
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmpowering boronic acids as hydroxyl synthons for aryne induced three-component coupling reactions†
Rong
Fan‡
a,
Shihan
Liu‡
 b,
Qiang
Yan
a,
Yun
Wei
a,
Jingwen
Wang
a,
Yu
Lan
*bc and
Jiajing
Tan
*a
b,
Qiang
Yan
a,
Yun
Wei
a,
Jingwen
Wang
a,
Yu
Lan
*bc and
Jiajing
Tan
*a
aDepartment of Organic Chemistry, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: tanjj@mail.buct.edu.cn
bChongqing Key Laboratory of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, China. E-mail: lanyu@cqu.edu.cn
cZhengZhou JiShu Institute of AI Science, Zhengzhou 450000, China
First published on 14th March 2023
Abstract
Boronic acids have become one of the most prevalent classes of reagents in modern organic synthesis, displaying various reactivity profiles via C–B bond cleavage. Herein, we describe the utilization of a readily available boronic acid as an efficient surrogate of hydroxide upon activation via fluoride complexation. The hitherto unknown aryne induced ring-opening reaction of cyclic sulfides and three-component coupling of fluoro-azaarenes are developed to exemplify the application value. Different from metal hydroxides or water, this novel hydroxy source displays mild activation conditions, great functionality tolerance and structural tunability, which shall engender a new synthetic paradigm and in a broad context offer new blueprints for organoboron chemistry. Detailed computational studies also recognize the fluoride activation mode, provide in-depth insights into the unprecedented mechanistic pathway and elucidate the reactivity difference of ArB(OH)xFy complexes, which fully support the experimental data.
Introduction
Boronic acids (BAs) have found myriad applications in modern organic synthesis as they can be readily converted to various functionalities.1 Aside from versatile transformations, their synthetic merit is further emphasized by bench stability, low toxicity and operational simplicity. Specifically, the relatively weak C–B bonds (323 kJ mol−1) in BAs have played a major synthetic role as carbon nucleophiles (Scheme 1).2 For instance, the Suzuki–Miyaura coupling reaction, a “household” name in both academic and industrial settings, employed aryl halides and BAs to forge C–C bonds with high efficiency and great robustness.3 Other transformations including Chan–Lam,4 Matteson type5 and Petasis reaction6 have also been extensively investigated and advanced to applications. The recent resurgence of visible-light photochemistry was also fueled by the progress that BAs and their derivatives could serve as radical precursors through C–B bond homolysis.7 In particular, these remarkable achievements relied on the addition of a suitable Lewis base (LB) to activate BAs toward C–B bond cleavage through the formation of anionic boron-centered “ate” complexes.8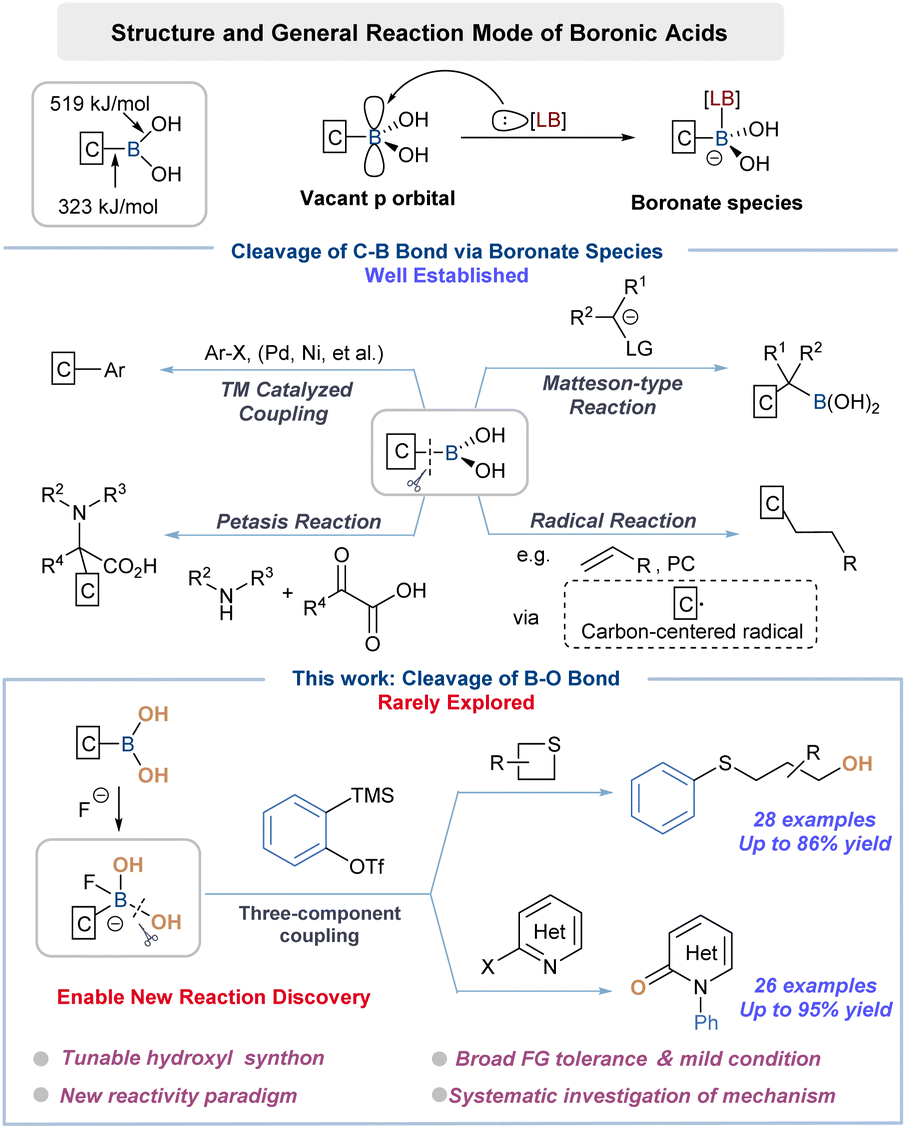 | ||
| Scheme 1 Reactivity of boronic acids: past and present. | ||
Despite the aforementioned achievements, the strategic use of BA through B–O bond cleavage has been very rare, which might be attributed to the much higher B–O bond energy than that of the C–B bond (519 vs. 323 kJ mol−1).2a In consideration of the synthetic advantage of BAs, a systematic investigation on employing BAs as a –OH source would be therefore of particular interest, which ought to expand the reactivity profile as well as the application value of such reagents, and even advance the fundamental knowledge of organoboron chemistry. In an attempt to fill the apparent synthetic gap, we recognized that identifying a suitable activator was crucial to switch the reactivity trend of BAs. From a conceptual standpoint, the leveraged activator ought to form a stronger bond with the boron center to elicit target behavior and not compete with desired hydroxylation reactions while displaying good functional group tolerance. Indeed, our research plan was initially guided by the recognition that aromatic BAs occasionally served as the affinity handle of fluorescent probes for fluoride ions.9 Naturally, we then questioned if the strong complexation of fluoride with boron (BDE B–F 646 kJ mol−1) would necessitate our design, ensuring activation while forcing the hydroxylation reactivity from the “ate” complex. The only available example, nonetheless, focused solely on looking for similarities in reactivities between BA and water rather than differences, letting alone detailed mechanistic studies.10 Hence, another ultimately significant task associated with our proposition was to fully understand the chemical reactivity character, mechanism of action and structure–activity relationship (SAR) of such boronate species.
