfonylimino phospholes have a more extended π system than their corresponding oxides. The transformation leads to a reduction of the HOMO–LUMO energy gaps resulting in longer wavelength photons (Fig. S5.1, ESI†). Herein, the chlorine derivatives 6a–9a show the highest bathochromic shift due to the electron-withdrawing properties of the sulfonyl chloride group. The respective fluorescence quantum yields in fluid solutions at RT (ΦF(IS)) were obtained in acetonitrile using a calibrated integrating sphere system and showed a strong dependence on the molecular structure. These ΦF(IS) values were additionally confirmed by a relative method and the use of DPHT as a standard reference.
In MeCN solutions at RT, dibenzophosphole derivatives 6a and 6b have ΦF values of 16% and 10%, respectively, which are significantly higher than that of the corresponding DBPO 1 (ΦF = 4.2%).22 Similar observations were made with the phosphindole derivatives 7a and 7b. The ΦF values for both derivatives are 5%, which is considerably higher than that of their precursor TPPIO 2 (ΦF = 1.2%).12 The higher knr values of compounds 7a-b (if compared with 6a-b) explain their lower quantum yields, which can be attributed to their reduced rigidity. The lowest ΦF values are shown by derivatives 8 and 9. This behavior can be explained by comparison of the corresponding rates, where higher radiationless constants knr were observed for derivatives 8 and 9 (Table S4.1, ESI†). This illustrates that these compounds, as well as the corresponding oxides 3 and 4, are weak emitters in solution with ΦF values < 2-3%. For these molecules, high rates of vibronic radiationless relaxation can be inferred, due to the rotation of the exocyclic phenyl rings, which favors a non-radiative decay and ultimately leads to a decrease in the emission intensity. 6a and 6b have significantly more rigid and planar configurations and thus show a stronger emission in solution. The rotation of the exocyclic phenyl rings and non-radiative decays can be hindered in frozen matrices, meaning that the emission can be strongly enhanced.51 For this purpose, the compounds were dissolved in butyronitrile and cooled down to 77 K. All derivatives show a significant increase in emission intensity at 77 K; this can be confirmed by ΦF(77 K), whose values are in a range of 62–98%. Moreover, the excited state lifetimes (τav_amp(77 K)) are longer than at RT. 8a has a τav_amp(77 K) of 6.46 ns, which means a lifetime extension of more than 60 times compared to RT. Except for 6a, the kr values for all compounds are comparable to RT when cooling down to 77 K. However, an important decrease of until two orders of magnitude in the knr values is observed for all derivatives in frozen glassy matrices, if compared with RT. This behavior is explained by the lack of vibronic radiationless relaxation in the frozen matrices. Interestingly, derivatives 8 and 9, which are those with the largest number of phenyl substituents, are the ones with the most significant drop in the knr value. These results are in agreement with the detected ΦF(77 K) and τav_amp(77 K).
AIEgens are characterized by weak fluorescence in solution. By adding a solvent in which the compound is not soluble, they aggregate. The resulting intermolecular interactions restrict the rotational freedom of the exocyclic rotor units, which leads to a significant increase in fluorescence intensity. We have shown this typical AIE behavior for the examples 8b and 9b in Fig. 6. The MeCN solutions of compounds 8b and 9b show no (9b) or weak (8b) fluorescence (Fig. 6B). When the water content is increased, a drastic increase in emission intensity is observed at about 80% water content, which illustrates the AIE nature of this novel class of compounds.
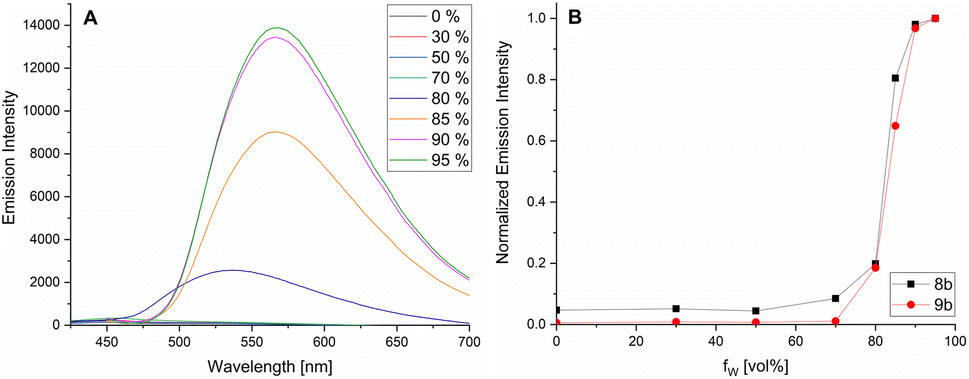 | ||
| Fig. 6 Photoluminescence spectra of 9b in different MeCN/water mixtures (λex = 398 nm) (A) and emission intensity of 8b and 9b in MeCN/water for increasing water content (fw) (B). | ||
Despite sulfonylimino phospholes showing weak emission in solution, they are strong emitters in the solid state (Fig. 7). The locations of the emission maxima are comparable to those in solution (Table 5). Exceptions are represented by the dibenzophosphole derivatives 6a and 6b, which show a strong bathochromic shift of 29–31 nm that can be attributed to the fact that planar molecular structures are capable of forming stronger interactions by π–π stacking in the solid state.52 The ΦF values in the solid state (ΦF(solid)) as well as τav_amp show characteristic differences between chlorine and tosyl derivates. 9b has a ΦF(solid) value of 75%, which is more than two-fold higher than that of the corresponding oxide PPPO 4 (ΦF(solid) = 36%), whereas 9a shows less emission with a ΦF(solid) of 10%. Furthermore, 7b shows intense blue fluorescence at 479 nm and a ΦF(solid) of 82%, which represents a significant increase if compared to TPPIO 2 (ΦF(solid) = 72%). Interestingly, for all the compounds studied, a trend was observed for the tosyl derivatives 6b–9b, which have larger ΦF(solid) and longer τ in the solid state than the comparable Cl derivatives 6a–9a. These differences in τav_amp and ΦF(solid) can be explained by analyzing and comparing the average radiative decay rate constants (kr) and the corresponding non-radiative decay rate constants (knr).
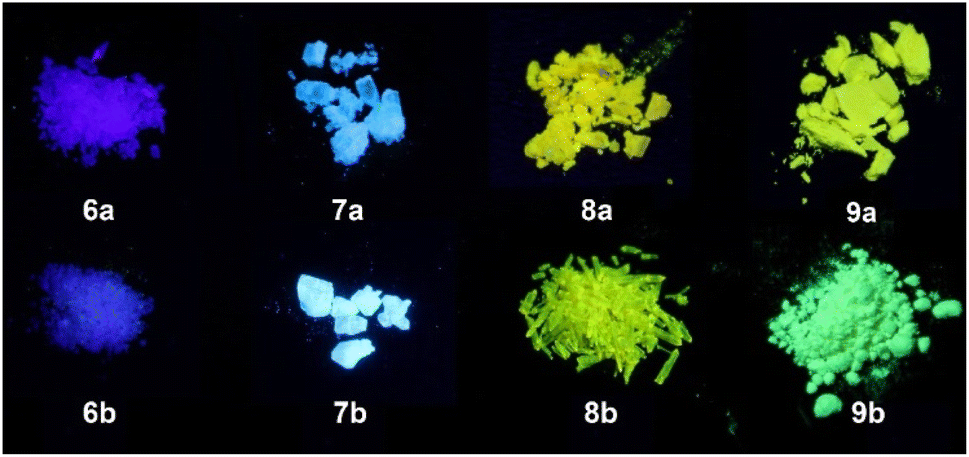 | ||
| Fig. 7 Solid-state fluorescence of phospholes 6–9 under irradiation of UV light (395 nm). | ||
| Compound | λ em [nm] | τ av_amp [ns] | Φ F(solid) [± 2%] |
|---|---|---|---|
| a Solid state at RT. b Amplitude-weighted average lifetimes at RT. c Absolute ΦF of solids at RT was obtained using a calibrated integrating sphere with a suitable sample holder. | |||
| 6a | 417 | 8.4 ± 0.4 | 34 |
| 6b | 407 | 14.2 ± 0.3 | 54 |
| 7a | 491 | 4.4 ± 0.1 | 46 |
| 7b | 479 | 8.5 ± 0.5 | 82 |
| 8a | 557 | 2.3 ± 0.1 | 18 |
| 8b | 548 | 9.2 ± 0.5 | 55 |
| 9a | 555 | 2.5 ± 0.1 | 10 |
| 9b | 528 | 8.1 ± 0.1 | 75 |
In 6a–9a, larger knr than kr values are observed. This is clearly demonstrated by phospholes 8a and 9a, which have a knr of 36 × 107 s−1 and kr of 8.0 × 107 s−1 and 3.8 × 107 s−1, respectively (Fig. 8). We could observe in the crystal structures that the Cl substituent of the sulfonyl group has no contact with neighboring molecules and can undergo rotation and vibration, leading to higher rates of radiationless vibrational relaxation, and thus, an increased radiationless decay rate. Moreover, Caryl–H⋯Cl interactions occur in 6a and 7a, resulting in increased rigidity, relatively low knr values and moderate ΦF(solid) of 34 and 46%, respectively (additional details are given in the ESI†). On the other hand, there are multiple Caryl–H⋯π and SO⋯H interactions in 6b–9b between the tosyl group and neighboring molecules. The resulting restriction of rotations and motions due to the fixation of the substituent is clearly reflected in the lower knr values.
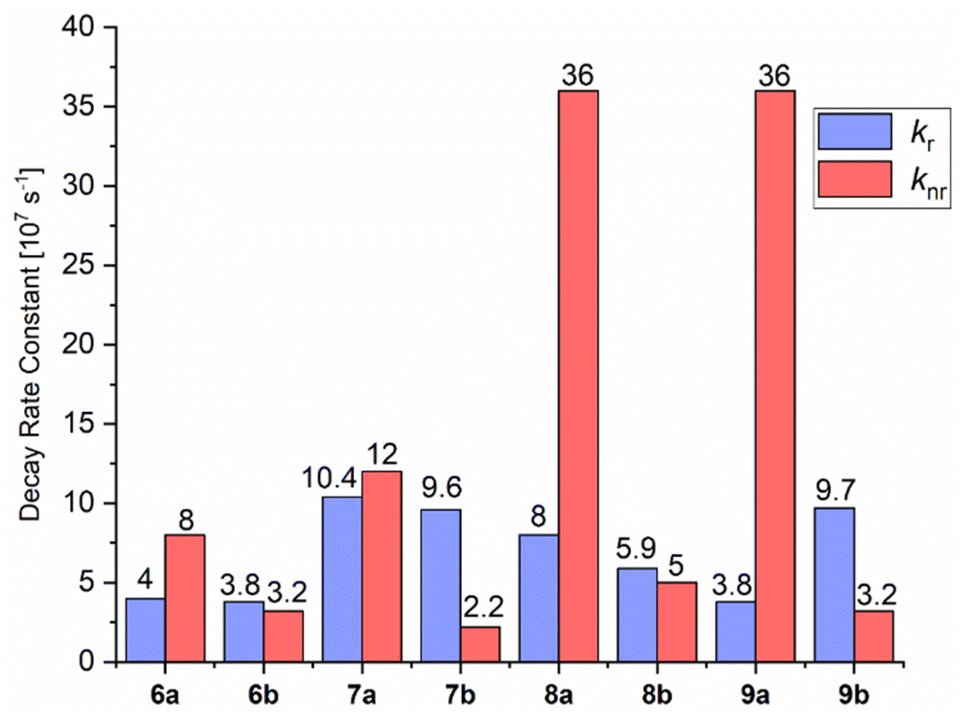 | ||
| Fig. 8 Radiative (kr) and non-radiative (knr) decay rate constants of compounds 6–9 in the solid state. The decay rate constants are determined according to kr = 1/τav_amp = ΦF/τav_amp and knr = 1 − ΦF/τav_amp by using amplitude-weighted average lifetimes, according to Engelborghs et al.50 The uncertainty analysis is not presented in this diagram for clarity (for kr and knr in MeCN solutions at RT and a frozen glassy matrix in butyronitrile at 77 K, see Chapter 4 in the ESI†). | ||
Also, additional intramolecular π–π interactions between the tosyl group and the phoshindole framework or the exocyclic phenyl rings in the α position can be noticed (Fig. 9). This type of through-space conjugation has been invoked to explain the enhanced photophysical properties by increased rigidity.53–56 The distances of the intramolecular π stacking are in the range of 3.219–3.501 Å, indicating strong interactions and agreeing with the observations of Tang et al.57 Except for 7a-b and 9b having the highest radiative rate constants, the knr values are generally comparable for all compounds in solid phases.
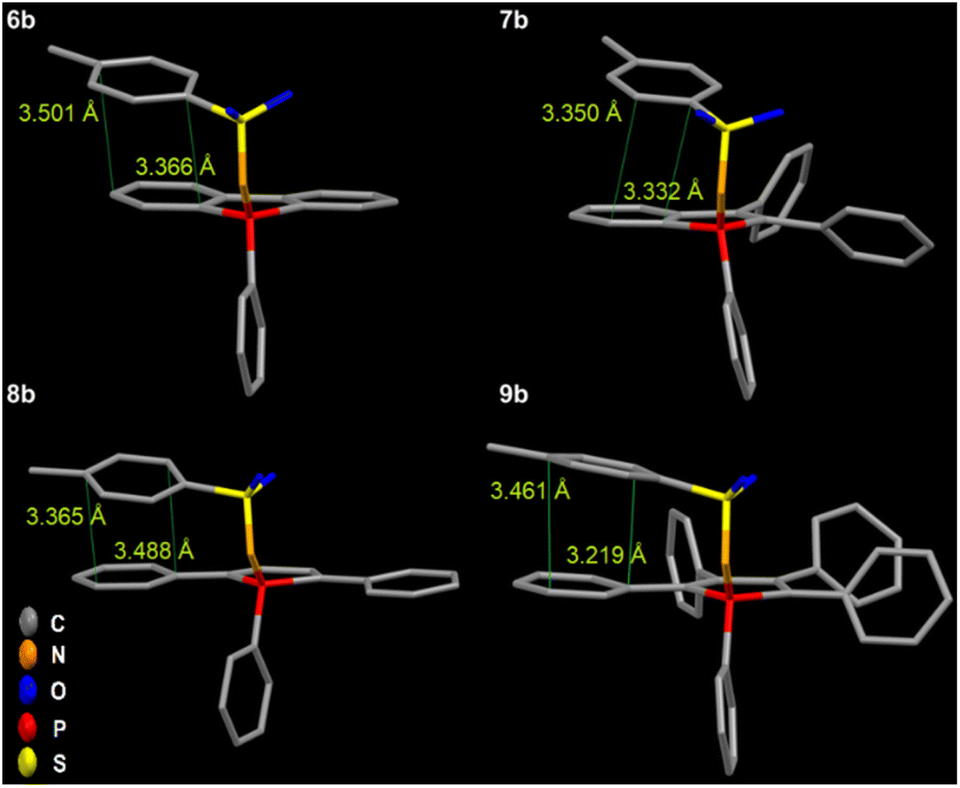 | ||
| Fig. 9 Intramolecular π–π interaction with the tosyl group in 6b–9b leads to through-space conjugation, additional restriction of motions, and increased rigidity. | ||
In general, the derivatisation of phosphole oxides with sulfonyl isocyanates results in a bathochromic shift of the absorption and emission maxima. All synthesized phospholes 6–9 show a weaker emission in solution, whereas 6a,b, and 7a,b have higher ΦF values than the corresponding oxides 1 and 2. Due to the different restrictions in motions and intramolecular interactions, tosyl derivatives 6b–9b have significantly higher ΦF(solid) values than chlorine derivatives 6a–9a. The high density of rotovibrational states make oxides 3-4 and derivatives 8-9 weak emitters in fluid solutions. This highlights the importance of the reaction of phosphole oxides with TSI 5b as a promising derivatisation method for the synthesis of high-emissive phosphole-based AIEgens.
Conclusion
A facile synthetic strategy for selectively converting phosphole oxides 1–4 into their corresponding sulfonylimines 6–9 a,b by using sulfonyl isocyanates 5a,b is reported. The reaction is characterized by high chemoselectivity and excellent yields. Theoretical calculations suggest an addition–elimination mechanism and show that this reaction method is particularly suitable for phosphole oxides. In addition to X-ray structure analysis, the photophysical properties of the phospholes 6–9 a,b were studied under different conditions, namely in solution, frozen glassy matrices, and solid state. In solution, all compounds show a bathochromic shift of absorption and emission maxima compared to the phosphole oxides 1–4, but low ΦF(IS) values were observed. At 77 K in glassy matrices, however, the ΦF values were significantly improved. The novel compounds are characterized by strong fluorescence in the solid state, although there are significant differences in quantum yields and lifetimes between chloride 6a–9a and tosyl derivates 6b–9b. In general, a trend towards considerably higher ΦF(solid) values and longer τ for tosyl derivatives 6b–9b is observed, due to better restriction of intramolecular rotations and vibrations in the solid state. Intriguingly, the transformation of phosphole oxides into their tosylimines significantly increased the quantum yield. These observations show that derivatisation with TSI 5b is an essential and facile synthetic strategy for the preparation of highly emissive phosphole-based luminophores and its potential will now be investigated further.Data availability
All experimental procedures, spectroscopic data, photophysical properties, information on the theoretical calculations and crystallographic data can be found in the ESI.†Author contributions
N. K. performed the experimental work and wrote the original draft. Y. G.-L., C. L. and F. Y. analyzed the photophysical properties. P. L. performed single crystal X-ray analysis and interpreted the structural data. M. L. performed the DFT calculations. E. H.-H. and C. A. S. supervised the project. All authors have corrected the final manuscript. All authors have read and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. is grateful for a doctoral fellowship provided by the Deutsche Bundesstiftung Umwelt. We thank Prof. Dr C. Schneider for providing the computational infrastructure for DFT calculations. C. A. S. gratefully acknowledges the generous financial support for the acquisition of an “Integrated Confocal Luminescence Spectrometer with Spatiotemporal Resolution and Multiphoton Excitation” (DFG/Land NRW: INST 211/915-1 FUGG; DFG EXC1003: “Berufungsmittel”). We thank Prof. Dr B. Abel for providing access to the fluorescence lifetime image microscope for the kinetical fluorescence studies.References
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- B. Liu and B. Z. Tang, Chem. –J. Mater. Chem. B Asian J., 2019, 14, 672–673 CAS.
- R. Zhan, Y. Pan, P. N. Manghnani and B. Liu, Macromol. Biosci., 2017, 17, 1600433 CrossRef PubMed.
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404–1414 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- L. Chen, C. Zhang, G. Lin, H. Nie, W. Luo, Z. Zhuang, S. Ding, R. Hu, S.-J. Su, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2775–2783 RSC.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- F. Bu, E. Wang, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 4440–4449 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Z. Zhao, S. Chen, J. W. Y. Lam, C. K. W. Jim, C. Y. K. Chan, Z. Wang, P. Lu, C. Deng, H. S. Kwok, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 7963–7972 CrossRef CAS.
- M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932–1937 RSC.
- Y. Ren and T. Baumgartner, Dalton Trans., 2012, 41, 7792–7800 RSC.
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, W.-T. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 11504–11508 CrossRef CAS PubMed.
- M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC.
- D. Joly, D. Tondelier, V. Deborde, B. Geffroy, M. Hissler and R. Réau, New J. Chem., 2010, 34, 1603–1611 RSC.
- C. Hay, M. Hissler, C. Fischmeister, J. Rault-Berthelot, L. Toupet, L. Nyulászi and R. Réau, Chem. – Eur. J., 2001, 7, 4222–4236 CrossRef CAS PubMed.
- H.-C. Su, O. Fadhel, C.-J. Yang, T.-Y. Cho, C. Fave, M. Hissler, C.-C. Wu and R. Réau, J. Am. Chem. Soc., 2006, 128, 983–995 CrossRef CAS PubMed.
- C. Hay, C. Fave, M. Hissler, J. Rault-Berthelot and R. Réau, Org. Lett., 2003, 5, 3467–3470 CrossRef CAS PubMed.
- J. Crassous and R. Réau, Dalton Trans., 2008, 6865–6876 RSC.
- F. Mathey, Chem. Rev., 1988, 88, 429–453 CrossRef CAS.
- P. R. Schleyer, H. Jiao, B. Goldfuss and P. K. Freeman, Angew. Chem., Int. Ed. Engl., 1995, 34, 337–340 CrossRef CAS.
- A. Dransfeld, L. Nyulászi and P. R. Schleyer, Inorg. Chem., 1998, 37, 4413–4420 CrossRef CAS PubMed.
- Y. Ren, J. Gao, A. K. Hailey, T. Baumgartner and Y.-L. Loo, Chem. Mater., 2016, 28, 8407–8414 CrossRef CAS.
- D. Klintuch, A. Kirchmeier, C. Bruhn and R. Pietschnig, Dyes Pigm., 2020, 180, 108443 CrossRef CAS.
- R. Kabe, V. M. Lynch and P. Jr Anzenbacher, CrystEngComm, 2011, 13, 5423–5427 RSC.
- M. Matsumura, M. Yamada, A. Muranaka, M. Kanai, N. Kakusawa, D. Hashizume, M. Uchiyama and S. Yasuike, Beilstein J. Org. Chem., 2017, 13, 2304–2309 CrossRef CAS PubMed.
- E. Takacs, A. Escande, N. Vanthuyne, C. Roussel, C. Lescop, E. Guinard, C. Latouche, A. Boucekkine, J. Crassous, R. Réau and M. Hissler, Chem. Commun., 2012, 48, 6705–6707 RSC.
- M. Hissler, C. Lescop and R. Réau, C. R. Chim., 2008, 11, 628–640 CrossRef CAS.
- K. Fourmy, D. H. Nguyen, O. Dechy-Cabaret and M. Gouygou, Catal. Sci. Technol., 2015, 5, 4289–4323 RSC.
- O. Fadhel, D. Szieberth, V. Deborde, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, Chem. – Eur. J., 2009, 15, 4914–4924 CrossRef CAS PubMed.
- H. Hoffmann, H. Förster and G. Tor-Poghossian, Monatsh. Chem., 1969, 100, 311–315 CrossRef CAS.
- C. R. Hall and D. J. H. Smith, Tetrahedron Lett., 1974, 15, 1693–1696 CrossRef.
- Y. Chemam, Z. Aouf, A. Amira, H. K’tir, H. Bentoumi, R. Ghodbane, R. Zerrouki and N.-E. Aouf, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 197, 777–787 CrossRef CAS.
- J. K. Rasmussen and A. Hassner, Chem. Rev., 1976, 76, 389–408 CrossRef CAS.
- Y. Matano, H. Ohkubo, Y. Honsho, A. Saito, S. Seki and H. Imahori, Org. Lett., 2013, 15, 932–935 CrossRef CAS PubMed.
- U. V. Monkowius, S. Nogai and H. Schmidbaur, Dalton Trans., 2004, 1610–1617 RSC.
- G. Wittig and A. Maercker, Chem. Ber., 1964, 97, 747–768 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- H. Folkerts, D. Nußhär, F. Weller, K. Dehnicke, J. Magull and W. Hiller, Z. Anorg. Allg. Chem., 1994, 620, 1986–1991 CrossRef CAS.
- E. Böhm, K. Dehnicke, J. Beck, W. Hiller, J. Strähle, A. Maurer and D. Fenske, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 138–148 CrossRef.
- A. F. Cameron, N. J. Hair and D. G. Morris, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 221–225 CrossRef CAS.
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 CAS.
- G. Tan, I. Maisuls, F. Strieth-Kalthoff, X. Zhang, C. Daniliuc, C. A. Strassert and F. Glorius, Adv. Sci., 2021, 8, 2101814 CrossRef CAS PubMed.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 1573–1579 RSC.
- J. Li, P. Shen, Z. Zhao and B. Z. Tang, CCS Chem., 2019, 1, 181–196 CAS.
- W. Luo, H. Nie, B. He, Z. Zhao, Q. Peng and B. Z. Tang, Chem. – Eur. J., 2017, 23, 18041–18048 CrossRef CAS PubMed.
- H. Puntscher, P. Kautny, B. Stöger, A. Tissot, C. Hametner, H. R. Hagemann, J. Fröhlich, T. Baumgartner and D. Lumpi, RSC Adv., 2015, 5, 93797–93807 RSC.
- M. Yu, R. Huang, J. Guo, Z. Zhao and B. Z. Tang, PhotoniX, 2020, 1, 11 CrossRef.
- T. Han, H. Deng, Z. Qiu, Z. Zhao, H. Zhang, H. Zou, N. L. C. Leung, G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 5588–5598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, synthesis, crystallographic data, compound spectra, optical properties and theoretical calculations. CCDC 2219165–2219173. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00308f |
| This journal is © The Royal Society of Chemistry 2023 |