
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOH radical-initiated single-electron transfer for accelerated degradation via carbocation intermediates†
Xiyang
Ge
,
Yiyan
Yin
,
Jianghui
Sun
,
Jin
Ouyang
 and
Na
Na
*
and
Na
Na
*
Key Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: nana@bnu.edu.cn
First published on 26th January 2023
Abstract
Single electron transfer (SET) has made great contributions to a broad range of chemical processes, whose radical cation and carbocation intermediates are important for mechanism studies. Herein, hydroxyl radical (˙OH)-initiated SET was revealed in accelerated degradations, via the online examination of radical cations and carbocations by electrosonic spray ionization mass spectrometry (ESSI-MS). In the green and efficient non-thermal plasma catalysis system (MnO2-plasma), hydroxychloroquine was efficiently degraded upon SET via carbocations. In the plasma field full of active oxygen species, ˙OH was generated on the MnO2 surface to initiate SET-based degradations. Furthermore, theoretical calculations revealed that ˙OH preferred to withdraw the electron from the N atom that was conjugated to the benzene ring. This facilitated the generation of radical cations through SET, which was followed by the sequential formation of two carbocations for accelerated degradations. Transition states and energy barriers were calculated to study the formation of radical cations and subsequent carbocation intermediates. This work demonstrates an ˙OH-initiated SET for accelerated degradation via carbocations, providing a deeper understanding and the potential for the wider application of SET in green degradations.
Introduction
Single electron transfer (SET) has made great contributions to reaction acceleration, the functionalization of intermediates and the breaking of bonds in a broad range of chemical processes.1–3 Through the SET process, substrate molecules are initially oxidized to form radical cation species by losing electrons with high stoichiometric efficiency and oxidation selectivity.4,5 Subsequently, activated radical cations are generated and spontaneously converted into carbocation intermediates through α-induced cleavage to participate in a series of reactions.6 Therefore, the formation of radical cations and carbocations can be considered as the crucial steps of SET, and their characterizations would be helpful for its examination.The capture and monitoring of activated radical cations are challenging and interesting topics owing to the high reactivity and poor thermodynamic stability of these cations. Furthermore, carbocation intermediates normally exist under harsh conditions (such as in the presence of high-energy UV irradiation, strong Lewis/Brønsted acids, solvolysis, and high temperatures),7,8 which makes real-time characterization difficult. Beyond instantaneous molecular information, the continuous monitoring of dynamic changes or transformations of different reactive intermediates (radical cations and carbocations) would also offer significant benefits to in-depth SET studies.9,10 Taking green and efficient degradation by plasma catalysis as an example, the SET process was predicated on the coupling of heterogeneous catalysts with non-thermal plasma. Generated by air discharge, the plasma normally contains active species. These active species would be catalytically converted into reactive oxygen species in water to facilitate degradation via SET.11–13 This makes the conversion of pollutants and reactive intermediates quite complicated, employing alternative and unpredicted pathways. Therefore, the examination of radical cation and carbocation intermediates of SET is a challenging and interesting topic, requiring both technical improvements and mechanistic explorations.
Herein, with MnO2 as the catalyst, hydroxychloroquine (HCQ) was selected as an example pollutant for studying the SET-initiated degradation in the plasma-catalysis system. Recently, many ambient mass spectrometry techniques have been proposed to monitor dynamic changes of various reactions.14–16 Given the advantages of electrosonic spray ionization mass spectrometry (ESSI-MS) in the real-time and continuous monitoring of reactions, short-lived radical cation and carbocation intermediates have been successfully observed.17–19 This facilitated the exploration of SET-initiated accelerated degradation without any sample pre-treatment. Based on multiple characterizations of catalysts, the hydroxyl radical (˙OH) was confirmed to be generated on the MnO2 surface to initiate SET processes. Therefore, via the dynamic monitoring of intermediates by ESSI-MS, SET was proposed as the key process for accelerated degradation. The ˙OH-initiated SET for degradation acceleration was further confirmed by density functional theory (DFT) calculations, which revealed the transition states and energy barriers step-by-step. This work not only provides a deeper understanding of the SET process but also initiates the improvement of the degradation efficiency in a green pathway.
Results and discussion
Configurations of MnO2-plasma degradation system and ESSI-MS monitoring device
With heavy usage in disease treatment, HCQ is present in wastewater and highly likely to bioaccumulate in organisms and be converted to more toxic forms.20 To minimize the hazard before discharge into the environment, HCQ was selected as the model for degradation via a SET process. As shown in Scheme 1A, the plasma was generated by dielectric barrier discharge (DBD) based on our previous works.21–23 Simply with air as the discharge gas in a quartz tube (i.d. 3.0 mm, o.d. 6.0 mm), the plasma was generated between two copper electrodes at a high alternating voltage (3–5 kV). The plasma contained large amounts of active species (O3), which were catalytically converted into reactive oxygen species (˙OH) in water to facilitate the degradation. The presence of O3 in the plasma was verified by monitoring the characteristic absorption of O3 (at about 254 nm) in the plasma-treated water by UV-vis spectroscopy (Fig. S1†).24,25 Then, upon introducing plasma into the HCQ solution with dispersed MnO2 catalyst (200 mg L−1), the degradation of HCQ occurred simply and easily in the MnO2-plasma system.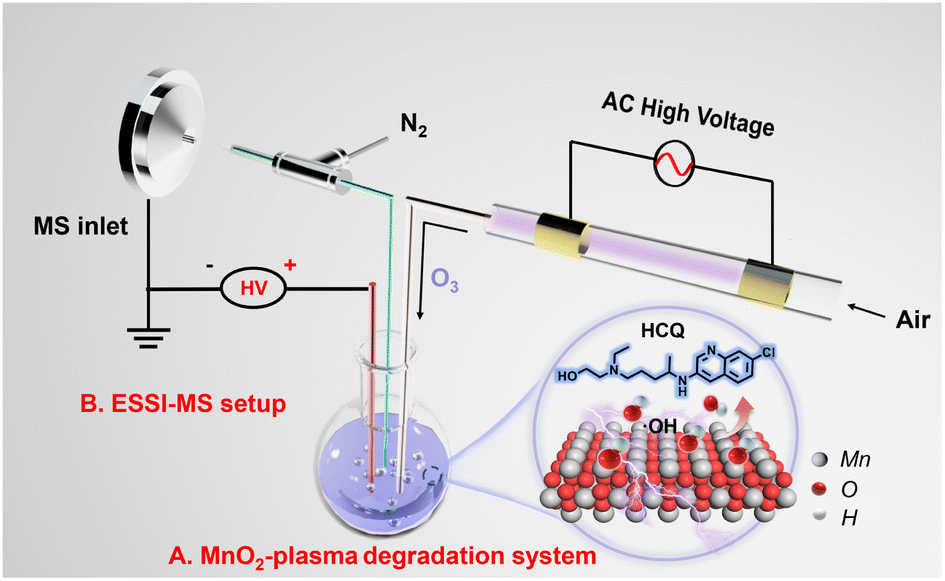 | ||
| Scheme 1 Schematic diagram of HCQ degradation in the MnO2-plasma system. (A) MnO2-plasma degradation system and (B) the ESSI-MS setup for capture and monitoring of the reaction system. | ||
For the online and continuous monitoring of the degradation without sample pre-treatment, ESSI-MS was used to couple with the MnO2-plasma system. As shown in Scheme 1B, a quartz capillary (i.d. 0.5 mm, o.d. 0.62 mm) was inserted into the degradation system. Then, the sample was extracted from the solution into the MS inlet, under the self-pumping of a high-velocity N2 flow from a coaxial outer quartz tube (i.d. 0.86 mm, o.d. 1.5 mm). Simultaneously, a high DC voltage of 5 kV was applied to the solution, and the electrospray of extracted samples in an N2 flow was realized. Therefore, bursts of small charged droplets from the degradation system were obtained for MS detection, enabling the online monitoring of the degradation. Notably, because of the supersonic injection in the ESSI-MS, the interface of the complicated matrix of the heterogeneous system can be avoided without sample pre-treatment or isolation.26,27 Therefore, the potential intermediates could be captured and monitored to examine the degradation in the MnO2-plasma system full of reactive oxygen species.