In addition to the aforementioned considerations, our group's systematic research on exploring zwitterion chemistry to engage in a multicomponent reaction eventually inspired us to choose such a platform to thoroughly investigate our working hypothesis.11 Note that we have established the nucleophilicity scale on these transformations, which ought to serve as a precise tool to map the whole reactivity profile of a B–F “ate” complex through comparative studies with other counterparts.12 Besides being the “tailor-made” activator for BAs, fluorides fulfilled an additional task in facilitating the generation of aryne species from Kobayashi precursors.13,14 Despite recent efforts, we herein would like to report the successful execution of these ideas, and present a proof-of-concept use of BAs as a tunable hydroxy source in unprecedented aryne-based multicomponent coupling reactions. DFT calculations fully support the experimental data and shed light on the activation mode along with distinct mechanistic underpinning involving a B–F ate complex. In addition to classic C–B bond cleavage manifolds, a variety of new discovery can be envisaged through the expansion of BA chemistry and zwitterion synthesis toolkits.
Results and discussion
To test the hypothesis, our investigation commenced with the model reaction of o-(trimethylsilyl)phenyl triflates (1a) and thietane (2a) with phenyl boronic acid (3a). To our delight, the ring-opening hydroxylation product 4a was obtained in moderate yield without engaging any extra reagents to assist the B–O bond cleavage (Table 1A, entries 1–2). Meanwhile, ether (4a′) was observed as the side product albeit in a small amount. We speculated that it was probably formed via the ring opening of electrophilic aryne activated 4a with the in situ generated alkoxide.15 To our delight, the addition of cesium carbonate (Cs2CO3) and varying the temperature could significantly inhibit such side products. Various mixtures of fluoride sources and cesium carbonate were then screened, among which a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of CsF and Cs2CO3 improved the overall yield of 4a to 74% (Table 1A, entries 3–6). Lowering the temperature to −10 °C completely suppressed the formation of side product 4a′, and the yield of 4a was improved to 87% yield (Table 1A, entry 7). Finally, other known hydroxy sources were examined, and the obtained results demonstrated the distinct reactivity of BAs (Table 1B). In the presence of water as the 3rd component, a ring-opening fluorination product11d was obtained as the major one through sulfonium intermediates under otherwise identical conditions.16,17 Clearly, this platform reaction served as an evaluation criterion of the nucleophilic scale to aid our understanding of the nature of such boronate complexes. When employing potassium hydroxide, lithium hydroxide or cesium hydroxide monohydrate as another option, we observed a significant loss of yield, which could be possibly attributed to the decomposition caused by their strong basicity.
:2 ratio of CsF and Cs2CO3 improved the overall yield of 4a to 74% (Table 1A, entries 3–6). Lowering the temperature to −10 °C completely suppressed the formation of side product 4a′, and the yield of 4a was improved to 87% yield (Table 1A, entry 7). Finally, other known hydroxy sources were examined, and the obtained results demonstrated the distinct reactivity of BAs (Table 1B). In the presence of water as the 3rd component, a ring-opening fluorination product11d was obtained as the major one through sulfonium intermediates under otherwise identical conditions.16,17 Clearly, this platform reaction served as an evaluation criterion of the nucleophilic scale to aid our understanding of the nature of such boronate complexes. When employing potassium hydroxide, lithium hydroxide or cesium hydroxide monohydrate as another option, we observed a significant loss of yield, which could be possibly attributed to the decomposition caused by their strong basicity.
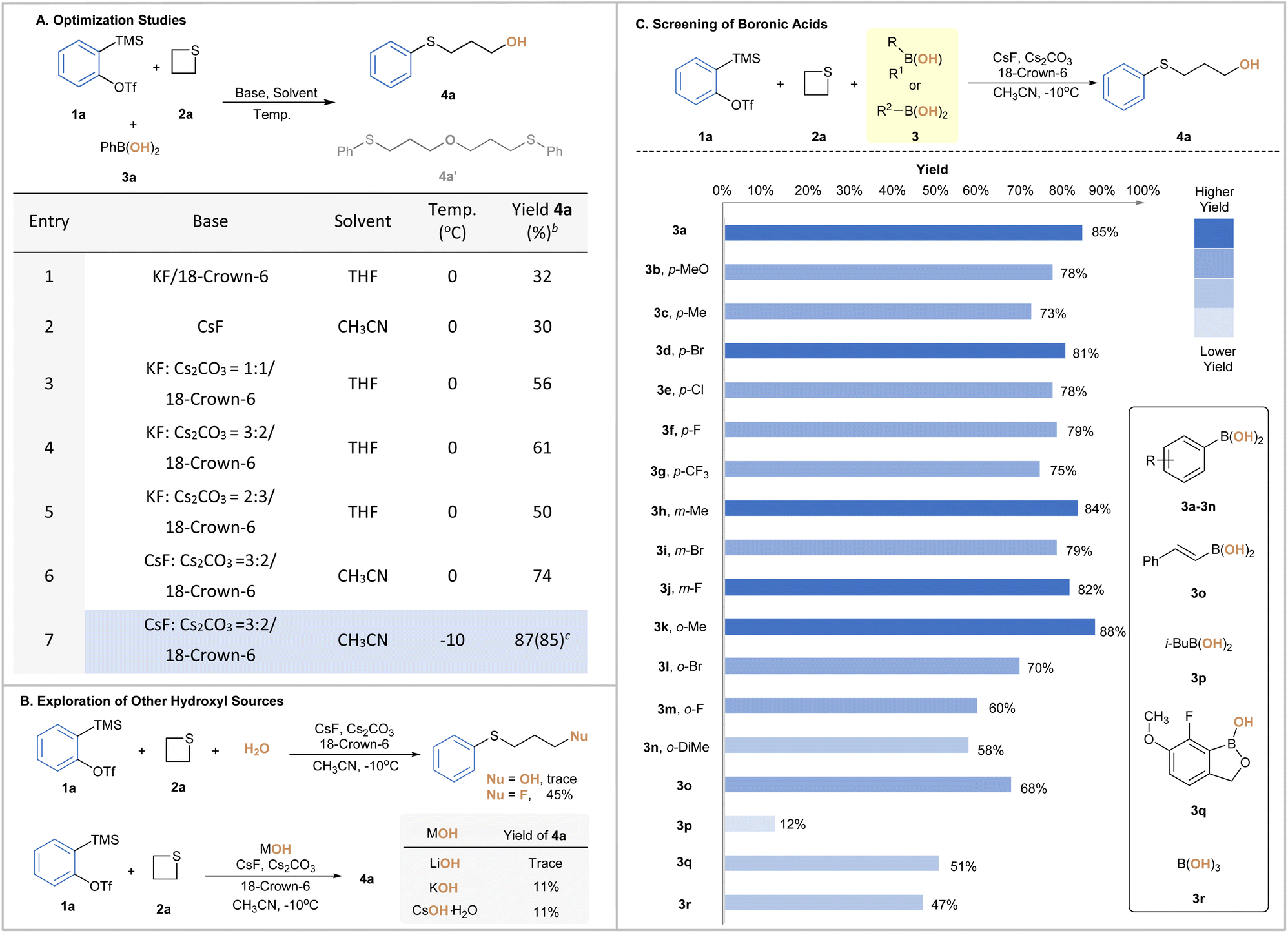
To unveil the SAR, a diverse array of BAs was then screened. As shown in Table 1C, aromatic BAs bearing either electron-donating or -withdrawing groups with different substitution patterns were all viable, among which 2-methylphenylboronic acids (3k) offered optimal results.18 BAs with either alkenyl or aliphatic backbones proved to be less suitable under similar reaction conditions, which was presumably due to the poor affinity of the boron center at different levels. Finally, the use of benzoxaborole (3q) and boric acid (3r) also gave 4a in 51% and 47% yield, respectively. Clearly, varying electronic and steric parameters around the boron center could effectively tune the reactivity of BAs, providing further benefits to our strategy. In consideration of the compromise between the reaction outcome and cost effectiveness, 3a was eventually chosen as the hydroxy-transfer precursors in the subsequent investigations.
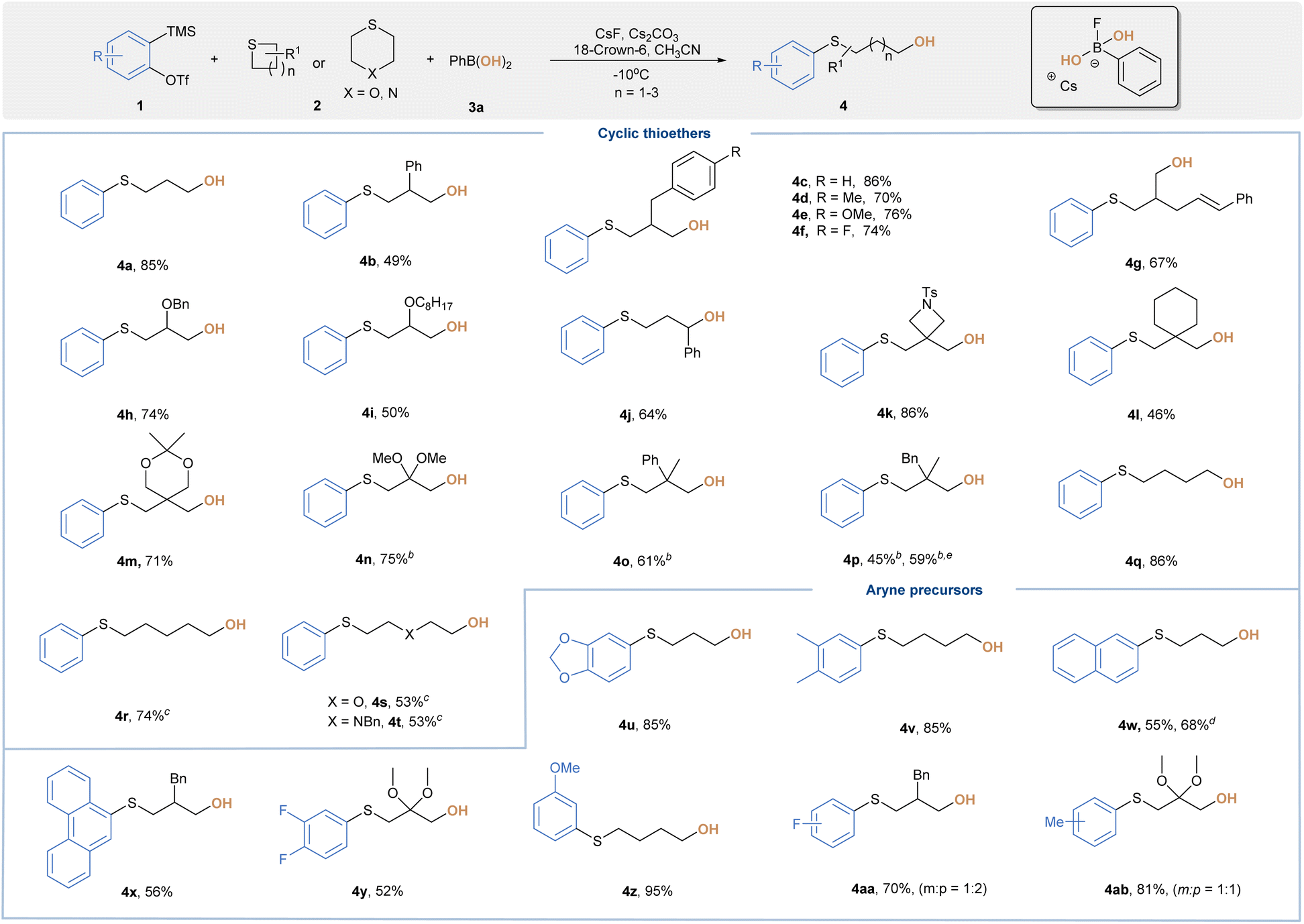
With the optimal conditions in hand, we then assessed the substrate scope of this transformation (Table 2). Three-component reactions of 2 or 3-monosubstituted thietanes with aryne and 3a readily afforded ring-opening hydroxylation products (4b–4j) in good yields. A wide range of functional groups were discovered to be suitable, including phenyl, benzyl, cinnamyl and alkoxy substitutions. Thietanes bearing all quaternary centers at the 3-position were also well-tolerated, leading to 4k–4p in 45–86% yields. It is noteworthy that the transformation of 6-tosyl-2-thia-6-azaspiro[3.3]heptane (2k) occurred chemoselectively at the more nucleophilic sulfur center to provide 4k in 86% yield, which could be further derived through ring opening of azetidines. For substrates bearing ketal moieties, the reaction also proceeded well to deliver 4m and 4n in 71% and 75% yield, respectively. We were also delighted to find that 5 or 6-membered cyclic sulfides with less ring strain were amenable to provide 4q–4t in moderate yields, although elevated reaction temperature was required. Encouraged by these results, we further examined versatile aryne precursors. Symmetrical and unsymmetrical arynes bearing various functional groups all underwent the title reaction smoothly to deliver target products (4u–4ab) in good yields. When a 3-methoxybenzyne precursor was subjected to standard conditions, we obtained 4z as a single regioisomer, which was consistent with the model proposed by Garg and Houk.19 As a further benefit, we were able to improve the overall reaction outcome for certain substrates by simply screening aryl BAs with different scaffolds (4p and 4w). Of note, neither ring-opening fluorination products nor direct C–O coupling side reactions were observed during our investigations.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), 3a (0.3 mmol), CsF (0.6 mmol), Cs2CO3 (0.4 mmol), 18-crown-6 (0.4 mmol), and CH3CN (2 mL) for 12 hours at −10 °C, and isolated yield. b CsF (0.15 mmol), Cs2CO3 (0.6 mmol), 18-crown-6 (0.6 mmol), and 40 °C. c 40 °C. d 2-MePhB(OH)2 used instead. e 3-MePhB(OH)2 used instead. |
|---|
|
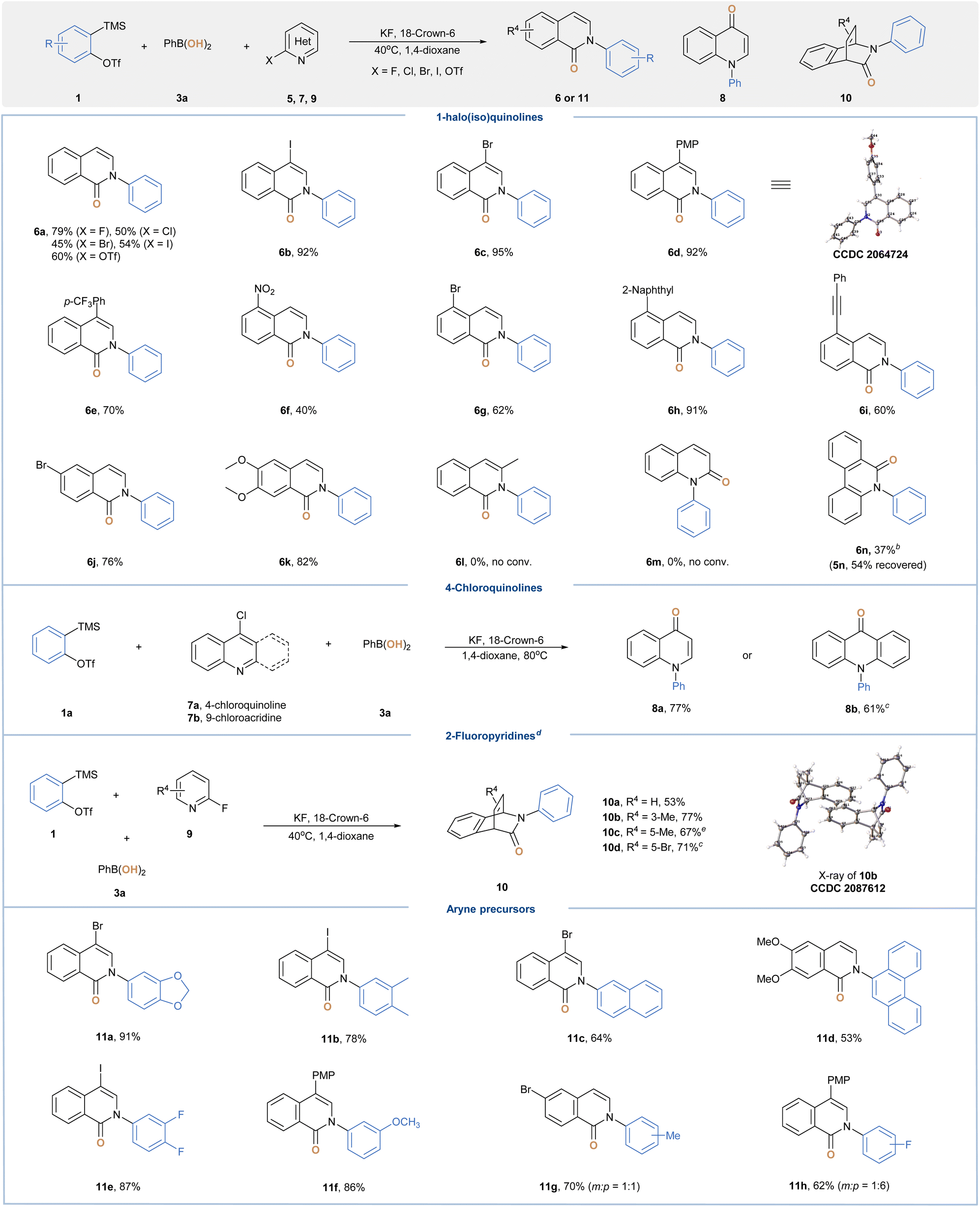
Having demonstrated the ability of aryl BAs as efficient nucleophilic hydroxy sources, we next ventured to explore other multicomponent coupling cascades in order to construct structurally complex and medicinally relevant heterocycles.20 To this end, we sought to incorporate insights gained vide supra to investigate the aryne-induced electrophilic activation-nucleophilic substitution of halo-azaarenes with phenylboronic acids (Table 3). Once optimal conditions were identified,15 a series of 1-haloisoquinolines and their pseudo-halo counterparts were explored. Among them, 1-fluoroisoquinoline (5a) resulted in a better yield of 2-phenylisoquinolin-1-one (6a), and the observed reactivity trend was consistent with the leaving group effect for SNAr.21 Next, substrates bearing various substitution groups were examined, and the desired 2-arylisoquinolin-1-one products (6b–6k) were all obtained in good yields. Notably, bromo and iodo groups as potential synthetic handles for further derivatization were