
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorocarbyne complexes via electrophilic fluorination of carbido ligands†
Richard A.
Manzano
 and
Anthony F.
Hill
*
and
Anthony F.
Hill
*
Research School of Chemistry, Australian National University, Canberra, Australian Capital Territory ACT 2601, Australia. E-mail: a.hill@anu.edu.au
First published on 7th March 2023
Abstract
The new fluorocarbynes [M(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CF)(CO)2(Tp*)] (M = Mo, W; Tp* = tris(dimethylpyrazolyl)borate) arise from electrophilic fluorination of the lithiocarbynes [M(CLi)(CO)2(Tp*)] with FN(SO2Ph)2. The reactions of [W(CF)(CO)2(Tp*)] with [AuCl(SMe2)] and PhICl2 afford the first μ2-fluorocarbyne complex [WAu(μ-CF)Cl(CO)2(Tp*)] and the first high oxidation state fluorocarbyne [W(CF)Cl2(Tp*)], respectively.
CF)(CO)2(Tp*)] (M = Mo, W; Tp* = tris(dimethylpyrazolyl)borate) arise from electrophilic fluorination of the lithiocarbynes [M(CLi)(CO)2(Tp*)] with FN(SO2Ph)2. The reactions of [W(CF)(CO)2(Tp*)] with [AuCl(SMe2)] and PhICl2 afford the first μ2-fluorocarbyne complex [WAu(μ-CF)Cl(CO)2(Tp*)] and the first high oxidation state fluorocarbyne [W(CF)Cl2(Tp*)], respectively.
Introduction
Isolable terminal fluorocarbyne complexes remain exceedingly rare,1 being limited to those reported by Hughes2 and Ozerov3 (Scheme 1). In both cases, access exploits the enhanced nucleofugacity of fluorine bound to a carbon α-to a transition metal.4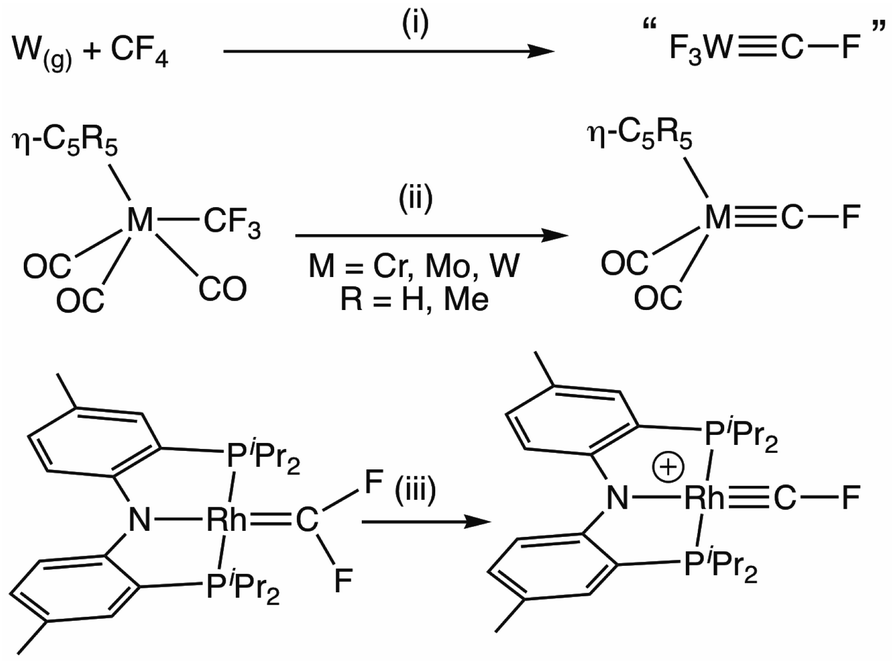 | ||
| Scheme 1 Synthesis of fluorocarbyne complexes via fluoride abstraction from CF4, trifluoromethyl and difluorocarbene ligands. (i) Ar(s), 8K.1 (ii) 2KC8, –2F−, –CO.2 (iii) [H(SiEt3)2][HCB11Cl11], –Et3SiF.3 | ||
Beyond the addition of cobalt carbonyl to [Mo(CF)(CO)2(η5-C5Me5)] to afford a μ3-fluorocarbyne capped cluster [MoCo2(μ3-CF)(CO)8(η5-C5Me5)],2a no subsequent reactivity of fluorocarbyne ligands has been reported. This is in marked contrast to the heavier halocarbynes [M(CX)(CO)2(Tp*)] (M = Mo, W; X = Cl, Br; Tp* = hydrotris(dimethylpyrazolyl)borate),5 the synthetic utility of which has been convincingly demonstrated.6–9 This utility centres on nucleophilic substitution of the halogen that may be spontaneous6–9 or palladium-mediated,10 the latter via the intermediacy of μ-carbido complexes [MPd(μ-C)Br(CO)2(PPh3)2(Tp*)].11 Alternatively, lithium/halogen exchange with nBuLi affords, in situ, the lithiocarbynes [M(CLi)(CO)2(Tp*)] (M = Mo, W) that allow the introduction of carbyne substituents in electrophilic form.9,12 This latter approach has allowed the synthesis of carbyne ligands that bear substituents based on boron and all the elements of groups 14–16 as well as a range of transition metals.11,12
Missing from the otherwise complete series of molybdenum and tungsten halocarbyne complexes are the lightest members [M(CF)(CO)2(Tp*)] (M = W 1a, Mo 2a) that might be seen as analogues of Hughes' fluorocarbynes [M(CF)(CO)2(η5-C5R5)]. These are not available via Lalor's original halocarbyne synthesis due to a range of very specific factors peculiar to that system,5b thereby preventing a complete comparative analysis beyond computational interrogation of the hypothetical complexes [Mo(CX)(CO)2(Tp)] (X = F, Cl, Br, I; Tp = hydrotris(pyrazolyl)borate).13 Herein, we now describe (i) the synthesis of the low-valent fluorocarbyne complexes 1a and 2avia a novel strategy; (ii) the reaction of 1a to afford the first example of a μ2-fluorocarbyne complex 3; (iii) oxidation to afford the first isolable1 high-valent fluorocarbyne complex [W(CF)Cl2(Tp*)](4).
Results and discussion
In the absence of the requisite but yet to be described complexes [M(CF3)(CO)3(Tp*)], Hughes' fluorocarbyne synthetic strategy via 2-electron reduction of trifluoromethyl complexes (Scheme 1) is not applicable. Although thermodynamically feasible, bromide substitution of, e.g., [W(CBr)(CO)2(Tp*)] (1c) by fluoride ([nBu4N]F or AgF) fails under the conditions we have so far explored. We therefore turned to an alternative strategy employing the lithiocarbyne [W(CLi)(CO)2(Tp*)] originally described by Templeton,14 but now more conveniently accessible via a simple lithium/halogen exchange reaction of 1c with nBuLi (THF, −78 °C, Scheme 2).10a,12 Templeton has previously accessed the iodocarbyne [W(C–I)(CO)2(Tp*)] (1d)5bvia iodination of [W(CLi)(CO)2(Tp*)].14 We therefore considered whether such a strategy might also afford fluorocarbyne complexes.
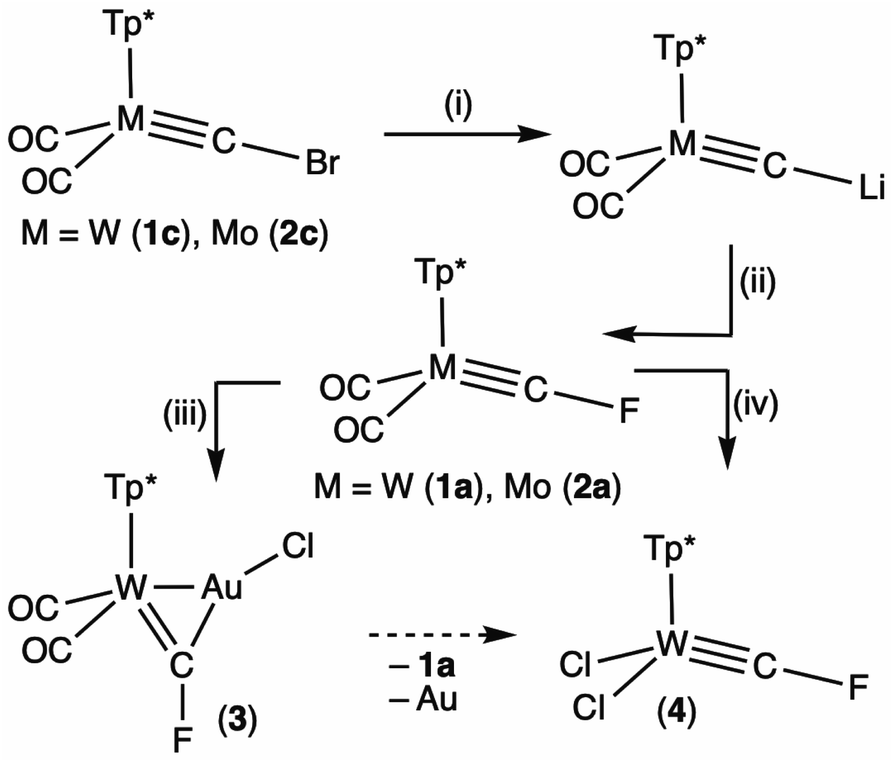 | ||
| Scheme 2 Synthesis of fluorocarbyne complexes via electrophilic fluorination of lithiocarbynes. (i) nBuLi, –nBuBr. (ii) F–N(SO2Ph)2. (iii) [AuCl(SMe2)]. (iv) PhICl2. | ||
Treating the lithiocarbyne [W(CLi)(CO)2(Tp*)], generated in situ, with an electrophilic source of fluorine, viz. FN(SO2Ph)2,15 affords the new fluorocarbyne complex [W(CF)(CO)2(Tp*)] (1a, 59%). Similar treatment of [Mo(CBr)(CO)2(Tp*)] (2c) affords the molybdenum analogue [Mo(CF)(CO)2(Tp*)] (2a, 77%, Scheme 2).
In addition to spectroscopic and electrochemical data (Table 1) 1a was characterised crystallographically to confirm connectivity, however detailed geometric analysis is precluded by positional disorder of isosteric CO and CF ligands (one contributor shown in Fig. 1 inset), as also recognised for Hughes' complexes [M(CF)(CO)2(η5-C5R5)] (M/R = Mo/Me, W/Me, W/H).2 Accordingly, and not surprisingly, crystals of 1a were found to be essentially isomorphous with those of the radical [W(CO)3(Tp*)].16 Crystals of the molybdenum complex 2a, however, led to a precise structural model free of disorder, geometric data for which indicate that the CF ligand exerts a pronounced trans influence on the unique pyrazolyl donor that is reproduced in the computationally optimised geometries of 1a, 2a and the simpler model complex [W(CF)(CO)2(Tp)] (see ESI†). It is a useful feature of the Tp* ligand that in contrast to cyclopentadienyls for which analogies are entertained, the more clearly octahedral geometry allows both structural and computational interrogation of bonding and implications, e.g., the trans influence. Comparative data for F–C(sp) bonds are surprisingly limited to Hughes' (in all but one case disordered) fluorocarbynes2,3 and the unique fluoroethynyl complex [Ru(CCF)(dppe)(η5-C5Me5)] (C–F = 1.324(4) Å).15a Amongst the spectroscopic data of interest, the carbyne gives rise to a doublet resonance in the 13C{1H} NMR spectrum at δC = 200.8 showing coupling to both 19F (1JCF = 528 Hz) and 183W (1JCW = 274.6 Hz) nuclei, the latter being somewhat larger than typically found for other carbyne complexes of pseudo-octahedral tungsten.5c The 19F{1H} NMR spectrum comprises a single resonance (δF = 45.5) straddled by a doublet due to the 183W isotopomer (2JWF = 111.1 Hz).
CR)(CO)2(Tp*)] (M = W, Mo)
| M | R | ν CO cm−1 | k CO N cm−1 | δ WC ppm | 1 J WC Hz | E 0 V |
|---|---|---|---|---|---|---|
| a Measured in CH2Cl2. b k CK = Cotton–Kraihanzel CO force constant. c Measured in CDCl3. d Relative to ferrocene (E1/2 = 0.460 V cf. Ag/Ag+ = 0), measured in CH2Cl2 with 1.0 M [NnBu4][PF6] at 100 mV s−1; see ESI for cyclic and square-wave voltammograms. n.r. = not reported. | ||||||
| W | F (1a) | 1988, 1888 | 15.15 | 200.8 | 275 | 0.795 |
| W | Cl (1b) | 1991, 1902 | 15.29 | 205.7 | 259 | 0.830 |
| W | Br (1c) | 1994, 1905 | 15.33 | 198.0 | 254 | 0.820 |
| W | I (1d)14 | 1992, 1907 | 15.33 | 183.2 | n.r. | — |
| Mo | F (2a) | 2002, 1910 | 15.44 | 193.5 | — | 0.865 |
| Mo | Cl (2b) | 2005, 1921 | 15.54 | 208.7 | — | 0.825 |
| Mo | Br (2c) | 2008, 1924 | 15.59 | 202.5 | — | 0.830 |
| Mo | I (2d)5b | 2009, 1927 | 15.62 | n.r. | — | — |
| W | H14 | 1986, 1891 | 15.16 | 280.6 | 192 | n.r. |
| W | CH3 (ref. 7d and 17) | 1968, 1867 | 14.86 | 289.3 | n.r. | +0.510 |
| W | Ph18 | 1969, 1876 | 14.91 | 277.9 | 187 | n.r. |
| W | CCtBu10c,19 |
1977, 1886 | 15.05 | 250.6 | 199 | +0.761 |
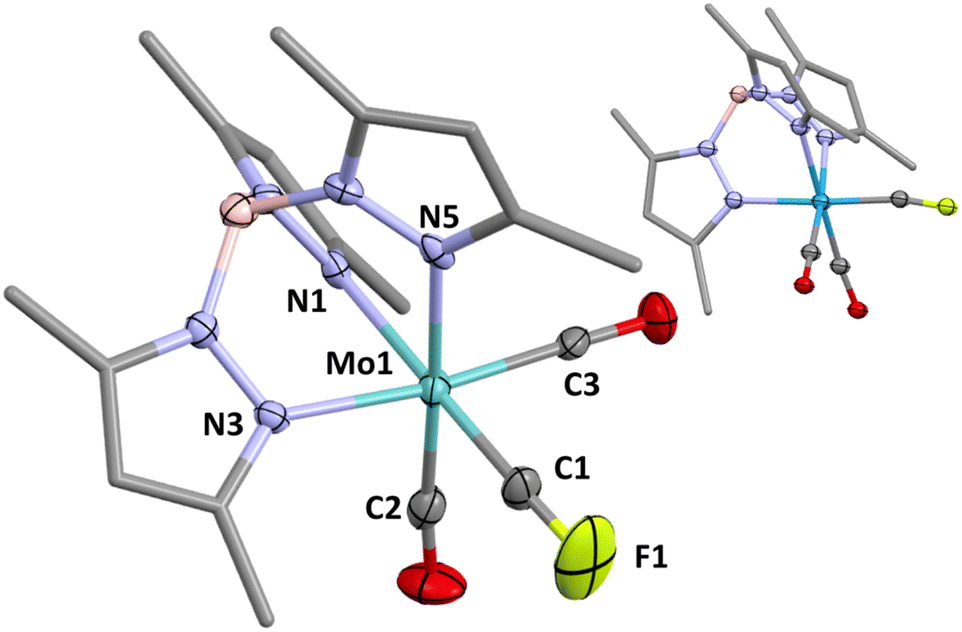 | ||
| Fig. 1 Molecular structure of [Mo(CF)(CO)2(Tp*)] (2a) in a crystal (50% displacement ellipsoids, pyrazolyl groups simplified). Selected bond lengths (Å) and angles (°): Mo1–N1 2.247(5), Mo1–N3 2.221(5), Mo1–N5 2.210(5), Mo1–C1 1.886(7), Mo1–C2 1.938(8), Mo1–C3 1.959(7), F1–C1 1.214(8), F1–C1–Mo1 168.4(7), C1–Mo1–N3 100.3(3), C1–Mo1–N5 98.2(3). Inset = less precise structural model for 1a (positional disorder of CF and CO ligands, one contributor shown). | ||
With the complete series of halocarbynes now in hand, a host of physico-chemical data is available to benchmark the nature of the fluorocarbyne ligand against other halocarbynes and more conventional carbyne ligands in the complexes [W(CR)(CO)2(Tp*)] (R = F, Cl, Br, I, H,14 CH3,17 Ph,18 CCPh,19Table 1) and to validate recent predictions based on the model complexes [Mo(CX)(CO)2(Tp)] (X = F, Cl, Br, I).13
Descending group 17 for the halocarbyne series, the metal centre becomes marginally more electron-poor and less π-basic (increase in νCO and kCO). In the absence of 1a it might seem that the carbyne 13C resonance moves to higher frequency down group 17 showing a normal halogen dependence arising from relativistic spin–orbit coupling.20 The non-conforming position of 1a in the series therefore most likely reflects a competing inverse halogen effect arising from paramagnetic shielding due to magnetic coupling of occupied and unoccupied orbitals, e.g., σ*(C–F) and π*(WC). The oxidation potential is remarkably insensitive to the nature of the halogen, as might be expected from computational studies on the hypothetical series [Mo(CX)(CO)2(Tp)] that show the energy of the metal-based HOMO (orthogonal to the C–X vector) to be essentially in variant.
Whilst μ3-fluorocarbyne complexes are well-known2a,21 there are no previous examples of doubly bridging fluorocarbyne ligands. The addition of the ‘AuCl’ fragment to terminal carbyne complexes has on numerous occasions been shown to afford heterobimetallic μ2-carbynes22 including the recent isolation of bromo- and chlorocarbyne examples.13 Accordingly, the reaction of 1a with [AuCl(SMe2)] was investigated and found to afford the μ2-fluorocarbyne complex [WAu(μ-CF)Cl(CO)2(Tp*)] (3). The resonance for the fluorocarbyne carbon appears at δC = 236.6, to higher frequency of the precursor and displays a reduced coupling to tungsten-183 (1JCW = 143.8 Hz) and fluorine-19 nuclei (1JCF = 454 Hz), consistent with a decrease in s-character for the sp2vs. sp carbon hybridisation and the increase in W–C bond length (1.876(6) Å). The molecular structure (Fig. 2) reveals that the fluorocarbyne ligand adopts an unsymmetrical semi-bridging mode (W1–C1–F1 = 148.7(6)°), as is commonly encountered for carbyne (and carbonyl) ligands bridging between tungsten and d10 metal centres. Therein, the carbyne may be viewed as a Z-type σ-acceptor ligand, i.e., donation from gold(I) to a π-hole on the carbonyl ligand. Similar geometric features are reproduced upon geometry optimisation (ωB97X-D/6-31G*/LANL2Dζ, see ESI†) of the model complex [WAu(μ-CF)Cl(CO)2(Tp)] (3′: W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C = 1.893 Å; Au–C = 2.042 Å, W–Au = 2.829 Å, W–C–Au = 91.8° W–C–F = 149.7°) for which Löwdin bond orders (WC = 1.80, AuC = 0.72) suggest that a dimetallacyclopropene description is useful.
C = 1.893 Å; Au–C = 2.042 Å, W–Au = 2.829 Å, W–C–Au = 91.8° W–C–F = 149.7°) for which Löwdin bond orders (WC = 1.80, AuC = 0.72) suggest that a dimetallacyclopropene description is useful.
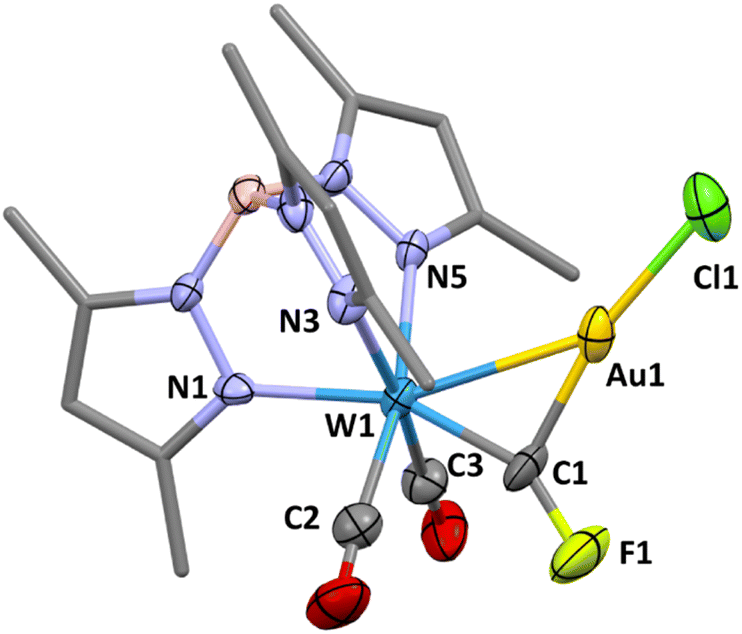 | ||
| Fig. 2 Molecular structure of [WAu(μ-CF)Cl(CO)2(Tp*)] (3) in a crystal (50% displacement ellipsoids, pyrazolyl groups simplified). Selected bond lengths (Å) and angles (°): Au1–W1 2.7974(4), Au1–Cl1 2.268(2), Au1–C1 2.007(8), W1–C1 1.876(6), F1–C1 1.318(7), Cl1–Au1–W1 150.49(6), C1–Au1–W1 42.08(18), C1–Au1–Cl1 167.42(19), W1–C1–Au1 92.1(3), F1–C1–Au1 119.2(5), F1–C1–W1 148.7(6). | ||
The complex 3 is unstable in solution, gradually depositing elemental gold (accompanied by some reformation of 1a) to provide a new purple species identified as the high-valent fluorocarbyne complex [W(CF)Cl2(Tp*)] (4). Once recognised as such, complex 4 could be more expediently and directly prepared from 1a and iodobenzene dichloride. Key spectroscopic features of note for 4 include a resonance for the carbyne carbon (δC = 224.9) that is moved to higher frequency of that for 1a, with a similar 1JCF coupling (512.8 Hz) but augmented 1JWC coupling (316 Hz). The 2JWF coupling (105.1 Hz) observed in the 19F{1H} NMR spectrum is similar to that observed for 1a. Taken together, these suggest retention of octahedral geometry at tungsten and sp-hybridisation at the carbyne carbon. Oxidative interconversion of ‘Fischer’ and ‘Schrock’ classes of carbyne is well established23 but in this case is gratifying in that 4 represents a connection between the exotic matrix-isolated ‘Schrock-type’ carbynes X3MCF (X = F, Cl)1 and tractable low-valent compounds such as 1a, 2a and Hughes' [M(CF)(CO)2(η5-C5R5)] (M = Cr, Mo, W; R = H, Me). For simplicity, the frontier molecular orbitals of interest for the model complexes [W(CF)(CO)2(Tp)] (1a′) and [W(CF)Cl2(Tp)] (4′) are shown in Fig. 3 (those for the full and sterically congested molecules 1a and 4 are provided in the ESI† alongside those for the matrix molecules X3WCF, X = F, Cl, calculated at the same level of theory). In the case of 1a, the HOMO is primarily associated with metal carbonyl back-donation (‘dxy’ taking ‘z’ as the W⋯F vector), explaining why the oxidation potentials (Table 1) are rather insensitive to changes in the carbyne substituent. The HOMO − 1 and HOMO − 2 comprise the two orthogonal π-components of the WC multiple bond for 1a′. For 4′, the manifold of frontier orbitals is rather similar with the notable exception that the ‘dxy’ orbital is now vacant and available for nucleophilic attack. This leaves the near degenerate pair of WC π-bonding orbitals as the highest in energy, as is also the case for the Schrock-type molecules X3WCF. Notably, for 1a′, while the LUMO has both metal and carbyne–carbon character, the real complex 1a is not especially susceptible to hydrolysis (cf. difluorocarbene complexes), easily surviving chromatographic purification without any identifiable formation of the anticipated hydrolysis product [WH(CO)3(Tp*)]. The situation for the high-valent 4′ is intriguing in that, by analogy with chloro- and bromo-carbynes, nucleophilic substitution of the fluoride would seem plausible. Furthermore, oxidation state has been shown to profoundly enhance the susceptibility of difluorocarbene ligands towards nucleophilic attack, as demonstrated for the zero- and divalent ruthenium complexes [Ru0(CF2)(CO)2(PPh3)2]24cf. [RuIICl2(CF2)(CO)(PPh3)2], respectively.25 High-valent Schrock-type carbyne complexes of the form [W(CR)X2(Tp′)] (X = Cl, Br; Tp′ = Tp, Tp*)23c,26 are generally prone to hydrolysis via nucleophilic attack (H2O/HO−) at the metal to provide [W(CHR)Cl(O)(Tp′)]. It is therefore noteworthy and somewhat surprising that complex 4 is comparatively robust, air stable and not readily hydrolysed.
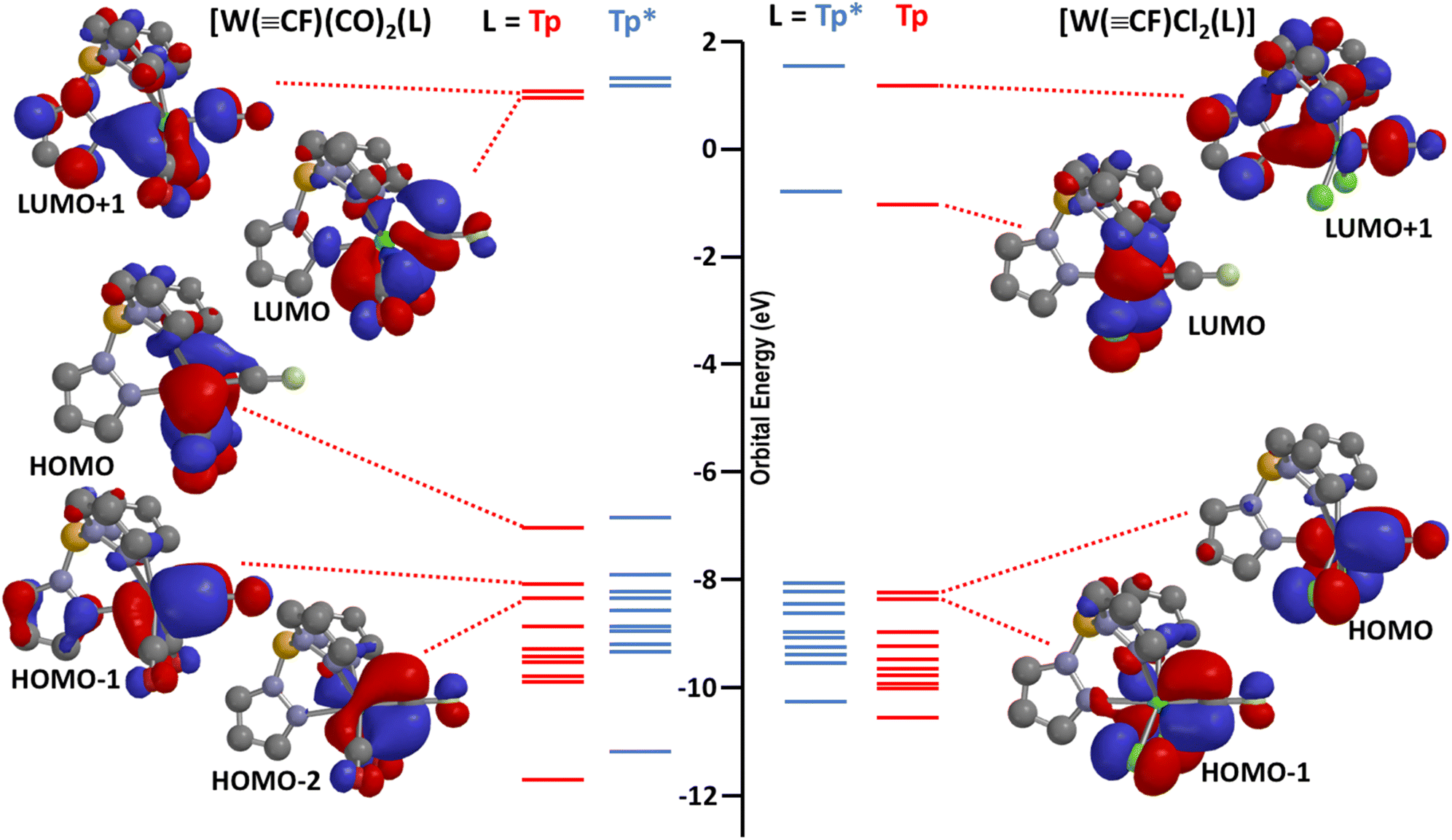 | ||
| Fig. 3 Frontier orbitals (ωB97X-D/6-31G*/LANL2Dζ/gas phase) of interest for the complexes [W(CF)(CO)2(L)] and [W(CF)Cl2(L)] (L = Tp, red, Tp* blue). | ||
In conclusion, electrophilic fluorination of carbido ligands provides a viable route to fluorocarbyne complexes, building upon previous examples of the iodination of terminal carbido ligands14,27 and the chlorination or bromination of bridging μ2-carbido complexes.28 The ‘Fischer-type’ fluorocarbyne [W(CF)(CO)2(Tp*)] may be oxidised to a rare example23d of a high oxidation state halocarbyne [W(CF)Cl2(Tp*)] that is best viewed as a ‘Schrock-type’ carbyne.
Experimental
General experimental details and instrumentation, synthetic methods, spectroscopic data, selected spectra, Cartesian coordinates and computational details are provided in the accompanying ESI.†Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Crystallographic data have been deposited at the CCDC under 2226627 and 2226628 and can be obtained from http://ccdc.cam.ac.uk.Author contributions
RAM was responsible for the design and execution of the experimental research, the acquisition and critical analysis of the characterising data. AFH was responsible for funding acquisition, project conceptualisation and administration, validation and compilation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Australian Research Council (DP190100723 and DP200101222) for funding.Notes and references
- Though not isolable, infrared spectroscopic evidence has been presented for the generation inter alia of complexes of the form F3MCF (M = Cr, Mo, W) in argon matrices:
(a) J. T. Lyon and L. Andrews, Inorg. Chem., 2006, 45, 9858–9863 CrossRef CAS PubMed;
(b) J. T. Lyon, H.-G. Cho and L. Andrews, Organometallics, 2007, 26, 6373–6387 CrossRef CAS;
(c) H.-G. Cho and L. Andrews, Organometallics, 2010, 29, 2211–2222 CrossRef CAS.
- (a) H. Huang, R. P. Hughes, C. R. Landis and A. L. Rheingold, J. Am. Chem. Soc., 2006, 128, 7454–7455 CrossRef CAS PubMed; (b) H. Huang, R. P. Hughes and A. L. Rheingold, Dalton Trans., 2011, 40, 47–55 RSC.
- C. J. Pell, Y. Zhu, R. Huacuja, D. E. Herbert, R. P. Hughes and O. V. Ozerov, Chem. Sci., 2017, 8, 3178–3186 RSC.
- (a) D. L. Reger and M. D. Dukes, J. Organomet. Chem., 1978, 153, 67–72 CrossRef CAS; (b) G. R. Clark, S. V. Hoskins and W. R. Roper, J. Organomet. Chem., 1982, 234, C9–C12 CrossRef CAS; (c) A. M. Crespi and D. F. Shriver, Organometallics, 1985, 4, 1830–1835 CrossRef CAS; (d) S. Anderson, A. F. Hill, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Chem. Commun., 1993, 266–267 RSC; (e) R. P. Hughes, R. B. Laritchev, J. Yuan, J. A. Golen, A. N. Rucker and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 15020–15021 CrossRef CAS PubMed; (f) P. J. Brothers and W. R. Roper, Chem. Rev., 1988, 88, 1293–1326 CrossRef CAS; (g) D. J. Harrson, A. L. Daniels, I. Korobkov and R. T. Baker, Organometallics, 2015, 34(24), 5683–5686 CrossRef.
- (a) T. Desmond, F. J. Lalor, G. Ferguson and M. Parvez, J. Chem. Soc., Chem. Commun., 1983, 457–459 RSC; (b) F. J. Lalor, T. J. Desmond, G. M. Cotter, C. A. Shanahan, G. Ferguson, M. Parvez and B. Ruhl, J. Chem. Soc., Dalton Trans., 1995, 1709–1726 RSC; (c) L. M. Caldwell, Adv. Organomet. Chem., 2008, 56, 1–94 CrossRef CAS.
- (a) T. Desmond, F. J. Lalor, G. Ferguson and M. Parvez, J. Chem. Soc., Chem. Commun., 1984, 75–77 RSC; (b) S. Chaona, F. J. Lalor, G. Ferguson and M. M. Hunt, J. Chem. Soc., Chem. Commun., 1988, 1606–1608 RSC; (c) F. J. Lalor and S. A. O'Neill, J. Organomet. Chem., 2003, 684, 249–265 CrossRef CAS.
- (a) M. Etienne, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1991, 113, 2324–2325 CrossRef CAS; (b) G. M. Jamison, P. S. White and J. L. Templeton, Organometallics, 1991, 10, 1954–1959 CrossRef CAS; (c) B. E. Woodworth and J. L. Templeton, J. Am. Chem. Soc., 1996, 118, 7418–7419 CrossRef CAS; (d) B. E. Woodworth, D. S. Frohnapfel, P. S. White and J. L. Templeton, Organometallics, 1998, 17, 1655–1662 CrossRef CAS; (e) K. C. Stone, P. S. White and J. L. Templeton, J. Organomet. Chem., 2003, 684, 13–19 CrossRef CAS; (f) K. C. Stone, G. M. Jamison, P. S. White and J. L. Templeton, Organometallics, 2003, 22, 3083–3095 CrossRef CAS; (g) K. C. Stone, G. M. Jamison, P. S. White and J. L. Templeton, Inorg. Chim. Acta, 2002, 330, 161–172 CrossRef CAS; (h) A. E. Enriquez and J. L. Templeton, Organometallics, 2002, 21, 852–863 CrossRef CAS.
- (a) L. Weber, I. Schumann, T. Schmidt, H.-G. Stammler and B. Neumann, Z. Anorg. Allg. Chem., 1993, 619, 1759–1764 CrossRef CAS; (b) L. Weber, I. Schumann, H.-G. Stammler and B. Neumann, Chem. Ber., 1994, 127, 1349–1353 CrossRef CAS; (c) L. Weber, I. Schumann, M. H. Scheffer, H.-G. Stammler and B. Neumann, Z. Naturforsch., B: J. Chem. Sci., 1997, 52, 655–662 CrossRef CAS; (d) L. Weber, G. Dembeck, R. Boese and D. Bläser, Chem. Ber., 1997, 130, 1305–1308 CrossRef CAS; (e) L. Weber, G. Dembeck, H.-G. Stammler and B. Neumann, Eur. J. Inorg. Chem., 1998, 579–582 CrossRef CAS; (f) L. Weber, G. Dembeck, H.-G. Stammler, B. Neumann, M. Schmidtmann and A. Müller, Organometallics, 1998, 17, 5254–5259 CrossRef CAS; (g) L. Weber, G. Dembeck, P. Lönneke, H.-G. Stammler and B. Neumann, Organometallics, 2001, 20, 2288–2293 CrossRef CAS; (h) L. Weber, G. Dembeck, R. Boese and D. Bläser, Organometallics, 1999, 18, 4603–4607 CrossRef CAS.
- (a) B. J. Frogley, A. F. Hill and C. S. Onn, Dalton Trans., 2019, 48, 11715–11723 RSC; (b) B. J. Frogley, T. L. Genet, A. F. Hill, C. S. Onn and S. Chee, Dalton Trans., 2019, 48, 7632–7643 RSC; (c) L. M. Caldwell, A. F. Hill, A. D. Rae and A. C. Willis, Organometallics, 2009, 27, 341–345 CrossRef.
- (a) M. I. Bruce, M. L. Cole, M. Gaudio, B. W. Skelton and A. H. White, J. Organomet. Chem., 2006, 691, 4601–4614 CrossRef CAS; (b) R. L. Cordiner, P. A. Gugger, A. F. Hill and A. C. Willis, Organometallics, 2009, 28, 6632–6635 CrossRef CAS; (c) R. A. Manzano and A. F. Hill, Dalton Trans., 2019, 48, 6596–6610 RSC; (d) L. K. Burt, R. L. Cordiner, A. F. Hill, R. A. Manzano and J. Wagler, Chem. Commun., 2020, 56, 5673–5676 RSC; (e) A. L. Colebatch and A. F. Hill, J. Am. Chem. Soc., 2014, 136, 17442–17445 CrossRef CAS PubMed; (f) B. J. Frogley and A. F. Hill, Chem.–Eur. J., 2020, 26, 12125–12128 CrossRef CAS PubMed; (g) A. F. Hill and R. A. Manzano, Angew. Chem., Int. Ed., 2019, 58, 15354–15357 CrossRef CAS PubMed; (h) A. F. Hill and R. A. Manzano, Angew. Chem., Int. Ed., 2019, 58, 7357–7360 CrossRef CAS PubMed.
- L. K. Burt and A. F. Hill, Dalton Trans., 2020, 49, 8143–8161 RSC.
- (a) R. L. Cordiner, A. F. Hill and J. Wagler, Organometallics, 2008, 27, 5177–5179 CrossRef CAS; (b) B. J. Frogley, A. F. Hill, R. Shang, M. Sharma and A. C. Willis, Chem.–Eur. J., 2020, 26, 8819–8827 CrossRef CAS PubMed; (c) B. J. Frogley and A. F. Hill, Chem. Commun., 2018, 54, 2126–2129 RSC; (d) B. J. Frogley and A. F. Hill, Chem. Commun., 2019, 55, 12400–12403 RSC; (e) A. L. Colebatch, B. J. Frogley, A. F. Hill and C. S. Onn, Chem.–Eur. J., 2021, 27, 5322–5343 CrossRef CAS PubMed; (f) B. J. Frogley, A. F. Hill and L. J. Watson, Chem.–Eur. J., 2020, 26, 12706–12716 CrossRef CAS PubMed.
- (a) L. K. Burt and A. F. Hill, Dalton Trans., 2022, 51, 12080–12099 RSC . For further computational studies of fluorocarbyne complexes see ref. 1 and 2 and ; (b) X. Gong, X. Zhang, Q.-S. Li, Y. Xie, R. B. King and H. F. Schaefer, Dalton Trans., 2010, 39, 5242–5253 RSC; (c) X.-M. Liu, C.-Y. Wang, Q.-S. Li, Y. Xie, R. B. King and H. F. Schaefer, Chem.–Eur. J., 2009, 15, 5520–5530 CrossRef CAS PubMed.
- A. E. Enriquez, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 2001, 123, 4992–5002 CrossRef CAS PubMed.
- Selected applications of FN(SO2Ph)2 in organometallic contexts: (a) L. M. Hall, D. P. Tew, N. E. Pridmore, A. C. Whitwood, J. M. Lynam and J. M. Slattery, Angew. Chem., Int. Ed., 2017, 56, 7551–7556 CrossRef CAS PubMed; (b) K. Sünkel, S. Weigand, A. Hoffmann, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy and N. W. Mitzel, J. Am. Chem. Soc., 2015, 137, 126–129 CrossRef PubMed; (c) G. E. Herberich, U. Englert and T. Wirth, Eur. J. Inorg. Chem., 2005, 4924–4935 CrossRef CAS.
- (a) M. B. Hursthouse, B. Piggott and M. Harman, CSD Communication, CCDC 216027, ASIFUD, 2003 Search PubMed; (b) C. C. Philipp, P. S. White and J. L. Templeton, Inorg. Chem., 1992, 31, 3825–3830 CrossRef CAS.
- D. S. Frohnapfel, B. E. Woodward, H. H. Thorp and J. L. Templeton, J. Phys. Chem. A, 1998, 102, 5665–5669 CrossRef CAS.
- K. C. Stone, G. M. Jamison, P. S. White and J. L. Templeton, Inorg. Chim. Acta, 2002, 330, 161–172 CrossRef CAS.
- (a) B. Schwenzer, J. Schleu, N. Burzlaff, C. Karl and H. Fischer, J. Organomet. Chem., 2002, 641, 134–141 CrossRef CAS; (b) A. F. Hill and R. A. Manzano, Adv. Organomet. Chem., 2019, 72, 103–171 CrossRef.
- (a) J. Autschbach, in High Resolution Nuclear Magnetic Resonance Parameters for Understanding Molecules and their Electronic Structure, Vol. 3 of Science & Technology of Atomic, Molecular, Condensed Matter & Biological Systems, ed. R. H. Contreras, Elsevier, Amsterdam, 2013, pp. 69–117 Search PubMed; (b) R. V. Viesser, L. C. Ducati, C. F. Tormena and J. Autschbach, Phys. Chem. Chem. Phys., 2018, 20, 11247–11259 RSC.
- (a) D. Lentz, I. Brudgam and H. Hartl, Angew. Chem., Int. Ed. Engl., 1985, 24, 119–120 CrossRef; (b) W. Schulze, H. Hartl and K. Seppelt, J. Organomet. Chem., 1987, 319, 77–85 CrossRef CAS; (c) D. Lentz and H. Michael, Chem. Ber., 1988, 121, 1413–1416 CrossRef CAS; (d) R. Vilar, S. E. Lawrence, D. M. P. Mingos and D. J. Williams, Chem. Commun., 1997, 285–286 RSC; (e) D. Lentz and H. Michael-Schulz, Inorg. Chem., 1990, 29, 4396–4401 CrossRef CAS.
- (a) G. R. Clark, C. M. Cochrane, W. R. Roper and L. J. Wright, J. Organomet. Chem., 1980, 199, C35–C38 CrossRef CAS; (b) G. A. Carriedo, J. C. Jeffery and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1984, 1597–1603 RSC; (c) G. A. Carriedo, V. Riera, G. Sánchez and X. Solans, J. Chem. Soc., Dalton Trans., 1988, 1957–1962 RSC; (d) C. E. Strasser, S. Cronje and H. G. Raubenheimer, New J. Chem., 2010, 34, 458–469 RSC; (e) A. L. Colebatch and A. F. Hill, Dalton Trans., 2017, 46, 4355–4365 RSC; (f) B. J. Frogley, A. F. Hill and C. S. Onn, Dalton Trans., 2019, 48, 11715–11723 RSC; (g) B. J. Frogley, A. F. Hill and L. J. Watson, Chem. Commun., 2019, 55, 14450–14453 RSC; (h) B. J. Frogley and A. F. Hill, Dalton Trans., 2020, 49, 12390–12400 RSC; (i) X. Zhou, Y. Li, Y. Shao, Y. Hua, H. Zhang, Y.-M. Lin and H. Xia, Organometallics, 2018, 37, 1788–1794 CrossRef CAS.
- (a) A. Mayr and G. A. McDermott, J. Am. Chem. Soc., 1986, 108, 548–549 CrossRef CAS PubMed; (b) A. C. Filippou, C. Wagner, E. O. Fischer and C. Vlilkl, J. Organomet. Chem., 1992, 438, C15–C22 CrossRef CAS; (c) S. Anderson, D. J. Cook, A. F. Hill, J. M. Malget, A. J. P. White and D. J. Williams, Organometallics, 2004, 23, 2552–2557 CrossRef CAS; (d) A. F. Hill and R. Y. Kong, Chem. Commun., 2017, 53, 759–762 RSC.
- G. R. Clark, S. V. Hoskins, T. C. Jones and W. R. Roper, J. Chem. Soc., Chem. Commun., 1983, 719–721 RSC.
- G. R. Clark, S. V. Hoskins and W. R. Roper, J. Organomet. Chem., 1982, 234, C9–C12 CrossRef CAS.
- (a) A. R. Delaney, B. J. Frogley and A. F. Hill, Dalton Trans., 2019, 48, 13674–13684 RSC; (b) A. C. Filippou, P. Hofmann, P. Kiprof, H. R. Schmidt and C. Wagner, J. Organomet. Chem., 1993, 459, 233–247 CrossRef CAS; (c) J. C. Jeffery, J. A. McCleverty, M. D. Mortimer and M. D. Ward, Polyhedron, 1994, 13, 353–356 CrossRef CAS; (d) S. Ahn and A. Mayr, J. Am. Chem. Soc., 1996, 118, 7408–7409 CrossRef CAS.
- A. Reinholdt, T. Vosch, A. Jos and J. Bendix, Angew. Chem., Int. Ed., 2016, 55, 12484–12487 CrossRef CAS PubMed.
- (a) H. J. Barnett and A. F. Hill, Chem. Commun., 2020, 56, 7738–7740 RSC; (b) H. J. Barnett and A. F. Hill, Chem. Commun., 2019, 55, 1734–1737 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic, computational and crystallographic data. CCDC 2226627 and 2226628. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00261f |
| This journal is © The Royal Society of Chemistry 2023 |