Accelerated degradation in the MnO2-plasma system
To explore the degradation of HCQ in the MnO2-plasma system, ESSI-MS was applied to detect the reaction intermediates during the degradation. No significant conversions were observed for the HCQ blank (Fig. S2(A)†) and the sample with MnO2 added (Fig. S2(B)†). The only significant signal was [HCQ + H]+ (m/z 336) at 5 min. When introducing plasma into the HCQ solution, in addition to the main peak at m/z 336, other adduct ions were also recorded, including [HCQ + H + O]+ (m/z 352) and [HCQ – H + 2O]+ (m/z 366) (Fig. 1A). This indicated the oxidation of HCQ by the reactive oxygen species in the plasma (O3). When the MnO2 catalyst was added into the plasma degradation system (MnO2-plasma, Fig. 1B), the intensity of the reactant ion peak at m/z 336 decreased dramatically, and ions at m/z 320 and 292 became the dominant ions. As characterized by collision-induced dissociation (CID) experiments and high-resolution mass spectrometry tests (Fig. S3 and S4†), both ions were assigned to the carbocation intermediates [C1]+ and [C2]+. The corresponding structures are shown in Fig. 1B. Notably, weak peaks at m/z 366 and 352 also appeared in the MnO2-plasma system, and the structures of these ions are similar to those in the single plasma system (Fig. S5†). In addition, the carbocation generation was not affected by the high voltage of the ESSI. This was confirmed by the similar MS spectrum with both [C1]+ (m/z 320) and [C2]+ (m/z 292) recorded without the application of the high voltage of the ESSI (Fig. S6†). Therefore, HCQ was efficiently degraded in the MnO2-plasma system with carbocations generated.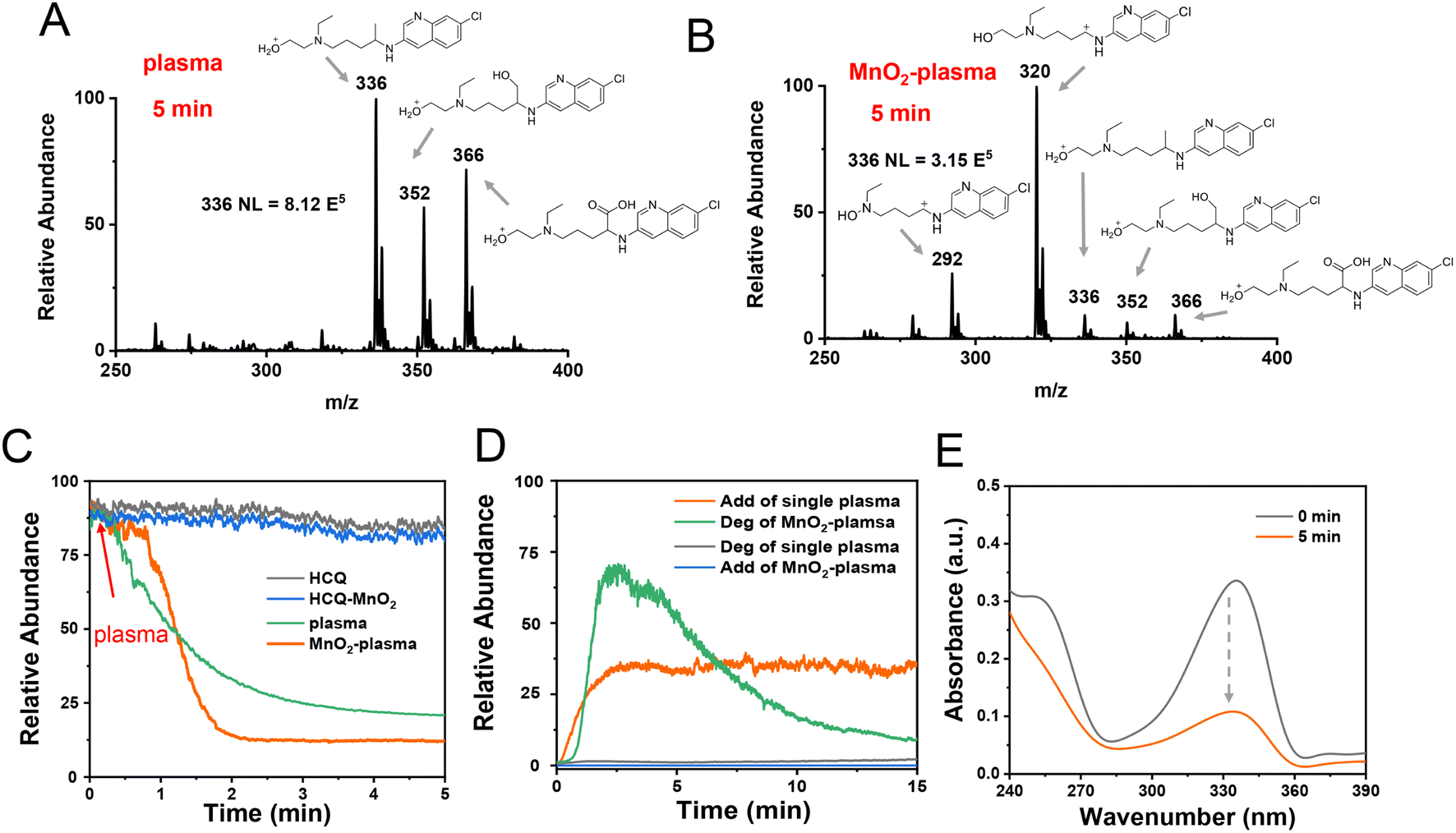 | ||
| Fig. 1 Confirmation of HCQ degradation in the MnO2-plasma system. (A) Mass spectrum of the HCQ solution after 5 min of plasma treatment without MnO2. (B) Mass spectrum of the HCQ solution after 5 min of treatment in the MnO2-plasma system. (C) Monitoring of [HCQ + H]+ (m/z 336) by ESSI-MS in the HCQ blank, MnO2, single plasma and MnO2-plasma systems. (D) Monitoring of Add (higher than m/z 336) and Deg (lower than m/z 336) by ESSI-MS in the single plasma and MnO2-plasma systems. (E) UV-vis absorptions of HCQ at 0 and 5 min degradation time in the MnO2-plasma system. | ||
To better evaluate the degradation behaviours of the different systems, the reaction ion of [HCQ + H]+ at m/z 336 was monitored. As shown in Fig. 1C, no remarkable change was recorded for the blank HCQ (black line) and the HCQ with MnO2 added (blue line). Meanwhile, upon introducing plasma into the HCQ solution, the intensity of the peak of the [HCQ + H]+ ion decreased gradually (green line, Fig. 1C). When MnO2 was introduced into the plasma (orange line, Fig. 1C), the rapid and significant decrease of [HCQ + H]+ at 1 min was recorded, which confirmed the efficient degradation in the MnO2-plasma system. Furthermore, the total ion intensities of the adduct species (add, with m/z values higher than m/z 336) and the degraded species (deg, lower than m/z 336) exhibited different dynamic conversions (Fig. 1D). As indicated, without MnO2 added, the signals were mainly attributed to the adduct species with the higher m/z values (orange line, Fig. 1D). Meanwhile, when MnO2 was added into the plasma system, higher signal intensities of the degraded ions (with lower m/z values) were recorded (green line, Fig. 1D). This was in accordance with the obvious decrease of UV-vis absorptions (at 250 nm and 330–340 nm) after the large conjugate structures of HCQ had been destroyed by MnO2-plasma treatment (Fig. 1E). Therefore, with MnO2 present, the MnO2-plasma exhibited efficient behaviour for HCQ degradation.
Characterization of active ˙OH on the surface of the catalyst
As demonstrated, when the MnO2 was added into the plasma system, the degradation efficiency was greatly increased to generate more degradation species. Therefore, it could be predicted that MnO2 makes a great contribution to the efficient degradation in the plasma field full of O3. The MnO2 present exhibited a morphology of nanorods 1-2 μm in length and with diameters of ∼50 nm (Fig. S7†), which was assigned to β-MnO2 crystals (Fig. S8†). As shown in Fig. 2A(a), a large number of active species of ˙OH would be generated via the conversion of O3 on the MnO2 surface. The obtained ˙OH would therefore attack pollutant molecules to facilitate efficient catalytic degradation.28,29 Meanwhile, insufficient ˙OH was provided without MnO2 present (Fig. 2A(b)), and oxidation reactions were mainly carried out to generate oxygen-adducts. Interestingly, in addition to using Mn–O vibration peaks of MnO6 octahedra in Fourier Transform Infrared Spectroscopy (FTIR) spectra (710 and 540 cm−1), the absorption of hydroxyl groups on the surface was also determined based on the bending vibrations of hydroxyl groups (1627 and 3400 cm−1) (Fig. S9†).30 Therefore, the MnO2 catalyst would facilitate the generation of ˙OH for efficient degradation.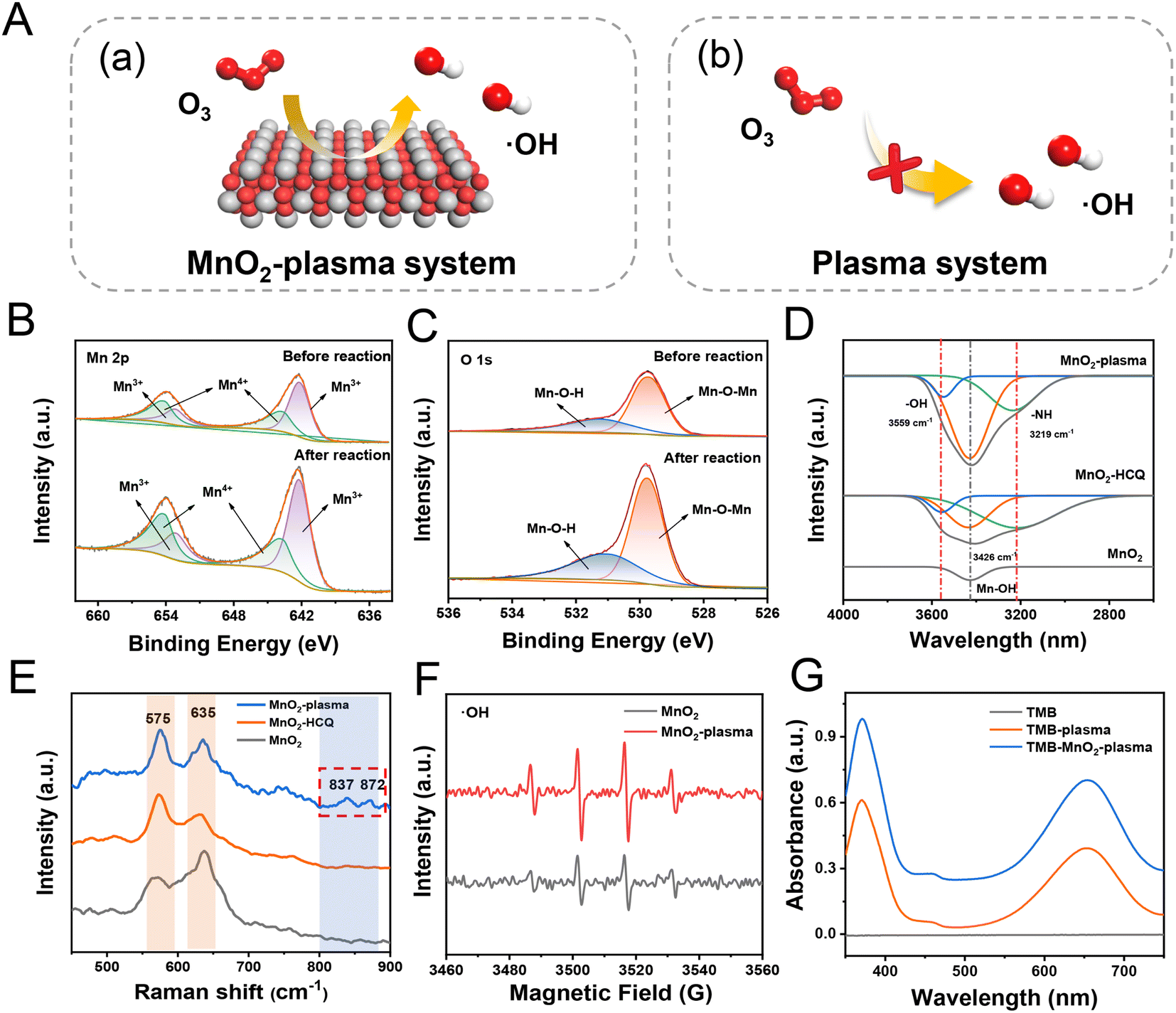 | ||
| Fig. 2 Examination of active OH radicals on the surface of the MnO2 catalyst. (Mn, O and H are shown in grey, red and white, respectively.) (A) Diagram showing the conversion of O3 in the MnO2-plasma (a) and the single plasma (b) systems. XPS spectra of Mn 2p (B) and O 1s (C) of MnO2 before and after degradation in the MnO2-plasma system. FTIR spectra (D) and Raman spectra (E) of HCQ solutions treated by the MnO2-plasma, MnO2-HCQ and single MnO2 systems. (F) DMPO-˙OH signals for the MnO2-plasma and single MnO2 systems. (G) UV-vis absorption of TMB solutions in the MnO2-plasma and single plasma systems. Blank TMB was used for comparison. | ||
To further clarify the roles of MnO2 in the degradation, the characterization of the catalysts before and after the MnO2-plasma treatment was carried out. As demonstrated by X-ray photoemission spectroscopy (XPS) (Fig. 2B), no obvious shift was observed for either Mn4+ (643.7 and 654.2 eV) or Mn3+ (642.2 and 653.2 eV) in the Mn 2p spectrum. Similarly, the peaks of the lattice oxygen (Mn–O–Mn, 529.8 eV) and surface hydroxyl oxygen (Mn–O–H, 531.0 eV) in the O 1s spectra of MnO2 (Fig. 2C) exhibited no significant shift after the plasma catalysis.31 This indicated the good stability of the MnO2 catalyst during the surface conversions in the degradation system. Notably, the Mn–O–Mn and Mn–O–H peaks were enhanced after the reaction (O 1s, Fig. 2C). This indicated the significant interaction between the catalysts and reactive species/intermediates on MnO2. This was in accordance with the emergence of new IR absorptions (–O–H at 3559 cm−1 and –N–H at 3219 cm−1, Fig. 2D) as well as new Raman peaks (837 cm−1 and 872 cm−1, the peroxide species for generating ˙OH) after MnO2-plasma treated (Fig. 2E).32,33 Therefore, reactive oxygen species of ˙OH were confirmed to be absorbed on the surface of MnO2, enhancing the degradation.
Next, the active species in the MnO2-plasma system were captured and characterized through electron paramagnetic resonance (EPR) analysis with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent. The characteristic quartet line of DMPO-˙OH (at an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1)34 was recorded for MnO2. As demonstrated, the EPR signals of ˙OH were enhanced after the treatment, which indicated the generation of more ˙OH to facilitate efficient pollutant degradation (Fig. 2F). The increase of ˙OH in the MnO2-plasma system was also confirmed by the increased absorption of the oxidized 4,4′-diamino-3,3′,5,5′-tetramethyl biphenyl (oxTMB, ˙OH marker) after treatment (Fig. 2G).35 Therefore, a large amount of active ˙OH was generated in the MnO2-plasma system, which would initiate the efficient degradation of pollutants via the SET process.
:2:2:1)34 was recorded for MnO2. As demonstrated, the EPR signals of ˙OH were enhanced after the treatment, which indicated the generation of more ˙OH to facilitate efficient pollutant degradation (Fig. 2F). The increase of ˙OH in the MnO2-plasma system was also confirmed by the increased absorption of the oxidized 4,4′-diamino-3,3′,5,5′-tetramethyl biphenyl (oxTMB, ˙OH marker) after treatment (Fig. 2G).35 Therefore, a large amount of active ˙OH was generated in the MnO2-plasma system, which would initiate the efficient degradation of pollutants via the SET process.
Examination of SET in the MnO2-plasma degradation system
Considering that SET could be the initial step for degradation in the MnO2-plasma system, the intermediates were examined by ESSI-MS at the beginning of the HCQ degradation. Different degradation systems, including MnO2-plasma, single plasma and MnO2-plasma systems with ˙OH quencher added, were examined for comparison. For HCQ degradation in the MnO2-plasma system (Fig. 3A), intermediate ions at m/z 335 (HCQ˙+), m/z 334 ([C1′]+) and 320 ([C1]+) were observed. Notably, HCQ˙+ (at m/z 335) was obtained by the extraction of an electron from the HCQ molecule via SET, and was the important initial intermediate of SET. The abundant ion at m/z 320 was defined as a carbocation ([C1]+) rather than an intermediate with a double bond, due to the easy oxidation of the double bond by ˙OH. The corresponding structure of [C1]+ was determined by collision-induced dissociation (CID) experiments (Fig. S2†) and high-resolution mass spectrometry (HR-MS) (Fig. S3, Table S1†). The C1 carbocation could be the intermediate generated by the removal of ·CH3 from HCQ˙+ through α-induced cleavage. Alternatively, the loss of ˙H through α-induced cleavage of HCQ˙+ could also occur, indicated by the observation of a low abundance C1′ carbocation (at m/z 334). The low signal of C1′ was because of the relatively poor stability of C1′ compared to C1 (Fig. S10†).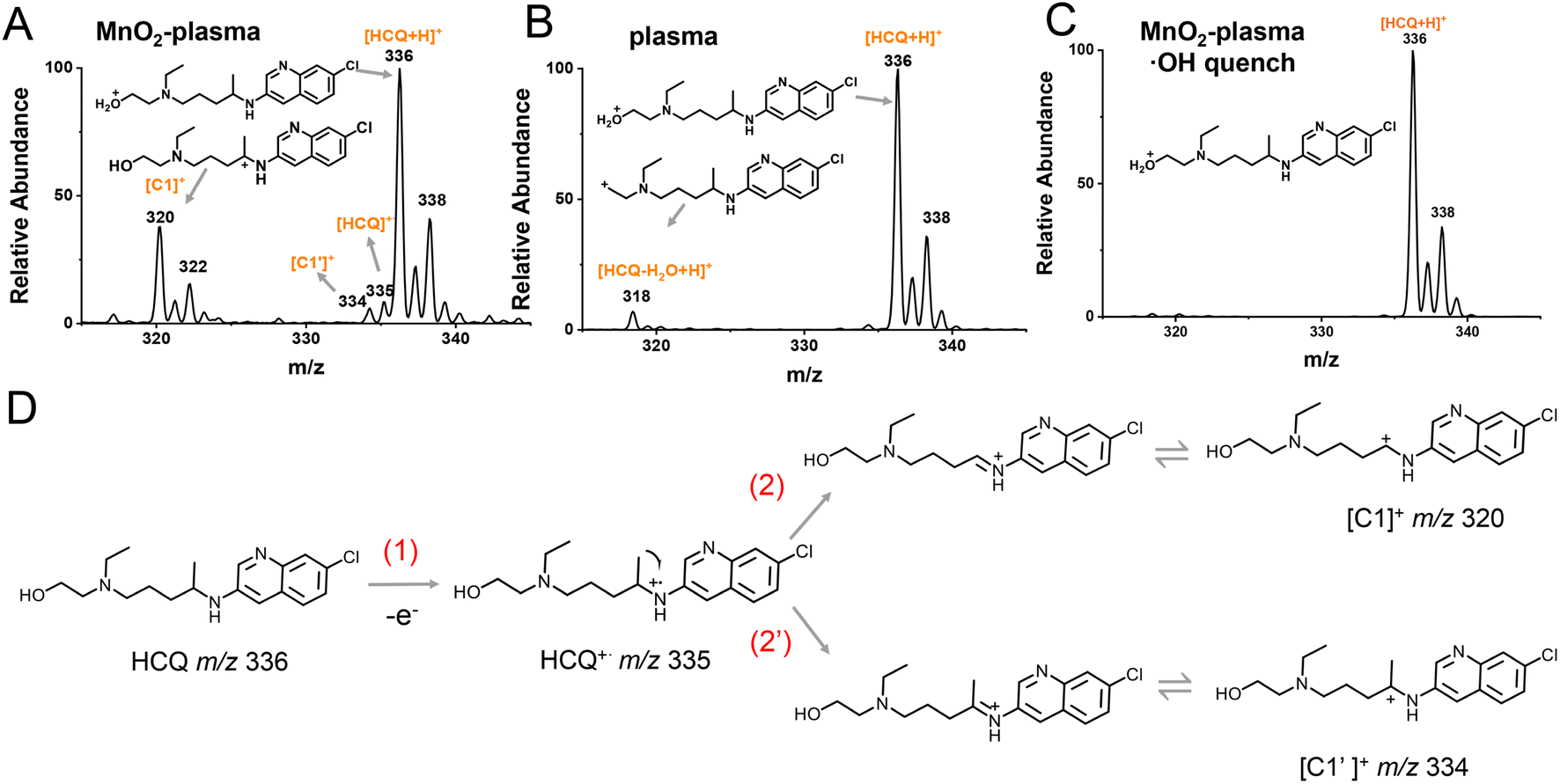 | ||
| Fig. 3 The examination of the SET process by ESSI-MS. Mass spectra of the HCQ solution in (A) the MnO2-plasma system, (B) the single plasma system and (C) the MnO2-plasma system with ˙OH quencher added. Degradation time: 1 min. (D) Proposed mechanism of SET accelerated degradation via carbocation intermediates. | ||
In addition, the active ˙OH played a crucial role in this SET-initiated conversion. This was confirmed by the failure of HCQ degradation (Fig. 3C and S11†) after the addition of the ˙OH radical quencher isopropanol. This was also in accordance with the absence of HCQ˙+, [C1]+ and [C1′]+ signals without MnO2 (Fig. 3B), because MnO2 was the main factor in ˙OH generation (Fig. 2A). Considering that the formation of HCQ˙+ (m/z 335), [C1′]+ (m/z 334) and [C1]+ (m/z 320) were the crucial steps for SET, it can be demonstrated that SET cannot be employed without ˙OH. Therefore, in the MnO2-plasma system, ˙OH-initiated SET process can facilitate efficient HCQ degradation with ˙OH as the active species. As shown in Fig. 3D, upon initiation by ˙OH with a strong oxidation capacity, an electron was initially withdrawn from HCQ to generate HCQ˙+ through SET. Subsequently, via the α-induced cleavage of HCQ˙+, the carbocations of both C1 and C1′ were generated for the following degradation.
On-line monitoring of the MnO2-plasma degradation
To further examine the ˙OH-initiated SET for the degradation of HCQ in the MnO2-plasma system, the degradation process was continuously monitored by ESSI-MS. As proposed (Fig. 4A), an electron was pulled out from HCQ to generate HCQ˙+via SET upon catalysis by the plasma-treated MnO2. Subsequently, the degradation of the HCQ occurred via (1) the rapid formation of carbocation and other intermediates, (2) dechlorination, and (3) generation of the final products of CO2 or NO3−. During the degradation, a series of important intermediates were generated: the carbocation C1 (at m/z 320) was produced by the α-induced cleavage of HCQ˙+; another carbocation (C2) was obtained by removing ˙CH2CH2OH from C1 and adding an OH group (confirmed by HR-MS, as shown in Table S1†); the ion at m/z 159 was attributed to the alkyl ketone-based intermediate product ion [KP + H]+, generated by the HCQ cleavage and dechlorination. The structure of [KP + H]+ is shown in Fig. 4B. The dechlorination was confirmed by the absence of the Cl isotope peak of [M + 2]+ for [KP + H]+, in contrast to the observation of the Cl isotope peak of C2 at m/z 294 (Fig. S12†).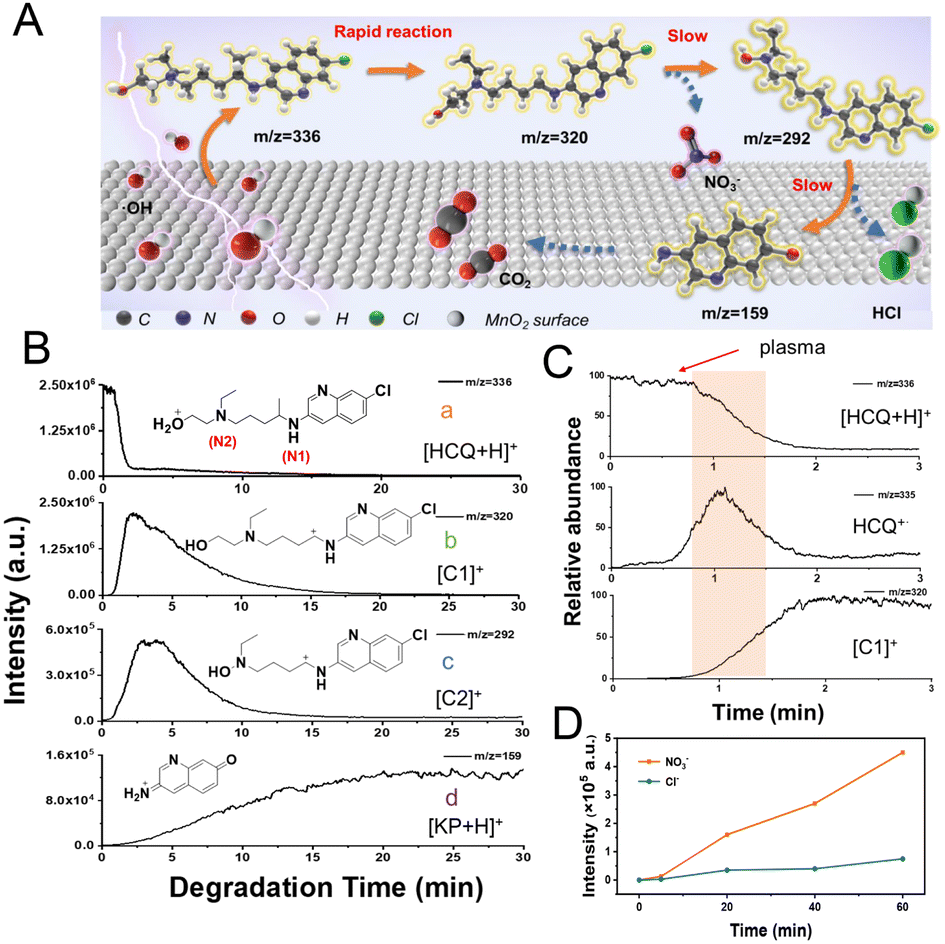 | ||
| Fig. 4 The online monitoring of SET accelerated degradation by ESSI. (A) Schematic representation of the degradation on the MnO2 surface. (B) EICs of [HCQ + H]+ at m/z 336 (a), carbocations C1 at m/z 320 (b) and C2 at m/z 292 (c), and intermediate [KP + H]+ at m/z 159 (d). (C) Magnified EICs of [HCQ + H]+, HCQ˙+ and C1 in the first 3 min. (D) Changes of Cl− and NO3− intensities during the degradation in the MnO2-plasma system. | ||
Ions at m/z 336, 335, 320, 292 and 159 were monitored by ESSI-MS for 30 min during the degradation. The online extracted ion chromatograms (EICs) of the corresponding reactant ions and carbocations were collected to examine the dynamic changes. As shown in Fig. 4B(a), upon introducing the plasma into the MnO2 system, a dramatic decrease of the reactant signal of [HCQ + H]+ (m/z 336) was recorded within 2.0 min. This indicated the potential outstanding degradation efficiency of the MnO2-plasma system for HCQ degradation. While followed by a rapid decrease of [HCQ + H]+, the signal of the carbocation C1 (m/z 320) increased almost immediately (at about 2.0 min) and then gradually decreased, and had vanished after about 20 min (Fig. 4B(b)). The zoomed-in EICs of important species ([HCQ + H]+, HCQ˙+ and [C1]+) in the first 3 min further confirmed the initial generation and rapid vanishing of HCQ˙+, along with the generation of C1 (Fig. 4C). Immediately after C1 generation (Fig. 4B(c)), the signal of another carbocation intermediate C2 (at m/z 292) increased and reached a peak value a little later than C1. This could indicate the conversion of intermediate C1 to C2 by decomposition. Interestingly, the generation of C1 from HCQ˙+via SET was much more rapid than the generation of C2 through decomposition. This further verified the initiation of the accelerated degradation by the rapid SET.
Additionally, the signal of [KP + H]+ at m/z 159 gradually increased during the entire monitoring time (Fig. 4B(d)), which demonstrated that KP could be an important intermediate product. In addition, the intensities of the signals of small species (such as Cl− and NO3−) increased along with the degradation (Fig. 4D), also demonstrating the dechlorination and advanced degradation in this MnO2-plasma system. Therefore, the ˙OH-initiated SET facilitated the accelerated degradation via the formation of carbocations in an efficient pathway.
Theoretical calculations for the ˙OH-initiated SET process
Based on experimental examination, the ˙OH-initiated SET would facilitate the initial generation of HCQ˙+via the withdrawal of an electron from N1 of the HCQ. The obtained HCQ˙+ would spontaneously rupture to form carbocation C1. Then, carbocation C2 is generated via electrophilic attack of the N2 atom of C1 by ˙OH. Herein, to further determine the reaction sites and explore the SET-based formation of the aforementioned intermediates (radical cation and carbocations), theoretical calculations were performed using DFT with the Gaussian 09 program package.36 At the M06-2X/DEF2TZVP level, geometrical parameters of the reactants, radical cation and carbocation intermediates, and transition states (TS) were optimized. The energy diagram of SET for intermediate formation (Fig. 5A) and the orbital-weighted double descriptor isosurface of the Fukui function of the important species (Fig. 5B) were determined by the Gaussian 09 program package and the Multiwfn program.37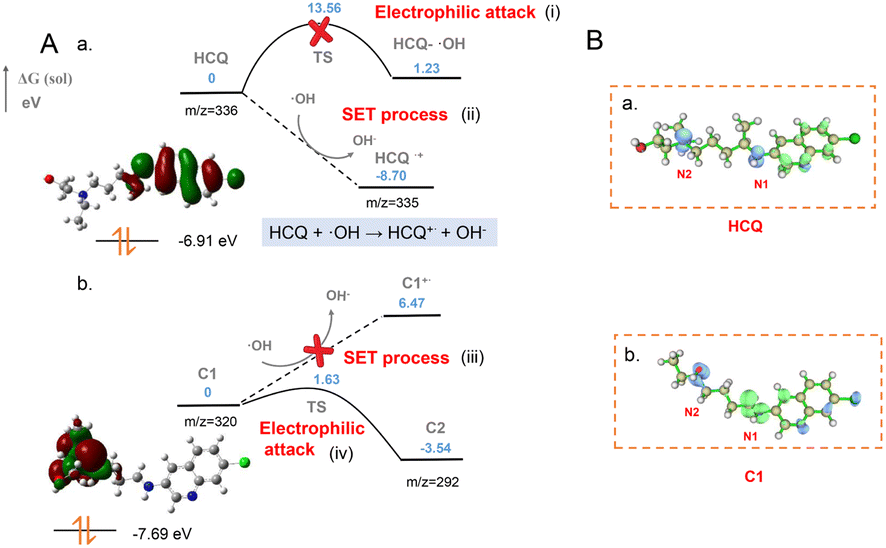 | ||
| Fig. 5 Mechanism of ˙OH-initiated SET. (A) The energy diagram of the SET process and carbocation formation. (B) The orbital-weighted double descriptor isosurface of the Fukui function for HCQ and the carbocation intermediate C1 (m/z = 320). The blue colour indicates negative regions and the green colour indicates positive regions. | ||
As indicated by the HOMO orbital of the HCQ molecule (Fig. 5A(a)), the electrons in the N1 atom of HCQ were rich and easily withdrawn by ˙OH to produce HCQ˙+. This was in accordance with the Fukui function and Fukui function indices of the HCQ molecule (Fig. 5B(a), Table S2†)38 as well as the much lower energy of HCQ˙+ (−8.70 eV) than the HCQ reactant (0 eV) (Fig. 5A(a-ii)). Furthermore, the energy barrier to electrophilic attack of the HCQ molecule by OH radicals was 1.23 eV (Fig. 5A(a-i)). This was much higher than that to the withdrawal of an electron from HCQ by ˙OH (−8.70 eV). This confirmed the initial generation of HCQ˙+via ˙OH-initiated SET for the subsequent degradation. Then, the HCQ˙+ facilitated spontaneous α cleavage to form C1, which resulted in the high intensity of the [C1]+ signal. Therefore, C1 was generated through ˙OH-initiated SET for the HCQ degradation. Subsequently, the C1 carbocation was able to be attacked by ˙OH at the active site of N2, which was confirmed by the HOMO orbital and Fukui function (Fig. 5B(b) and Table S3†). As shown in Fig. 5A(b), two potential pathways including SET and electrophilic attack could occur for the subsequent C1 conversion. The calculations show that, unlike for the N1 atom, there is no conjugated structure adjacent to N2, resulting in a decreased orbital energy of the lone pairs on the N2 atom. Thus, the electrons on N2 are stable and not easily withdrawn by ˙OH because of a very high energy barrier. This indicated that the SET would not occur to produce C1˙+ by loss of an electron from N2 of C1 (Fig. 5A(b-iii)). In contrast, the energy of the electrophilic attack of the N2 atom by ˙OH was much lower than that of the SET process, indicating that the second step of the degradation process could be an electrophilic attack step (Fig. 5A(b-iv)). Therefore, carbocation C2 was generated by the loss of the ·CH2CH2OH radical upon electrophilic attack. This process was further confirmed by the calculated heat release of 3.54 eV (from 0 to −3.54 eV) via a transition state with an energy barrier of 1.63 eV. In addition, the energy of the HOMO orbital of HCQ (−6.91 eV) was higher than that of C1 (−7.69 eV). As indicated by the HOMO orbital energy, C1 was generated by the ˙OH-initiated SET process, while C2 was formed by the ˙OH electrophilic attack. This was also in accordance with the slightly later generation of C2 than C1 (Fig. 4B). Consequently, the different pathways for carbocation generation and the easily ˙OH-initiated SET for HCQ˙+ generation were confirmed. Therefore, upon ˙OH attack, the SET-initiated generation of HCQ˙+ and the subsequent sequential formation of two carbocations were theoretically demonstrated, in accordance with the experiments.
In addition, a step-by-step examination of the degradation was carried out using calculations to propose the degradation mechanism (Fig. S14†). Attacked by ˙OH, the HCQ˙+ would subsequently cleave into the C1 carbocation (at m/z 320) by losing ˙CH3 through the transition state of TS1 (Fig. S14(ii)†). The corresponding TS structures are shown in Fig. S13.† Subsequently, an intermediate carbocation C2 (at m/z 292) and radical fragment ˙CH2CH2OH were generated through the transition state TS2 (Fig. S14(iii)†). Then, via the transition state TS3, another intermediate IM3 was formed by the further attack by ˙OH of the C2 carbocation (Fig. S14(iv)†). This process was confirmed by the formation of a short C–O bond of TS3 (2.01 Å) with a small energy barrier of 0.28 eV (from −14.96 to −14.68 eV). Subsequently, HCl could be rapidly removed from IM3 to generate IM4 via an exothermic process (Fig. S14(v)†). Thus, KP was obtained by the cleavage of IM4 (Fig. S14(vi)†). Although another intermediate, IM5, could be generated by the attack of ˙OH at C10 in IM4 (Fig. S14(vi)†), it was still unfeasible to obtain KP via an endothermic pathway (from −19.04 to −18.02 eV). Therefore, ˙OH-initiated SET occurred to generate HCQ˙+ in the MnO2-plasma system, which facilitated the following generation of carbocation intermediates for efficient degradation.
Mechanism of SET accelerated degradation via carbocation intermediates
The mechanism of HCQ degradation via ˙OH-initiated SET is proposed in Fig. 6. Initially, a large amount of ˙OH was generated on the MnO2 surface treated by the plasma. This facilitated the subsequent efficient and rapid degradation of pollutants. Then, upon ˙OH attack, SET occurred to obtain HCQ˙+via the withdrawal of an electron from N1 of HCQ. Subsequently, HCQ˙+ spontaneously ruptured to form the carbocation C1 (at m/z 320) by removal of a ˙CH3 group. Then, supported by the electron-rich and low orbital energy of the lone pair of N2, ˙OH electrophilically attacked N2 of C1 rather than withdrawing electrons. This resulted in the formation of C2 by the removal of a ˙CH2CH2OH group and the addition of a ˙OH group at the N2 of C1. Subsequently, the alkyl ketone-based intermediate product (KP) was generated via ˙OH attack and the corresponding cleavages. Finally, some small molecules resulting from advanced degradation (HCl, CO2, H2O and NO3−) were generated when the degradation reaction was prolonged.39,40 This was confirmed by the generation of precipitates via the Ca(OH)2 precipitation test as well as the recording of nitric acid (NO3−) and chloride (Cl−) ions by MS (Fig. S15†). Therefore, in the MnO2-plasma degradation system, the SET process occurred upon ˙OH attack, resulting in efficient degradation with carbocations as important intermediates.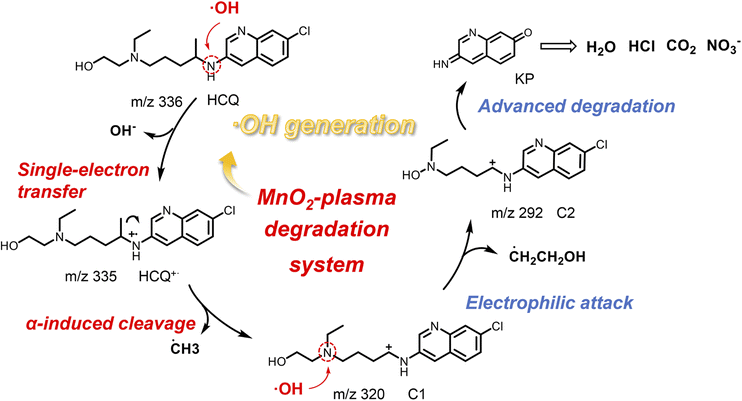 | ||
| Fig. 6 The mechanism of ˙OH-initiated SET for accelerated degradation with carbocations as intermediates. | ||
Conclusion
In conclusion, radical cations, carbocations and other reaction intermediates were successfully captured during ˙OH-initiated SET in the MnO2-plasma system. Characterization showed that a large amount of ˙OH was generated on the MnO2, which was demonstrated to be important for initiating SET-based degradation. Upon the attack by ˙OH, the radical cation HCQ˙+ was initially formed through SET. Simultaneously, two carbocations were sequentially generated for efficient degradation. Significantly, based on dynamic changes of the intermediates, the SET-accelerated degradation via the formation of carbocations was proposed. Through theoretical calculations, transition states, energy barriers of the formation of radical cations via SET, and subsequent carbocation formations were revealed step-by-step. Additionally, the generation of advanced degradation products (CO2, NO3− and H2O) was demonstrated as the degradation proceeded. By demonstrating ˙OH-initiated SET via carbocations, this work not only supports the understanding of SET but also develops efficient pathways for green degradation.Data availability
All relevant data is presented in the manuscript and ESI.† Raw data is available upon request by email to the corresponding author.Author contributions
X. Ge and N. Na conceived and designed the project. X. Ge performed the experiments. Y. Yin contributed to the calculations. J. Sun supported figure preparations and J. Ouyang supported the characterizations. N. Na directed the whole research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (NNSFC 22274012), the National Key Research and Development Program of China (No. 2019YFC1805600) and NNSFC 21974010.Notes and references
- A. J. Musacchio, L. Q. Nguyen, G. H. Beard and R. R. Knowles, J. Am. Chem. Soc., 2014, 136, 12217–12220 CrossRef CAS PubMed.
- A. Hazra, J. A. Kephart, A. Velian and G. Lalic, J. Am. Chem. Soc., 2021, 143, 7903–7908 CrossRef CAS PubMed.
- X. Mu, Y. Li, N. Zheng, J. Long, S. Chen, B. Liu, C. Zhao and Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 11211–11216 CrossRef CAS PubMed.
- L. Lu, R. Shi and A. Lei, Trends Chem., 2022, 4, 179–190 CrossRef CAS.
- J. M. Ganley, P. D. Murray and R. R. Knowles, ACS Catal., 2020, 10, 11712–11738 CrossRef CAS PubMed.
- Q. Zhu, E. C. Centry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS PubMed.
- X. Tang, W. Chen, X. Yi, Z. Liu, Y. Xiao and Z. Chen, Angew. Chem. Int. Ed., 2021, 60, 4581–4587 CrossRef CAS PubMed.
- A. Kumar, S. Mondal, Sandeep, P. Venugopalan, A. Kumar and S. Banerjee, J. Am. Chem. Soc., 2022, 144, 3347–3352 CrossRef CAS PubMed.
- A. Kumar, S. Mondal and S. Banerjee, J. Am. Chem. Soc., 2021, 143, 2459–2463 CrossRef CAS PubMed.
- A. Kumar, S. Mondal, M. Mofidfar, R. N. Zare and S. Banerjee, J. Am. Chem. Soc., 2022, 144, 7573–7577 CrossRef CAS PubMed.
- H. Ma, R. K. Sharma, S. Welzel, M. Sanden and M. N. Tsampas, Nat. Commun., 2021, 13, 402 CrossRef PubMed.
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef CAS PubMed.
- P. Mehta, P. Barboun, D. B. Go, J. C. Hicks and W. F. Schneider, ACS Energy Lett., 2019, 4, 1115–1133 CrossRef CAS.
- Y. Meng, E. Gnanamani and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 19709–19713 CrossRef CAS PubMed.
- J. He, N. Li, D. Zhang, G. Zheng, H. Zhang, K. Yu and J. Jiang, Environ. Sci.: Water Res. Technol., 2020, 6, 181–188 RSC.
- K. Yu, H. Zhang, J. He, R. N. Zare, Y. Wang, L. Li, N. Li, D. Zhang and J. Jiang, Anal. Chem., 2018, 90, 7154–7157 CrossRef CAS PubMed.
- J. Sun, Y. Yin, J. Li, J. Ouyang and N. Na, Mass Spectrom. Rev., 2022, 41, 70–99 CrossRef CAS PubMed.
- X. Zhong, H. Chen and R. N. Zare, Nat. Commun., 2020, 11, 1049 CrossRef CAS PubMed.
- J. Ghosh, J. Mendoza and R. G. Cooks, Angew. Chem., Int. Ed., 2022, e202214090 CAS.
- N. Bensalaha, S. Midassi, M. I. Ahmad and A. Bedoui, Chem. Eng. J., 2020, 402, 126279 CrossRef PubMed.
- N. Zeng, Z. Long, Y. Wang, J. Sun, J. Ouyang and N. Na, Anal. Chem., 2019, 91, 15763–15768 CrossRef CAS PubMed.
- Y. Wang, J. Sun, J. Qiao, J. Ouyang and N. Na, Anal. Chem., 2018, 90, 14095–14099 CrossRef CAS PubMed.
- H. Lu, Y. Yin, J. Sun, W. Li, X. Shen, X. Feng, J. Ouyang and N. Na, Chin. Chem. Lett., 2021, 32, 3457–3462 CrossRef CAS.
- M. Fiedrich, R. Kurtenbach, P. Wiesen and J. Kleffmann, Atmos. Environ., 2017, 165, 57–61 CrossRef CAS.
- X. Wang, P. Wamg, X. Liu, L. Hu, Q. Wang, P. Xu and G. Zhang, Chem. Eng. J., 2020, 389, 124381 CrossRef CAS.
- Y. Wang, M. Sun, J. Qiao, J. Ouyang and N. Na, Chem. Sci., 2018, 9, 594–599 RSC.
- J. Sun, X. Fan, H. Lu, H. Tan, Y. Zhang, Y. Wang, Y. Zhao, J. Ouyang and N. Na, Chem. Commun., 2021, 57, 3921 RSC.
- R. Song, H. Chai, Q. Ma, D. Li, X. Wang, W. Gao, H. Wang, X. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2021, 143, 13664–13674 CrossRef CAS PubMed.
- X. Tan, Y. Wan, Y. Huang, C. He, Z. Zhang, Z. He, L. Hu, J. Zeng and D. Shu, J. Hazard. Mater., 2017, 321, 162–172 CrossRef CAS PubMed.
- G. Venkateswalu, D. Madhu and J. V. Rani, Int. J. Energy Res., 2020, 44, 10238–10250 CrossRef.
- R. Yang, S. Peng, B. Lan, M. Sun, Z. Zhou, C. Sun, Z. Gao, G. Xing and L. Yu, Small, 2021, 17, 2102408 CrossRef CAS PubMed.
- L. Kankate, T. Hamann, S. Li, L. V. Moskaleva, A. Turchanin and P. Swiderek, Phys. Chem. Chem. Phys., 2018, 20, 29918 RSC.
- J. Jia, P. Zhang and L. Chen, Appl. Catal., B, 2016, 189, 210–218 CrossRef CAS.
- X. Yang, X. Xu, J. Xu and Y. Han, J. Am. Chem. Soc., 2013, 135, 16058–16061 CrossRef CAS PubMed.
- X. Meng, D. Li, L. Chen, H. He, Q. Wang, C. Hong, J. He, X. Gao, Y. Yang, B. Jiang, G. Nie, X. Yan, L. Gao and K. Fan, ACS Nano, 2021, 15, 5735–5751 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Q. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Zhang, J. Chen, J. Wang, C. Cui, B. Wang and Y. Zhang, Water, 2021, 13, 128 CrossRef CAS.
- W. Wu, Y. Song, L. Bai, Z. Chen, H. Sun, G. Zhen, R. Zhan, Y. Shen, J. Qian, Q. Yuan and Z. Sun, ACS Appl. Nano Mater., 2020, 3, 9363–9374 CrossRef CAS.
- S. Yang, H. Yang, J. Yang, H. Qi, J. Kong, Z. Bo, X. Li, J. Yan, K. Cen and X. Tu, Chem. Eng. J., 2020, 402, 126154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06915f |
| This journal is © The Royal Society of Chemistry 2023 |