compatible with our conditions.22 Unfortunately, the transformations of 3-methyl-1-fluoroisoquinoline (5l) and 2-fluoroquinoline (5m) failed to deliver the corresponding products, whereas most of the starting materials were recovered. We speculated that the steric hindrance around the nitrogen center suppressed the initial nucleophilic addition step. In line with such rationalization, the subjection of 6-fluorophenanthridine (5n) to standard conditions gave 5-phenylphenanthridin-6-one (6n) in 37% yield along with 54% of 5n recovered. Subsequently, we assessed the applicability of the current method with respect to other electron-deficient azaarenes. To our delight, both 4-chloroquinoline and 9-chloroacridine were amenable to give 1-phenylquinolin-4-one (8a) and 10-phenylacridin-9-one (8b) in 77% and 61% yield, respectively. A unique reaction cascade comprising such three-component coupling and [4 + 2] cycloaddition was observed for 2-fluoropyridine substrates. In the presence of excess arynes, three-dimensional, bicyclic products (10a–10d) were readily acquired in 53–77% yields from planar heteroaromatics, which clearly demonstrated the potential “escape-from-flatland” value of our approach.23 Finally, we extend this protocol to a similar set of aryne components and in all cases obtained desired heterocyclic products (11a–11h) in moderate to good yields.
To demonstrate the synthetic utility, scale-up experimentation was conducted under standard conditions, affording 4u and 6c at the same level of isolated yields (Scheme 2A). Next, we sought to present exemplary downstream derivatization of the obtained three-component coupling products (Scheme 2B). Sulfide 4u could be easily converted to sulfoxide 12 and sulfone 13 in 92% and 98% yield respectively, depending on the reaction time and amount of m-CPBA used. An FeCl3-promoted intramolecular Friedel–Crafts reaction of 4j proceeded smoothly to give the annulated product 4-phenylthiochromane (14) in 81% isolated yield. The treatment of 6g with Lawesson's reagent furnished 5-bromoisoquinoline-1(2H)-thione 15 in 89% yield, while the palladium-catalyzed C–N coupling gave 16 in 83% yield. In addition, product 8a was subjected to both copper catalyzed conjugate addition with p-tolylmagnesium bromide and N-iodosuccinimide (NIS) promoted iodination reactions, leading to 17 and 18 in 82% and 98% yield, respectively. Finally, our protocol proved to be applicable for late-stage modification of the active pharmaceutical ingredient, giving drug-like fasudil derivative (6o) in 50% yield (Scheme 2C). The showcased reactivity spectrum further signified the value of our protocol for facile preparation of highly versatile synthetic building blocks.
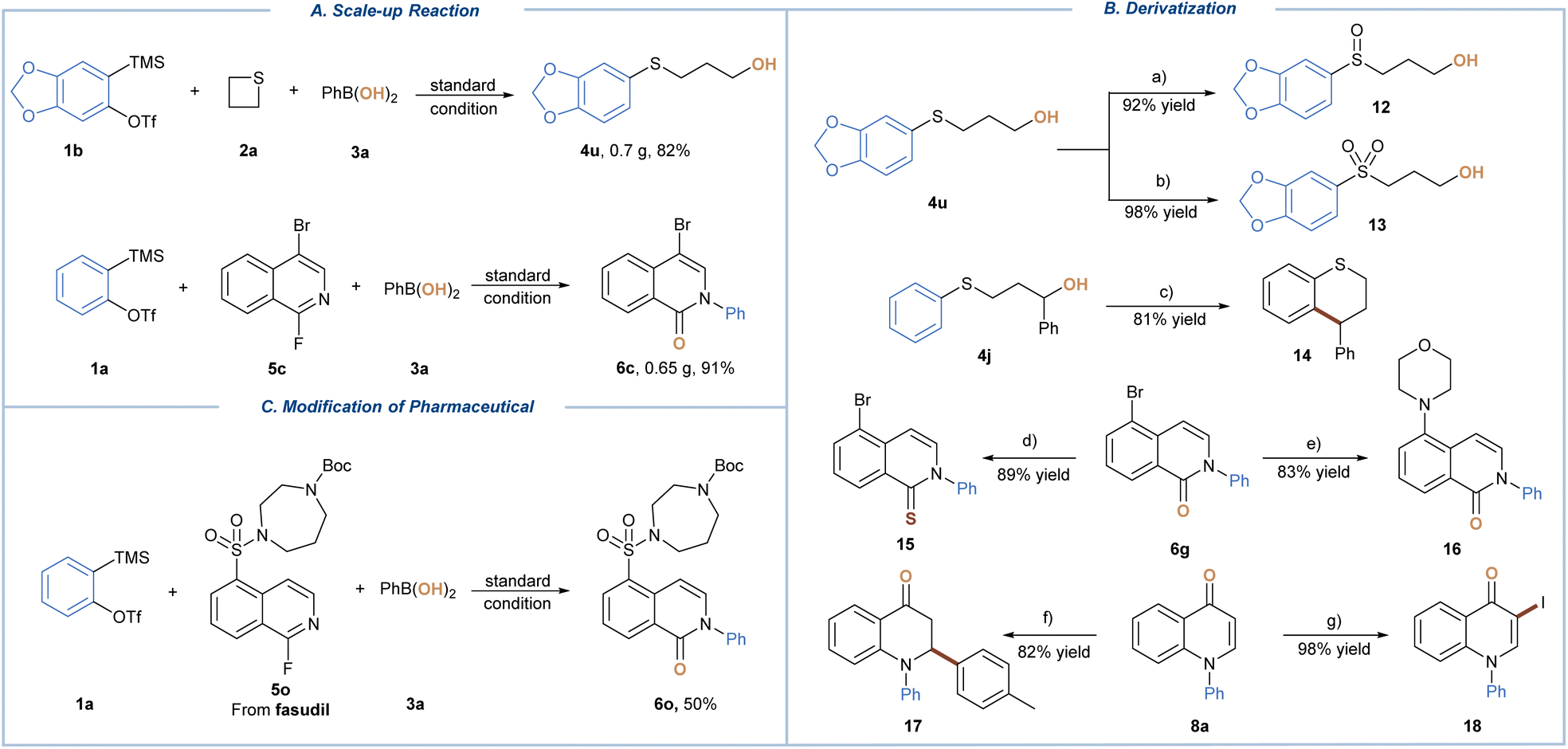 | ||
| Scheme 2 Synthetic application. Reaction conditions: (a) m-CPBA (1.1 equiv), DCM, and 0 °C. (b) m-CPBA (3.2 equiv), DCM, and 0 °C to r.t. (c) FeCl3 (1 equiv), DCE, and 70 °C. (d) Lawesson's reagent (4 equiv), toluene, and 120 °C. (e) Morpholine (2 equiv), Pd2(dba)3 (10 mol%), BINAP (10 mol%), t-BuOK (3 equiv), toluene, and 110 °C. (f) CuI (2 equiv), TMSCl (3 equiv), p-TolMgBr (2 equiv), THF/DCM, and −78 °C. (g) NIS (1.5 equiv), AcOH, and r.t. | ||
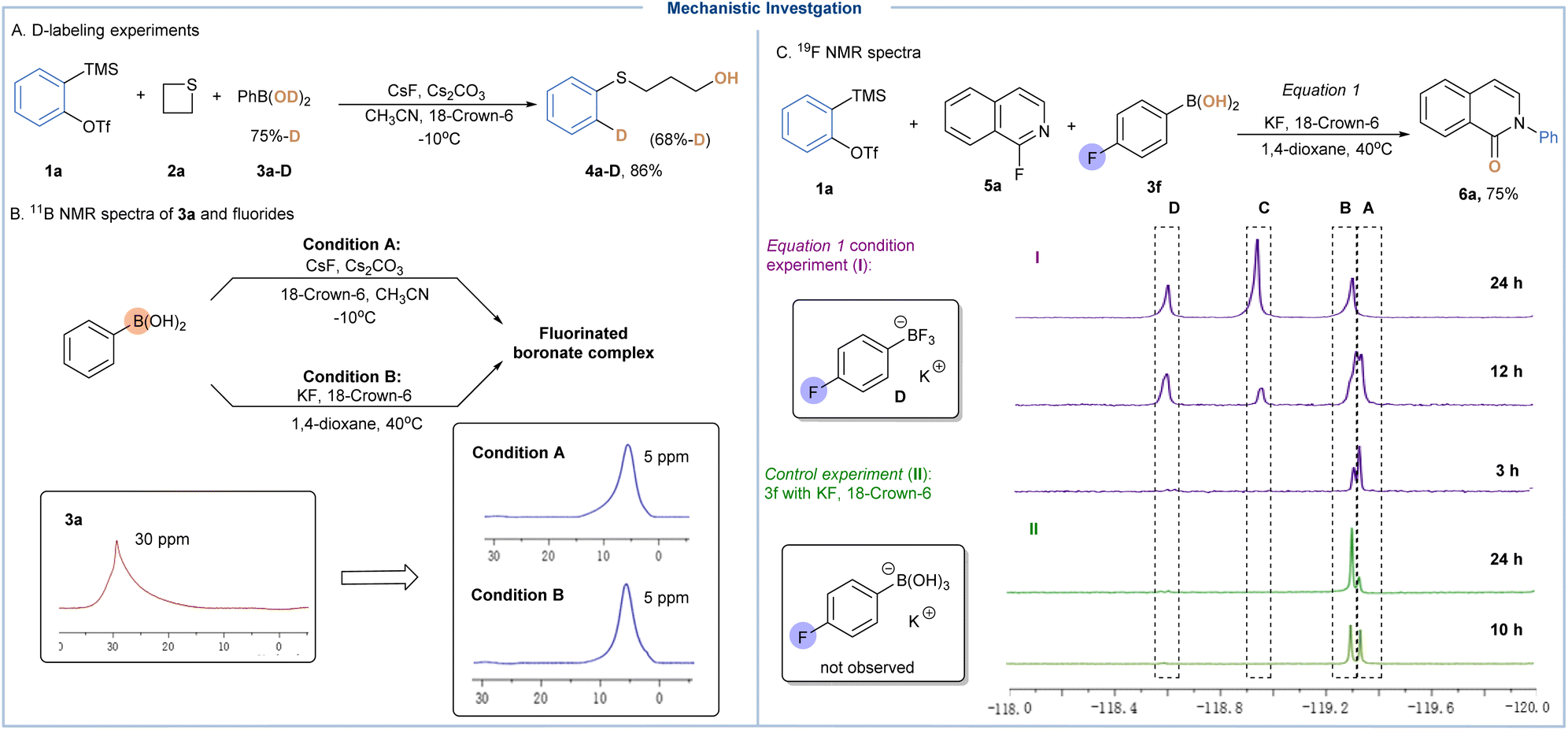
To shed light on the mechanistic underpinnings of these methods, both experimental and theoretical studies have been performed. First, a deuterium-labeling study was conducted employing PhB(OD)2 (75% D-incorporation) under standard conditions, which gave an ortho-deuterated sulfide product (4a-D) in 86% yield and 68% deuterium incorporation, which not only indicated that the proton came from aryl BAs, but also ruled out the possibility of involving cyclic sulfonium ylide type intermediates (Scheme 3A).24 In the absence of arynes, neither isoquinolin-1-one or 1-hydroxyisoquinoline was observed under otherwise identical conditions, thereby excluding the possible pathway involving hydroxylation (SNAr) and N-arylation cascades.15 With the subjection of 3a to CsF or the combination of KF and 18-crown-6, a new upfield peak at 5 ppm could be observed in the 11B spectrum whereas the peak of 3a disappeared, offering direct evidence on the formation of tetrahedral boronate complexes (Scheme 3B). Next, the reaction progress of 4-fluorophenylboronic acid (3f) with 1a was monitored by the 19F NMR technique. Upon treatment with KF and 18-crown-6, we immediately observed two signals at −119.34 ppm (peak A) and −119.36 ppm (peak B), which should refer to the formation of a B–F ate complex as supported by the control experiment (Scheme 3C).25 As the reaction proceeded, peak C (−118.97 ppm) and peak D (−118.63 ppm) gradually appeared, and the former was successfully identified as 4-fluorophenyltrifluoroborate by control experiments. Note that this observation suggested that our “fluoride activation-nucleophilic hydroxylation” strategy could drive the equilibrium on B–F coordination of BAs through the consumption of hydroxyl groups. Additionally, high-resolution mass spectrometry (HRMS) was employed to support the direct evidence on the in situ formed B–F complex.15
 | ||
| Scheme 3 Mechanistic study. | ||
Density functional theory (DFT) calculations at the M06 level of theory were then conducted to explore the role of the B–F complex in both unprecedented three-component protocols. As shown in Scheme 4A, in situ generated aryne 1 was chosen as the relative zero in the calculated free energy profiles, which could react with thietane 2avia nucleophilic addition transition state TS-1 to afford a sulfonium zwitterion intermediate Int-1. The calculated activation free energy for this step proved to be only 7.2 kcal mol−1, indicating an easily reachable process. After that, a ring-opening process could occur through the nucleophilic attack of the hydroxyl group on boronate via transition state TS-2 with an energy barrier of 14.4 kcal mol−1, leading to the cleavage of the B–O bond and formation of aryl anion intermediate Int-2. Subsequently, an intramolecular proton transfer via transition state TS-3 through a barrier-less process led to the formation of alkoxide Int-3, which could be subsequently protonated during work up to yield the target product 4a. It is noteworthy that this boronate based hydroxylation system showed a unique pathway where instead of protonation to form sulfonium ions, the direct hydroxylation of sulfonium zwitterions occurred at the first place.
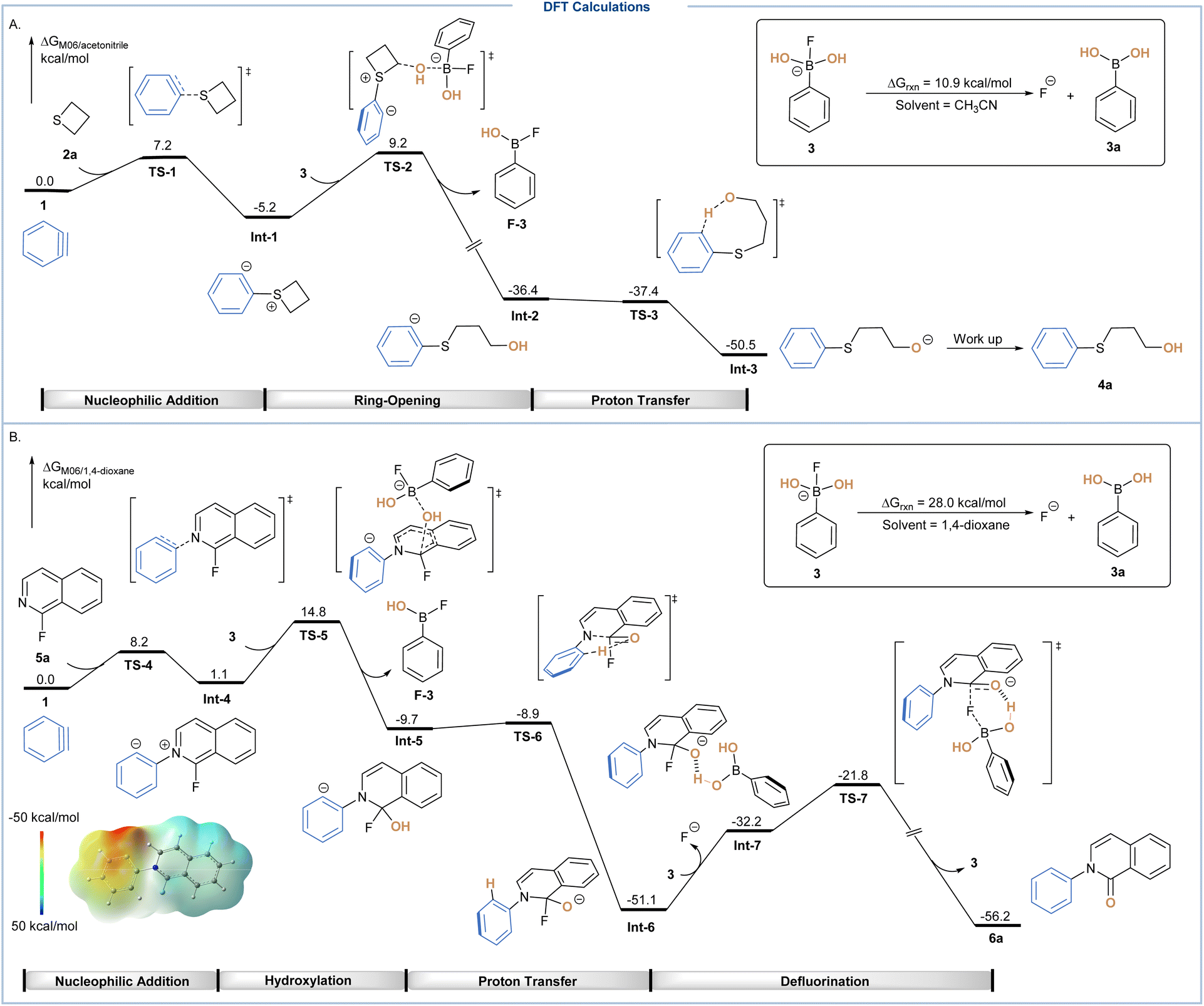 | ||
| Scheme 4 DFT calculations. (A) Free energy profile for the ring-opening hydroxylation of aryne 1 and thietane 2a with phenyl boronic acid 3a. The energy values are in kcal mol−1 and represent the relative free energies calculated at the M06/def2-TZVP/SMD(acetonitrile)//B3LYP/def2-SVP/SMD(acetonitrile) level of theory in acetonitrile solvent. (B) Free energy profile for the three-component reaction of aryne 1 and 1-fluoroisoquinoline 5a with phenyl boronic acid 3a. The energy values are in kcal mol−1 and represent the relative free energies calculated at the M06/def2-TZVP/SMD(1,4-dioxane)//B3LYP/def2-SVP/SMD(1,4-dioxane) level of theory in 1,4-dioxane solvent. | ||
For the three-component coupling of 1-fluoroisoquinoline, in situ generated aryne 1 was again chosen as the relative zero in the calculated free energy profiles, which was nucleophilically attacked by 1-fluoroisoquinoline (5a) to form the zwitterionic isoquinolinium Int-4 (the energy of activation is 8.2 kcal mol−1) (Scheme 4B). For Int-4, the electrostatic potential surface clearly revealed that the isoquinoline moiety was still electron-deficient. Therefore, rather than protonation to form isoquinolinium salt, it could directly react with boronate 3 through an electrophilic substitution via transition state TS-5 with an energy barrier of 13.7 kcal mol−1. Afterwards, a dearomatized intermediate Int-5 was formed, which then underwent a rapid intramolecular 1,5-proton transfer to form the stable isoquinolinolate Int-6. As the generation of 3a was endergonic by 28.0 kcal mol−1 from the corresponding boronate 3, the combination of such a process and conversion of intermediate Int-6 to Int-7 was endergonic by 18.9 kcal mol−1. Therefore, the overall activation free energy for the BA-assisted defluorination to yield target product 6a was 29.3 kcal mol−1, proceeding via a six-member ring transition state TS-7.
Alternatively, we considered the possible participation of phenyl(difluoro)(hydroxy)borate 19 as a hydroxy source via transition state TS-8, the relative free energy of which was only 3.0 kcal mol−1 higher than its counterpart TS-5 (see Fig. S6†).15 Accordingly, potassium phenyltrifluoroborate could be formed after the consumption of the hydroxyl group, which was consistent with the 19F NMR study (Scheme 3C). In the case of the fluoride dissociation step, other pathways involving either phenyl(hydroxy)(difluoro)borate 19 or phenyltrifluoroborate 21 were also taken into account, both of which displayed much higher reaction energies, making the corresponding pathway less favorable (see Fig. S7†).15
Conclusions
In summary, we have demonstrated that BAs can be employed as novel tunable hydroxide surrogates. The synthetic value was presented by hitherto unknown aryne triggered ring opening of cyclic sulfides and three-component synthesis of N-aryl (iso)quinolinones. Fluorides played a “tailor made” role in generating aryne species, activating BAs via complexation and facilitating B–O bond cleavage. The B–F “ate” complex displayed a unique nucleophilic paradigm that water or a metal hydroxide base couldn't convey. Furthermore, DFT calculation suggested that the unique hydroxylation of sulfur/nitrogen-containing zwitterions with boronate complexes occurred in favor of the formation of sulfonium/isoquinolinium intermediates via protonation. Experimental and theoretical studies also shed light on both the activation mode and reactivity trend of ArB(OH)xFy. Overall, we are strongly confident that the systematic knowledge gained in this investigation can be broadly translated to other reaction manifolds, thereby providing new synthetic blueprints for organoboron chemistry.Data availability
The data supporting this study is available within the main text and the associate ESI.† The X-ray crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 2064724 (6d) and 2087612 (10b).Author contributions
R. F. and J. T. conceived the project; R. F. performed the experiments; Q. Y. and J. W. prepared some starting materials; Y. W. discussed the results; S. L. performed the DFT calculation; Y. L. supervised the DFT calculations. R. F., S. L., Y. L. and Y. W. prepared this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22271010 and 21702013) and the Fundamental Research Funds for the Central Universities (XK1802-6) in BUCT. We also acknowledges Beijng PARATERA Technology Co., Ltd. for providing HPC resources.This work is dedicated to Professor Hisashi Yamamoto on the occasion of his 80th birthday.Notes and references
- (a) J. Plescia and N. Moitessier, Eur. J. Med. Chem., 2020, 195, 112270–112289 CrossRef CAS PubMed; (b) J. P. M. António, R. Russo, C. P. Carvalho, P. M. S. D. Cal and P. M. Gois, Chem. Soc. Rev., 2019, 48, 3513–3536 RSC; (c) W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed; (d) N. D. J. Hiller, N. A. do Amaral, E. Silva, T. A. Tavares, R. X. Faria, M. N. Eberlin and D. de Luna Martins, Eur. J. Org. Chem., 2020, 2020, 4841–4877 CrossRef CAS.
- (a) D. G. Hall, in Boronic Acids. Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2006, ch. 1, pp. 1–99 Search PubMed; (b) C. Williams, M. Luis, I. Lopez and A. Coca, Chem. Rev., 1995, 95, 2457–2483 CrossRef.
- (a) I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS; (b) I. D. Kostas, Suzuki-Miyaura Cross-Coupling Reaction and Potential Applications, MDPI, Basel, 2017 Search PubMed; (c) J. Tan, Y. Chen, H. Li and N. Yasuda, J. Org. Chem., 2014, 79, 8871–8876 CrossRef CAS PubMed . and references therein..
- For selected references, see: (a) J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS; (b) I. Munir, A. F. Zahoor, N. Rasool, S. A. R. Naqvi, K. M. Zia and R. Ahmad, Mol. Diversity, 2019, 23, 215–259 CrossRef CAS PubMed; (c) P. Y. S. Lam, in Synthetic Methods in Drug Discovery, ed. D. C. Blakemore, P. M. Doyle and Y. M. Fobian, Royal Society of Chemistry, Cambridge, 2016, ch. 7, pp. 242–273 Search PubMed; (d) M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed.
- For selected reports, see: (a) Z. He, F. Song, H. Sun and Y. Huang, J. Am. Chem. Soc., 2018, 140, 2693–2699 CrossRef CAS PubMed; (b) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed; (c) H. Wang, C. Jing, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 16859–16872 CrossRef CAS PubMed; (d) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed; (e) D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed; (f) S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Chem. Rec., 2009, 9, 24–39 CrossRef CAS PubMed; (g) D. P. Hari, R. Madhavachary, V. Fasano, J. Haire and V. K. Aggarwal, J. Am. Chem. Soc., 2021, 143, 7462–7470 CrossRef CAS PubMed; (h) S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 16766–16775 CrossRef CAS PubMed.
- (a) P. Wu, M. Givskov and T. E. Nielsen, Chem. Rev., 2019, 119, 11245–11290 CrossRef CAS PubMed; (b) P. Wu and T. E. Nielsen, Drug Discovery Today: Technol., 2018, 29, 27–33 CrossRef PubMed; (c) W. Masamba, Molecules, 2021, 26, 1707 CrossRef CAS PubMed.
- For selected reports, see: (a) F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed; (b) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed; (c) G. Yan, M. Yang and X. Wu, Org. Biomol. Chem., 2013, 11, 7999–8008 RSC; (d) Y. Iwata, Y. Yanaka, S. Kubosaki, T. Morita and Y. Yoshimi, Chem. Commun., 2018, 54, 1257–1260 RSC.
- (a) D. Roscales and A. G. Csáky, Chem. Soc. Rev., 2020, 49, 5159–5177 RSC; (b) V. S. Sadu, H. R. Bin, D. M. Lee and K. I. Lee, Sci. Rep., 2017, 7, 242 CrossRef PubMed; (c) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed; (d) W.-D. Li, Y. Wu, S.-J. Li, Y.-Q. Jiang, Y.-L. Li, Y. Lan and J.-B. Xia, J. Am. Chem. Soc., 2022, 144, 8551–8559 CrossRef CAS PubMed.
- (a) C. R. Cooper, N. Spencer and T. D. James, Chem. Commun., 1998, 1365–1366 RSC; (b) B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 13028–13044 CrossRef CAS PubMed.
- A. Bouziane, M. Helou, B. Carboni, F. Carreaux, B. Demerseman, C. Bruneau and J.-L. Renaud, Chem.–Eur. J., 2008, 14, 5630–5637 CrossRef CAS PubMed.
- (a) J. Tan, B. Liu and S. Su, Org. Chem. Front., 2018, 5, 3093–3097 RSC; (b) T. Zheng, J. Tan, R. Fan, S. Su, B. Liu, C. Tan and K. Xu, Chem. Commun., 2018, 54, 1303–1306 RSC; (c) A. Saputra, R. Fan, T. Yao, J. Chen and J. Tan, Adv. Synth. Catal., 2020, 362, 2683–2688 CrossRef CAS; (d) R. Fan, B. Liu, T. Zheng, K. Xu, C. Tan, T. Zeng, S. Su and J. Tan, Chem. Commun., 2018, 54, 7081–7084 RSC.
- For selected examples on the nucleophilicity–electrophilicity scale, see: (a) H. Mayr and A. R. Ofial, J. Phys. Org. Chem., 2008, 21, 584–595 CrossRef CAS; (b) R. Loos, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2003, 125, 14126–14132 CrossRef CAS PubMed; (c) H. F. Schaller, A. A. Tishkov, X. Feng and H. Mayr, J. Am. Chem. Soc., 2008, 130, 3012–3022 CrossRef CAS PubMed; (d) N. F. Fine Nathel, L. A. Morrill, H. Mayr and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 10402–10405 CrossRef CAS PubMed.
- For representative examples on aryne chemistry, see: (a) M. Bürger, N. Ehrhardt, T. Barber, L. T. Ball, J. C. Namyslo, P. G. Jones and D. B. Werz, J. Am. Chem. Soc., 2021, 143, 16796–16803 CrossRef PubMed; (b) E. Picazo, S. M. Anthony, M. Giroud, A. Simon, M. A. Miller, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2018, 140, 7605–7610 CrossRef CAS PubMed; (c) S. Bhattacharjee, S. Deswal, N. Manoj, G. Jindal and A. T. Biju, Org. Lett., 2021, 23, 9083–9088 CrossRef CAS PubMed; (d) L. Garve and D. B. Werz, Org. Lett., 2015, 17, 596–599 CrossRef CAS PubMed; (e) M. Scherübl, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 711–715 CrossRef PubMed; (f) T. Yao, Z. Hui, T. Liu and T. Li, J. Org. Chem., 2021, 86, 5477–5488 CrossRef CAS PubMed; (g) J. Liu, J. Li, B. Ren, Y. Zhang, L. Xue, Y. Wang, J. Zhao, P. Zhang, X. Xu and P. Li, Adv. Synth. Catal., 2020, 362, 2683–2688 CrossRef; (h) J. Shi, L. Li, C. Shan, Z. Chen, L. Dai, M. Tan, Y. Lan and Y. Li, J. Am. Chem. Soc., 2021, 143, 10530–10536 CrossRef CAS PubMed; (i) L. Li, C. Shan, J. Shi, W. Li, Y. Lan and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202117351 CAS; (j) S. Bhattacharjee, A. Guin, R. N. Gaykar and A. T. Biju, Org. Lett., 2020, 22, 9097–9101 CrossRef CAS PubMed; (k) M. Pawliczek, L. K. B. Garve and D. B. Werz, Org. Lett., 2015, 17, 1716–1719 CrossRef CAS PubMed; (l) R. N. Gaykar, S. Deswal, A. Guin, S. Bhattacharjee and A. T. Biju, Org. Lett., 2022, 24, 4145–4150 CrossRef CAS PubMed; (m) J. Shi, L. Li, C. Shan, J. Wang, Z. Chen, R. Gu, J. He, M. Tan, Y. Lan and Y. Li, J. Am. Chem. Soc., 2021, 143, 2178–2184 CrossRef CAS PubMed; (n) S. Liu, Y. Li and Y. Lan, Eur. J. Org. Chem., 2017, 2017, 6349–6353 CrossRef CAS.
- For reviews, see: (a) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892–4044 CrossRef CAS PubMed; (b) H. Yoshida, in Multicomponent Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M.-X. Wang, Wiley-VCH, Weinheim, 2015, ch. 3, pp. 39–72 Search PubMed; (c) J. Chen and J. Tan, in Comprehensive Aryne Synthetic Chemistry, ed. H. Yoshida, Elsevier, Amsterdam, 2022, ch. 2.3, pp. 125–221 Search PubMed; (d) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658–1670 CrossRef CAS PubMed; (e) D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2020, 59, 3385–3398 CrossRef CAS PubMed; (f) L. L. Fluegel and T. R. Hoye, Chem. Rev., 2021, 121, 2413–2444 CrossRef CAS PubMed; (g) J. Chen, R. Fan, Z. Liu and J. Tan, Adv. Synth. Catal., 2021, 363, 657–667 CrossRef CAS; (h) A. T. Biju, Modern Aryne Chemistry, Wiley-VCH, Weinheim, 2021 Search PubMed; (i) S. M. Anthony, L. G. Wonilowicz, M. S. McVeigh and N. K. Garg, JACS Au, 2021, 1, 897–912 CrossRef CAS PubMed; (j) D. Pérez, D. Peña and E. Guitián, Eur. J. Org. Chem., 2013, 2013, 5981–6013 CrossRef; (k) J. He, D.-C. Qiu and Y. Li, Acc. Chem. Res., 2020, 53, 508–519 CrossRef CAS PubMed; (l) T. Matsuzawa, S. Yoshida and T. Hosoya, Tetrahedron Lett., 2018, 59, 4197–4208 CrossRef CAS; (m) Q. Yan, R. Fan, B. Liu, S. Su, B. Wang, T. Yao and J. Tan, Chin. J. Org. Chem., 2021, 41, 455–470 CrossRef CAS; (n) R. Zhang, X. Peng and J. Tan, Synthesis, 2022, 54, 5064–5076 CrossRef CAS.
- For details, please see the ESI.†.
- For selected reports on sulfonium chemistry, see: (a) X. Xiao, J. Zeng, J. Fang, J. Sun, T. Li, Z. Song, L. Cai and Q. Wan, J. Am. Chem. Soc., 2020, 142, 5498–5503 CrossRef CAS PubMed; (b) R. Oost, J. D. Neuhaus, A. Misale, R. Meyrelles, L. F. Veiros and N. Maulide, Chem. Sci., 2018, 9, 7091–7095 RSC; (c) X.-X. Nie, X.-Y. Chen, Y. Wu and P. Wang, Org. Lett., 2020, 22, 7716–7720 CrossRef CAS PubMed; (d) F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Mateos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, J. Am. Chem. Soc., 2021, 143, 16041–16054 CrossRef PubMed; (e) L. Zhang, W. Bao, Y. Liang, W. Pan, D. Li, L. Kong, Z.-X. Wang and B. Peng, Angew. Chem., Int. Ed., 2021, 60, 11414–11422 CrossRef CAS PubMed; (f) M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163–169 CrossRef CAS; (g) D. J. Wang, K. Targos and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 21503–21510 CrossRef CAS PubMed; (h) L. Shang, Y. Chang, F. Luo, J.-N. He, X. Huang, L. Zhang, L. Kong, K. Li and B. Peng, J. Am. Chem. Soc., 2017, 139, 4211–4217 CrossRef CAS PubMed; (i) Y. Li, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2016, 55, 14435–14438 CrossRef CAS PubMed; (j) W. Guo, Y. Luo, H. H.-Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed; (k) D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed.
- For selected reviews on sulfonium chemistry, see: (a) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (b) S. Otsuka, K. Nogi and H. Yorimitsu, Top. Curr. Chem., 2018, 376, 13 CrossRef PubMed; (c) H. Yorimitsu, Chem. Rec., 2021, 21, 3356–3369 CrossRef CAS PubMed; (d) R. Fan, C. Tan, Y.-G. Liu, Y. Wei, X.-W. Zhao, X.-Y. Liu, J.-J. Tan and H. Yoshida, Chin. Chem. Lett., 2021, 32, 299–312 CrossRef CAS; (e) L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed; (f) Z.-Y. Tian, Y. Ma and C.-P. Zhang, Synthesis, 2022, 54, 1478–1502 CrossRef CAS; (g) S. I. Kozhushkov and M. Alcarazo, Eur. J. Inorg. Chem., 2020, 2020, 2486–2500 CrossRef CAS PubMed.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC.
- J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798–15805 CrossRef CAS PubMed.
- (a) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed; (b) K. C. Majumdar and S. K. Chattopadhyay, Heterocycles in Natural Product Synthesis, Wiley-VCH, Weinheim, 2011 CrossRef; (c) T. Shiro, T. Fukaya and M. Tobe, Eur. J. Med. Chem., 2015, 97, 397–408 CrossRef CAS PubMed; (d) R. Hua, Catalysts, 2021, 11, 620 CrossRef CAS.
- N. A. Senger, B. Bo, Q. Cheng, J. R. Keeffe, S. Gronert and W. Wu, J. Org. Chem., 2012, 77, 9535–9540 CrossRef CAS PubMed.
- (a) J. C. Badenock and G. W. Gribble, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scirven and C. A. Ramsden, Academic Press, New York, 2016, ch. 4, pp. 99–136 Search PubMed; (b) C. Jin, K. Xu, X. Fan, C. Liu and J. Tan, Chin. Chem. Lett., 2020, 31, 91–94 CrossRef CAS; (c) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed . and references therein..
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) F. Lovering, Med. Chem. Comm., 2013, 4, 515–519 RSC.
- For selected reports, see: (a) J. Hen, V. Palani and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 4318–4321 CrossRef PubMed; (b) S.-J. Li, Y. Wang, J.-K. Xu, D. Xie, S.-K. Tian and Z.-X. Yu, Org. Lett., 2018, 20, 4545–4548 CrossRef CAS PubMed.
- (a) M. Buttersm, J. H. Harvey, J. Jover, A. J. J. Lennox, G. C. Lloyd-Jones and P. M. Murray, Angew. Chem., Int. Ed., 2010, 49, 5156–5160 CrossRef PubMed; (b) A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2012, 51, 9385–9388 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2064724 and 2087612. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00072a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |