
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple and scalable electrosynthesis of 1H-1-hydroxy-quinazolin-4-ones†
Olesja
Koleda‡
 ac,
Tobias
Prenzel‡
a,
Johannes
Winter‡
a,
Tomoki
Hirohata
ad,
María
de Jesús Gálvez-Vázquez
a,
Dieter
Schollmeyer
a,
Shinsuke
Inagi
d,
Edgars
Suna
c and
Siegfried R.
Waldvogel
*ab
ac,
Tobias
Prenzel‡
a,
Johannes
Winter‡
a,
Tomoki
Hirohata
ad,
María
de Jesús Gálvez-Vázquez
a,
Dieter
Schollmeyer
a,
Shinsuke
Inagi
d,
Edgars
Suna
c and
Siegfried R.
Waldvogel
*ab
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10–14, 55128 Mainz, Germany. E-mail: waldvogel@uni-mainz.de; Web: https://www.aksw.uni-mainz.de/
bInstitute of Biological and Chemical Systems –Functional Molecular Systems (IBCS-FMS) Hermann-von-Helmholtz-Platz 1 76344 Eggenstein-Leopoldshafen, Germany
cLatvian Institute of Organic Synthesis, Aizkraukles 21, LV-1006 Riga, Latvia
dDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8502, Japan
First published on 16th February 2023
Abstract
Cathodic synthesis provides sustainable access to 1-hydroxy- and 1-oxy-quinazolin-4-ones from easily accessible nitro starting materials. Mild reaction conditions, inexpensive and reusable carbon-based electrode materials, an undivided electrochemical setup, and constant current conditions characterise this method. Sulphuric acid is used as a simple supporting electrolyte as well as a catalyst for cyclisation. The broad applicability of this protocol is demonstrated in 27 differently substituted derivatives in high yields of up to 92%. Moreover, mechanistic studies based on cyclic voltammetry measurements highlight a selective reduction of the nitro substrate to hydroxylamine as a key step. The relevance for preparative applications is demonstrated by a 100-fold scale-up for gram-scale electrolysis.
Introduction
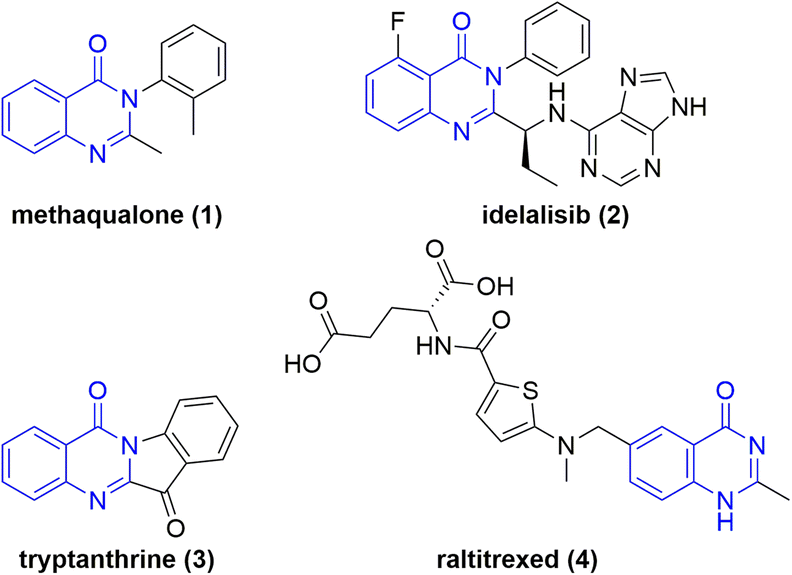
Nitrogen containing heterocycles are ubiquitous in pharmaceuticals1 and biomolecules as core motifs.2 One particular example is quinazolin-4-ones. Derivatives are present in a variety of drugs and biomolecules with anticancer, diuretic, anti-inflammatory, anticonvulsant, and antihypertensive properties.3Methaqualone (1) (Chart 1), known by its brand name Quaalude, is a hypnotic sedative that increases GABAA receptor activity.4 The drug was withdrawn from the U.S. market in 1985, primarily because of its psychologically addictive potential, widespread abuse, and illicit recreational use.5 The medication idelalisib (2) (Chart 1) is used to treat certain types of blood cancer and acts as a phosphoinositide 3-kinase inhibitor.6 This drug alone amounted to a $72 million annual revenue for Gilead Sciences, Inc.7 The naturally occurring plant alkaloid tryptanthrine (3) exhibits a broad spectrum of biological and pharmaceutical activity including antimicrobial, antiviral, anticancer, and antiparasitic properties.8 In addition, the AstraZeneca anti-metabolite and cytotoxic drug raltitrexed (4) is used in chemotherapy treatments. The folic acid analogue acts as a selective inhibitor of thymidylate synthase.9 Notably, N-oxides and N-hydroxy derivatives of these pharmaceuticals were found to be metabolites of these drugs.10,11 However, the therapeutic effects of endocyclic N-hydroxy and N-oxy compounds have not been researched to the same degree. Considering their metabolic stability and the unique features of the N–O bond, this class of novel compounds are the subject of current pharmaceutical research.12
 | ||
| Chart 1 Important biologically active substances with quinazolin-4-one motif. | ||
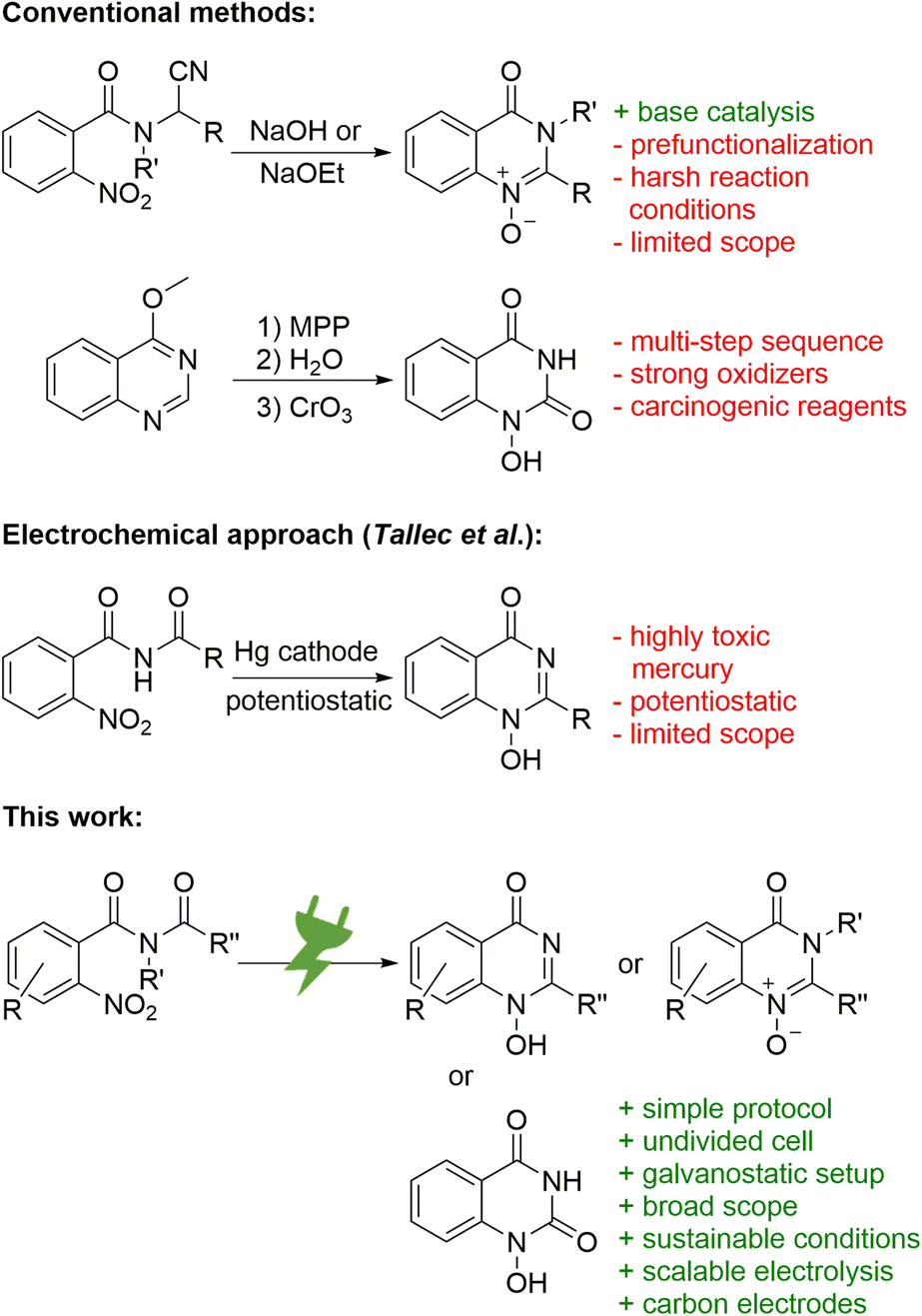
The synthesis of N-oxides and N-hydroxy derivatives of quinazolin-4-ones is scarce in the literature and the few published examples present various challenges. The synthesis of 1-oxy-2-alkylquinazolin-4-ones was described by Tennant starting from N-(1-cyano-alkyl)-2-nitro benzamides (Scheme 1, top).13 Its base-catalysed cyclisation approach does not require additional reagents, but does use harsh reaction conditions.13 The synthesis of 1-hydroxyquinazoline-2,4-diones by Yamanaka comprises a multi-step sequence from 4-methoxyquinazoles and involves two oxidizers: hazardous monoperoxyphthalate for the generation of the 1-oxide and highly toxic chromium(VI) oxide for the oxidation of the C-2 (Scheme 1, top).14 Reductive cyclisation of nitro precursors is also known, typically via hydrogenation with expensive palladium catalysts.15 In addition, Tallec described an electrochemical synthesis within polarographic studies (Scheme 1, middle). However, highly toxic mercury electrodes were required in a sophisticated potentiostatic reaction setup that was run on a small scale with a limited scope.16
 | ||
| Scheme 1 Synthetic approaches to 1H-1-hydroxy-quinazolin-4-ones. MPP = monoperoxyphthalate. | ||
Herein, a versatile, scalable, and high-yielding electrochemical reductive cyclisation of widely available, easy to prepare, and inexpensive nitro arenes17 into 1-hydroxy-quinazolin-4-ones is presented. The method uses a simple constant current setup and applies conditions that consider sustainable and environmental aspects (Scheme 1, bottom). In particular, this methodology can easily pay off for high value-added products.18 The field of electrosynthesis is experiencing a renaissance as an alternative to conventional synthesis protocols19–21 and is emerging as a key discipline for future synthetic applications.22 The use of electric current as a reagent enables inherently safe processes by precise control of the reaction. Practically, turning off the electricity immediately stops the conversion and, unlike with traditional reagents, thermal runaway reactions are not possible. The absence of toxic and hazardous reagents and the use of sustainable electricity makes these methods almost waste- and pollutant-free, especially when solvents and supporting electrolytes are reused.23,24 However, several parameters and counter reactions seem to play a crucial role for success.25,26 Carbon-based electrode materials such as graphite, glassy carbon (GC), and boron-doped diamond (BDD) are sustainable and widely available.27 In particular, these are superior in the synthesis of pharmaceuticals or APIs where trace metal impurities must be avoided.22,28
Results and discussion
Optimisation of the electrolysis conditions
Benzamide 5a was chosen as a test substrate for cathodic reduction of a nitro group, and it was easily synthesised in a single step from commercially available 2-nitrobenzamide by treatment with acetic anhydride in a microwave-assisted acylation reaction or in a pressured vessel approach, both in high yields (see ESI† for detailed description).29 Based on previous work by the Waldvogel lab, a foundation for the electrochemical conditions for nitro reduction were initially tested (Table 1, entry 1).30–32|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield 6ab/% |
a Concentration of sulphuric acid in the electrolyte, obtained by using methanol and 1 M aqueous sulphuric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)).
b Yield of 6a was determined by 1H NMR spectroscopy using 2,2-dimethylmalonic acid as internal standard.
c Isolated yield.
d 0.5 M AcOH/AcONa was prepared with 90 mmol acetic acid and 10 mmol sodium acetate in 100 mL of distilled water and 100 mL methanol. BDD = boron-doped diamond; GC = glassy carbon. :1 (v/v)).
b Yield of 6a was determined by 1H NMR spectroscopy using 2,2-dimethylmalonic acid as internal standard.
c Isolated yield.
d 0.5 M AcOH/AcONa was prepared with 90 mmol acetic acid and 10 mmol sodium acetate in 100 mL of distilled water and 100 mL methanol. BDD = boron-doped diamond; GC = glassy carbon.
|
||
| 1 | None | 91% (91%)c |
| 2 | 2.7 mA cm−2 | 78% |
| 3 | 5.7 mA cm−2 | 78% |
| 4 | EtOH instead of MeOH | 84% |
| 5 | MeCN instead of MeOH | 71% |
| 6 | 0.5 M acetate bufferd | 66% |
| 7 | Pb cathode | 27% |
| 8 | CuSn7Pb15 cathode | 0% |
| 9 | Pt cathode | 0% |
| 10 | Graphite cathode | 84% |
| 11 | GC cathode | 90% |
| 12 | 0.06 M 5a | 85% |
| 13 | 0.10 M 5a | 73% |
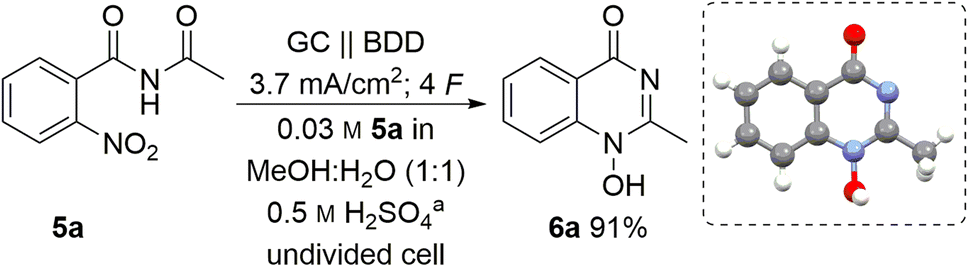
The starting conditions used a water–methanol mixture (1:1 (v/v)) as a green solvent capable of dissolving the nitro compound 5a. A moderate concentration of sulphuric acid (0.5 M) was used as the supporting electrolyte based on previous investigations into the concentration effect of the acidic component on the electrochemical reduction.16,33 Moreover, it was envisioned that sulphuric acid may play a dual role in the reaction since acidic media should be beneficial for cyclisation after a nitro group reduction. Taking this into account, we examined constant current electrolysis (3.7 mA cm−2 current density) of benzamide 5a in an undivided cell in water–methanol media, using a glassy carbon anode and a boron-doped diamond (BDD) cathode. BDD as a carbon-based material offers unique reactivity towards electrochemical conversion of a multitude of substrates and can be manufactured in a sustainable manner by utilizing methane as carbon source.34
To our delight, the desired 1-hydroxyquinazolinone 6a was isolated in 91% yield. Furthermore, the theoretical amount of charge required for this process (4 F) was applied and the high yield obtained shows that this process has a high current efficiency (Table 1, entry 1). The molecular structure of heterocycle 6a was confirmed by X-ray analysis of a suitable single crystal.
Deviation from the starting electrolysis conditions obtained by electrosynthetic screening resulted in lower yields.20,21,24,35 Both lower or higher current densities led to a decreased yield of 1-hydroxyquinazolinone 6a to 78% (Table 1, entries 2 and 3). Replacement of methanol with other solvents such as ethanol and acetonitrile (Table 1, entries 4 and 5) afforded the desired heterocycle 6a in slightly lower yields of 84% and 71%, respectively. Acetate buffer (Table 1, entry 6) was used as a weaker and biogenic alternative to sulphuric acid; however, the yield of 6a decreased to 66%. Substitution of BDD as cathode material for lead significantly decreased the yield of 6a to 27% (Table 1, entry 7). Furthermore, cathodic corrosion was observed resulting in the precipitation of lead salts.
More stable alternatives to lead cathodes such as leaded bronzes36 (Table 1, entry 8) failed entirely in the formation of 6a. Platinum was equally unsuccessful as cathode material (Table 1, entry 9), completely avoiding the desired reaction likely due its low overpotential for the hydrogen evolution side-reaction.37
Besides BDD, other carbon-based cathode materials such as graphite and glassy carbon provided product 6a in comparable yields of 84% and 90% (Table 1, entries 10 and 11). Nevertheless, we decided to proceed with BDD due to its sustainability and chemical durability.38 Electrolysis at higher benzamide 5a concentrations resulted in lowered yields by up to nearly 20% (Table 1, entries 12 and 13). It is likely that higher concentrations of starting material result in the formation of high molecular weight side products which were observed as a brown plaque after the electrolysis was finished.
Scope of the reductive cyclisation
The broad applicability of the optimised reaction conditions was demonstrated on a versatile scope of substrates (Chart 2). First, we explored the effect of substitution pattern at the C-2 position of quinazolin-4-ones 6b–i (Chart 2, top). The influence of primary, secondary, and tertiary alkyl substitutions was investigated in the synthesis of heterocycles 6a–c, resulting in good yields of 78–91%. Here, the tert-butyl derivative 6c with its sterically demanding substituent gave the lowest yield. Furthermore, 1H-2-heptyl-1-hydroxy-quinazolin-4-one (6d) with a hydrophobic chain was obtained in a moderate yield of 52%.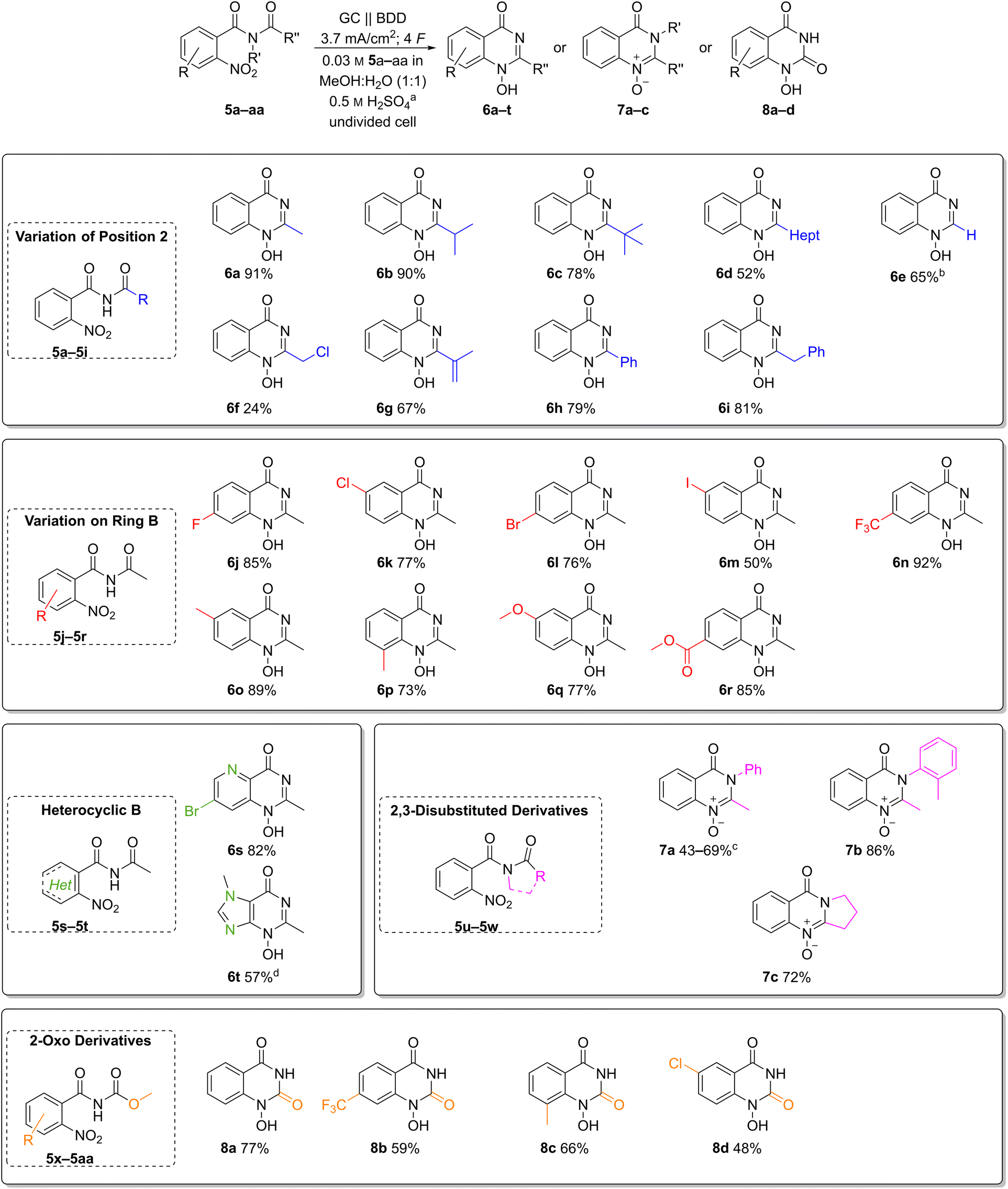 | ||
| Chart 2 Scope of electrochemical reductive cyclisation and isolated yields. a Concentration of sulphuric acid in the electrolyte, obtained by using 12.5 mL methanol and 12.5 mL 1 M aqueous sulphuric acid (1 : 1 (v:v)), undivided 25 mL beaker-type glass cell; b isolated yield as formate adduct; c variation in yield with repeated electrolysis; d isolated yield as acetate adduct. | ||
Both the 2-unsubstituted quinazolin-4-one 6e and the 2-phenyl analogue 6h were obtained in moderate to good yields (65% and 79%, respectively). Alkene as well as a benzylic moieties, usually prone to anodic oxidation, were well tolerated in the electrochemical reduction, and the desired heterocycles 6g and 6i were obtained in 67% and 81% yield. Interestingly, even the 2-chloromethyl substituted product 6f was isolated in 24% yield despite its inherent instability.
Next, various functional groups in the aromatic subunit of 1-hydroxyquinazolin-4-ones 6j–r were tested (Chart 2, middle). In all cases, the products were obtained in good to excellent yields regardless of the substituent's electronic nature. Benzamides with an electron-donating methoxy group and an electron-withdrawing ester moiety afforded the corresponding heterocycles 6q and 6r in comparable yields (77% and 85%, respectively). Likewise, the trifluoromethyl derivative 6n and methyl quinazolin-4-ones 6o and 6p were obtained in 73–92% yield.
The 6-methyl derivative 6o is an N-hydroxy analogue of a precursor to raltitrexed (4), thus its successful formation adds industrial relevance to this transformation.39 Halides are redox-active groups in electrochemical reactions. To our delight, fluoro-, chloro- and bromo-substituted quinazolin-4-ones 6j–l were obtained in 76–85% yield. Moreover, the developed electrochemical method also afforded the iodo-substituted product 6m, albeit in moderate yield (50%) likely due to its susceptibility to oxidation.
