
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide valence-induced breaks in plasmonic coupling†
Yu-Ci
Chang‡
 a,
Zhicheng
Jin‡
b,
Ke
Li
c,
Jiajing
Zhou
b,
Wonjun
Yim
a,
Justin
Yeung
d,
Yong
Cheng
b,
Maurice
Retout
b,
Matthew N.
Creyer
b,
Pavla
Fajtová
e,
Tengyu
He
a,
Xi
Chen
f,
Anthony J.
O’Donoghue
e and
Jesse V.
Jokerst
*abg
a,
Zhicheng
Jin‡
b,
Ke
Li
c,
Jiajing
Zhou
b,
Wonjun
Yim
a,
Justin
Yeung
d,
Yong
Cheng
b,
Maurice
Retout
b,
Matthew N.
Creyer
b,
Pavla
Fajtová
e,
Tengyu
He
a,
Xi
Chen
f,
Anthony J.
O’Donoghue
e and
Jesse V.
Jokerst
*abg
aMaterials Science and Engineering Program, University of California San Diego, La Jolla, California 92093, USA. E-mail: jjokerst@ucsd.edu
bDepartment of NanoEngineering, University of California San Diego, La Jolla, California 92093, USA
cInstitute of Materials Research and Engineering, Agency for Science, Technology and Research, Singapore 138634, Singapore
dDepartment of Bioengineering, University of California San Diego, La Jolla, California 92093, USA
eSkaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, USA
fSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore
gDepartment of Radiology, University of California San Diego, La Jolla, California 92093, USA
First published on 7th February 2023
Abstract
Electrostatic interactions are a key driving force that mediates colloidal assembly. The Schulze-Hardy rule states that nanoparticles have a higher tendency to coagulate in the presence of counterions with high charge valence. However, it is unclear how the Schulze–Hardy rule works when the simple electrolytes are replaced with more sophisticated charge carriers. Here, we designed cationic peptides of varying valencies and demonstrate that their charge screening behaviors on anionic gold nanoparticles (AuNPs) follow the six-power relationship in the Schulze–Hardy rule. This finding further inspires a simple yet effective strategy for measuring SARS-CoV-2 main protease (Mpro) via naked eyes. This work provides a unique avenue for fundamental NP disassembly based on the Schulze–Hardy rule and can help design versatile substrates for colorimetric sensing of other proteases.
Introduction
Control of nanoparticle (NP) assembly is a popular bottom-up strategy for fabricating tailored functional ensembles in many fields including optics, catalysis, and biomedicine.1–3 Such colloidal assemblies have been widely modulated via chemical linking (e.g., covalent and hydrogen bonds) and physical factors (e.g., ionic strength, solvent polarity, and ligand hydrophilicity).4,5 Electrostatic interactions play a particularly vital role in mediating the colloidal stability of NPs.6,7 For example, in the presence of high-concentration counterions, charged NPs easily aggregate due to the decreased Debye length and double-layer potential. Moreover, the critical coagulation concentration (CCC) highly depends on the charge valency (Z) of counterions, i.e., CCC ∝ Z−6, which has been summarized in the Schulze–Hardy rule.8,9In recent decades, the Schulze–Hardy rule has been validated using simple ionic additives (e.g., Na+, Ca2+, and Mg2+) to promote supramolecular processes such as nanocrystal formation, nanotube coagulation, and macromolecular gelation.7,10–18 However, minor efforts are devoted to studying electrostatic interactions mediated by ionic additives beyond simple metallic ions due to the limited choice of modular counterions. Synthetic peptides have a diverse set of chemical functions, relatively easy chemical synthesis and modification, and remarkable selectivity toward enzymes.19,20 Several peptide coatings have been exploited to provide NPs with enhanced colloidal stability while preserving their photo-physical features.21,22 The charge valence of peptides can significantly alter the colloidal stability.23–25 Thus, this work asked whether these peptides follow a similar trend to electrolytes during charge screening of colloids. We rationally designed oligopeptides of various charge valencies and then studied their role in plasmonic coupling of electrostatic-stabilized gold nanoparticles (AuNPs).
Here, we demonstrate that the aggregation and dispersion behavior of AuNPs driven by charged peptides is governed by the Schulze–Hardy rule (Fig. 1a). Modular cationic oligopeptides five amino acids long were designed with increasing arginine residues, and thus increasing the charge valency. The AuNP coagulation concentration increased more than 1000-fold as the ratio of positive charges in the oligopeptide slightly increased from 0.2 to 1.0. The fit in the plot of the CCC against charge valence indicated a six-power correlation similar to the Schulze–Hardy rule. We further translated this finding for plasmonic sensing application and validated the visual detection for main protease (Mpro) implicated in SARS-CoV-2.26 This sensing platform via the Schulze–Hardy rule provides an emerging approach for mediating NP assembly/disassembly and can be repurposed for probing other bioanalytical targets.
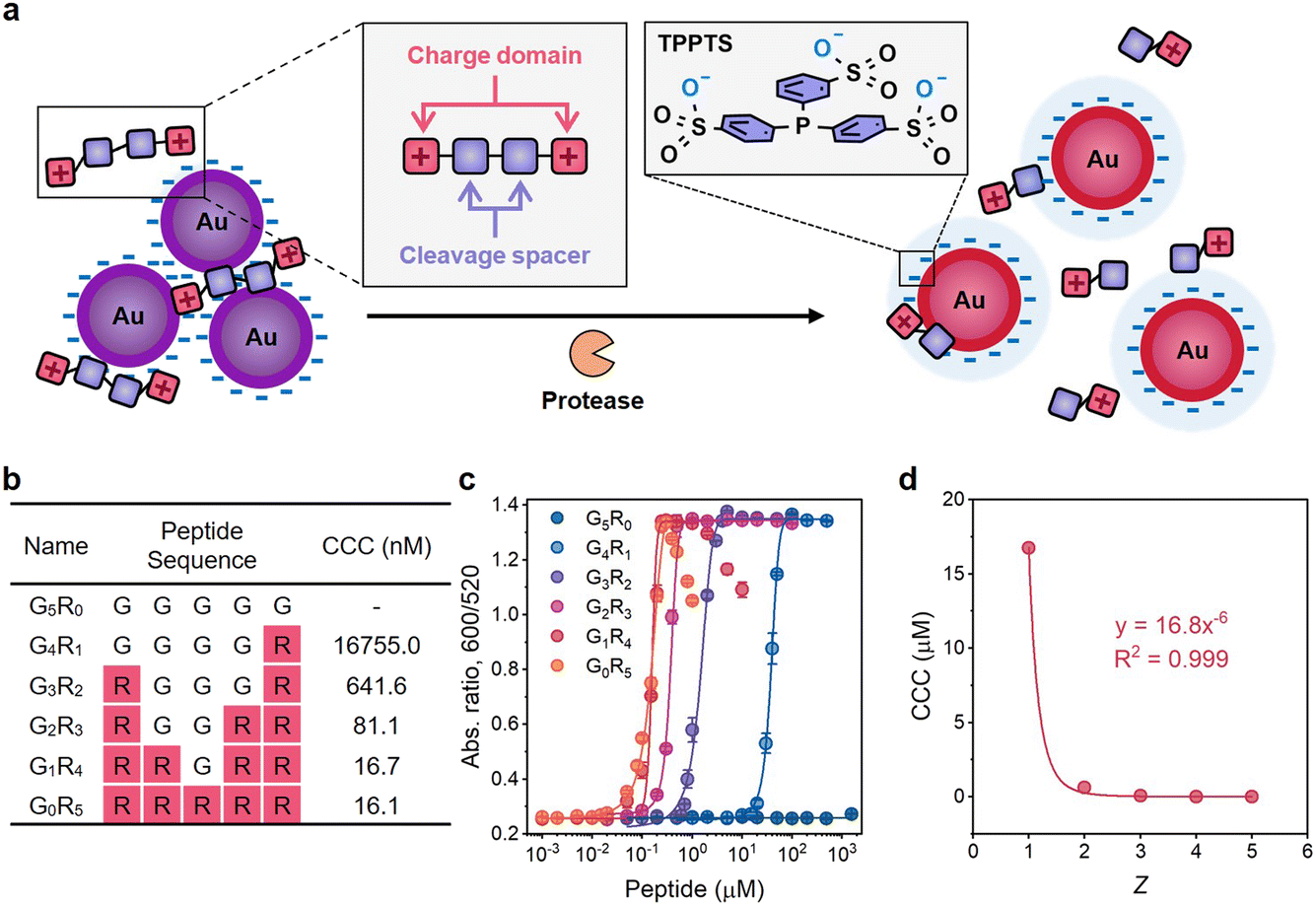 | ||
| Fig. 1 Mechanism of the plasmonic sensing system and examination of the Schulze–Hardy rule on charged peptides. (a) Schematic illustration of plasmonic sensing based on the Schulze–Hardy rule. The orange cartoon represents Mpro; tandem squares represent positively charged peptides with Mpro recognition sites. (b) Sequence design and molecular weights of G5−xRx (x = 0–5) peptides. CCC was determined based on the results of titration experiments. (c) Titration curves of the G5−xRx peptide on TPPTS-AuNPs. Error bar = standard deviation of the two samples. (d) The fitting curve of the CCC of G5−xRx peptides against Z. | ||
Results and discussion
Interactions between oppositely charged nanoparticles and ions/peptides
We used triphenylphosphine-3,3,3-trisulfonate modified AuNPs (TPPTS-AuNPs) to justify the six-power relationship of CCC and Z in the classical colloid-simple ion system. The sulfonate-rich TPPTS ligand (Fig. S1c†) gives the AuNPs strong negative charges, i.e., the zeta potential of TPPTS-AuNPs (ζ) was −31.0 ± 2.1 mV. We titrated TPPTS-AuNPs with simple metallic cations of different charge valences in water, including Na+, K+, Ca2+, Mg2+, Cu2+, Fe2+, Fe3+, Gd3+, and Er3+. The CCC of AuNP aggregation induced by different cations is summarized in Table 1. The CCC calculation uses the statistical method previously reported in the literature (eqn (1) and Fig. S2†).27| Na+ | K+ | Cu2+ | Mg2+ | Fe2+ | Ca2+ | Fe3+ | Gd3+ | Er3+ | |
|---|---|---|---|---|---|---|---|---|---|
| CCC (μM) | 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 938 938 |
29736 |
216 | 215 | 206 | 114 | 11.8 | 11.0 | 6.4 |
Table 1 shows that the CCC of cations with +2 and +3 charge fit with the Z−6 relationship, whereas the CCC of monovalent ions is underpredicted by the Schulze–Hardy rule. Our results closely fit with the ∼11-fold reduction as the valency increases from +2 to +3. In contrast, the CCC deviates (i.e., at least 138 times) from the 64-fold increment predicted by the Schulze–Hardy rule when the valency decreases from +2 to +1. This inconsistency might contribute to other interactions such as coordination bonding and hydration forces.16 Nevertheless, our results generally follow the Schulze–Hardy guidelines: the charge number has a much bigger impact on the CCC than the total charge distribution across colloids. A higher charge number makes it easier for the nanoparticles to aggregate.
Next, we studied the Schulze–Hardy rule using a charged peptide system that is much larger and less spherically shaped than electrolytes. We synthesized G5−xRx (x = 0–5) peptides whose charge numbers ranged from 0 to +5 (Fig. 1b and Fig. S3†). Glycine (G) was used as the neutral spacer, and arginine (R) with positive guanidine groups changed the charge. We then titrated TPPTS-AuNPs with these peptides in water to find the CCC (Fig. S4c–g†). Fig. 1c shows the optical measurements (Abs600/Abs520) at 10 min of titration. There is an obvious difference in the CCC from 16755.0 nM to 16.1 nM when the charge number of the peptide increased from +1 to +5. G1R4 and G0R5 peptides can further disassemble these aggregates and lead to a change in optical absorption: these data suggest that highly charged peptides can indeed restore colloidal stability when present at sufficiently high concentration.
A higher charge number implies less of a decrease in CCC and vice versa. For instance, the CCC of the G1R4 peptide is almost the same as that of the G0R5 peptide. However, the CCC of the G4R1 peptide has a 26-fold increase relative to that of the G3R2 peptide. AuNPs do not aggregate even at high concentration of neutral G5R0 peptide (1.5 mM) suggesting that the aggregation behavior is only controlled by the electrostatic force resulting from guanidine groups of R. The CCC of these peptides were plotted against the peptidic charge valency, and the fitting curve showed that CCC was inversely proportional to the sixth power of charge numbers with R2 = 0.999 (Fig. 1d), i.e., CCC ∝ Z−6. This result strongly suggested that the prevalence of the Schulze–Hardy rule is not only restricted to the simple ions—it also applies to more complex molecules.
Dynamics of charged peptides within nanoparticle aggregates
To better understand the above results, we performed molecular dynamics (MD) simulations of TPPTS-AuNPs interacting with G4R1 and G3R2 peptides as model singly and doubly charged molecules. We studied the ability of G4R1 and G3R2 peptides to mediate attractive interactions between two TPPTS-AuNPs. In these MD simulations, 900 G4R1 or 450 G3R2 (to neutralize TPPTS-AuNPs) was randomly distributed in the simulation box (25 nm × 15 nm × 15 nm), and AuNPs (d = 5 nm, used instead of 13 nm for computational efficiency) were randomly decorated with 150 TPPTS ligands to ensure high surface charge density (Fig. S5†). Fig. 2a and b show snapshots from MD simulations of two TPPTS-AuNPs neutralized by the corresponding number of G4R1 or G3R2 peptides (900 and 450, respectively). While both molecules exhibited a strong affinity to NPs, only G3R2 peptides had the ability to bring particles together during the simulation. We evaluated the inter-particle space between the two AuNPs, and the G3R2 peptides bind NPs together like a “dynamic ionic glue”,7 thus promoting the structural transformation of NP aggregates (Fig. 2c). These results are also shown in Fig. 2d where we plotted the distance between the centres of masses of the two AuNPs as a function of the MD simulation time. The distance between the two TPPTS-AuNPs remained at 12–14 nm when interacting with G4R1. G3R2 peptides drove TPPTS-AuNPs together, and the distance between centers of AuNPs remained at ∼8.5 nm even after 50 ns. These data suggest that molecules with a high charge ratio have stronger charge screening ability and can mediate NP aggregation, which is consistent with our experimental results (Fig. 1b). These findings also have great implications for colorimetric sensor design involving electrostatic interactions. A proof-of-concept sensor design for protease detection is shown next.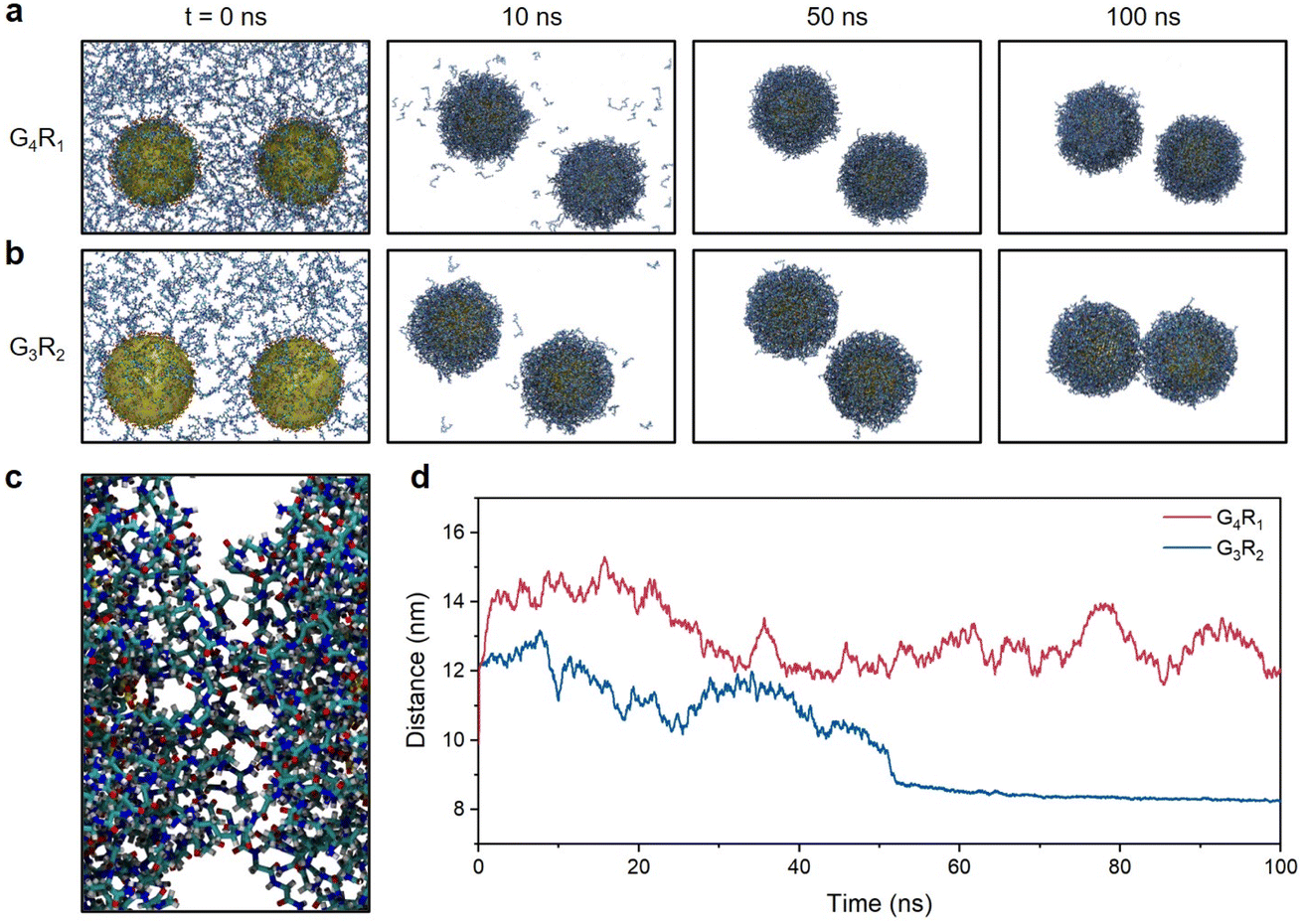 | ||
| Fig. 2 MD simulations of electrostatic interactions between TPPTS-AuNPs and charged peptides. (a) Snapshots from MD simulations of two TPPTS-AuNPs in the presence of G4R1. (b) Snapshots from MD simulations of two TPPTS-AuNPs in the presence of G3R2. (c) The snapshot from the MD simulation of the space between two TPPTS-AuNPs in the presence of G3R2 peptides at t = 100 ns. Ligands in the figure represent G3R2 peptides or TPPTS, where carbon is in cyan, hydrogen is in white, nitrogen is in blue, sulfur is in yellow, and oxygen is in red. (d) Distance between the centres of two TPPTS-AuNPs in the presence of G4R1 (red) or G3R2 (blue) as a function of the MD simulation time. | ||
Peptide design and characterization for plasmonic sensing
Coagulation behavior is strongly dependent on the charge number of molecules. Thus, we hypothesized that the AuNP aggregation induced by highly charged peptides would reverse when the charged peptide was enzymatically cleaved (Fig. 1a): while the identity of the amino acids is the same, the charge screening ability of cleaved peptides is much lower than that of intact peptides. In turn, the color would change from purple to red. Here, we selected the main protease (Mpro) of SARS-CoV-2 as the enzyme to test peptides and AuNPs. Fig. 3a illustrates sequences of R-series peptides of varying charge valence, which flank the centered Mpro cleavage sequence (AVLQ↓SGF) with different numbers of R residues. The synthesis of intact peptides and the cleavage by Mpro were confirmed by HPLC and ESI-MS (Fig. 3d and e, and S6†).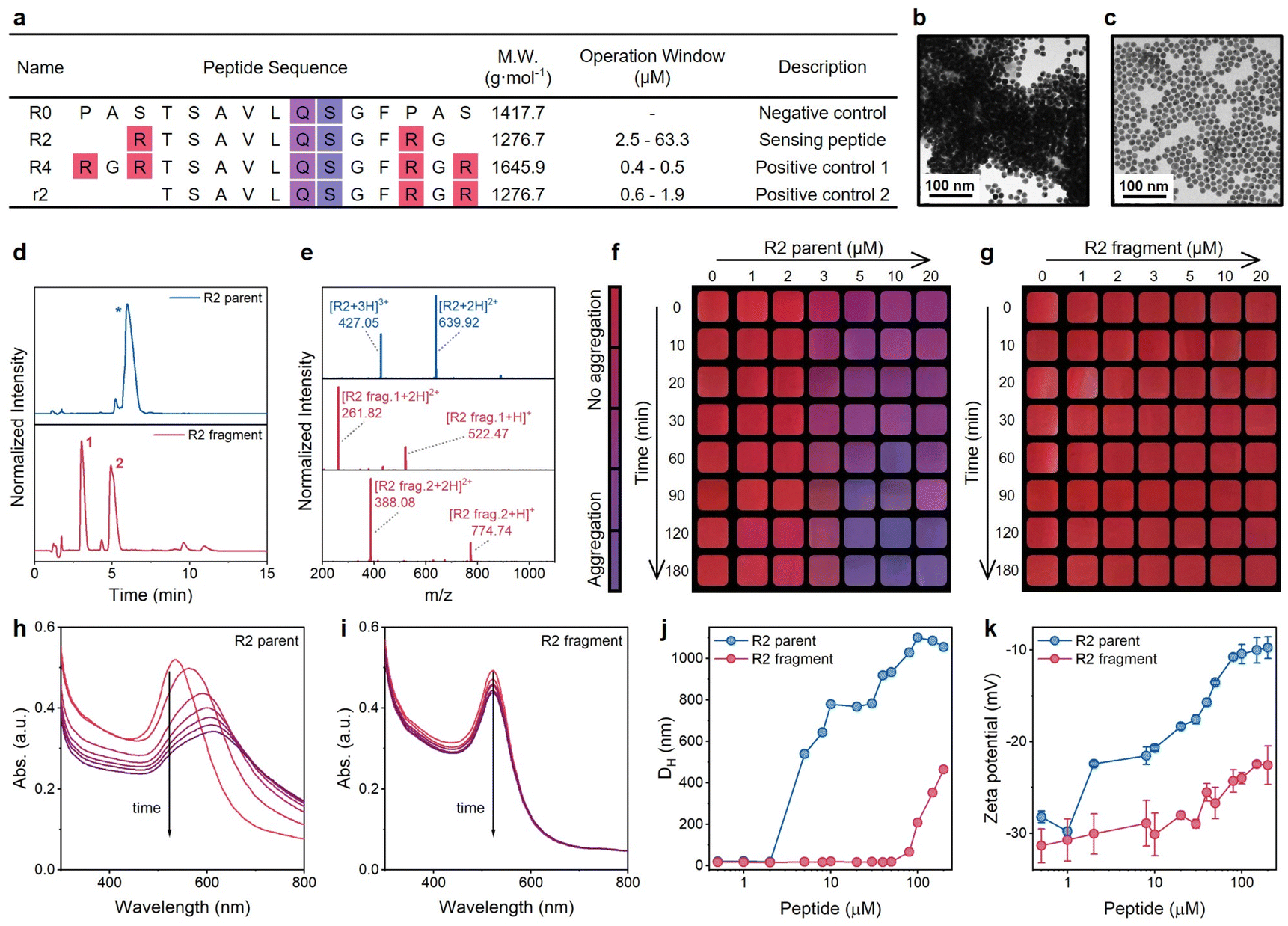 | ||
| Fig. 3 Examination of the plasmonic sensing system. (a) Sequence design of R series peptides for the plasmonic sensing system. All peptides contain a free N-terminus (–NH2) and an amidated C-terminus (–CONH2). The number after R implies the amount of arginine residues, and the lowercase r implies a different arrangement of arginine compared with the uppercase R. Mpro cleaves the peptide at Gln↓Ser (i.e., Q↓S). The operation window with TPPTS-AuNPs was determined in Tris-buffer at 10 min readout time. (b) TEM image of aggregated TPPTS-AuNPs induced by R2 intact peptides. (c) TEM image of monodispersed TPPTS-AuNPs mixed with proteolytic fragments of the R2 peptide. (d and e) HPLC (d) and ESI-MS (e) data confirm the cleavage of the R2 peptide by Mpro. (f and g) The color evolution of TPPTS-AuNPs in the presence of R2 intact peptides (f) or R2 fragments (g). Cropped images are shown with a color bar. (h and i) The time progression of optical absorption of AuNPs when incubated with an R2 parent peptide (h) and R2 fragments (i). Curves from red to purple were recorded every 5 min for 30 min (j) DLS profiles of TPPTS-AuNPs incubated with increasing concentrations of R2 parent peptide (blue) and its fragments (red). (k) Zeta potential measurements of TPPTS-AuNPs incubated with increasing concentrations of R2 parent peptide (blue) and its fragments (red). Error bars represent triplicate measurements for one sample. | ||
We first studied the impact of peptide charge on colloidal stability by incubating TPPTS-AuNPs with an R2 parent peptide or its pre-digested fragments. Transmission electron microscopy (TEM) images confirm the morphological difference between AuNPs incubated with the parent peptide (Fig. 3b) and Mpro pre-cleaved fragments (Fig. 3c). Fig. 3f and g show the color change as a function of the peptide concentration and time. The addition of 1 to 20 μM of R2 parent peptides (net charge = +2, Fig. 3f and S7†) to the TPPTS-AuNP dispersion caused an instant color shift from ruby red to purple. The change in color correlated with increasing parent concentration and reaction time. In comparison, TPPTS-AuNPs coexisted with R2 fragments (net charge = +1, Fig. 3g) showing a consistent ruby red color. We quantitatively assessed the color change through analyses of the UV-vis spectra. As shown in Fig. 3h, the time-lapsed absorbance profiles of TPPTS-AuNPs with R2 parents lead to a fast and sizable decrease in the surface plasmon resonance (SPR) at 520 nm with a noticeably increasing band at 600 nm in 10 min. Meanwhile, the same SPR of R2 fragments indicates that the AuNP dispersion was maintained (Fig. 3i). We thus defined the ratiometric signal, Abs600/Abs520, to quantify the aggregation and the color change. Fig. 3h shows relatively low absorption of aggregated particles above ∼750 nm; however, 2D assemblies could lead to near-IR absorption and may be investigated in future work.
We next characterized AuNP aggregation and R2 peptides for mechanistic studies. Dynamic light scattering (DLS) profiles (Fig. 3j) monitored AuNP aggregation in the presence of the R2 parent peptide, and zeta potentials of TPPTS-AuNPs mixed with an increasing number of intact peptides showed a sizable alternation for 20 mV (Fig. 3k). In contrast, TPPTS-AuNPs with Mpro pre-cleaved R2 fragments remained dispersed until high peptide concentration (Fig. 3j); the zeta potential data suggested no significant interactions between the two species with only a 10 mV increment (Fig. 3k). These data confirmed that AuNPs agglomerate because of intact peptides but are monodisperse in the presence of proteolytic fragments. The number of positive R groups in every single peptide is halved due to the Mpro cleavage, thus strongly increasing the peptide concentration required for AuNP aggregation. Moreover, the aggregation state of AuNPs titrated with R2 fragments is still not comparable to the case of R2 parent peptides even if highly concentrated fragments were added; the coagulated particle size of AuNPs titrated with 100 μM R2 fragments is less than half that of AuNPs incubated with 5 μM R2 parent peptide (Fig. 3j). This suggests that the charge screening ability is strongly suppressed by the slightly lower charge number of the peptide (Fig. 3k) as predicted by the Schulze–Hardy rule.
The limit of detection (LoD) for Mpro in our sensing system is related to the concentration and charge of the peptide as well as the NP stability in the assay. To obtain the best sensing performance, we tested a combination of peptide/AuNPs by adjusting the surface chemistry, the peptide charge density, and concentration. We first synthesized different negatively charged AuNPs modified with diphenylphosphinobenzene sulfonate (DPPS), bis(p-sulfonatophenyl)phenylphosphine (BSPP), or TPPTS.22 These ligands differ in the number of sulfonated groups (Fig. S1†) and thus, the negative surface charge density on AuNPs. DPPS-AuNPs (one sulfonate group) has the lowest zeta-potentials: −24.1 ± 1.4 mV. The zeta-potential of BSPP-AuNPs (two sulfonate groups) was −25.0 ± 1.2 mV, and that of TPPTS-AuNPs (three sulfonate groups) was −31.0 ± 2.1 mV.
These three AuNPs (3.6 nM) were incubated with the R2 peptide or fragments of varying concentrations, and the optical measurements (Abs600/Abs520) were recorded at 10 min after AuNP addition. DPPS-AuNPs have the narrowest operation window, i.e., 0.1 to 4.6 μM, while TPPTS-AuNPs have the widest one, i.e., 2.5 to 63.3 μM (Table S2† and Fig. 4a–c). Derjaguin–Landau–Verwey–Overbeek (DLVO) theory explains this observation. The Debye lengths of colloids with low surface charge density are compressed due to the domination of the van der Waals force, while highly charged colloids have more electrostatic repulsion that reduced the van der Waals attraction: charged colloids can thus maintain long Debye lengths because of the double-layer repulsion.28,29 Therefore, DPPS-AuNPs can easily aggregate due to the intrinsic short Debye length regardless of whether the screening agent is intact peptides or pre-cleaved fragments. The TPPTS-AuNPs stay dispersed until very high concentrations of fragments are used. The zeta potentials of ligand-AuNPs confirm that the dispersion stability of ligand-AuNPs is TPPTS-AuNPs > BSPP-AuNPs > DPPS-AuNPs. Due to the widest operation window, we picked up TPPTS-AuNPs for the following operations.
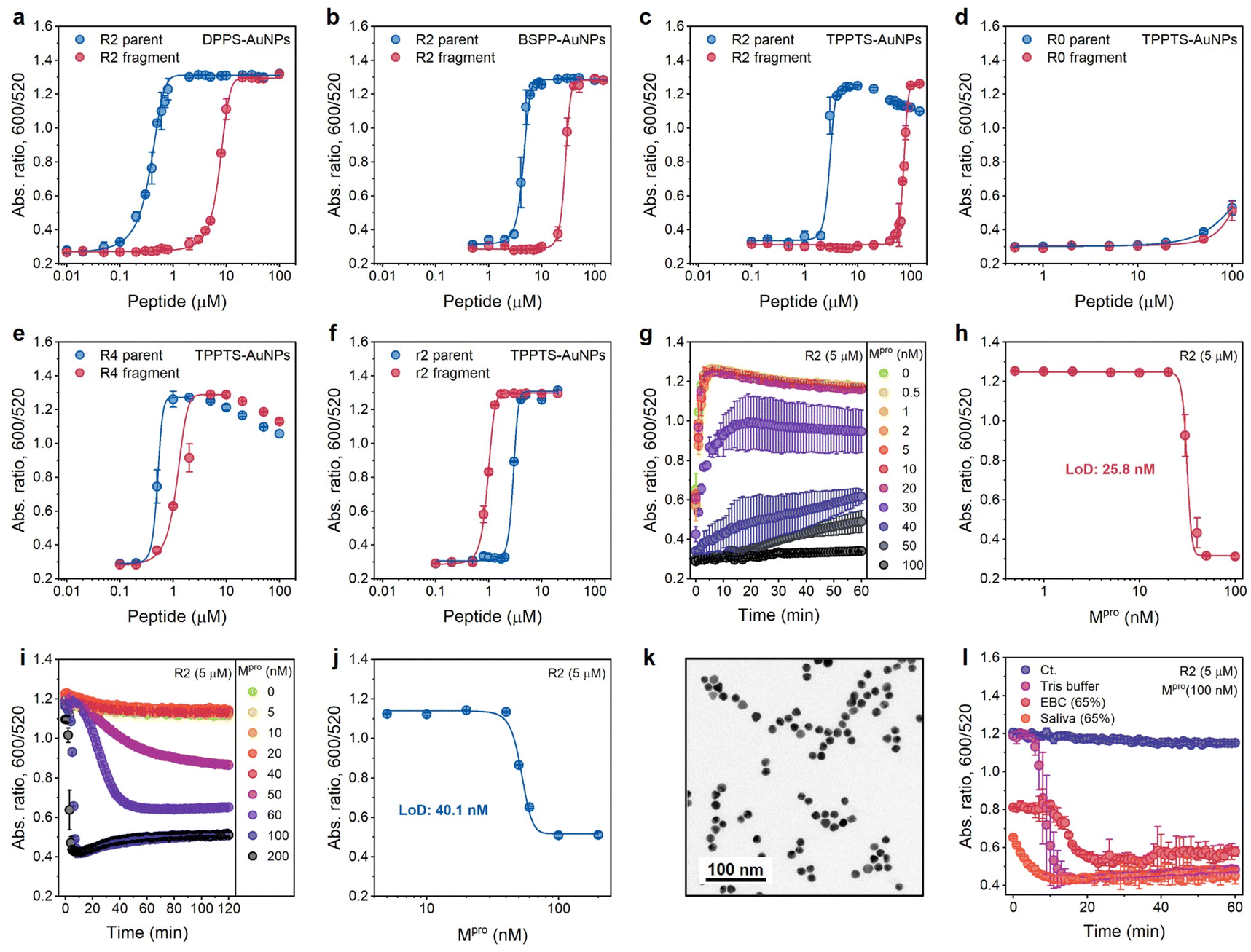 | ||
| Fig. 4 Optimization of sensing performance. (a–c) Operation windows of Mpro sensors based on the ratiometric signal (Abs600/Abs520) collected from DPPS-AuNPs (a), BSPP-AuNPs (b), and TPPTS-AuNPs (c) incubated with various amounts of R2 parent (blue) and fragments (red), respectively. (d–f) Operation windows of Mpro sensors based on R0, R4, and r2 peptides, respectively. Data in (a–f) were collected at readout time = 10 min. Error bar = standard deviation of the two samples. (g) Time progression of ratiometric absorbance (Abs600/Abs520) in the enzyme assay where a fixed amount of R2 substrate was incubated with increasing concentrations of Mpro (0−100 nM) in Tris buffer. Data points were read every 1 min for 1 h. The experiments were performed in triplicate. (h) The absorbance ratio as a function of Mpro concentration in the enzyme assay. Data were collected at 10 min. Error bar = standard deviation of the three samples. The linear region used to calculate LoD can be found in Fig. S9a.† (i) One-pot protease assays. Curves assaying an increasing Mpro concentration (cfinal = 0–200 nM) in the presence of constant TPPTS-AuNPs and R2 peptide. Data were collected every 1 min for 2 h. The experiments were performed in triplicate. (j) The absorbance ratio as a function of Mpro concentration in one-pot assays. The data were collected at readout time = 2 h. Error bar = standard deviation of the three samples. The linear region used to calculate the LoD can be found in Fig. S9b.† (k) TEM image of TPPTS-AuNPs in the one-pot assay. (l) Time progression of ratiometric absorbance (Abs600/Abs520) in the one-pot protease assay with different media. The control curve (Ct.) designates Tris buffer without the addition of Mpro. Error bar = standard deviation of the three samples. | ||
Here, we explored the effect of tuning the number of R from 0, 2 (one R and one R after Mpro cleavage), 2 with different arrangements (no R and two Rs after Mpro cleavage), and 4 (two Rs and two Rs after Mpro cleavage), i.e., R0, R2, r2, and R4 peptides, respectively (Fig. 3a). These peptides or fragments were again incubated individually with TPPTS-AuNPs (3.6 nM), and the optical measurements (Abs600/Abs520) were recorded at 10 min after AuNP addition. The aggregating sequence containing zero guanidine side chains in the R0 peptide produced no optical signal change (Fig. 4d). This non-aggregating system has a very low ionic valence and attenuated electrostatic interaction according to the Schulze–Hardy rule.
Compared to R2 fragments consisting of one R residue each, the inclusion of four R in R4 strongly interferes with double-layer potentials of nanoparticles: both the parent peptide and fragments trigger AuNP aggregation at low concentrations (Fig. 4e). We predicted that both r2 parents and fragments should behave similarly with the R2 parent peptide because there are +2 net charges before and after Mpro cleavage. The CCC for the r2 parent peptides induces TPPTS-AuNPs almost the same as that of R2 parents (2.5 μM vs. 1.9 μM). However, r2 fragments show a stronger Debye length reduction at an even lower concentration (Fig. 4f), which may be attributed to the apparently short fragmental sequence, SGFRGR, which is only half the length of the R2 parent peptide. Previous reports assumed that the charge screening ability of higher valence ions was usually overpredicted due to more obvious charge dispersion for a larger ion size.18 In our system, two Rs concentrated on a small fragment result in higher charge density on the peptide, thus contributing stronger electrostatic force to AuNPs. Nevertheless, the operation window of r2 is too narrow to use practically since the color difference disappears at a relatively low peptide concentration (Fig. 3a).
We also verified that this charge screening behavior is not restricted to arginine. Here, a K2 peptide replaced the R in the R2 peptide with lysine (K, Table S1†). This system reproduced the operation window with a similar range when used with TPPTS-AuNPs (Fig. S4h†). Nevertheless, the R2 peptide at 5 μM has the widest operation window and was used subsequently.
Protease detection based on colorimetric sensing
We set 10 min as the readout time for rapid protease detection. Fig. 4g shows the ratio of Abs600/Abs520 as a function of Mpro concentration. The concentration of R2 peptide was 5 μM. The LoD for Mpro was determined to be 25.8 nM in Tris buffer (Fig. 4h). The protease LoD of our sensor is either similar to or lower than that of other nanoparticle-based protease assays (i.e., in low nanomolar ranges) or much less time-consuming (i.e., t = 10 min) versus previous reports.24,30–33Positively charged peptides bind to TPPTS-AuNPs in a dynamic manner,7 and we hypothesized that the one-step reaction leading to enzymatic cleavage and particle disassembly can be applied to our system. We incubated R2 parents with TPPTS-AuNPs, and then added Mpro in the concentration varied from 0 to 200 nM. The LoD for Mpro, 40.1 nM in Tris buffer, was successfully obtained at the readout time = 2 h (Fig. 4i and j). Moreover, the TEM image showed that TPPTS-AuNPs were deeply monodispersed (Fig. 4k) compared to pre-incubated R2 fragments (Fig. 3c). We further applied this procedure to different media such as exhaled breath condensate (EBC, 65%) and pooled saliva (65%) to see if this assay would be interrupted by materials in the other matrices. Even though the aggregation state of TPPTS-AuNPs was slightly interrupted by both EBC and saliva when mixing with R2 parent peptides, the absorbance quickly decreased to a lower level within 20 min after Mpro addition (Fig. 4l). Proteins in saliva modify AuNP surfaces with protein corona, thus stabilizing particles and making it difficult to induce aggregation of NPs in typical colorimetric assays.22,34–36 By disassembling AuNPs, we can avoid this problem and obtain a color change. These results demonstrate that our sensor based on the electrostatic interaction can provide a very simple and effective one-step colorimetric assay without pre-incubation requirements, compared to a conventional multistep procedure.24,30–32,37
To confirm that the action of Mpro was indeed the cause of AuNP dispersion, control experiments were performed in the presence of a known competitive inhibitor (GC376).38 GC376 is a covalent inhibitor against viruses with 3C protease (3Cpro) or 3C-like protease (3CLpro) such as picornaviruses, noroviruses, and coronaviruses.39,40Fig. 5a shows results of assaying an increasing molarity of GC376 (i.e., 0–1 μM) in the presence of a constant amount of Mpro (50 nM) and R2 substrate (5 μM). Note that the inhibitor itself did not affect the dispersity of AuNPs as the control line. The aggregation kinetics were strongly retarded because the activity of Mpro was suppressed by the inhibitor. Examination of the absorbance ratio at 10 min yields a typical inhibitor titration curve (Fig. 5b). A linear form of the Morrison equation derived by Henderson (eqn (2) and Fig. S9c†) was applied to evaluate the titrated Mpro concentration ([E]0 = 74.1 nM) and the potency of the GC376 inhibitor (inhibitory constant Ki(app.) = 0.23 nM, IC50 = 37.3 nM).41 This half maximal inhibitory concentration (IC50) is lower than the majority of reported values,26,42–44 thus demonstrating that our sensing system can be employed for rapid screening of anti-Mpro therapeutic agents.
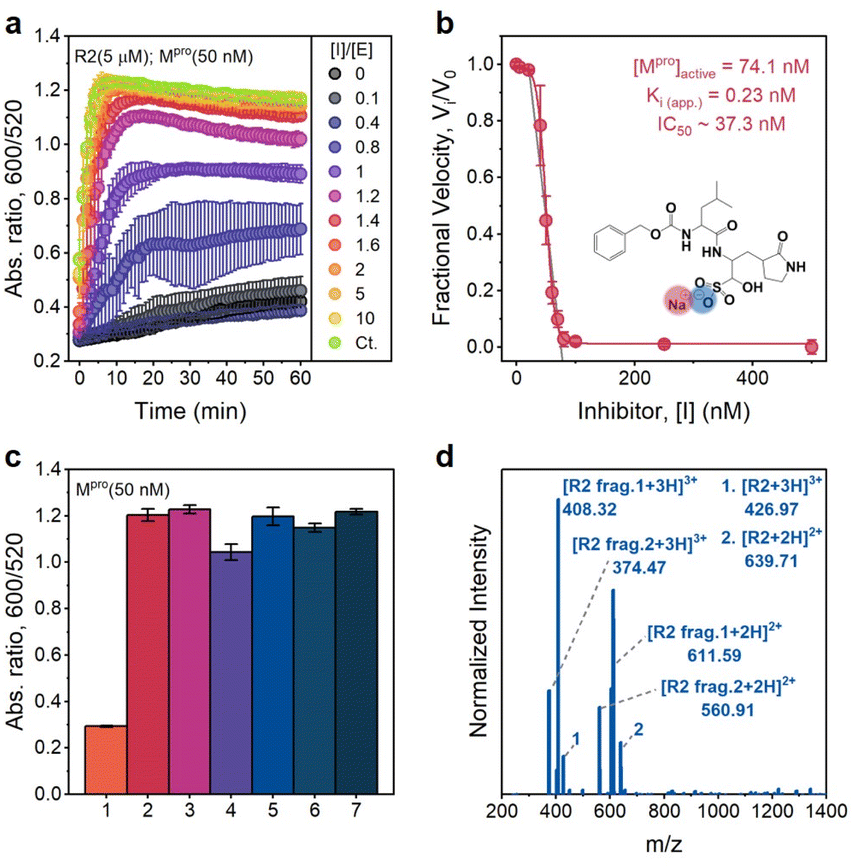 | ||
| Fig. 5 Inhibition and selectivity assays. (a) Time progression of the ratiometric signal (Abs600/Abs520) in inhibitor assays. An increasing molar ratio of [inhibitor]/[Mpro] from 0 to 10 was employed. The control curve (Ct.) designates an inhibitor only without an Mpro additive. Error bar = standard deviation of the three samples. (b) A typical inhibition titration curve fitted with the Morrison equation (eqn. (2)) is shown for the competitive inhibitor, GC376. The inset shows the chemical structure of the GC376 inhibitor. Error bar = relative standard deviation of the three samples. (c) Sensor activation by mammalian proteins (50 nM). 1 = positive control only with Mpro, 2 = trypsin, 3 = thrombin, 4 = hemoglobin, 5 = BSA, 6 = α-amylase (50 U per mL), and 7 = negative control without Mpro, Error bar = standard deviation of the three samples. (d) ESI-MS data confirm the cleavage of the R2 peptide by trypsin. | ||
We further cross-tested whether other related analytes such as bovine serum albumin (BSA), hemoglobin, trypsin (cleaving C-terminus of R),45 thrombin (cleaving R–G bonds in human proteinase-activated receptor 4),46 and α-amylase (digesting α-1,4-glucosidic bonds in starch) can activate our sensing system off-site.47Fig. 5c reveals that only the positive control (with 50 nM Mpro) does not have a distinguishable optical signal change due to the AuNP dispersion resulting from low charged R2 fragments. No changes in the optical signal were measured in the presence of 50 nM mammalian proteins (e.g., BSA and hemoglobin) or other enzymes (e.g., amylase, thrombin, and trypsin). Normally, R-containing substrate probes have limited stability in the presence of trypsin because R could be easily cleaved.24,45,48 However, in our case, TPPTS-AuNPs aggregate, and the ratiometric signal behaves almost the same as the negative control.
To investigate this phenomenon further, we incubated R2 parent peptides with trypsin and purified products by conducting HPLC (Fig. S9d†). ESI-MS data in Fig. 5d confirm that trypsin cleaves the C-terminus of R at P7 and P4′ sites, thus releasing TSAVLQSGFRG (net charge = +1) and RTSAVLQSGFR (net charge = +2), respectively. Here, trypsin reacts with R at the P4′ site, and RTSAVLQSGFR with 2 Rs thus becomes the majority in the R2 mixture. There are also some parent peptides in the solution. Therefore, the dispersion stability of TPPTS-AuNPs is destroyed due to electrostatic interactions induced by the overall highly charged peptide, resulting in aggregation and color change. This result indicates the remarkable selectivity and specificity of our sensor to the SARS-CoV-2 virus.
Sensing kit for SARS-CoV-2 protease
The value of this approach is its simplicity: no complex bioconjugation techniques or readout instrumentation are required. This allows for the large-scale implementation of qualitative diagnostics for protease markers. Various approaches have been developed for protease detection, including electrochemical sensors, colorimetric sensors, and fluorescence probes.22,49–51 Here, we report a portable and user-friendly biosensor based on the Schulze–Hardy rule. Fig. 6 shows the scheme of a simple colorimetric detection tool. TPPTS-AuNP solution and R2 intact peptides were stored in a small tube, respectively, and a nasal swab was made with polyester (PE) heads with a plastic handle. TPPTS-AuNPs were first mixed with 6 μM R2 peptide in the small tube to obtain purple aggregates, anf then the PE swab with Mpro attachment was immersed in the mixture. As Mpro was released from the swab, it cleaved the R2 peptide and unveiled low charged fragments, thus redispersing TPPTS-AuNPs. After 15 h room temperature incubation, the initial purple color was transformed into pink or red depending on the concentration of Mpro (Fig. S10†). The color of the solution starts to appear pink at 20 nM Mpro; or LoD = 20 nM. Future work may integrate these results into wearable colorimetric sensors as in our prior work.48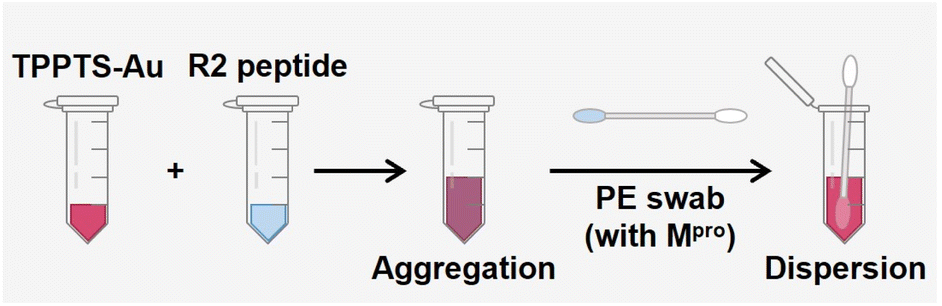 | ||
| Fig. 6 Schematic illustration of the sensing kit. The ruby red TPPTS-AuNPs were mixed with transparent R2 peptide solution and they transformed into purple. Mpro released from the PE swab restores the color of TPPTS-AuNPs from purple to red. | ||
Conclusions
In this study, we successfully demonstrated that the Schulze–Hardy rule can be used beyond simple ions, e.g., charged peptide. The CCC of AuNPs is inversely proportional to the power of six of the peptide charge valence. Molecular dynamics simulations also demonstrated that the highly charged peptide presents as “ionic glue” to drive AuNP assembly. Therefore, by tuning the net charge of the peptide, the colloidal stability can be carefully tuned via the concentration of the peptide. We applied this rule in a plasmonic sensing system. The R2 peptide with a high charge number was designed to collapse the dispersion stability and induce the aggregation of negatively charged TPPTS-AuNPs. After enzymatic cleavage of by Mpro, R2 fragments with low charge number are released. The Debye length of the AuNPs is thus restored, and the purple aggregation recovers to a ruby red dispersion. By quantifying the color change with a measurable absorbance ratio, we obtained the LoD for Mpro in Tris buffer under the two-step reaction condition, that is, 25.8 nM. Notably, we successfully demonstrated that this system can be operated under one-pot reaction. This rapid color change provides a simple, efficient, and specific platform for inhibitor-screening targeting at Mpro, and we therefore designed a sensing kit for simple protease detection.Materials and methods
Gold nanoparticle synthesis
Citrate-stabilized AuNPs (∼13 nm diameter, TEM) were prepared using the Turkevich method by rapidly injecting an aqueous solution of sodium citrate tribasic dihydrate (150 mg, 5 mL) into an aqueous solution of HAuCl4·3H2O (45 mg, 300 mL) under boiling conditions and vigorous stirring. The reaction mixture was left boiling while stirring for 20 min, and then cooled down to room temperature. Then, TPPTS-AuNPs were prepared by the ligand exchange method. A citrate-AuNP dispersion in water (150 mL, 3.6 nM) was vigorously stirred overnight with TPPTS (150 mg, 5 mL) in a round bottom flask. The mixture was purified via syringe filtration (hydrophilic PTFE, 0.45 mm) followed by centrifugation at 18000 g for 40 min. The supernatant was removed and pellets of TPPTS-AuNPs were redispersed in DI water followed by sonication for 20 min. The optical density of the final solution was 1.45 (c ∼3.6 nM, ε520 = 4.0 × 108 M−1 cm−1) and it was stored at 4 °C for long-term use. DPPS-AuNPs and BSPP-AuNPs were made by the same procedure and their concentrations were brought to ∼3.6 nM.
Titration of metal cations/charged peptides on TPPTS-AuNPs
The stock cation (Na+, K+, Cu2+, Mg2+, Fe2+, Ca2+, Fe3+, Gd3+, Er3+; and counterions were Cl−)/peptide solution of the desired final concentration was prepared by dissolving the electrolyte in H2O, and then injection into a 96-well plate. H2O and TPPTS-AuNPs (100 μL, 3.6 nM, optical density ∼1.45) were added to reach the final volume of 120 μL. At least two replicates of each experiment were measured. Absorbance at 600 nm and 520 nm at room temperature was measured every 1 min for 1 h. The ratiometric signal (λ600/520) at 10 min was extracted for analyses. H2O was used as the blank. The critical coagulation concentration (CCC) was calculated using a statistical method previously reported in the literature.27 In this study, the value of the limit of detection (LoD) gives a close approximation to CCC:| CCC ≡ LoDint. = meanblank + 1.645 × (SDblank) + 1.645 × (SDlow concentration sample) | (1) |
Classical molecular dynamics simulation
The TPPTS-AuNP model was composed of two parts, including the surficial TPPTS molecules and the AuNP. The AuNP was constructed through the InorganicBuilder module within the VMD52 package. Considering the computational cost, the diameter of the AuNP was set to be 5 nm. 150 TPPTS were then randomly distributed at the surface of the AuNP using the Packmol package,53 forming one TPPTS-AuNP. Two simulation boxes (25 nm × 15 nm × 15 nm) were constructed. System one contains 2 TPPTs-AuNPs and 900 G4R1 peptides and was then solvated with 153575 water molecules (Fig. S5a†). System two contains 2 TPPTS-AuNPs and 450 G3R2 peptides and was then solvated with 160386 water molecules (Fig. S5b†). The initial distance between the two AuNPs was set to 10 nm for the two systems. The general Amber force field (GAFF)54 was applied to describe the dynamic behavior between peptides and TPPTS. For the AuNP, the force field for the AuNP developed by Heinz et al.55 was applied. The restricted electrostatic potential (RESP) atomic charge56 was used for peptides and TPPTS molecules throughout the simulation. The water model used in these simulations was TIP3P, and the SHAKE algorithm was used to constrain the bond lengths and bond angles in the water molecules. All simulations were first energy minimized and then equilibrated in an NPT (temperature of 298.15 K and pressure of 1 bar) ensemble for 100 ns via a velocity rescaling thermostat and Berendsen barostat. All the molecular dynamics simulations were carried out using the Gromacs57 package under periodic boundary conditions with a 2 fs time step using Ewald, specifically PME, to account for long-range electrostatics.
Operation window measurement
The stock of the parent Mpro substrate was prepared by dissolving the intact peptide in Tris buffer (20 mM, pH 8.0, with 150 mM NaCl and 1 mM DTT). The solution of peptide fragments was prepared by incubating the parent peptide solution (∼1.2 mg mL−1) with Mpro (∼152 nM) in Tris buffer at 37 °C for 3 hours. After incubation, parent/fragment peptide stock of the desired final concentration was injected into the 96-well plate. Then, Tris buffer and DPPS/BSPP/TPPTS-AuNPs (100 μL, 3.6 nM, optical density ∼1.45) were added to reach the final volume of 120 μL. At least two replicates of each experiment were measured. Absorbance at 600 nm and 520 nm at room temperature was measured every 1 min for 1 h. The ratiometric signal (λ600/520) at 10 min was extracted for analyses. Tris buffer was used as the blank.LoD measurement
R2 peptide and TPPTS-AuNPs were used to detect Mpro. The Mpro enzymes of desirable amount were mixed with the parent R2 peptide (cfinal = 5 μM) in Tris buffer and incubated at 37 °C for 3 hours. At least two replicates of each experiment were made. After 3 hours, the assay was transferred into a 96-well plate and incubated with TPPTS-AuNPs (100 μL, 3.6 nM). The absorbance of the mixtures at 600 and 520 nm were readout in a microplate reader at 25 °C every 1 min for 1 hour. The ratiometric signal (λ600/520) at 10 min was extracted for analyses. The limit of detection was calculated.Inhibitor assay
The desired amount of GC376 was pre-incubated with Mpro protease (cfinal = 50 nM) at room temperature for 10 min. Then, the parent R2 peptide (cfinal = 5 μM) and Tris buffer were added and incubated at 37 °C for 20 min. After incubation, the inhibitor assay was transferred into a 96-well plate, and incubated with TPPTS-AuNPs (100 μL, 3.6 nM). The microplate reader read the absorbance at 600 and 520 nm at 25 °C every 1 min for 1 h. The experiments were conducted in triplicate. A linear form of the Morrison equation derived by Henderson was applied to calculate the active enzyme concentration and apparent inhibitory constant, Ki (app.):41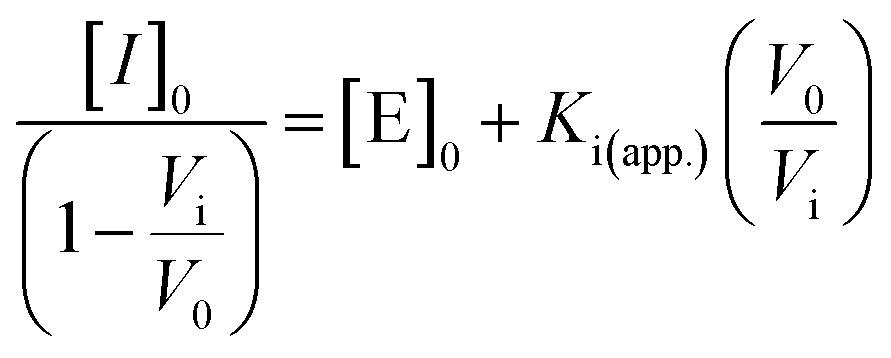 | (2) |
Specificity test
Desirable amounts of α-amylase, trypsin, thrombin, bovine serum albumin (BSA), and hemoglobin of were spiked into Tris buffer to reach a final concentration of 50 nM (the concentration of amylase was 50 U per mL), respectively, which were equivalent to the same amount in a 120 μL solution. Then, the parent R2 peptide in Tris buffer (cfinal = 5 μM) was added to the above mixtures in microtubes and incubated at 37 °C for 3 hours. Then, the mixtures were transferred into the 96-well plate and incubated with TPPTS-AuNPs (100 μL, 3.6 nM). The experiments were performed in triplicate. The absorbance of the mixtures at 600 and 520 nm were readout in the microplate reader at 25 °C every 1 min for 1 hour. The ratiometric signal (λ600/520) at 10 min was extracted for analyses.Sensing kit design for protease detection
The parent R2 peptide (cfinal = 6 μM) in 60 μL Tris buffer was added to TPPTS-AuNP (300 μL, 3.6 nM) solution for 15 min incubation at room temperature to reach a stable aggregation state. Then, Mpro enzymes in 40 μL Tris buffer were absorbed into the polyester swab, followed by immersion of the swab in a solution of TPPTS-AuNPs and the R2 peptide. The mixtures were mildly shaken and incubated at room temperature for 15 h. Photographic images were taken by using a smartphone in a lightbox with a white background.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Y.-C. C., Z. J., and J. V. J. designed the research. Y.-C. C. and Z. J. designed and performed the experiments. K. L. and X. C. performed the computer simulations. P. F. and A. O. contributed the Mpro. Y.-C. C., Z. J., K. L., J. Z., W. Y., J. Y., Y. C., M. R., M. N. C., T. H., and J. V. J. analyzed and interpreted the data. Y.-C. C., Z. J., K. L., and J. V. J. wrote the manuscript. All authors critically revised the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Institutes of Health (R01 DE031114; R21 AG065776-S1; R21 AI157957) for financial support. The electron microscopy work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of the University of California, San Diego, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542148). M. N. C. acknowledges fellowship support from T32 CA153915.References
- M. Grzelczak, L. M. Liz-Marzan and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- M. Ha, J. H. Kim, M. You, Q. Li, C. Fan and J. M. Nam, Chem. Rev., 2019, 119, 12208–12278 CrossRef CAS.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS.
- Z. H. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS.
- Y. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS.
- M. Alafeef, P. Moitra, K. Dighe and D. Pan, Nat. Protoc., 2021, 16, 3141–3162 CrossRef CAS.
- T. Bian, A. Gardin, J. Gemen, L. Houben, C. Perego, B. Lee, N. Elad, Z. Chu, G. M. Pavan and R. Klajn, Nat. Chem., 2021, 13, 940–949 CrossRef CAS.
- H. Schulze, J. Prakt. Chem., 1882, 25, 431–452 CrossRef.
- W. B. Hardy, Proc. R. Soc. London, 1900, 66, 110–125 Search PubMed.
- I. Coropceanu, E. M. Janke, J. Portner, D. Haubold, T. D. Nguyen, A. Das, C. P. N. Tanner, J. K. Utterback, S. W. Teitelbaum, M. H. Hudson, N. A. Sarma, A. M. Hinkle, C. J. Tassone, A. Eychmuller, D. T. Limmer, M. O. de la Cruz, N. S. Ginsberg and D. V. Talapin, Science, 2022, 375, 1422 CrossRef CAS.
- Y. Suzuki, M. Endo and H. Sugiyama, Nat. Commun., 2015, 6, 8052 CrossRef CAS.
- F. Gobeaux, N. Fay, C. Tarabout, C. Meriadec, F. Meneau, M. Ligeti, D. A. Buisson, J. C. Cintrat, K. M. Nguyen, L. Perrin, C. Valery, F. Artzner and M. Paternostre, J. Am. Chem. Soc., 2012, 134, 723–733 CrossRef CAS.
- S. Roy, V. S. S. Adury, A. Rao, S. Roy, A. Mukherjee and P. P. Pillai, Angew. Chem., Int. Ed. Engl., 2022, e202203924 CAS.
- M. Sano, J. Okamura and S. Shinkai, Langmuir, 2001, 17, 7172–7173 CrossRef CAS.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2010, 44, 2412–2418 CrossRef CAS PubMed.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- H. Rapaport, H. Grisaru and T. Silberstein, Adv. Funct. Mater., 2008, 18, 2889–2896 CrossRef CAS.
- M. R. Caplan, P. N. Moore, S. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomacromolecules, 2000, 1, 627–631 CrossRef CAS PubMed.
- M. Muttenthaler, G. E. King, D. J. Adams and P. E. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- C. D. Spicer, C. Jumeaux, B. Gupta and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574–3620 RSC.
- Z. C. Jin, J. Yeung, J. J. Zhou, Y. Cheng, Y. Li, Y. Mantri, T. Y. He, W. Yim, M. Xu, Z. H. Wu, P. Fajtova, M. N. Creyer, C. Moore, L. Fu, W. F. Penny, A. J. O'Donoghue and J. V. Jokerst, Chem. Mater., 2022, 34, 1259–1268 CrossRef CAS.
- N. Xia, X. Wang, X. Wang and B. Zhou, Materials, 2016, 9, 857 CrossRef PubMed.
- W. Xue, G. Zhang and D. Zhang, Analyst, 2011, 136, 3136–3141 RSC.
- W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- D. A. Armbruster and T. Pry, Clin. Biochem. Rev., 2008, 29(Suppl 1), S49–S52 Search PubMed.
- H. Ohshima, Electrical Phenomena at Interfaces and Biointerfaces: Fundamentals and Applications in Nano-, Bio-, and Environmental Sciences, John Wiley & Sons, 1st edn, 2012 Search PubMed.
- J. Israelachvili and H. Wennerström, Nature, 1996, 379, 219–225 CrossRef CAS PubMed.
- C. Guarise, L. Pasquato, V. De Filippis and P. Scrimin, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 3978–3982 CrossRef CAS PubMed.
- X. Ding, D. Ge and K.-L. Yang, Sens. Actuators, B, 2014, 201, 234–239 CrossRef CAS.
- W. Zhao, W. Chiuman, J. C. Lam, M. A. Brook and Y. Li, Chem. Commun., 2007, 3729–3731 RSC.
- M. Sabela, S. Balme, M. Bechelany, J. M. Janot and K. Bisetty, Adv. Eng. Mater., 2017, 19, 1700270 CrossRef.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, Chem. Soc. Rev., 2015, 44, 6094–6121 RSC.
- D. Srinivasan, W. H. Phue, K. Xu and S. George, Nanoimpact, 2020, 19 Search PubMed.
- M. Retout, Y. Mantri, Z. C. Jin, J. J. Zhou, G. Noel, B. Donovan, W. Yim and J. V. Jokerst, Acs Nano, 2022, 16, 6165–6175 CrossRef CAS PubMed.
- J. Oishi, Y. Asami, T. Mori, J. H. Kang, M. Tanabe, T. Niidome and Y. Katayama, Chembiochem, 2007, 8, 875–879 CrossRef CAS PubMed.
- H.-C. Hung, Y.-Y. Ke, S.-Y. Huang, P.-N. Huang, Y.-A. Kung, T.-Y. Chang, K.-J. Yen, T.-T. Peng, S.-E. Chang, C.-T. Huang, Y.-R. Tsai, S.-H. Wu, S.-J. Lee, J.-H. Lin, B.-S. Liu, W.-C. Sung, S.-R. Shih, C.-T. Chen and J. T. Hsu, Antimicrob. Agents Chemother., 2020, 64 Search PubMed.
- Y. Kim, S. Lovell, K. C. Tiew, S. R. Mandadapu, K. R. Alliston, K. P. Battaile, W. C. Groutas and K. O. Chang, J. Virol., 2012, 86, 11754–11762 CrossRef CAS PubMed.
- L. Fu, F. Ye, Y. Feng, F. Yu, Q. Wang, Y. Wu, C. Zhao, H. Sun, B. Huang, P. Niu, H. Song, Y. Shi, X. Li, W. Tan, J. Qi and G. F. Gao, Nat. Commun., 2020, 11, 4417 CrossRef CAS PubMed.
- P. J. Henderson, Biochem. J., 1972, 127, 321–333 CrossRef CAS PubMed.
- G. Macip, P. Garcia-Segura, J. Mestres-Truyol, B. Saldivar-Espinoza, G. Pujadas and S. Garcia-Vallvé, Int. J. Mol. Sci., 2021, 23, 259 CrossRef PubMed.
- C. Ma and J. Wang, Proc. Natl. Acad. Sci. U.S.A., 2021, 118 CAS.
- Z. Li, X. Li, Y. Huang, Y. Wu, R. Liu, L. Zhou, Y. Lin, D. Wu, L. Zhang, H. Liu, X. Xu, K. Yu, Y. Zhang, J. Cui, C.-G. Zhan, X. Wang and H.-B. Luo, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 27381–27387 CrossRef CAS PubMed.
- J. V. Olsen, S. E. Ong and M. Mann, Mol. Cell. Proteomics, 2004, 3, 608–614 CrossRef CAS PubMed.
- K. Mihara, R. Ramachandran, M. Saifeddine, K. K. Hansen, B. Renaux, D. Polley, S. Gibson, C. Vanderboor and M. D. Hollenberg, Mol. Pharmacol., 2016, 89, 606–614 CrossRef CAS PubMed.
- M. J. van der Maarel, B. van der Veen, J. C. Uitdehaag, H. Leemhuis and L. Dijkhuizen, J. Biotechnol., 2002, 94, 137–155 CrossRef CAS PubMed.
- Z. Jin, Y. Mantri, M. Retout, Y. Cheng, J. Zhou, A. Jorns, P. Fajtova, W. Yim, C. Moore, M. Xu, M. N. Creyer, R. M. Borum, Z. Wu, T. He, W. F. Penny, A. J. O'Donoghue and J. V. Jokerst, Angew. Chem., Int. Ed. Engl., 2022, 61, e202112995 CAS.
- S. S. Liew, Z. Zeng, P. Cheng, S. He, C. Zhang and K. Pu, J. Am. Chem. Soc., 2021, 143, 18827–18831 CrossRef CAS PubMed.
- Z. Jin, A. Jorns, W. Yim, R. Wing, Y. Mantri, J. Zhou, J. Zhou, Z. Wu, C. Moore, W. F. Penny and J. V. Jokerst, Anal. Chem., 2021, 93, 11025–11032 CrossRef CAS PubMed.
- Y. X. Feng, G. Liu, M. La and L. Liu, Molecules, 2022, 27, 615 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- L. Martinez, R. Andrade, E. G. Birgin and J. M. Martinez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. Cha, Biochem. Pharmacol., 1975, 24, 2177–2185 CrossRef CAS PubMed.
- D. R. Hurst, M. A. Schwartz, Y. Jin, M. A. Ghaffari, P. Kozarekar, J. Cao and Q. X. Sang, Biochem. J., 2005, 392, 527–536 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, synthesis and instrumentations, characterization studies, computational simulations, one-pot assays, table of peptide sequences and molecular weights, table of operation windows of R2 and K2 peptides, structures of ligands and R2 peptide, HPLC and ESI-MS data, CCC, operation windows, LoD, and specificity measurements, initial structures for molecular dynamics simulation, and the colorimetric map of the sensing kit. See DOI: https://doi.org/10.1039/d2sc05837e |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2023 |