DOI:
10.1039/D3SC00181D
(Edge Article)
Chem. Sci., 2023,
14, 2706-2712
Palladium-catalyzed addition of acylsilanes across alkynes via the activation of a C–Si bond†
Received
11th January 2023
, Accepted 12th February 2023
First published on 13th February 2023
Abstract
Palladium-catalyzed addition of a C–Si bond in acylsilanes across the triple bonds in an alkyne bearing a carbonyl group at one terminal is reported. The reaction proceeds with excellent regioselectivity, in which a silyl group is incorporated into the carbon α to the carbonyl group, allowing for straightforward access to a variety of functionalized alkenylsilane derivatives. Catalytic synthesis of indanones by annulation between acylsilanes and alkynes with an identical catalytic system is also reported.
Introduction
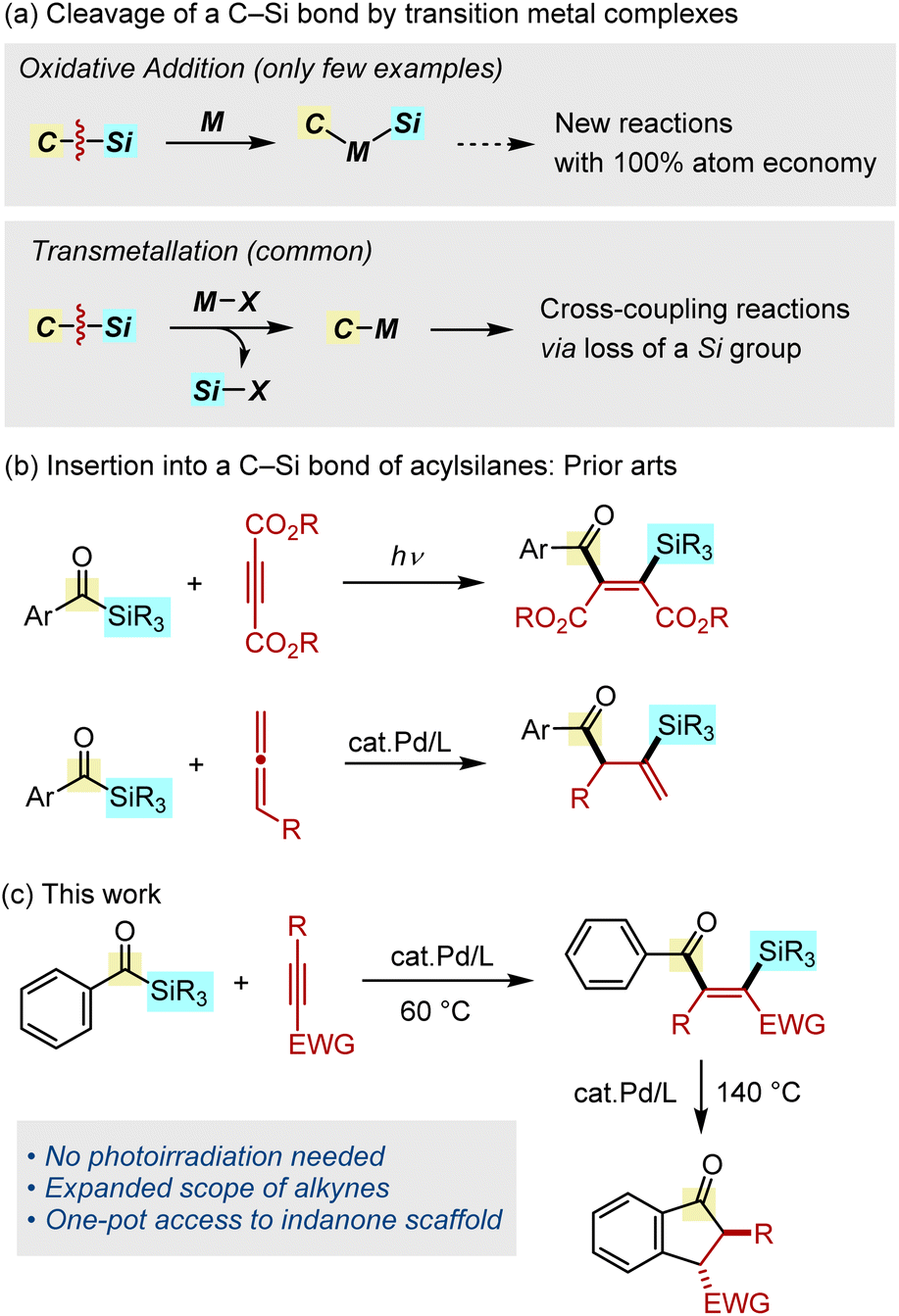
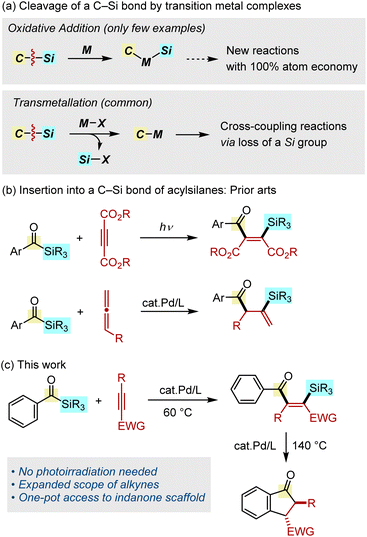
Organosilicon compounds are indispensable molecules that find widespread use in both academia and industry that are related to synthetic organics,1a–c polymers,1d,e materials,1f,g and bioorganic and medicinal chemistry.1h,i The most widely used method for the synthesis of organosilicon compounds is the transition metal-catalyzed hydrosilylation of unsaturated hydrocarbons, in which a H–Si bond is added across alkenes or alkynes.2 On the other hand, the transition metal-catalyzed carbosilylation of unsaturated hydrocarbons, in which a C–Si bond is added in a similar manner, would serves as a powerful method for the synthesis of more complex derivatives from simpler organosilicon feedstocks with an atom economy of 100%. However, the development of such carbosilylation reactions remains a challenging task because the requisite oxidative addition of a C–Si bond by a transition metal complex is an uncommon organometallic process (Fig. 1a, top).3 Although the cleavage of a C–Si bond can occur using complexes bearing an X-type ligand, the mechanism involves a transmetallation, during which the silicon group is eliminated, thus making it unsuitable for use in carbosilylation reactions (Fig. 1a, bottom).1b,c,4 Because of this difficulty, insertion reactions of unsaturated hydrocarbons into a C–Si bond have been limited to strained silacycles (i.e., silacyclopropanes5 and silacyclobutanes6), silyl cyanides7 and intramolecular reactions.8–11
 |
| | Fig. 1 Metal-mediated activation of a C–Si bond and its application to insertion reactions. | |
Our research program has focused on the addition reactions of a C–Si bond in acylsilanes.12 In an early study, Narasaka and coworkers reported on the palladium-catalyzed decarbonylative bis-silylation of dimethyl acetylenedicarboxylate using a bis-silyl ketone, although its application to acylsilanes was not mentioned.13 The photochemical addition of acylsilanes to activated alkynes was also reported, although the applicable alkynes are limited to acetylene dicarboxylates or intramolecular reactions (Fig. 1b, top).14 Although this photocatalytic process involves a formal insertion into a C–Si bond, the reaction proceeds through the generation of a siloxycarbene, cyclopropenation, followed by ring opening. We previously reported on the palladium-catalyzed addition of a C–Si bond in acylsilanes across allenes, in which a C–C double bond can be functionalized with both acyl and silyl groups (Fig. 1b, bottom).15 These studies led us to hypothesize that it might be possible to capture the postulated oxidative addition intermediate (acyl–Pd–SiR3) by alkynes, instead of allenes.16 We report herein on the palladium-catalyzed addition of acylsilanes across alkynes, allowing a C–C triple bond to be functionalized with both acyl and silyl groups (Fig. 1c). Moreover, we also found that the silylacylation products obtained in this insertion process could be directly transformed into indanone derivatives at higher temperature.
Results and discussion
Palladium-catalyzed silylacylation of alkynes
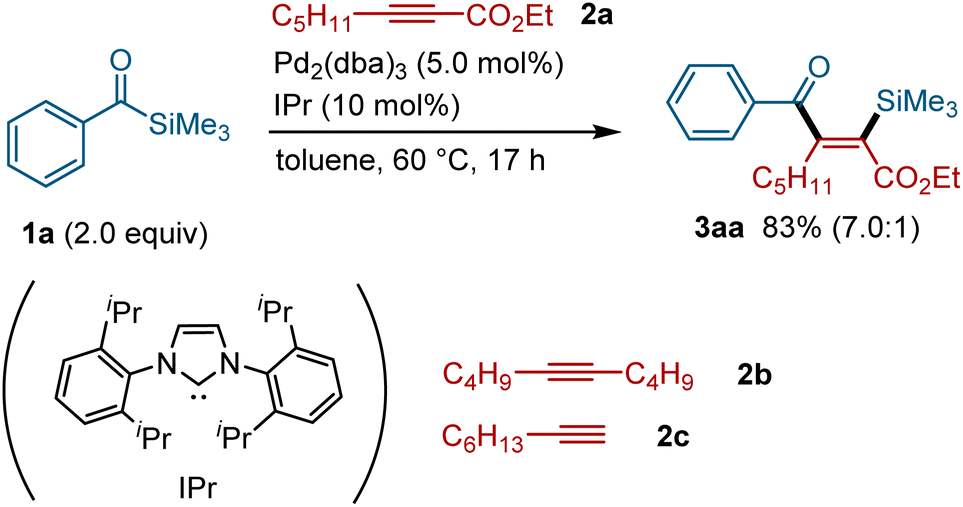
On the basis of our previous studies on the palladium-catalyzed silylacylation of allenes,15 we investigated the possibility of expanding the scope to the reaction with alkynes. After screening a series of alkynes (see the ESI for details†), internal alkynes bearing an ester group at one terminal were found to participate in the silyacylation reaction. Thus, the reaction of acylsilane 1a with alkyne 2a in the presence of Pd2(dba)3 (10 mol% [Pd]) and IPr (10 mol%) at 60 °C afforded the silylacylation product 3aa in 83% yield (Fig. 2). The reaction proceeded with excellent regio- and stereo-selectivity; a silyl group is incorporated into the carbon α to the ester group in 2a, while an acyl group is attached to the β-position with a high syn selectivity. Alkynes without an ester group, such as dec-5-yne (2b) and oct-1-yne (2c), failed to afford the corresponding silylacylation products under these conditions, indicating that an ester group is required for this reaction to proceed.
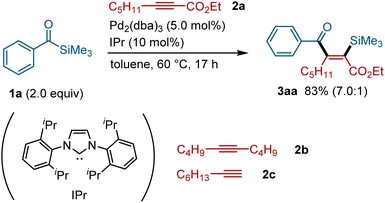 |
| | Fig. 2 Palladium-catalyzed silylacylation of alkynes using acylsilane 1a.a aIsolated yield with a ratio of 3aa and its isomers in the parentheses. | |
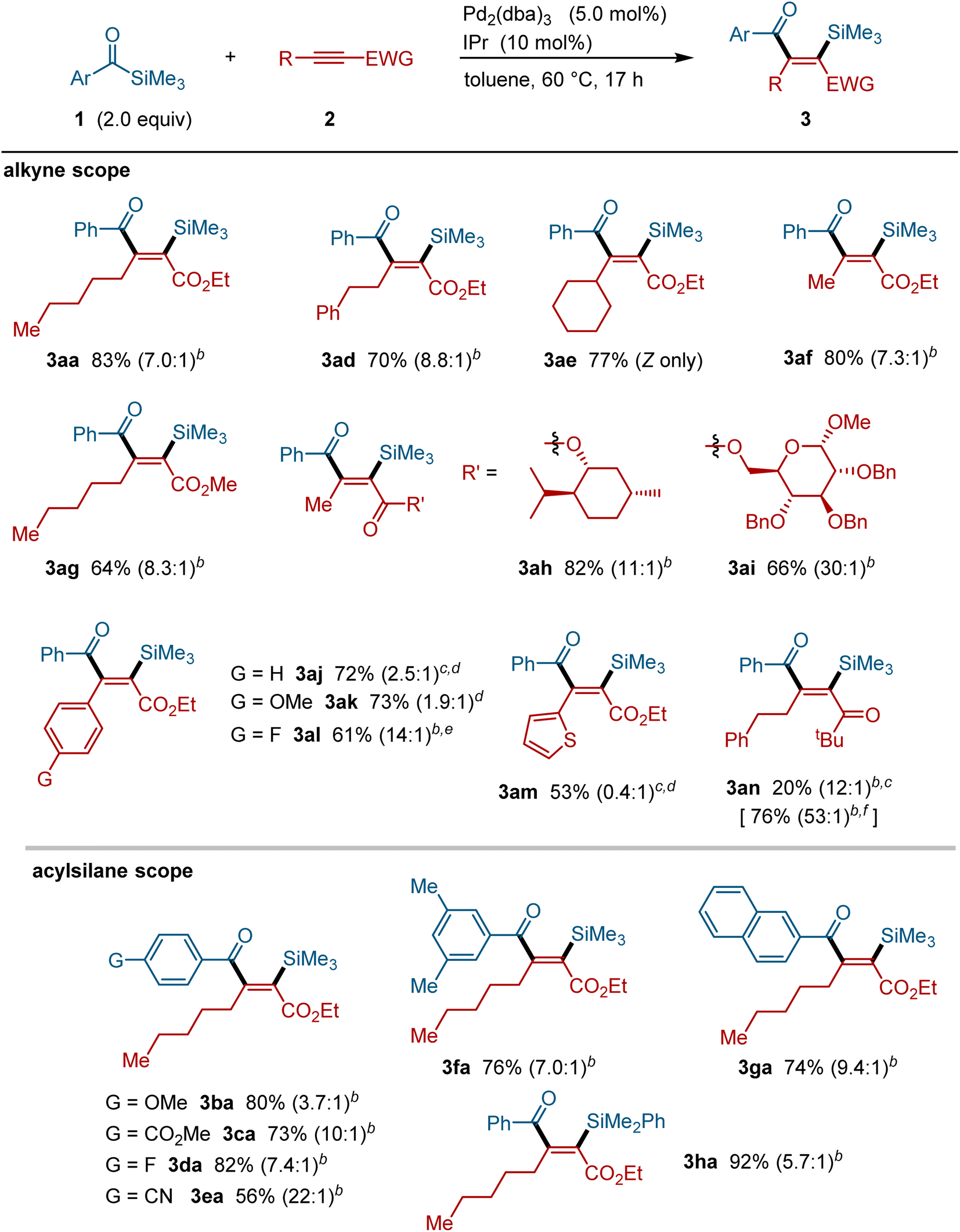
The palladium-catalyzed silylacylation reaction is applicable to a range of alkynes and acylsilanes (Fig. 3). Alkynes bearing alkyl groups, such as phenethyl (i.e., 2d), cyclohexyl (i.e., 2e) and methyl groups (i.e., 2f), successfully participated in this silylacylation reaction, providing the corresponding addition products in a regiospecific manner. Alkynes bearing more elaborate substructures, such as terpenes (i.e., 2h) and sugars (i.e., 2i), also underwent this silylacylation reaction. Moreover, alkynoates bearing aryl (i.e., 2j–2l) and heteroaryl groups such as thiophene (i.e., 2m) were also applicable to this reaction with the corresponding silylacylated products being formed in a regiospecific manner.17 In addition to an ester group, alkynes with a ketone group (i.e., 2n) could also be silylacylated under these palladium-catalyzed conditions. With respect to the acylsilane component, various benzoylsilane derivatives, including those bearing ether (1b), ester (1c), fluoride (1d), cyano (1e), and 2-naphthyl groups (1g), smoothly participated in the silylacylation reaction with a high regio- and stereo-selectivity. Acylsilanes bearing a PhMe2Si group (i.e., 1h) can also be used successfully to form the corresponding silylacylated products.
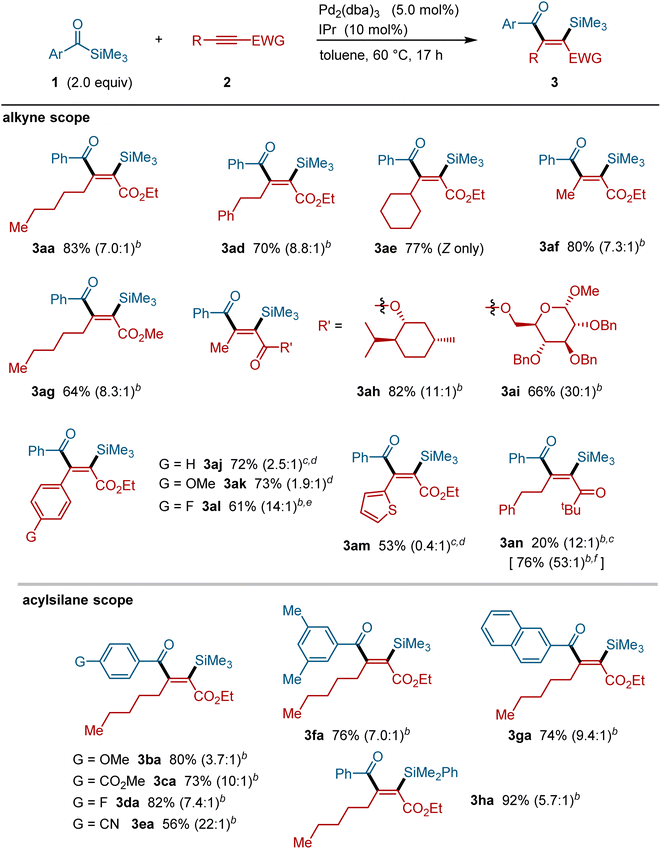 |
| | Fig. 3 Scope of the palladium-catalyzed silylacylation of alkynes.a aAcylsilane (0.40 mmol), alkyne (0.20 mmol), Pd2(dba)3 (0.010 mmol), IPr (0.020 mmol) and toluene (0.60 mL) in a sealed tube at 60 °C for 17 h. bRatios in parentheses refer to the ratios to their minor isomer(s). cPd2(dba)3 (0.0075 mmol) and IPr (0.015 mmol) were used. dRatios in parentheses refer to the ratios of Z/E isomers. ePd2(dba)3 (0.0050 mmol) and IPr (0.010 mmol) were used. fThe isolated yield of desilylated product 3an′. | |
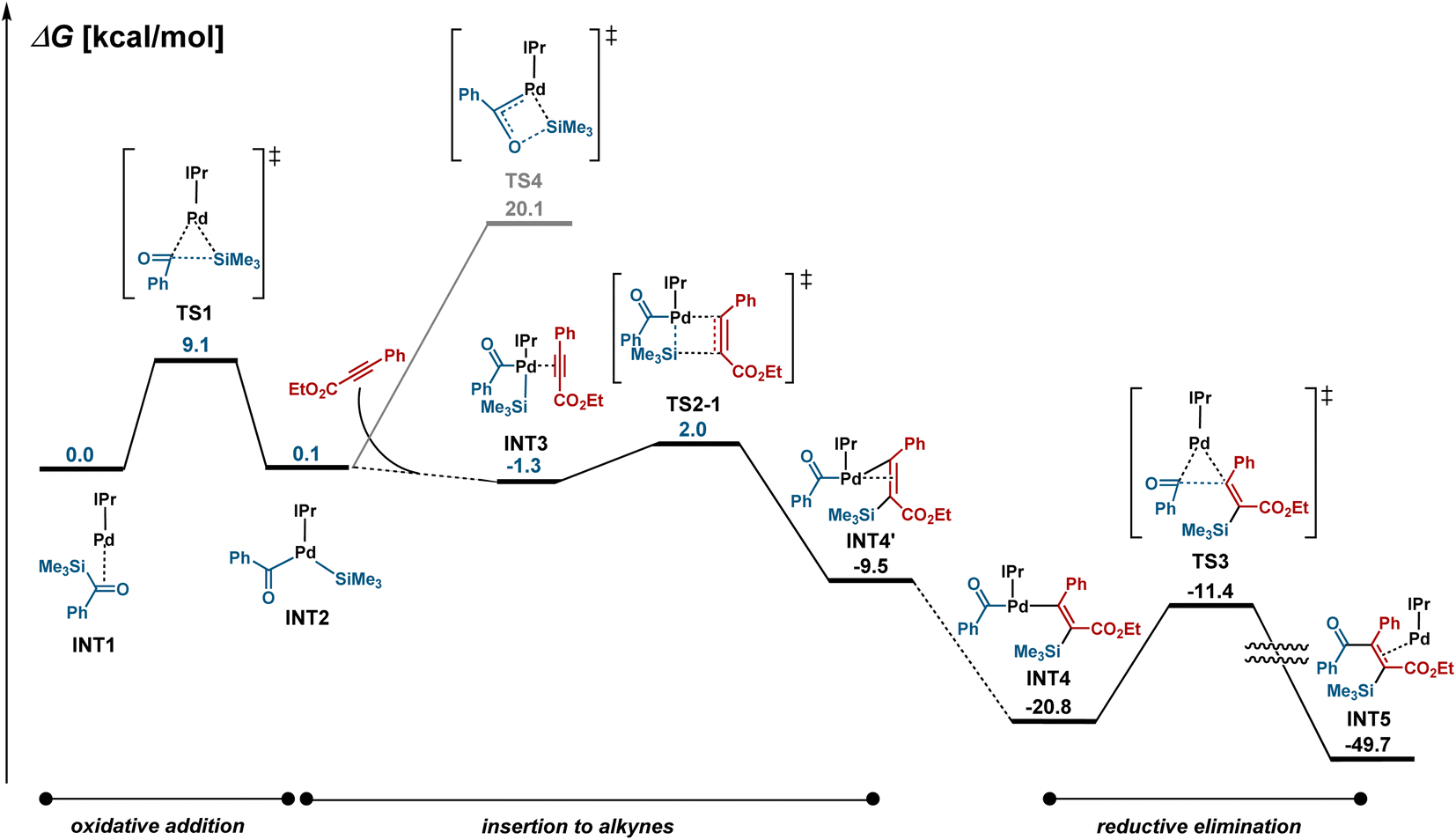
The mechanism for the palladium-catalyzed silylacylation of alkynes was investigated by DFT calculations (Fig. 4). The oxidative addition of acylsilane 1a to Pd–IPr proceeds via a three-centered transition state TS1 with a reasonable activation barrier of 9.1 kcal mol−1 to generate INT2. The subsequent insertion of alkyne 2j could proceed via eight different transition states, depending on which of the Pd–Si and Pd–acyl bonds are added, on the orientation of alkyne 2j, and on whether the silyl and benzoyl ligands are located in a cis or trans configuration. A comparison of all the possible transition states revealed that TS2-1, which leads to alkenylpalladium INT4, was the most stable (see Fig. S3 and S5† for details). Finally, C–C bond-forming reductive elimination viaTS3 from INT4 provides the silylacylated product, which is in agreement with the experimentally observed regio- and stereo-selectivity. We previously reported that the Pd/IPr system catalyzes siloxycyclopropanation reactions through the generation of a siloxycarbene–Pd intermediate when acylsilanes are reacted with terminal alkenes.16 In the case of the reaction with alkynes, the generation of the carbene complex viaTS4 is energetically less favorable by 18.1 kcal mol−1 compared to TS2-1.
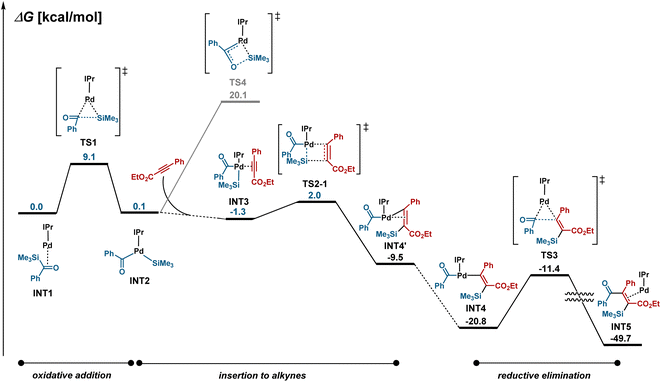 |
| | Fig. 4 DFT calculations.a aComputed energy profiles of the Pd-catalyzed silylacylation pathways of acylsilane 1a with alkyne 2j at the ωB97XD/SDD-6-311+G*//ωB97XD/LANL2DZ-6-31G* level of theory. | |
Palladium-catalyzed indanone synthesis from acylsilanes and alkynes
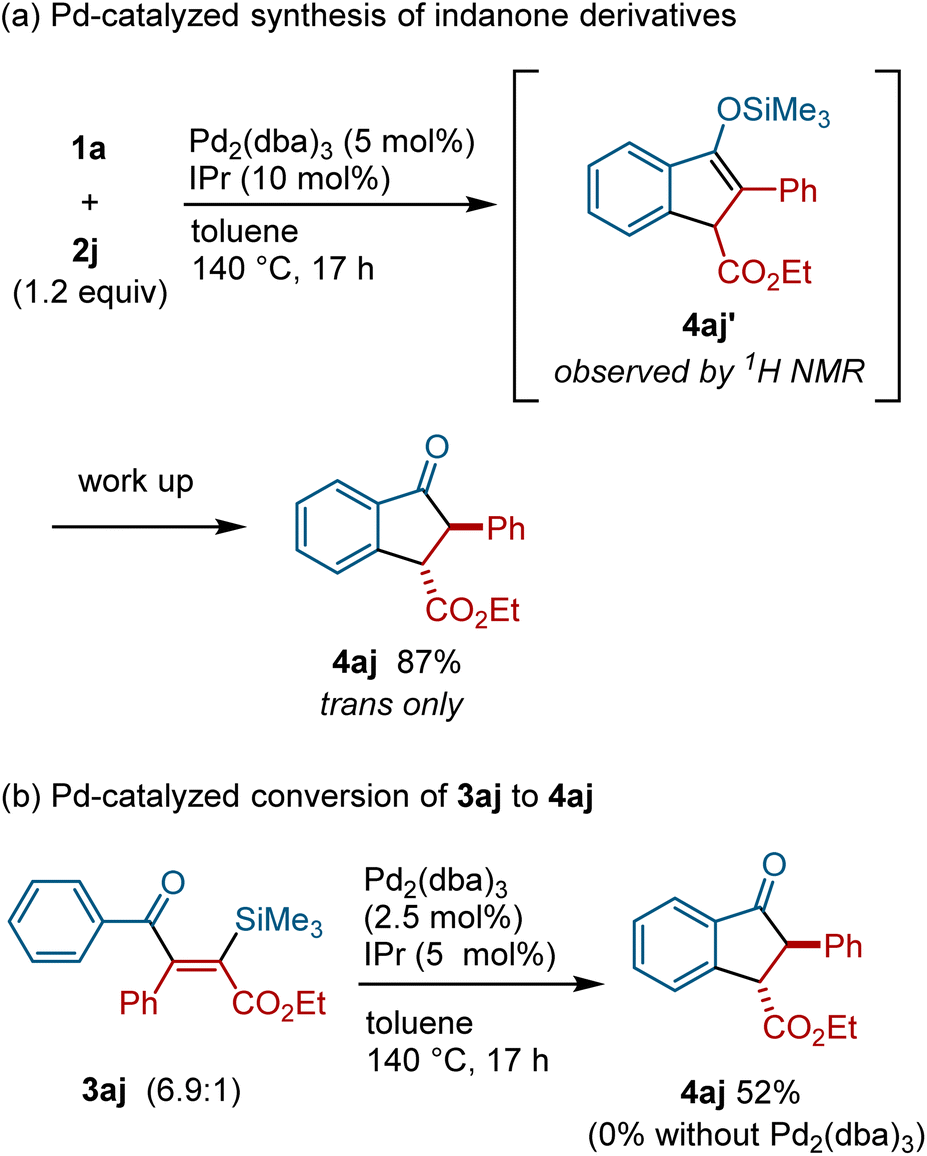
During the course of our investigations of silyacylation reactions of 2j, we discovered that indanone derivative 4aj was formed when the reaction was carried out at higher temperature (see the ESI for the details of optimization†). Since indanone derivatives are compounds of great value as pharmaceuticals and agrochemicals,18 we decided to optimize the reaction conditions that allow for the selective formation of indanone derivatives. Thus, the reaction of acylsilane 1a with alkyne 2j (1.2 equiv.) in the presence of Pd2(dba)3 (10 mol% [Pd]) and IPr (10 mol%) at 140 °C was found to afford the indanone derivative 4aj in 87% yield (Fig. 5a). The analysis of the crude reaction mixture by 1H and 13C NMR as well as GC/MS revealed that silyl enol ether 4aj′ was formed as the primary product, which was converted into 4aj during chromatographic isolation (see the ESI for details†). To examine the relevance of silylacylation product 3aj in the formation of indanone 4aj, 3aj was treated with Pd2(dba)3/IPr at 140 °C, which resulted in the formation of 4aj (Fig. 5b). It is important to note that 3aj was not converted into 4aj in the absence of a Pd(0) catalyst or in the presence of a Pd(II) catalyst, indicating that 4aj was formed from 3ajvia the action of a Pd(0) catalyst (see the ESI for details†).
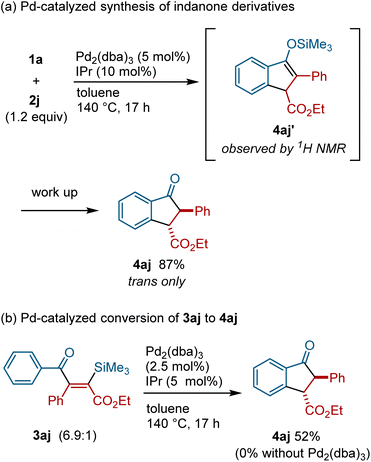 |
| | Fig. 5 The palladium-catalyzed synthesis of indanone derivatives. | |
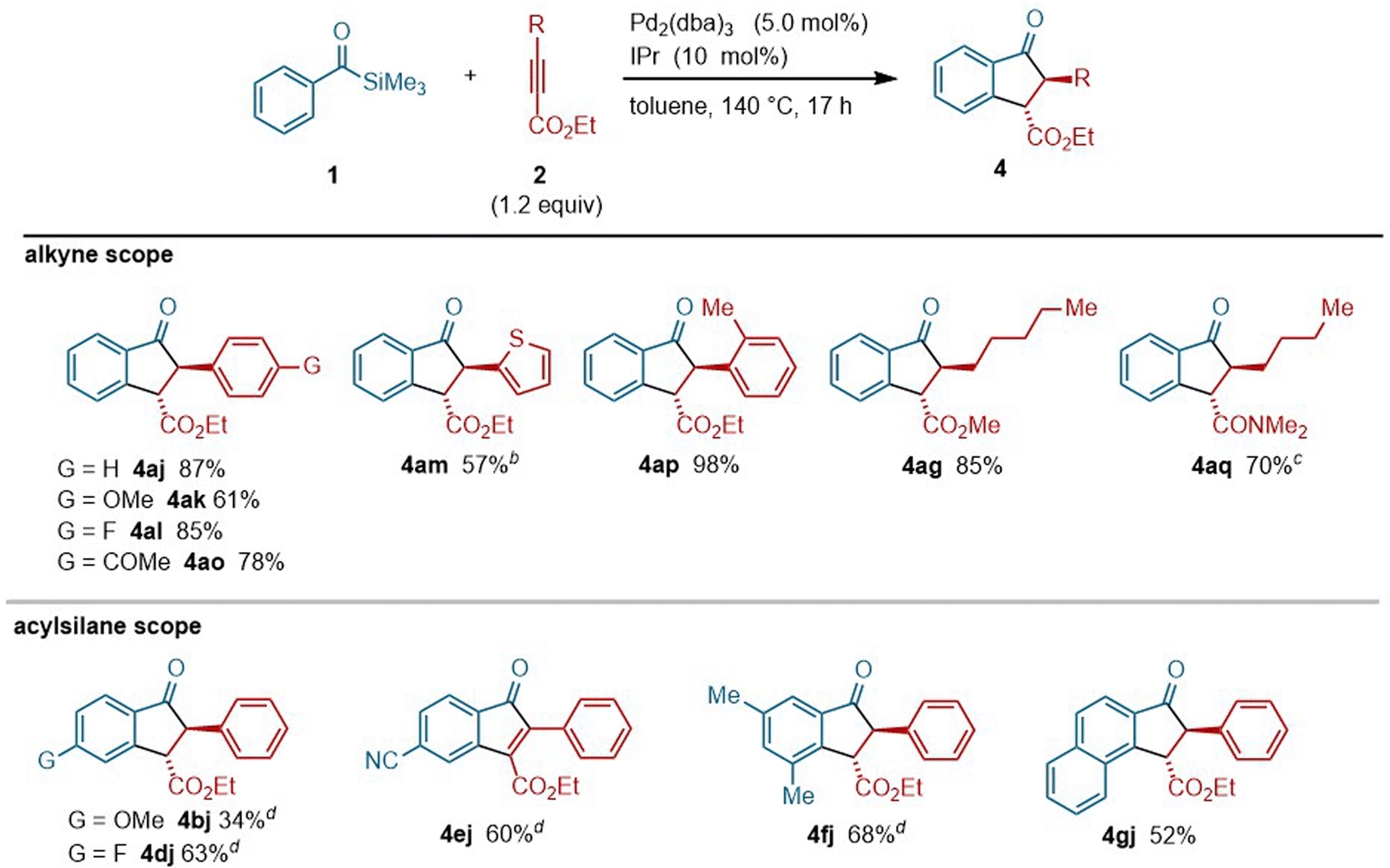
Given the fact that catalytic synthesis of indanones by annulation between acylsilanes and alkynes is unprecedented, the scope of this indanone synthesis was explored (Fig. 6). Aromatic alkynoates bearing electronically diverse substituents, including methoxy (i.e., 2k), fluoro (i.e., 2l) and acetyl groups (i.e., 2o), as well as heteroaryl alkynoate 2m, served as suitable coupling partners to provide the corresponding indanone derivatives. An alkyne bearing a sterically hindered o-tolyl group (i.e., 2p) was also tolerated. Alkyl-substituted alkyne 2g was also applicable to this reaction. Interestingly, an alkyne with an amide group (i.e., 2q) was efficiently converted into indanone 4aq even at 60 °C. Regarding the scope of acylsilanes, a range of derivatives including both electron-rich (1b) and electron-deficient (1d and 1e) acylsilanes successfully reacted with alkyne 2j to provide the corresponding indanone products. With respect to the acylsilane bearing a cyano group (i.e., 1e), indenone 4ej was obtained selectively, rather than an indanone derivative.19 An acylsilane with a 2-naphthyl group (i.e., 1g) was selectively cyclized at a more sterically hindered position of the naphthalene ring to afford the corresponding indanone derivative 4gj.
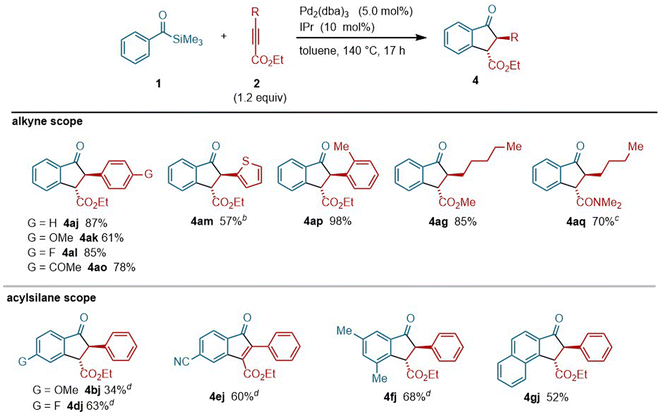 |
| | Fig. 6 Scope of the palladium-catalyzed synthesis of indanone derivatives using acylsilanes and alkynes.a aAcylsilane (0.20 mmol), alkyne (0.24 mmol), Pd2(dba)3 (0.010 mmol), IPr (0.020 mmol) and toluene (0.60 mL) in a sealed tube at 140 °C for 17 h. bIndenone 4am′ was also obtained in 27% yield. cAcylsilane (0.40 mmol), alkyne (0.20 mmol), Pd2(dba)3 (0.0075 mmol), IPr (0.015 mmol) and toluene (0.60 mL) in a sealed tube at 60 °C. A silylacylated Product 3aq was also obtained in 26% yield. dRun at 160 °C. | |
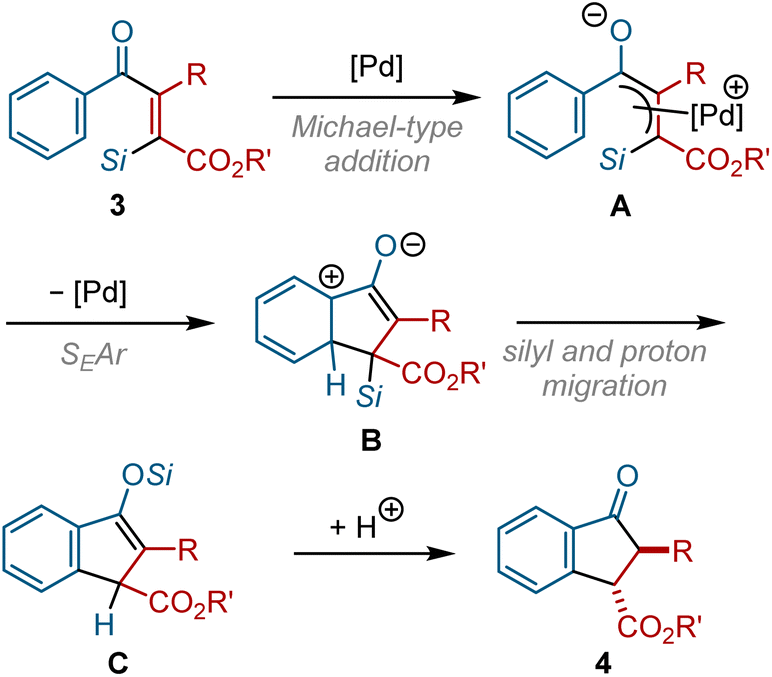
This transformation can be classified as a Nazarov-type cyclization, in which dialkenyl ketones are cyclized to form cyclopentenone derivatives via a pentadienyl cation intermediate.20 Although most Nazarov-type cyclization reactions are promoted by (Lewis) acid catalysts, Tius reported that the non-Lewis acidic Pd(0) can serve as a catalyst for the Nazarov-type cyclization of alkenyl diketoesters.21 Based on the study reported by Tius, a possible mechanism for our indanone synthesis is proposed in Fig. 7. The nucleophilic attack of Pd(0) on the β-carbon of the α,β-unsaturated ketone backbone of 3 generates π-allylpalladium intermediate A. A cationic π-allylpalladium moiety in A is susceptible to an intramolecular nucleophilic attack by a neighboring aromatic ring to form cyclized intermediate B. The subsequent migration of the silyl group and proton in B affords silyl enol ether C, which would then be transformed into indanone derivative 4 by hydrolysis.
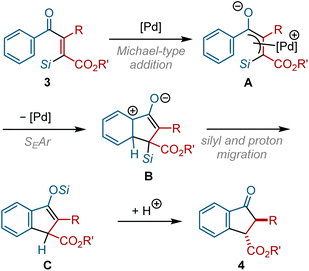 |
| | Fig. 7 Proposed mechanism for indanone synthesis. | |
Conclusions
In summary, we report on the palladium-catalyzed addition of a C–Si bond in acylsilanes across alkynes bearing a carbonyl group at one terminal, leading to the atom economical formation of functionalized alkenylsilanes. The elaborate organosilane derivatives synthesized by this silylacylation reaction can serve as versatile building blocks for use in the construction of more elaborate molecular architectures via silicon-based organic synthesis. Computational studies indicate that the reaction proceeds through the oxidative addition of a C–Si bond to Pd(0). We also report on the palladium-catalytic synthesis of indanones by annulation between acylsilanes and alkynes. Mechanistic studies revealed that Pd(0) can serve as a catalyst for converting the silylacylated products to indanone derivatives. These studies pave the way for the further development of catalytic transformations that proceed via the oxidative addition of a C–Si bond, which is the subject of our ongoing research.
Data availability
Additional data and spectra, NMR spectra, details on the DFT calculations and Cartesian coordinates of the DFT optimized structures can be found in the ESI.†
Author contributions
T. I., T. A., S. S., M. Y. and M. T. conceived the project. T. I., T. A., and S. S. performed the experiments. T. I. performed the quantum chemical calculations and M. Y. supervised it. T. I. and M. T. wrote the manuscript. All authors discussed the results and reviewed the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by KAKENHI (JP21H04682 for MT and JP22K05101 for MY) from MEXT, Japan. We also thank the Instrumental Analysis Center, Graduate School of Engineering, Osaka University, for their assistance with HRMS.
Notes and references
- Selected reviews:
(a) T. H. Chan and I. Fleming, Synthesis, 1979, 761–786 CrossRef CAS;
(b) W.-T. T. Chang, R. C. Smith, C. S. Regens, A. D. Bailey, N. S. Werner and S. E. Denmark, Org. React., 2011, 75, 213–745 CAS;
(c) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS;
(d) K. Tamao and S. Yamaguchi, J. Organomet. Chem., 2000, 611, 5–11 CrossRef CAS;
(e) V. B. Kumar and E. M. Leitao, Appl. Organomet. Chem., 2020, 34, e5402 CrossRef CAS;
(f) Y. Zuo, Z. Gou, W. Quan and W. Lin, Coord. Chem. Rev., 2021, 438, 213887 CrossRef CAS;
(g) Y. Zuo, X. Liang, J. Yin, Z. Gou and W. Lin, Coord. Chem. Rev., 2021, 447, 214166 CrossRef CAS;
(h) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed;
(i) E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- Recent reviews:
(a) L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed;
(b) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- Recent reviews on C–Si bond activation by transition metal complexes:
(a) L. Li, Y. Zhang, L. Gao and Z. Song, Tetrahedron Lett., 2015, 56, 1466–1473 CrossRef CAS;
(b)
M. Tobisu, T. Kodama and H. Fujimoto, Synthetic Applications of C–O and C–E Bond Activation Reactions, in Comprehensive Organometallic Chemistry IV, 2022, vol. 12, pp. 347–420 Search PubMed.
- Transmetallation with acylsilanes has been known:
(a) Y. Obora, Y. Ogawa, Y. Imai, T. Kawamura and Y. Tsuji, J. Am. Chem. Soc., 2001, 123, 10489–10493 CrossRef CAS PubMed;
(b) J. R. Schmink and S. W. Krska, J. Am. Chem. Soc., 2011, 133, 19574–19577 CrossRef CAS PubMed;
(c) S. D. Ramgren and N. K. Garg, Org. Lett., 2014, 16, 824–827 CrossRef CAS PubMed.
-
(a) M. Ishikawa, H. Sugisawa, O. Harata and M. Kumada, J. Organomet. Chem., 1981, 217, 43–50 CrossRef CAS;
(b) D. Seyferth, D. P. Duncan, M. L. Shannon and E. W. Goldman, Organometallics, 1984, 3, 574–578 CrossRef CAS;
(c) H. Saso and W. Ando, Chem. Lett., 1988, 17, 1567–1570 CrossRef;
(d) H. Saso and W. Ando, Tetrahedron, 1989, 45, 1929–1940 CrossRef CAS;
(e) Z. Nevárez and K. A. Woerpel, Org. Lett., 2007, 9, 3773–3776 CrossRef PubMed;
(f) K. M. Buchner and K. A. Woerpel, Organometallics, 2010, 29, 1661–1669 CrossRef CAS PubMed;
(g) G. C. Nandi, Eur. J. Org. Chem., 2021, 587–606 CrossRef CAS.
- Recent reviews:
(a) Q.-C. Mu, J. Chen, C.-G. Xia and L.-W. Xu, Coord. Chem. Rev., 2018, 374, 93–113 CrossRef CAS;
(b) J. Huang, F. Liu, X. Wu, J.-Q. Chen and J. Wu, Org. Chem. Front., 2022, 9, 2840–2855 RSC.
-
(a) N. Chatani, T. Takeyasu, N. Horiuchi and T. Hanafusa, J. Org. Chem., 1988, 53, 3539–3548 CrossRef CAS;
(b) P. Hansjacob, F. R. Leroux, V. Gandon and M. Donnard, Angew. Chem., Int. Ed., 2022, 61, e202200204 CrossRef CAS PubMed.
-
(a) R. Shintani, H. Kurata and K. Nozaki, Chem. Commun., 2015, 51, 11378–11381 RSC;
(b) Q. Yang, L. Liu, Y. Chi, W. Hao, W.-X. Zhang and Z. Xi, Org. Chem. Front., 2018, 5, 860–863 RSC.
- The Lewis acid-mediated carbosilylation of related activated silanes has been reported, but via a carbocation intermediate. Selected examples:
(a) N. Asao, E. Yoshikawa and Y. Yamamoto, J. Org. Chem., 1996, 61, 4874–4875 CrossRef CAS;
(b) E. Yoshikawa, V. Gevorgyan, N. Asao and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 6781–6786 CrossRef CAS;
(c) N. Asao, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 3797–3798 CrossRef CAS;
(d) N. Asao, T. Shimada, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 10899–10902 CrossRef CAS PubMed.
- Introduction of a carbon-based substituent and a silyl group into unsaturated hydrocarbons is also possible by three-component coupling with a silyl nucleophile and a carbon electrophile. Selected examples:
(a) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487–11490 CrossRef CAS PubMed;
(b) R. Shintani, H. Kurata and K. Nozaki, J. Org. Chem., 2016, 81, 3065–3069 CrossRef CAS PubMed;
(c) T. Iwamoto, T. Nishikori, N. Nakagawa, H. Takaya and M. Nakamura, Angew. Chem., Int. Ed., 2017, 56, 13298–13301 CrossRef CAS PubMed;
(d) Q. Chen, Z. Li and Y. Nishihara, Org. Lett., 2022, 24, 385–389 CrossRef CAS PubMed;
(e) W. Bernhard, I. Fleming and D. Waterson, J. Chem. Soc., Chem. Commun., 1984, 28–29 RSC;
(f) Y. Obora, Y. Tsuji and T. Kawamura, J. Am. Chem. Soc., 1995, 117, 9814–9821 CrossRef CAS;
(g) D. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858–1862 CrossRef CAS PubMed;
(h) W. Zheng, Y. Xu, H. Luo, Y. Feng, J. Zhang and L. Lin, Org. Lett., 2022, 24, 7145–7150 CrossRef CAS PubMed;
(i) J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CAS;
(j) M.-Y. Wu, F.-Y. Yang and C.-H. Cheng, J. Org. Chem., 1999, 64, 2471–2474 CrossRef CAS;
(k) F.-Y. Yang, M. Shanmugasundaram, S.-Y. Chuang, P.-J. Ku, M.-Y. Wu and C.-H. Cheng, J. Am. Chem. Soc., 2003, 125, 12576–12583 CrossRef CAS PubMed;
(l) Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706–17709 CrossRef CAS PubMed.
- Catalytic addition of a Si-heteroatom bond across unsaturated hydrocarbons is known. Selected reviews:
(a) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed;
(b) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS PubMed.
- Reviews on acylsilanes:
(a) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540–8571 RSC;
(b) D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS.
- H. Sakurai, M. Yamane, M. Iwata, N. Saito and K. Narasaka, Chem. Lett., 1996, 25, 841–842 CrossRef.
-
(a) H.-J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F.-F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157–2161 CrossRef CAS;
(b) P. Becker, D. L. Priebbenow, H.-J. Zhang, R. Pirwerdjan and C. Bolm, J. Org. Chem., 2014, 79, 814–817 CrossRef CAS PubMed.
- T. Inagaki, S. Sakurai, M. Yamanaka and M. Tobisu, Angew. Chem., Int. Ed., 2022, 61, e202202387 CrossRef CAS PubMed.
- When alkenes were used instead of allenes, siloxycyclopropanation proceeds, possibly via the formation of a palladium-siloxycarbene intermediate: S. Sakurai, T. Inagaki, T. Kodama, M. Yamanaka and M. Tobisu, J. Am. Chem. Soc., 2022, 144, 1099–1105 CrossRef CAS PubMed.
- The products derived from aromatic alkynoates were prone to undergo E/Z isomerization under the catalytic conditions. A higher Z/E selectivity can be obtained by shortening the reaction time at the expense of chemical yield. For example, 3aj was obtained in 53% yield with a Z/E ratio of 31/1, when the reaction was terminated at 4 h. See the ESI† for details.
- Recent reviews on 1-indanones:
(a) M. Turek, D. Szczęsna, M. Koprowski and P. Bałczewski, Beilstein J. Org. Chem., 2017, 13, 451–494 CrossRef CAS PubMed;
(b) S. A. Patil, R. Patil and S. A. Patil, Eur. J. Med. Chem., 2017, 138, 182–198 CrossRef CAS PubMed;
(c) S. Das and A. Dutta, New J. Chem., 2021, 45, 4545–4568 RSC.
-
1H NMR analysis of the crude reaction mixture after 17 h indicated that the corresponding indanone product was similarly formed. However, upon workup and chromatographic isolation, the indanone product had been completely converted into the indenone 4ej. A palladium residue might promote aerobic oxidation of this specific indanone product due to an increased acidity of the α-hydrogen of the ketone moiety. For related oxidation, see: T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef CAS PubMed.
- Selected recent reviews on Nazarov cyclization:
(a) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2017, 15, 8245–8269 RSC;
(b) A. V. Yadykov and V. Z. Shirinian, Adv. Synth. Catal., 2020, 362, 702–723 CrossRef CAS.
-
(a) N. Shimada, C. Stewart, W. F. Bow, A. Jolit, K. Wong, Z. Zhou and M. A. Tius, Angew. Chem., Int. Ed., 2012, 51, 5727–5729 CrossRef CAS PubMed;
(b) Z. Zhou and M. A. Tius, Angew. Chem., Int. Ed., 2015, 54, 6037–6040 CrossRef CAS PubMed;
(c) K. Kitamura, N. Shimada, C. Stewart, A. C. Atesin, T. A. Ateşin and M. A. Tius, Angew. Chem., Int. Ed., 2015, 54, 6288–6291 CrossRef CAS PubMed;
(d) T. A. Ateşin, G. M. Martinez and D. Flores, Organometallics, 2017, 36, 3589–35961 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Mamoru
Tobisu
*b and
Mamoru
Tobisu