It is noteworthy that heteroaromatic amides such as those derived from pyridines and imidazoles are applicable with the developed conditions (Chart 2, middle left). The pyrido pyrimidinone 6s was obtained in 82% yield and the bromo substituent offers availability for a variety of post-functionalization reactions. Even the N-hydroxy purine 6t was obtained in a good yield of 57%.
Furthermore, N-acetyl-N-aryl benzamides were converted into the corresponding N-oxy-quinazolin-4-ones 7a and 7b in moderate yields, likely due to their tendency to rearrangement reactions (Chart 2, middle right).40 Interestingly, 7b is a reported metabolite of methaqualone (1).10 Furthermore, tertiary amides are also suitable as substrates as exemplified by the synthesis of pyrrolidone-based N-oxy-quinazolin-4-one 7c (72%).
Finally, the presented methodology has also been applied to the synthesis of 1-hydroxy-quinazoline-2,4-diones 8a–d by electrochemical reduction of methyl-(2-nitrobenzoyl)carbamates, which gave up to 77% yield (Chart 2, bottom). Here, the unsubstituted derivative 8a had the best yield of 77%. The electron-withdrawing trifluoromethyl- and chloro-substituted products 8b and 8d were isolated in 59% and 48% yield. The methyl-substituted derivative 8c was obtained in moderate yield of 66%.
Scale-up of electrolysis
To demonstrate the synthetic utility and scalability of the developed method, the synthesis of 1H-1-hydroxy-2-methyl-quinazolin-4-one (6a) was successfully scaled up to 15 mmol in a single electrolysis batch (Table 2). The 5-fold scale-up was performed with BDD and GC cathodes to demonstrate the robustness and applicability of the described method. Encouragingly, 20-fold scale-up (3.00 mmol) in a 100 mL electrolysis cell afforded the desired product 6a without loss in yield or faradaic efficiency. Gram-scale electrolysis was also performed at 7.5 mmol and 15.0 mmol loading in a 250 mL electrolysis cell, affording 1.13 g and 2.19 g of the desired 1-hydroxy-quinazolin-2-one 6a, respectively. The trend of decreased yield at higher concentrations was also observed in the optimisation results (Table 1, entries 12 & 13).| Electrolysis cell | Scale/mmol | Current (electrolysis time) | Yield 6a |
|---|---|---|---|
| a Constant current conditions; surface areas of the electrodes: 5 mL Teflon™ cell (1.5 cm 2), 25 mL and 100 mL glass cell (6.0 cm2), 250 mL glass cell (29.7 cm2). b GC cathode. c 0.06 M 5a. | |||

|
0.15 | 5.6 mA (2.9 h) | 24 mg (91%) |

|
0.75 | 22.2 mA (3.6 h) | 120 mg (91%) |
| 0.75b | 118 mg (89%) | ||

|
3.00 | 22.2 mA (14.5 h) | 481 mg (91%) |

|
7.50 | 109.9 mA (7.3 h) | 1.13 g (86%) |
| 15.0c | 109.9 mA (14.6 h) | 2.19 g (83%) | |
Mechanistic studies
A plausible mechanism for the electrochemical cyclisation sequence is proposed based on earlier nitro reduction studies and cyclic voltammetry (CV) measurements of benzamide 5a (Scheme 2; for detailed information see ESI†).16,31–33 Accordingly, the reactions starts with the reduction of the nitro group on the BDD cathode which is evidenced by a single broad, irreversible wave (−0.94 V vs. FcH/FcH+) corresponding to the 4e−/4H+ reduction to the hydroxylamine. It has been previously suggested that the reaction proceeds with two 2e− steps via intermediates Int-I and Int-II. However, because only one broad wave was observed rather than two distinct or overlapping waves, this suggests one reduction event occurs and that 5a is reduced directly to Int-II The irreversible reduction to Int-II indicates a fast cyclisation step as no corresponding oxidative wave was observed. Further reduction of the N-hydroxy moiety was not observed for non-cyclised Int-II or the product 6a. The impact of the counter reaction on the electrochemical conversion must also be considered; the oxidation of water at the glassy carbon anode is possibly the main counter reaction.25 The corrosion of the glassy carbon anode was not observed after electrolysis. There was no indication of the oxidation of methanol, which was only observed under harsh basic conditions in previous studies.41 Overall, the high performance of the reported method was enabled by the electrochemical stability of the product, the selective reduction to the hydroxylamine, and the fast cyclisation process.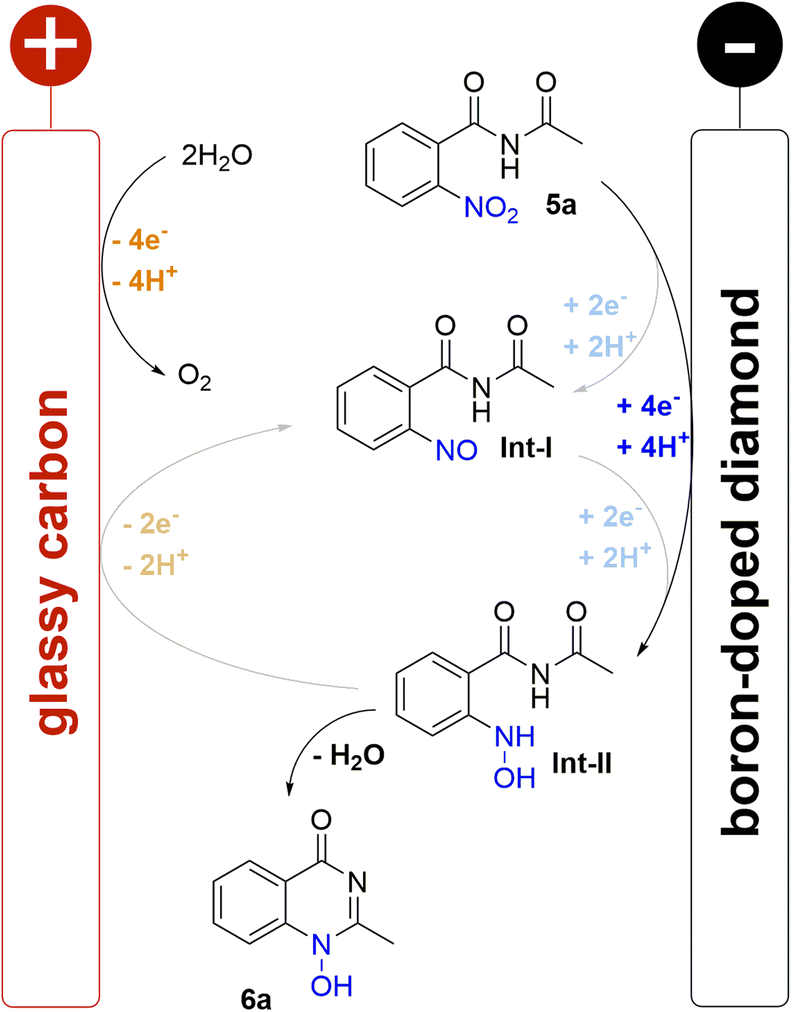 | ||
| Scheme 2 Proposed mechanism for the reductive cyclisation. | ||
Conclusions
In summary, the established method provides simple, direct, and sustainable electrochemical access to N-hydroxy- and N-oxy-quinazolin-4-ones by a cathodic reduction sequence. The electrolytic conditions allow for a reproducible transformation in commercially available experimental setups. The simplest undivided cell was operated under constant current with widely available and sustainable carbon-based electrodes and a water–methanol mixture as an environmentally benign solvent. The sulphuric acid additive served a dual role as supporting electrolyte and acidic catalyst. The broad applicability of this method was demonstrated by 27 examples with up to 92% isolated yield. The electrolysis tolerates various functional groups, including electron-withdrawing and -donating, sterically demanding, and redox labile moieties such as bromides and iodides. The N-hydroxy analogue precursor of the cytotoxic drug raltitrexed was obtained in 89% yield. Selective nitro reduction to the hydroxylamine was confirmed by CV measurements to be a key step in the mechanism. The scalability of this electrochemical protocol was demonstrated by multigram-scale electrolysis.Data availability
The ESI is available and contains experimental and analytical data.Author contributions
O. K., T. P. and J. W. contributed equally. T. P. and S. W. conceived the idea of the research. O. K., T. P., J. W. and T. H. designed and carried out the batch electrolysis experiments and analysed the data. M. G. and T. P. designed and analysed the cyclic voltammetry measurements. D. S. performed the X-ray analysis and structural elucidation of the synthesised test substrate. T. P., O. K., J. W., and S. W. wrote the manuscript. S. W. supervised the project and E. S. and S. I. reviewed and edited the manuscript. All authors discussed the results and agreed to the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge funding by the DFG (Wa1276/17-2 and Wa1276/26-1). Support by the profile area SusInnoScience (Forschungsinitiative Rheinland-Pfalz) is highly appreciated. This project was funded by Japan Science and Technology Agency’s SPRING program under the grant agreement no. JPMJSP2106 (T. H.). We would like to thank Dr R. Hamill for her assistance with editing the manuscript.References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed
.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed
- A. Masri, A. Anwar, N. A. Khan, M. S. Shahbaz, K. M. Khan, S. Shahabuddin and R. Siddiqui, Antibiotics, 2019, 8, 179 CrossRef CAS PubMed
- P.-F. Wang, A. A. Jensen and L. Bunch, ACS Chem. Neurosci., 2020, 11, 4362 CrossRef CAS PubMed
- K. Romanek, H. Fels, T. Dame, G. Skopp, F. Musshoff, H. Eiglmeier and F. Eyer, Subst. Abuse, 2021, 42, 503 CrossRef PubMed
- J. R. Somoza, D. Koditek, A. G. Villaseñor, N. Novikov, M. H. Wong, A. Liclican, W. Xing, L. Lagpacan, R. Wang, B. E. Schultz, G. A. Papalia, D. Samuel, L. Lad and M. E. McGrath, J. Biol. Chem., 2015, 290, 8439 CrossRef CAS PubMed
- San. Franc. Bus. Times, https://www.bizjournals.com/sanfrancisco/news/2022/01/21/blood-cancer-gilead-sciences-gild-zydelig-fl-sll.html, accessed January 2023.
- A. M. Tucker and P. Grundt, Arkivoc, 2012, 546 Search PubMed
- N. S. Gunasekara and D. Faulds, Drugs, 1998, 55, 423 CrossRef CAS PubMed
- C. N. Reynolds, K. Wilson and D. Burnett, Xenobiotica, 1976, 6, 113 CrossRef CAS PubMed
- M. H. Bickel, Xenobiotica, 1971, 1, 313 CrossRef CAS PubMed
- R. Rani and C. Granchi, Eur. J. Med. Chem., 2015, 97, 505 CrossRef CAS PubMed
- T. W. M. Spence and G. Tennant, J. Chem. Soc., Perkin Trans. 1, 1972, 97 RSC
- H. Yamanaka, Chem. Pharm. Bull., 1959, 7, 152 CrossRef CAS
- R. Fielden, O. Meth-Cohn and H. Suschitzky, J. Chem. Soc., Perkin Trans. 1, 1973, 702 RSC
- A. Chibani, R. Hazard and A. Tallec, Bull. Soc. Chim. Fr., 1991, 6, 814 Search PubMed
- A. Z. Halimehjani, I. N. N. Namboothiri and S. E. Hooshmand, RSC Adv., 2014, 4, 31261 RSC
-
(a) J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, 42 CrossRef
-
(a) D. Cantillo, Chem. Commun., 2022, 58, 619 RSC
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72 CrossRef CAS PubMed
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275 CrossRef
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386 RSC
-
(a) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed
- M. Klein and S. R. Waldvogel, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS PubMed
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750 CrossRef CAS PubMed
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866 CrossRef CAS PubMed
- T. Wirtanen, T. Prenzel, J.-P. Tessonnier and S. R. Waldvogel, Chem. Rev., 2021, 121, 10241 CrossRef CAS PubMed
-
(a) J. Lee, M. Hong, Y. Jung, E. J. Cho and H. Rhee, Tetrahedron, 2012, 68, 2045 CrossRef CAS
- E. Rodrigo, H. Baunis, E. Suna and S. R. Waldvogel, Chem. Commun., 2019, 55, 12255 RSC
- E. Rodrigo and S. R. Waldvogel, Green Chem., 2018, 20, 2013 RSC
- J. Winter, T. Prenzel, T. Wirtanen, D. Schollmeyer and S. R. Waldvogel, Chem.–Eur. J., 2022, e202203319 Search PubMed
- R. Hazard, M. Jubault, C. Mouats and A. Tallec, Electrochim. Acta, 1986, 31, 489 CrossRef CAS
- S. Lips and S. R. Waldvogel, ChemElectroChem, 2019, 6, 1649 CrossRef CAS
-
(a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415 CrossRef CAS PubMed
- C. Gütz, V. Grimaudo, M. Holtkamp, M. Hartmer, J. Werra, L. Frensemeier, A. Kehl, U. Karst, P. Broekmann and S. R. Waldvogel, ChemElectroChem, 2018, 5, 247 CrossRef
- A. Hickling and F. W. Salt, Trans. Faraday Soc., 1940, 36, 1226 RSC
-
(a) Y. Einaga, Acc. Chem. Res., 2022, 55, 3605 CrossRef CAS PubMed
-
(a) S.-L. Cao, Y.-P. Feng, Y.-Y. Jiang, S.-Y. Liu, G.-Y. Ding and R.-T. Li, Bioorg. Med. Chem. Lett., 2005, 15, 1915 CrossRef CAS PubMed
- F. P. Theil, H. Poehlmann, S. Pfeifer and P. Franke, Pharmazie, 1985, 40, 328 CAS
- T. Wirtanen, E. Rodrigo and S. R. Waldvogel, Chem.–Eur. J., 2020, 26, 5592 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2234739. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00266g |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |