
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompact hydrophilic electrophiles enable highly efficacious high DAR ADCs with excellent in vivo PK profile†
Philipp
Ochtrop
ac,
Jahaziel
Jahzerah
a,
Paul
Machui
c,
Isabelle
Mai
c,
Dominik
Schumacher
c,
Jonas
Helma
c,
Marc-André
Kasper
*abc and
Christian P. R.
Hackenberger
 *ab
*ab
aLeibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Department of Chemical Biology, Robert-Rössle-Strasse 10, 13125 Berlin, Germany. E-mail: hackenbe@fmp-berlin.de
bHumboldt Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str.2, 12489 Berlin, Germany
cTubulis GmbH, Am Klopferspitz 19 a, 82152 Planegg-Martinsried, Germany. E-mail: marc.kasper@tubulis.com
First published on 3rd January 2023
Abstract
The recent success of antibody–drug conjugates (ADC), exemplified by seven new FDA-approvals within three years, has led to increased attention for antibody based targeted therapeutics and fueled efforts to develop new drug-linker technologies for improved next generation ADCs. We present a highly efficient phosphonamidate-based conjugation handle that combines a discrete hydrophilic PEG-substituent, an established linker-payload and a cysteine-selective electrophile in one compact building block. This reactive entity provides homogeneous ADCs with a high drug-to-antibody ratio (DAR) of 8 in a one-pot reduction and alkylation protocol from non-engineered antibodies. The compact branched PEG-architecture introduces hydrophilicity without increasing the distance between antibody and payload, allowing the generation of the first homogeneous DAR 8 ADC from VC-PAB-MMAE without increased in vivo clearance rates. This high DAR ADC exhibits excellent in vivo stability and increased antitumor activity in tumour xenograft models relative to the established FDA approved VC-PAB-MMAE ADC Adcetris, clearly showing the benefit of the phosphonamidate based building-blocks as a general tool for the efficient and stable antibody-based delivery of highly hydrophobic linker-payload systems.
Introduction
The site-specific and chemoselective modification of proteins with unnatural functionalities like fluorescent dyes or cytotoxic drugs has become an indispensable strategy to advance the molecular life sciences and to develop biological therapeutics.1 In particular antibody–drug conjugates (ADCs) receive increasing attention for the targeted treatment of diverse malignant diseases, as exemplified by seven new FDA approvals within the past three years, accumulating to eleven marketed ADCs in total.2–4 Most of these ADCs are produced via cysteine-selective bioconjugation strategies that exploit the unique nucleophilicity of thiols, liberated by reduction of interchain disulfides of monoclonal antibodies, to yield chemically defined conjugates.5–7 Strikingly, 9 out of 11 marketed ADCs are manufactured using maleimide functionalized linkers despite their known instability towards retro-Michael additions and thiol–exchange reactions in the presence of external thiols.8–10 In particular the transfer of cytotoxic payload to endogenous serum proteins such as albumin was identified as the main pathway of these undesired decompositions.11 The resulting drug loss in early days of circulation increases off target toxicity and reduces the ADC efficacy. To address these stability issues in the process of developing safer and more efficacious next generation ADCs, several strategies have been pursued to improve maleimide-based bioconjugates by promoting hydrolysis of the thiosuccinimide linkage to yield stable thioethers.12,13 Beyond that, a set of new cysteine-selective conjugation platforms including α,β-unsaturated acrylamides,14–16 unsaturated bissulfones,17,18 sulfonamides,19 vinyl-20 and divinylpyrimidines,21 divinyltriazines,22 as well as disubstituted maleimides23–25 and pyridazinediones26,27 have been developed to produce stable bioconjugates. However, most of these conjugation platforms lack ultimate evaluation in an in vivo setting, thus falling short their ambition to enable next generation therapeutics.Recently, our laboratory contributed to this growing toolbox for cysteine-selective conjugation methods by introducing electrophilic vinyl- and ethynylphosphonamidates for the modification of peptides and proteins (Fig. 1a).28,29 Those unsaturated phosphonamidates exhibit excellent thiol selectivity and outstanding linkage stability against thiol exchange and retro addition. Along with these favored characteristics, ethynylphosphonamidates are conveniently accessible from various azides and diverse phosphonites through chemoselective Staudinger phosphonite reactions (SPhR) (Fig. 1b). Previously, we applied ethynyl phosphonamidates for the synthesis of ADCs employing commonly used payloads such as the tubulin inhibitors MMAF and MMAE to generate efficacious stably linked DAR 4 ADCs.30 In a head-to-head comparison to the analogous maleimide linked Adcetris, we showed that a phosphonamidate-based ADC displays a drastically improved serum stability which translated into a beneficial in vivo antitumor activity.30
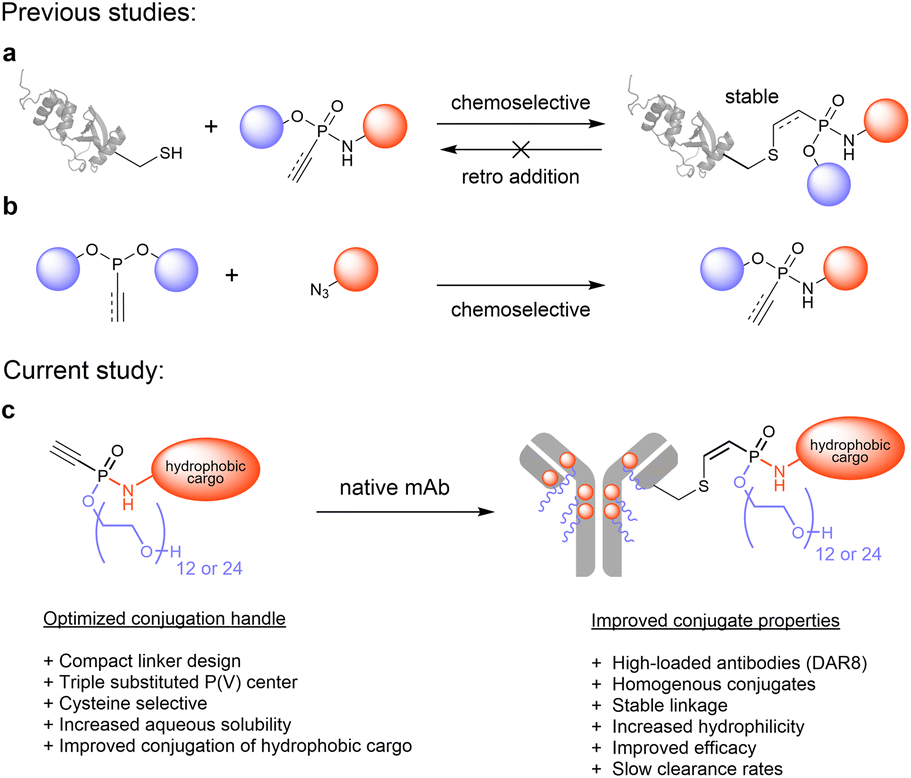 | ||
| Fig. 1 Cysteine-selective phosphonamidate electrophiles. (a): Thiol-selective bioconjugation reaction of ethynylphosphonamidates with cysteine containing proteins yield highly stable conjugates. (b): Synthesis of ethynylphosphonamidates by chemoselective Staudinger-phosphonite reactions from ethynylphosphonites and azides. (c): Compact hydrophilic phosphonamidate building blocks that allow convenient ADC generation from native antibodies. | ||
Aside from improving linkage stability, another strategy to enhance the in vivo efficacy of ADCs is to increase the drug loading to a maximum of eight drugs per antibody (DAR 8). Although such high-loaded ADCs usually exhibit greater potency in vitro compared to their lower modified analogues, they are often inferior in vivo owing to their increased overall hydrophobicity that triggers accumulation and aggregation in the liver.31,32 These processes ultimately lead to fast clearance from blood circulation and decreased payload delivery to the tumor. Therefore, the four marketed ADCs Adcetris, Polivy, Padcev and Tivdak, all conjugated with the hydrophobic linker-payload maleido-caproyl-valine-citrulline-PAB-MMAE (mc-VC-PAB-MMAE), are limited to an average DAR of 3.5 to 4. To overcome this limitation, efforts towards more hydrophilic linker that enable the generation of high DAR MMAE conjugates with acceptable pharmacokinetic profile are ongoing. It has been demonstrated that the replacement of the hydrophobic Val-Cit-PAB cleavage site with a hydrophilic glucuronide linker that contains branched PEG polymers or polysarcosine had a significant effect on clearance from circulation in vivo and improved the pharmacokinetic profile of DAR 8 MMAE ADCs.33–35
In our present study, we introduce a new phosphonamidate based hydrophilic conjugation handle that allows the straightforward modification of monoclonal antibodies with challenging hydrophobic payloads to yield highly modified antibody conjugates without impairing their favourable pharmacokinetic profiles (Fig. 1c). To that end, we exploited the intrinsic compact structure of ethynylphosphonamidates, and designed cysteine-selective conjugation handles with hydrophilic O-substituents that are directly incorporated into the phosphorous core of the electrophile. The resulting compact branched structures improve the aqueous solubility and conjugation efficiency of highly hydrophobic linker-payloads as the established Val-Cit-PAB-MMAE that is part of five marketed ADCs (Fig. 1c).
In the course of this study, we identified PEG12 and PEG24 as most suitable hydrophilic substituents that enable the generation of the first ethynylphosphonamidate-based ADCs with eight MMAE payloads per antibody. Most importantly, our strategy succeeds without changing the VC-PAB cleavage site to the much more polar glucuronide linker. These promising bioconjugates display improved in vitro and in vivo efficacy while maintaining exquisite PK profiles.
Results and discussion
We started our investigation by evaluating the impact of different water-soluble polar phosphonamidate-O-substituents to mediate solubility of a hydrophobic cargo. For this purpose, we chose the fluorescent dye cyanine 5 (Cy5) as non-toxic hydrophobic model and equipped the Cy5-phosphonamidate with O-substituents such as diethylene glycol (PEG2), dodecaethylene glycol (PEG12) or choline to obtain hydrophilic conjugation handles that can compensate for the hydrophobicity of the fluorophore. To that end, bis(diisopropylamino)-chlorophosphine 1 was alkylated with ethynylmagnesium bromide, followed by 1H-tetrazole mediated substitution of the diisopropylamino groups with the respective alcohol to prepare phosphonites 3a–c (Fig. 2a). The resulting highly hydrophilic phosphonites, were directly used in a subsequent SPhR with Cy5 azide 4 (for synthesis refer to ESI†) to give phosphonamidates 5a–c. To initially evaluate the solubility enhancing properties of the different O-substituents we set up a shake-flask solubility assay and determined the maximum solubility of phosphonamidates 5a–c in the aqueous system that is usually used for phosphonamidate bioconjugation reactions. For comparison we also included the previously reported Cy5 phosphonamidate 5d with ethyl- O-substituent to our evaluation.29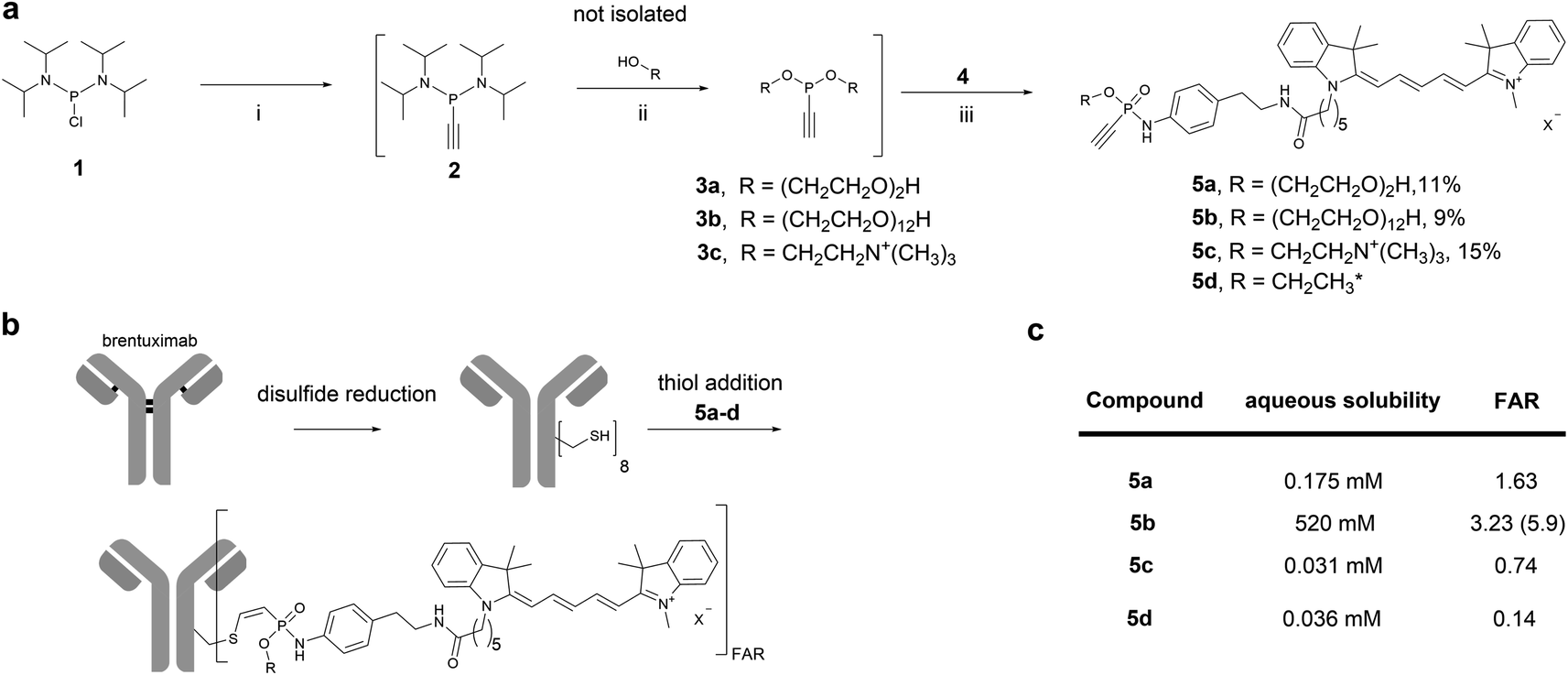 | ||
| Fig. 2 a): Sequential one-pot synthesis of differently O-substituted Cy5-phosphonamidates. (i): Ethynyl magnesium bromide, THF, 0°C-rt, 30 min.; (ii): R–OH, 1H tetrazole, MeCN, 0°C-rt, 2-3 h; (iii): Cy5-azide 4, DMF, rt, overnight. *Synthesized according to previous report.29 (b): Scheme for the sequential reduction and alkylation protocol for the modification of brentuximab with Cy5-phosphonamidates 5a–d. Reduction: DTT, 50 mM sodium borate in PBS (pH 8.0), 37 °C, 40 min. Thiol addition: 50 mM Tris–HCl, 1 mM EDTA, 100 mM NaCl, pH 8.5, 5% DMSO, 14 °C, 16 h. (c): Table summarizing properties of phosphonamidates 5a–d; FAR = fluorophore to antibody ratio. | ||
As expected, ethyl phosphonamidate 5d displayed low solubility under the tested conditions with a saturation concentration of 36 μM (Fig. 2c). Changing the substituent to the more polar diethylene glycol significantly increased the aqueous solubility of the Cy5 conjugate 5a to 175 μM. Interestingly, the cationic choline substituent did not significantly improve the solubility of the Cy5 phosphonamidate 5c, which is displayed by the lowest measured solubility of 31 μM. In contrast, the PEG12 substituent drastically increased the solubility of phosphonamidate 5b to 520 mM. This immense increase in solubility qualified longer polyethylene glycol substituted phosphonamidates as most promising candidates for improving the conjugation of hydrophobic cargo to antibodies.
Next, we evaluated the set of fluorescent phosphonamidates in conjugation experiments with the therapeutically relevant monoclonal antibody (mAb) brentuximab (Fig. 2b). After reduction of intramolecular disulfides, we incubated the mAb with 10 equivalents excess of Cy5 5a–d for 16 h at 14 °C, using only a limited amount of DMSO cosolvent. After purification we obtained brentuximab-Cy5 conjugates with fluorophore to antibody ratios (FAR) ranging from 0.1 to 2.8 (Fig. 2c). Under the tested conjugation conditions, we observed the expected trend of increasing FAR for differently substituted phosphonamidates from low soluble ethyl 5d and choline 5c over moderately soluble PEG25a to highly soluble PEG125b, confirming the expected correlation between aqueous solubility and conjugation efficiency. The low FAR of ethyl- (FAR 0.2) and choline (FAR 0.3) substituted conjugates 5d and 5c is a result of their low solubility (30 μM) in the reaction buffer, which reduces the effective concentration to only one equivalent labeling reagent per antibody. This unfavored stoichiometry is improved when the more polar PEG2-and PEG12 phosphonamidates 5a and 5b are used. Especially the PEG12O-substituent increases aqueous solubility of the Cy5-phosphonamidate in buffer which allows the use of 100 equivalent excess during conjugation reactions yielding in an improved FAR of 5.9.
Encouraged by the beneficial solubility and conjugation efficiency of the PEG12 substituted phosphonamidate, we decided to employ this structural motif to synthesize homogeneously modified ADCs from the challenging hydrophobic linker-payload Val-Cit-PAB-MMAE. In the linker-payload design of compound 7, depicted in Fig. 3a, the PEG-substituent is directly incorporated into the electrophilic phosphonamidate unit for the cysteine addition, which delivers a branched conjugation handle that introduces hydrophilicity to the hydrophobic linker-payload without increasing the distance between antibody and payload. This compact structure limits exposure of the hydrophobic linker-payload and should reduce unfavored accumulation and aggregation of highly conjugated ADCs. To synthesize the respective MMAE-phosphonamidate we used phosphonite 3b in a SPhR with azido benzoic acid succinimide ester and reacted the resulting NHS-phosphonamidate building block 6 with VC-PAB-MMAE (Fig. 3a). With the resulting compact drug–linker conjugate 7 in hand, we set up an initial screening experiment to identify optimal conditions for a one-pot reduction and alkylation protocol that yields a DAR 8 ADC from native antibodies (Fig. 3b). To that end, we co-incubated brentuximab at 10 mg mL−1 with 10 equivalents of 7 and increasing equivalents of TCEP. In this set of experiments, we observed that a maximum degree of modification with a DAR of 6.9 is reached with 8 equivalents of the reductant (Fig. 3c). Those 8 equivalents were carried over to a second experiment in which we increased the stoichiometry of 7 to 12 and 15 equivalents relative to the antibody. We identified that a slight excess of 12 equivalents of 7 is sufficient to generate a fully modified DAR 8 ADC in a one-pot reduction and alkylation protocol. Remarkably, this translates to only 1.5 equivalents 7 per cysteine, which allows a reagent efficient ADC production.
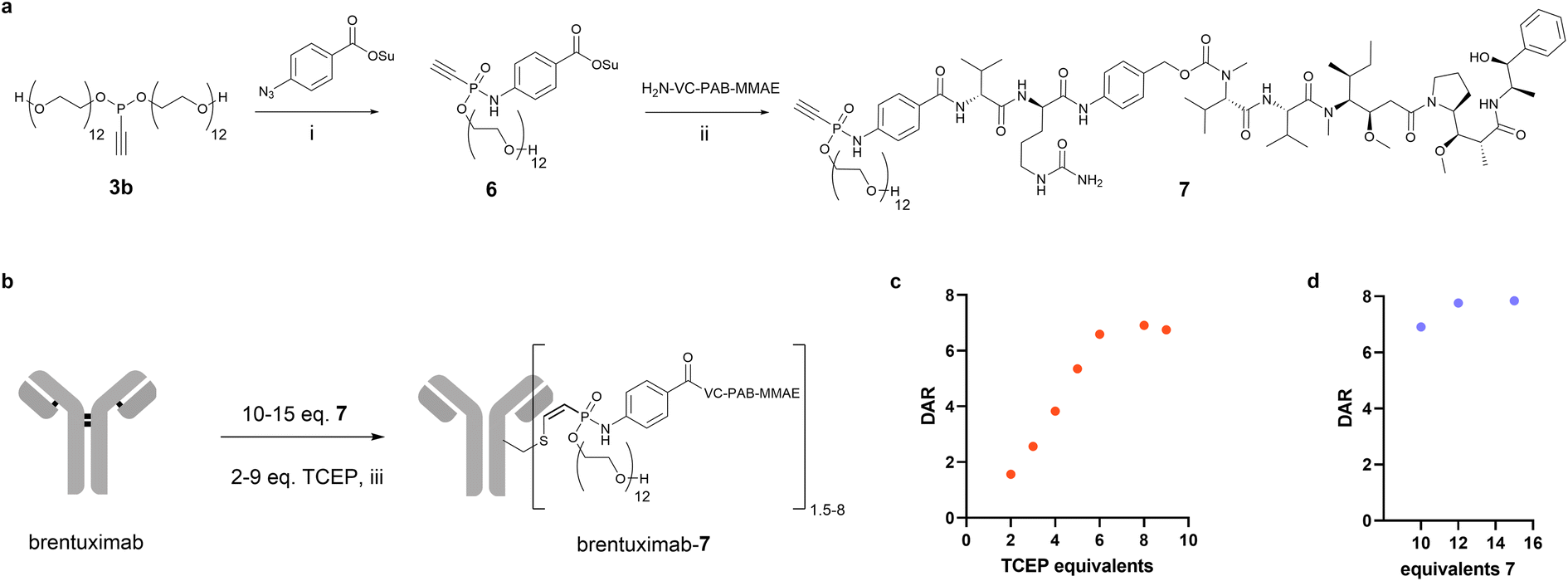 | ||
| Fig. 3 Synthesis of ethynylphosphonamidate 7 and optimization of conjugation conditions to brentuximab. (a): Synthesis scheme for the preparation of ethynylphosphonamidate 7. (i): 4-Azidobenzoic-acid-N-hydroxysuccinimide ester, DMF, rt, overnight, 68%; (ii): VC-PAB-MMAE, DIPEA, DMSO, overnight, 35%. (b): Equivalent screen of TCEP and 7 for one-pot reduction and alkylation protocol of brentuximab. (iii): 50 mM Tris–HCl, 1 mM EDTA, 100 mM NaCl, pH 8.5, 5% DMSO, 14 °C, 16 h. (c): DAR of brentuximab-7 depending on equivalents of TCEP with 10 equivalents 7; (d): DAR of brentuximab-7 depending on equivalents of 7 with 8 equivalents TCEP. | ||
Next, we started the evaluation of the DAR 8 brentuximab-7 by determining its in vitro potency in a cell-based viability assay with three CD30-overexpressing cell lines and compared the cell killing activity to a DAR 4 brentuximab-7 and Adcetris (Fig. 4a–c). Across all tested antigen positive cell lines, we observed nearly identical cell-killing behavior for Adcetris and the DAR 4 brentuximab-7. These results indicate that the modification of brentuximab with PEG12 phosphonamidate 7 has negligible effect on the recognition, binding, and subsequent internalization. Furthermore, both ADCs had no effect on the HL60 control cell line (Fig. 4d). When we further compared the IC50 values for the DAR 4 ADCs with the high loaded DAR 8 ADC, we observed a clear increase in potency, which is in-line with the higher payload loading of the DAR 8 brentuximab-7. To further evaluate cathepsin B cleavage and the traceless release of MMAE from the VC-PAB-linker after ADC internalization, we set up an indirect bystander assay for which we incubated CD30 positive L-540 cells with Adcetris, brentuximab-7 DAR4 or DAR8. In this setting, the ADC is internalized and exposed to intracellular cathepsin B activity. If the VC-peptide is recognized and cleaved by cathepsin B, free MMAE can passively diffuse across the cell membrane into the cell culture medium while uncleaved cysteine adducts would be trapped intracellularly. The supernatant of L-540 cells was then transferred to CD30 negative HL-60 cells and viability was assayed. As shown in Fig. 4e, we observed the same trend of potency for the three ADCs, indicating that PEG12 substituted phosphonamidate VC-PAB-MMAE does not inhibit the cathepsin B mediated payload release in an in vitro potency setting.
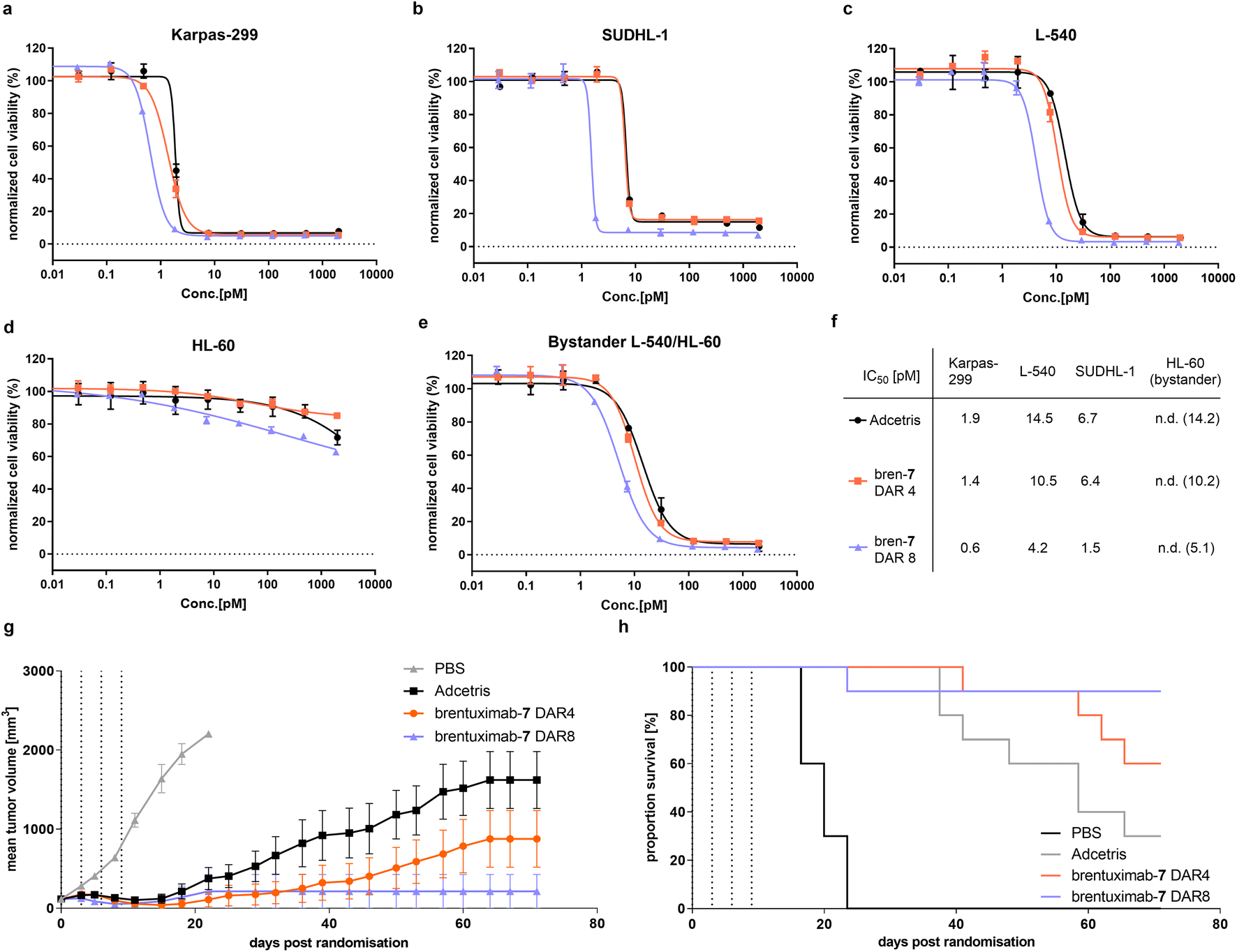 | ||
| Fig. 4 In vitro and in vivo efficacy evaluation of brentuximab-7. (a–c): In vitro cell viability of different CD30 overexpressing cell lines in response to increasing concentration of ADC brentuximab-7 DAR 4 (orange) and DAR 8 (violet) in comparison to Adcetris (black). (d): In vitro cell viability of CD30 negative control cell line HL-60 in response to increasing concentration of ADC brentuximab-7 DAR 4 (orange) and DAR 8 (violet) in comparison to Adcetris (black). (e): In vitro cell viability of CD30 negative cell line HL-60 in response to supernatants of L-540 cells pre-incubated with increasing concentration of ADCs brentuximab-7 DAR 4 (orange) and DAR 8 (violet) in comparison to Adcetris (black). (f): Table summarizing IC50 for cell viability of the tested ADCs. (g): Anti-tumor activity of ADC brentuximab-7 DAR 4 (orange) and DAR 8 (violet) in comparison to Adcetris (black) and vehicle control (PBS, grey) in Karpas 299 tumor xenograftmodel in SCID mice. Treatment of 0.5 mg kg−1 was administered 4 times on day 0, 3, 6 and 9 post randomizations. Data are mean of n = 10 animals per group. (h): Kaplan–Meier survival plot of anti-tumor activity study shown in (g). | ||
To investigate, if the improved in vitro cell killing of DAR 8 brentuximab-7 is also translated to more efficient tumor growth inhibition, we assessed the in vivo efficacy of brentuximab-7 in a Karpas 299 derived tumor xenograft model. To that end, we adapted a previously reported study design, which revealed high antitumor activity of a DAR 4 phosphonamidate linked MMAE brentuximab. In the latter study a dosage of two times 1 milligram per kilogram of body weight (mg kg−1) lead to complete tumor remission, while a reduced dosing of two times 0.5 mg kg−1 displayed decreased activity with only partial tumor growth regression.30 We assumed that the decreased antitumor activity, observed for the low dosing scheme, would substantially improve once the DAR is increased from 4 to 8, as long as the high DAR does not trigger accelerated ADC clearance. To challenge this assumption, we treated four groups of 10 mice with either brentuximab-7 (DAR 8 and DAR 4), Adcetris (DAR 4) or PBS as vehicle control. All groups received four doses of 0.5 mg kg−1 ADC every third day and tumor growth was monitored over an extended period of 74 days (Fig. 4g). Under these experimental settings we observed only temporary and incomplete tumor regression for Adcetris with 30% remission rate, which is in line with earlier reports.31 Remarkably, our DAR 4 brentuximab-7 displays improved efficacy compared to Adcetris with higher remission rate of 60%. This is also in line with our previous in vivo efficacy experiments and again can be explained with the high stability of phosphonamidate-linked conjugates that leads to prolonged payload delivery to the tumor when compared to less stable maleimide conjugates. The improved activity of brentuximab-7 is even more pronounced when the DAR is increased to 8 drugs per antibody. The DAR 8 treated group showed an excellent response to the treatment and was in tumor remission already a few days after ADC treatment. Only one mouse exhibited a tumor relapse and had to be sacrificed at day 25, while no relapse was observed for the remaining group over the whole observation period of 74 days, resulting in an excellent remission rate of 90%. This favored in vivo activity at a low dosing of 0.5 mg kg−1 of the high loaded brentuximab-7 indicates that its plasma clearance is not affected by the payload increase and that the here presented compact branched PEG12 phosphonamidate moderates the additional hydrophobicity to yield a sufficiently hydrophilic ADC. These results are in line with similar studies that investigated the effect of alternative hydrophilic linker on in vivo activity of high DAR ADCs.33–35 Notably, in these studies the highly validated but hydrophobic Val-Cit-PAB cleavage site had to be replaced with a glucuronide-cleavage site to achieve similar results. Thus, brentuximab-7 is the first example of a stable conjugated Val-Cit-PAB-MMAE based DAR 8 ADC with improved in vivo efficacy.
In the final part of our study, we focused on the pharmacokinetic behaviour of Val-Cit-PAB-MMAE based ADCs and set out to investigate if the here presented compact hydrophilic conjugation handle can compensate the additional hydrophobicity of a DAR 8 ADC to sufficiently ensure ADC circulation without enhanced clearance in comparison to the established DAR 4 Adcetris. As one can anticipate that the relative clearance of an ADC can be predicted from its overall hydrophobicity, we analyzed brentuximab-7 by hydrophobic interaction chromatography (HIC) and compared it to Adcetris (Fig. 5b). In contrast to the heterogeneously modified Adcetris, that is mainly comprised of zero-, two-, four- and six-times modified mAb, brentuximab-7 is a highly homogeneous ADC, eluting in a single narrow peak at a similar retention time as the DAR 6 Adcetris and thus significantly earlier than the Adcetris DAR 8. This reflects a reduced hydrophobicity of high loaded brentuximab-MMAE conjugates when the PEG12 phosphonamidate 7 is used for ADC preparation. To estimate if these beneficial properties of PEG12 phosphonamidates can be further optimized, we prepared another brentuximab-MMAE conjugate with a PEG24-substituted phosphonamidate 8. The HIC analysis of brentuximab-8 displays a similar narrow peak shape as observed for brentuximab-7 with a shift to even lower retention times, corresponding to a homogeneous and significantly more hydrophilic species in the range of a DAR 5 Adcetris. These results indicated that a brentuximab-8 might exhibit a similar PK profile as the marketed DAR 4 Adcetris.
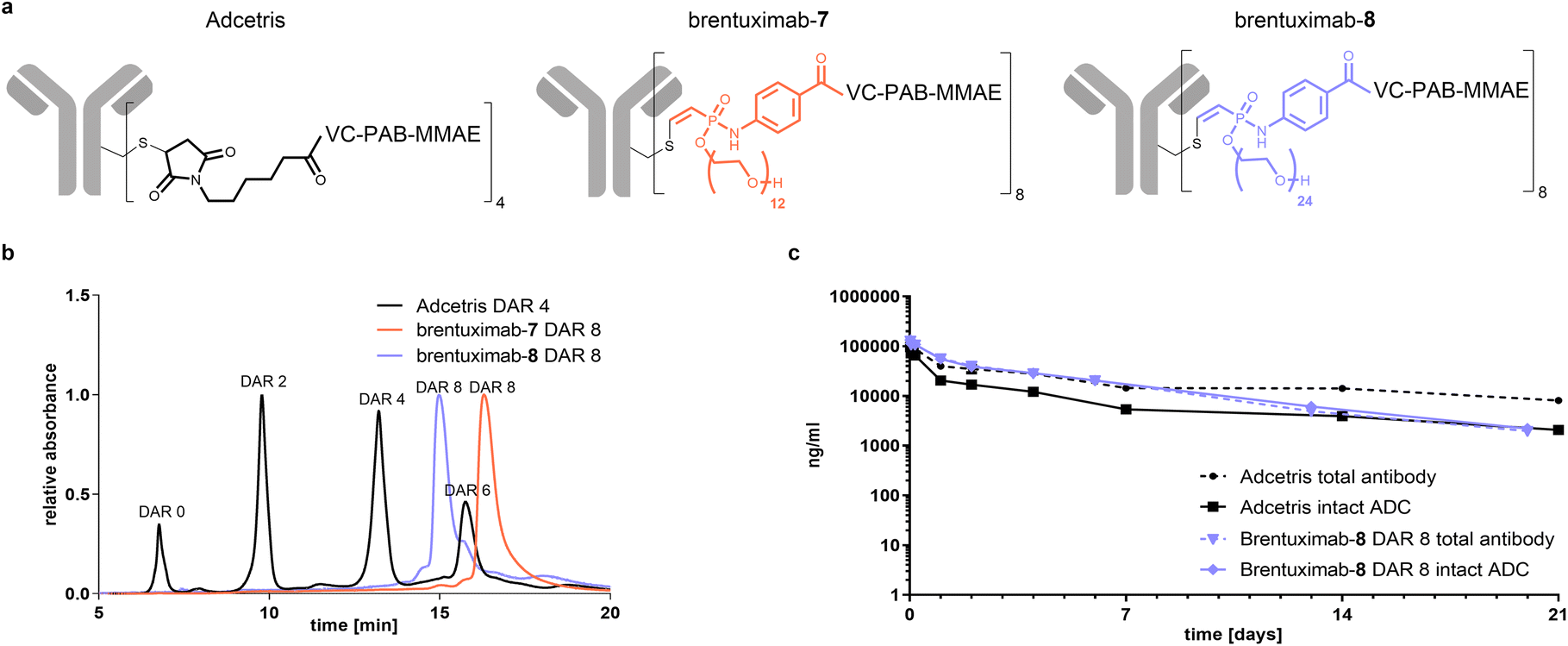 | ||
| Fig. 5 a): Chemical structures of the different ADCs and their linker architectures compared in this study. (b): UV-traches of analytical hydrophobic interaction chromatography (HIC) of homogenous DAR 8 ADCs brentuximab-7 (orange) and brentuximab-8 (violet) in comparison to the heterogeneously modified DAR4 ADC Adcetris (grey). (c): Plasma pharmacokinetics of brentuximab-8 in female Sprague-Dawley rats. Data are mean of n = 3 animals per group and time point. | ||
Finally, we wanted to investigate if the decreased hydrophobicity is sufficient to translate into an advantageous PK profile with slow plasma clearance. To that end, we focused on the most hydrophilic DAR 8 ADC discovered in this study and treated female Sprague-Dawley rats with 5 mg kg−1 of brentuximab-8 or Adcetris. The levels of total antibody and intact ADC were determined by ELISA at different time points. As displayed in Fig. 5c, both curves for total antibody show a slow decrease of blood concentration during the first 7 days of the study, pointing towards slow clearance and acceptable blood circulation. Remarkably, the high loaded brentuximab-8 shows very similar clearance compared to the total antibody of Adcetris. This slow clearance behavior indicates that the compact PEG24-phosphonamidate compensates for the additional hydrophobicity introduced by increased DAR and that our here presented conjugation handle can improve the pharmacokinetic profile of high loaded ADCs, even when challenging hydrophobic linker-payloads, such as Val-Cit-PAB-MMAE are used. Looking closer at the intact ADC fraction of both conjugates, we see a clear drop for Adcetris during the first 7 days of circulation, reflecting a loss of payload, while the curve for intact brentuximab-8 perfectly correlates with its total antibody curve. This observation again confirms that the phosphonamidate linkage is highly stable in vivo, leading to higher concentration of intact ADC for longer periods of time while maleimide linked conjugates suffer from early drug loss and lower concentration of intact ADC. From day 7 onwards until the end of our observation period, both curves for total antibody and intact ADC of brentuximab-7 keep decreasing slowly to the level of intact Adcetris. This is a remarkable PK profile for a stable conjugated Val-Cit-PAB-MMAE based DAR 8 ADC, especially when keeping in mind that at this time of circulation the remaining Adcetris has a DAR far less than 4 due to linker payload loss caused by retro Michael addition. Moreover, those results confirm the excellent stability of the phosphonamidite linkage that has been measured in ex vivo settings, previously.29
Conclusions
With this study, we demonstrate how the modular synthesis of ethynylphosphonamidates via chemoselective Staudinger-phosphonite reactions allows the straightforward preparation of hydrophilic labeling reagents that are suitable for the conjugation of challenging hydrophobic cargo molecules to non-engineered antibodies in a one-pot reduction and alkylation protocol. We identified ethynylphosphonamidates substituted with hydrophilic PEG12 as ideal conjugation handle that increases the aqueous solubility and conjugation efficiency of hydrophobic Cy5 dyes and employed the compact PEG12 substituted ethynylphosphonamidate for the streamlined synthesis of homogeneously modified ADCs with eight drugs per antibody. Here, we chose the established linker-payload VC-PAB-MMAE as challenging hydrophobic model drug and demonstrated that the branched PEG12 substituted ethynylphosphonamidate allows the generation of a DAR 8 brentuximab with only slight excess of 1.5 equivalents labelling reagent per cysteine. The resulting DAR 8 brentuximab-7 displays increased in vitro potency when compared to DAR 4 ADCs and most importantly maintains this advantage in an in vivo xenograft tumor model.Here, the DAR 8 brentuximab-7 outperformed its DAR 4 analogue and the DAR 4 Adcetris, displaying an excellent in vivo efficacy, which indicates that the introduced hydrophilicity compensates for the overall hydrophobicity of 8 VC-PAB-MMAE units and sufficiently inhibits accumulation and aggregation of the ADC during circulation. Finally, we showed that the hydrophilicity of phosphonamidate linked ADCs can be further increased when longer PEG is incorporated into the conjugation handle and showed that a PEG24 substituted VC-PAB-MMAE ethynylphosphonamidate delivers a DAR 8 brentuximab-8 that displays a remarkable pharmacokinetic profile with slow clearance from blood, which is in the range of lower modified Adcetris. In conclusion, the here presented PEG substituted compact phosphonamidate conjugation handle combines outstanding linkage stability and a branched hydrophilic architecture that enables the generation of highly efficacious DAR 8 ADCs from challenging hydrophobic payloads and native antibodies without impairing the favorable pharmacokinetic profile of monoclonal antibody conjugates. To the best to our knowledge, we here present the first example of a Val-Cit-PAB-based MMAE linker-payload that is applicable to generate DAR 8 ADCs without enhanced clearance in vivo, emphasizing the potential of branched PEG phosphonamidates to conjugate even more hydrophobic and therefore demanding payloads to antibodies in high DAR. Consequently, we highlight how adequate linker design opens synthetic pathways to well defined protein-conjugates with improved properties that may help to develop future biological therapeutics with beneficial therapeutic indices.
Data availability
The datasets supporting this article have been uploaded as part of the ESI material.†Author contributions
P. O., M.-A. K. and C. P. R. H. conceptualization; P. O., J. J., P. M., I. M., M.-A. K. and C. P. R. H. investigation; P. O., J. J., M.-A. K. formal analysis; P. O., M.-A. K. and C. P. R. H. writing-original draft; P. O., M.-A. K. and C. P. R. H. writing-review and editing; P. O., D. S., J. H., M.-A. K. and C. P. R. H. supervision and funding acquisition.Conflicts of interest
The technology described in the manuscript is part of a pending patent application by P. O., J. J., P. M., I. M., D. S., H. L., J. H., and M.-A. K, C. P. R. H. P. O., P. M., I. M., D. S., J. H., and M.-A. K are employers of Tubulis.Acknowledgements
We thank Dr Marcus Gerlach for assistance in the Brentuximab production and excellent analytical support. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) SPP1623, the Einstein Foundation Berlin (Leibniz–Humboldt Professorship), the Boehringer–Ingelheim Foundation (Plus 3 award), the Fonds der Chemischen Industrie, the Leibniz Association within the Leibniz Wettbewerb to C. P. R. H. (T18/2017); the German Federal Ministry of Education and Research (BMBF, 16GW0357K) to M.-A. K. and C. P. R. H.; the German Federal Ministry for Economic Affairs and Energy and the European Social Fund with grants to D. S. and J. H. (EXIST FT I); and the Bavarian Ministry of Economic Affairs, Regional Development and Energy with grants to D. S., J. H. and C. P. R. H. (m4-Award).Notes and references
- D. Schumacher and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2014, 22, 62–69 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed.
- J. Z. Drago, S. Modi and S. Chandarlapaty, Nat. Rev. Clin. Oncol., 2021, 18, 327–344 CrossRef PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- M. R. Lewis and J. E. Shively, Bioconjugate Chem., 1998, 9, 72–86 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- C. Wei, G. Zhang, T. Clark, F. Barletta, L. N. Tumey, B. Rago, S. Hansel and X. Han, Anal. Chem., 2016, 88, 4979–4986 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- S. Ariyasu, H. Hayashi, B. Xing and S. Chiba, Bioconjugate Chem., 2017, 28, 897–902 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- N. J. Smith, K. Rohlfing, L. A. Sawicki, P. M. Kharkar, S. J. Boyd, A. M. Kloxin and J. M. Fox, Org. Biomol. Chem., 2018, 16, 2164–2169 RSC.
- S. Shaunak, A. Godwin, J. W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS PubMed.
- H. Khalili, A. Godwin, J. W. Choi, R. Lever and S. Brocchini, Bioconjugate Chem., 2012, 23, 2262–2277 CrossRef CAS PubMed.
- R. Huang, Z. Li, Y. Sheng, J. Yu, Y. Wu, Y. Zhan, H. Chen and B. Jiang, Org. Lett., 2018, 20, 6526–6529 CrossRef CAS PubMed.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana, T. P. J. Knowles, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2019, 58, 6640–6644 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- A. J. Counsell, S. J. Walsh, N. S. Robertson, H. F. Sore and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4739–4743 RSC.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 2–10 Search PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- M.-A. Kasper, M. Glanz, A. Oder, P. Schmieder, J. P. von Kries and C. P. R. Hackenberger, Chem. Sci., 2019, 10, 6322–6329 RSC.
- M. A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef CAS PubMed.
- M. A. Kasper, A. Stengl, P. Ochtrop, M. Gerlach, T. Stoschek, D. Schumacher, J. Helma, M. Penkert, E. Krause, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11631–11636 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- P. Malik, C. Phipps, A. Edginton and J. Blay, Pharm. Res., 2017, 34, 2579–2595 CrossRef CAS PubMed.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-Laford, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- W. Viricel, G. Fournet, S. Beaumel, E. Perrial, S. Papot, C. Dumontet and B. Joseph, Chem. Sci., 2019, 10, 4048–4053 RSC.
- P. J. Burke, J. Z. Hamilton, S. C. Jeffrey, J. H. Hunter, S. O. Doronina, N. M. Okeley, J. B. Miyamoto, M. E. Anderson, I. J. Stone, M. L. Ulrich, J. K. Simmons, E. E. McKinney, P. D. Senter and R. P. Lyon, Mol. Cancer Ther., 2017, 16, 116–123 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05678j |
| This journal is © The Royal Society of Chemistry 2023 |