
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient Förster resonance energy transfer between an emissive tetraphenylethylene-based metal–organic cage and the encapsulated dye guest†
Danyang
Li
a,
Xin
Liu
 a,
Linlin
Yang
b,
Hechuan
Li
a,
Guoxu
Guo
a,
Xuezhao
Li
a and
Cheng
He
*a
a,
Linlin
Yang
b,
Hechuan
Li
a,
Guoxu
Guo
a,
Xuezhao
Li
a and
Cheng
He
*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116012, P. R. China. E-mail: hecheng@dlut.edu.cn
bXinxiang Key Laboratory of Forensic Science Evidence, School of Forensic Medicine, Xinxiang Medical University, Xinxiang 453003, P. R. China
First published on 31st January 2023
Abstract
The host–guest strategy presents an ideal way to achieve efficient Förster resonance energy transfer (FRET) by forcing close proximity between an energy donor and acceptor. Herein, by encapsulating the negatively charged acceptor dyes eosin Y (EY) or sulforhodamine 101 (SR101) in the cationic tetraphenylethene-based emissive cage-like host donor Zn-1, host–guest complexes were formed that exhibit highly efficient FRET. The energy transfer efficiency of Zn-1⊃EY reached 82.4%. To better verify the occurrence of the FRET process and make full use of the harvested energy, Zn-1⊃EY was successfully used as a photochemical catalyst for the dehalogenation of α-bromoacetophenone. Furthermore, the emission color of the host–guest system Zn-1⊃SR101 could be adjusted to exhibit bright white-light emission with the CIE coordinates (0.32, 0.33). This work details a promising approach to enhance the efficiency of the FRET process by the creation of a host–guest system between the cage-like host and dye acceptor, thus serving as a versatile platform for mimicking natural light-harvesting systems.
Introduction
Over the past few decades, coordination-driven assembled metal–organic cages have provided a sustainable way to echo the remarkable properties of natural enzymes, due to their facile synthesis, high solubility, and stability in common solvents. With their precisely controlled well-defined cavities, such supramolecular coordination hosts have been widely studied for selective guest encapsulation1 and recognition,2 using which highly efficient and stereoselective catalysis has been realized via substrate preorganization and the stabilization of reactive intermediates.3,4 Besides these exciting confinement effects, host–guest systems with their essential components forced close together within the inner space of the cage are also promising candidates to use to develop supramolecular charge/energy donor–acceptor assemblies, which lead to efficient enhancement in energy, electron, or substance transfer, based on short through-space and long through-bond pathways.5Förster resonance energy transfer (FRET) is a classic energy transfer process between different chromophores that proceeds via nonradiative dipole–dipole coupling, which has received considerable attention on account of its various applications in photocatalysis,6 biological imaging,7 fluorescence probes,8 and luminescent materials.9 In addition to matching the spectral overlap between the donor emission and acceptor excitation,10 effective FRET requires an appropriate distance and dipole orientation between the donor and acceptor.11 To date, various challenges have been faced using the reported FRET systems linked by covalent bonds (such as porphyrin arrays and dendrimers), such as their time-consuming, multistep synthetic processes, as well as the possibility of emission changes resulting from covalent functionalization.12 Alternatively, systems formed via non-covalent interactions could pave a facile and promising path to construct light-harvesting systems,13 for example, the chlorophyll–protein system for photosynthesis in green plants.14
Inspired by nature, significant advances have been made in mimicking light antenna harvesting systems via the FRET process. A wide variety of supramolecular assemblies with noncovalent interactions have been successfully synthesized by designing special scaffolds to accommodate chromophores,15 such as polymers16 and organic–inorganic hybrid materials.17 Challenges in constructing efficient supramolecular FRET systems remain as to how to densely assemble the donor with a high donor–acceptor ratio while avoiding the self-quenching effect induced by molecular aggregation.18 Therefore, the use of a metal–organic cage as a donor could be a promising approach to assemble light-harvesting systems with highly efficient FRET processes.6b,c,19 The encapsulation of a guest within the inner cavity of the cage would make the interaction between the donor and the acceptor stronger and more robust in close proximity.20 Moreover, a highly charged and rigid coordination cage provides an isolated microenvironment in which the donors are fixed and arranged with high local concentration, and decreases the possibility of self-quenching.21
Among the building blocks of supramolecular coordination complexes, tetraphenylethene (TPE) has been shown to exhibit unique aggregation-induced emission (AIE) properties, due to the restriction of the rotation of its phenyl ring by coordination bonds, which decreases the nonradiative decay.22,23 Moreover, quasi C4-symmetrical TPE-based ligands with multiple phenyl rings are good building blocks to construct face-driven assembled metal–organic cages with confined cavities that can encapsulate suitable guests rigidly. Therefore, TPE-based coordination cages that exhibit good emission would be ideal light-emitting donors for highly efficient energy transfer.
Herein, a highly emissive cage Zn-1 was constructed as an energy donor via face-driven assembly between TPE-based ligands and zinc ions. The cationic cage Zn-1 is capable of encapsulating negatively charged guests, such as eosin Y (EY) or sulforhodamine 101 (SR101), as suitable energy acceptors (Scheme 1). It was envisioned that a highly efficient FRET process could be realized between the face-capped host and the dye guests with a high local donor–acceptor ratio and close distance between them. Thus, a systematic investigation of the host–guest chemistry, as well as the FRET process between the donor cage Zn-1 and acceptor dye EY or SR101, was conducted. To better verify the occurrence of the FRET process and make full use of the harvested energy, the Zn-1⊃EY complex was applied as a photochemical catalyst in the dehalogenation of α-bromoacetophenone under ultraviolet (UV) light (365 nm) irradiation, and the Zn-1⊃SR101 complex was shown to function as a white light-emitting system.
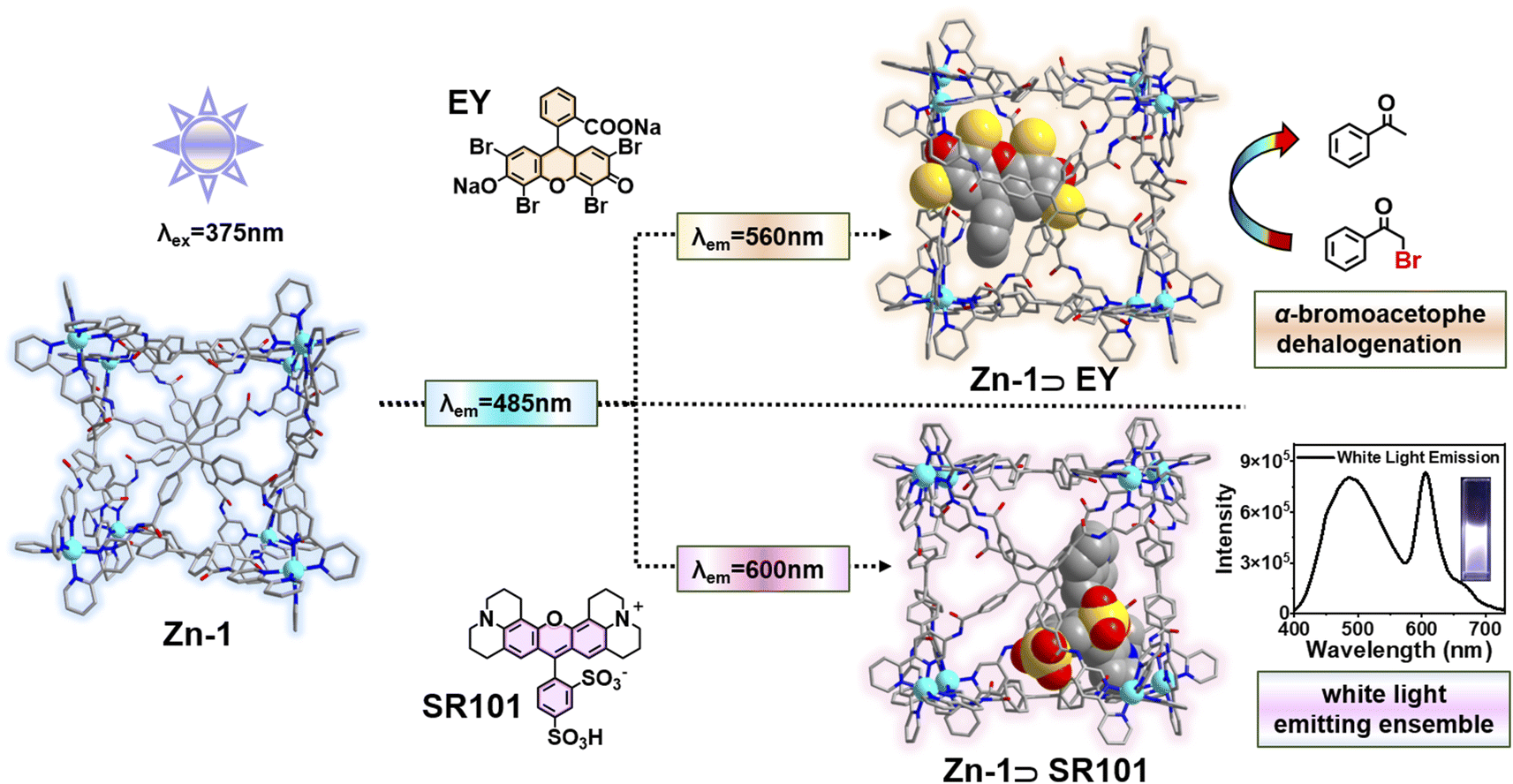 | ||
| Scheme 1 Illustration of the host–guest strategy between the cage Zn-1 and the encapsulated negatively charged acceptor dye (EY and SR101) that induces effective FRET. | ||
Results and discussion
Synthesis and characterization of cage Zn-1
Ligand L (L = 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)tetrakis(N-([2,2′-bipyridin]-5-yl) benzamide)) with four long arms containing bidentate bipyridine units was synthesized by reacting 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)tetrabenzoyl chloride with 2,2′-bipyridin-5-amine (Scheme 2). The amide groups were incorporated into the ligand as potential hydrogen bond sites for host–guest binding. Reaction of L (6 equiv.) with zinc tetrafluoroborate (Zn(BF4)2, 8 equiv.) in N,N-dimethylformamide (DMF) at 80 °C afforded compound Zn-1 in a yield of 61%. Electrospray ionization mass spectrometry (ESI-MS) measurements on the Zn-1 solution revealed the formation of a stable Zn8L6 complex with a characteristic sequence of signals associated with the combination of different numbers of anions that give the target species, which showed isotopically resolved peaks at m/z = 698.5301, 777.0846, 873.0788, 993.0855, and 1147.3845 (Fig. 1a). A simple comparison with the simulation results based on the natural isotopic abundances showed that the resulting peaks could be attributed to [Zn-1·n(BF4−)](16−n)+ (n = 5–9, with the correct fractional isotope spacings in the high-resolution signals in every case).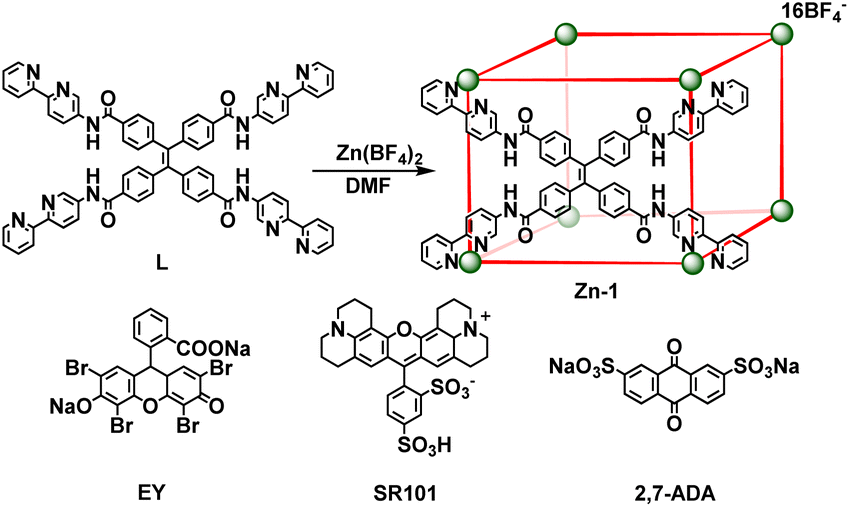 | ||
| Scheme 2 The self-assembly of cage Zn-1 and guests EY, SR101, and 2,7-ADA. | ||
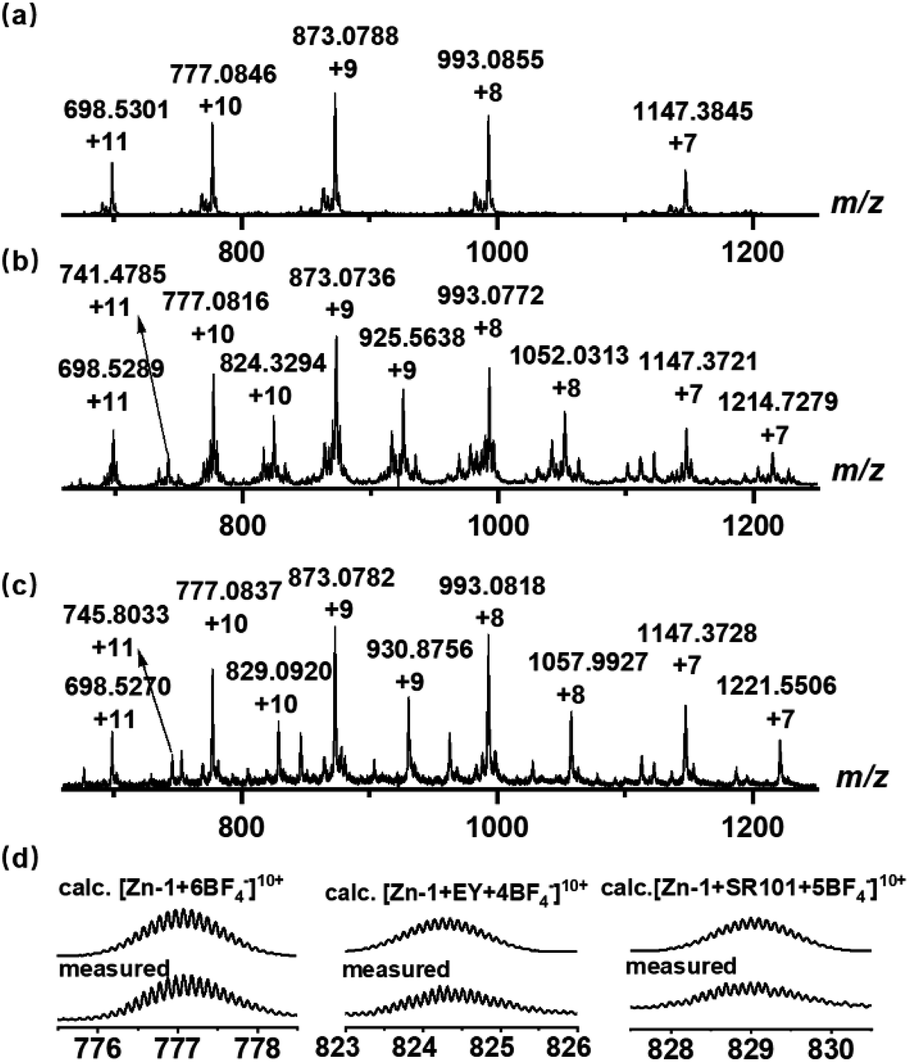 | ||
| Fig. 1 ESI-MS spectra of the synthesized (a) cage Zn-1 and host–guest complexes, (b) Zn-1⊃EY, and (c) Zn-1⊃SR101. (d) Experimental and calculated peaks. | ||
A crystal of Zn-1 suitable for the single-crystal X-ray diffraction study was obtained via the slow vapor diffusion of diethyl ether into a DMF/acetonitrile (CH3CN) solution of Zn-1 (ESI, Fig. S11a†). The crystal analysis of Zn-1 confirmed the formation of a face-capped slightly disorder cubic Zn8L6 cage with a large cavity (Fig. 2a), where each face of the cage is covered by one TPE-based ligand and each vertex is occupied by an octahedral coordination geometry Zn(II) ion chelated to three 2,2′-bipyridine groups from different TPE-based ligands. The four benzene rings in the TPE moiety are non-coplanar and slightly protrude into the cavity, and the Zn–Zn separations along the cage edges are in the range of 14.86–17.89 Å, giving an inner volume of approximately 4650 Å3, which could provide a big enough space for large guests, expanding the scope of host–guest chemistry.
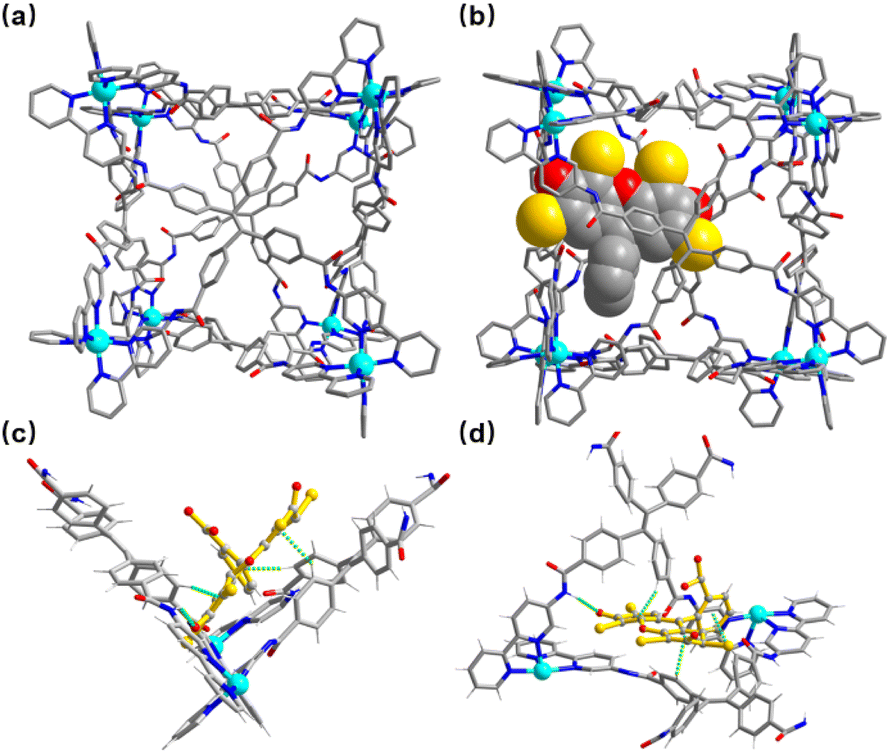 | ||
| Fig. 2 (a) View of the crystal structure of Zn-1; (b) view of the crystal structure of Zn-1⊃EY (the cavity-bound guest EY shows space-filling); (c) and (d) different views showing C–H⋯π and N–H⋯O interactions between an EY guest and Zn-1. Color coding: Zn = cyan; C = gray; N = blue; O = red; Br = yellow. Disorder, anions, and solvent molecules are omitted for clarity. | ||
The cage Zn-1 was further characterized by 1H, 13C, and 19F NMR spectroscopies (ESI, Fig. S7–S9†). Most of the 1H NMR signals of Zn-1 show a slight downfield shift with respect to those of the free ligand L. Furthermore, the protons of ligand L were split into multiple signals after coordination, indicating the formation of a low-symmetry cage. Additionally, the 1H diffusion-ordered (DOSY) NMR spectrum of Zn-1 showed the formation of a single species, with a diffusion coefficient (D) value of 3.16 × 10−9 m2 s−1 (ESI, Fig. S10†).
Host–guest complexes with encapsulated EY and SR101
The energy acceptors EY and SR101 with suitable absorptions were respectively introduced into the Zn-1 cage as guests. Electrostatic interactions were expected to be helpful for the binding of the two negatively charged dyes within the positively charged cage. Meanwhile, the planar π-conjugated moiety in EY and SR101 has the potential to form aromatic interactions with the ligand L of Zn-1. The ESI-MS data of the Zn-1 solution in the presence of EY exhibited a new set of intense peaks at m/z = 741.4785, 824.3294, 925.5638, 1052.0313, and 1214.7279, corresponding to [Zn-1·n(BF4−)·EY](14−n)+ (n = 3–7), respectively (Fig. 1b). Moreover, for the solution containing Zn-1 and SR101, new intense peaks appeared at m/z = 745.8033, 829.0920, 930.8756, 1057.9927, and 1221.5506, which were assigned to [Zn-1·n(BF4−)·SR101](15−n)+ (n = 4–8), respectively (Fig. 1c). The results thus suggested the possible formation of stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric host–guest complexes in both cases.
:1 stoichiometric host–guest complexes in both cases.
The formation of the host–guest complexes was also validated by conducting 1H NMR titration experiments between Zn-1 and the EY or SR101 guests (Fig. 3a and b, ESI, S13a and b†), and most of the NMR signals of the guests showed slight up-field shifts. Furthermore, H–H interactions between the aromatic rings of EY and the aromatic rings of Zn-1 could be observed in the nuclear Overhauser effect (NOESY) spectrum of Zn-1⊃EY (ESI, Fig. S15a†), which showed the guest EY to reside within the cavity of Zn-1. In the NOESY spectrum of Zn-1⊃SR101, the NOE contacts indicated strong interactions between the protons of the aromatic rings in ligand L and the methylene (–CH2–) protons of SR101 (ESI, Fig. S15b†). The results thus suggested the presence of potential weak interactions between the ligand and the guests, and indicated that the host and guest were close enough in space for efficient transfer of energy to occur. The binding constants of the guests (EY and SR101) with Zn-1 were detected by UV-Vis titration in DMF, using a cooperative 1:1 host–guest binding model, with association constants (K) of 2.2 × 106 M−1 and 1.6 × 106 M−1 for EY and SR101, respectively (Fig. 3c and d).
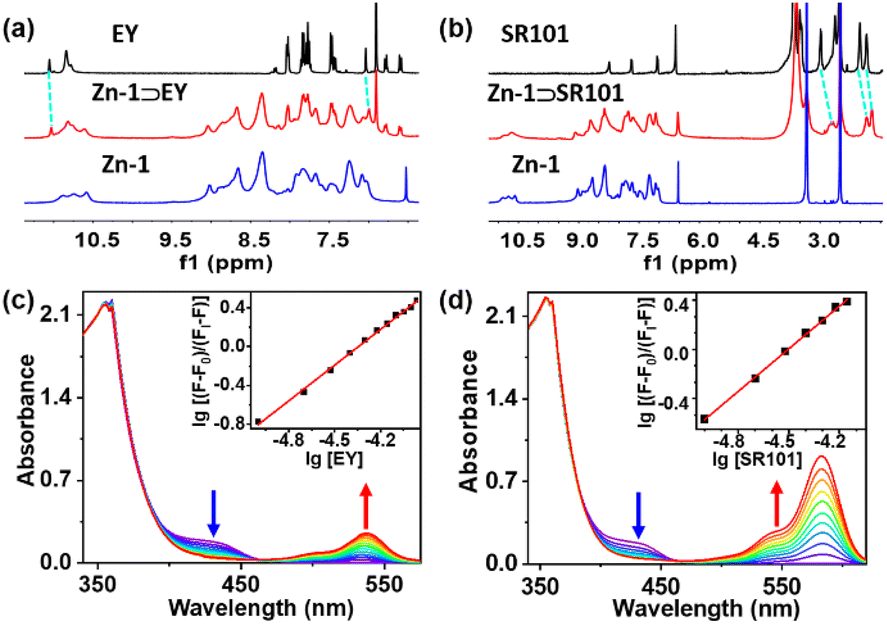 | ||
| Fig. 3 (a) Sections of the 1H NMR spectra of Zn-1, Zn-1⊃EY, and EY (in DMSO-d6); (b) sections of the 1H NMR spectra of Zn-1, Zn-1⊃SR101, and SR101 (in DMSO-d6); (c) absorbance spectra of Zn-1 ([Zn-1] = 1.0 × 10−5 M) in DMF upon the addition of EY. Inset: Hill plot of the titration curve showing the calculation of the associate constant (K = 2.2 × 106 M−1); (d) absorbance spectra of Zn-1 ([Zn-1] = 1.0 × 10−5 M) in DMF upon the addition of SR101. Inset: Hill plot of the titration curve showing the calculation of the associate constant (K = 1.6 × 106 M−1). | ||
When the guest EY or SR101 was added to a solution of Zn-1 in DMF in a 4:1 ratio and stirred at 80 °C overnight, the faint yellow color of the solution of Zn-1 turned orange (Zn-1⊃EY) or pink (Zn-1⊃SR101), respectively. Diffusing diethyl ether into the respective solutions, orange (Zn-1 with EY) or pink (Zn-1 with EY) crystal was obtained (ESI, Fig. S11†). The uniform distribution of EY (or SR101) in the fluorescence signals of Zn-1⊃EY (or Zn-1⊃SR101) was investigated by confocal fluorescence microscopy (Fig. 4), where the images suggested that the guest dye was encapsulated in the Zn-1 cage to form co-crystals rather than being adsorbed on the external surface of the crystals.
 | ||
| Fig. 4 Confocal images of Zn-1 (a and b), Zn-1⊃EY (c and d), and Zn-1⊃SR101 (e and f). Bright-field images (left) and dark-field images (right); (a, c and e) detected at λem = 440–500 nm through a 405 nm filter; (b) detected at λem = 440–500 nm; (d) detected at λem = 530–580 nm; (f) detected at λem = 580–630 nm, through a 488 nm filter. | ||
Although the crystal was low in quality, the crystal of encapsulated Zn-1⊃EY was still used in single-crystal X-ray diffraction measurements (Fig. 2b). Zn-1⊃EY crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with the structure of the cage being maintained by Zn–Zn distances in the range of 15.60–17.87 Å. The eosin Y anion with an average refined occupancy of 25% in the cavity of Zn-1 (ca. 1/4 cages formed the host–guest complexes) was located close to the corner of two adjacent TPE-based ligands to obtain a close distance between the donor host Zn-1 and acceptor EY, falling within the necessary range (generally <10 nm) to allow the FRET process to proceed. Although detailed analysis of the host–guest interactions was difficult due to the poor diffraction data as well as the high disorder of the components, some possible weak interactions could still be determined. The nearest C–H⋯π distances were found to be in the range of 3.05–3.30 Å between the benzene rings of the TPE-based ligand and the aromatic rings of the EY (Fig. 2c and d). Moreover, hydrogen bonding interactions seemed to be present between the N–H of the amine group in the ligand and the oxygen atom of EY, with a N⋯O distance of around 3.10 Å. These multiple weak interactions are helpful in maintaining the stability of the host–guest complex.
, with the structure of the cage being maintained by Zn–Zn distances in the range of 15.60–17.87 Å. The eosin Y anion with an average refined occupancy of 25% in the cavity of Zn-1 (ca. 1/4 cages formed the host–guest complexes) was located close to the corner of two adjacent TPE-based ligands to obtain a close distance between the donor host Zn-1 and acceptor EY, falling within the necessary range (generally <10 nm) to allow the FRET process to proceed. Although detailed analysis of the host–guest interactions was difficult due to the poor diffraction data as well as the high disorder of the components, some possible weak interactions could still be determined. The nearest C–H⋯π distances were found to be in the range of 3.05–3.30 Å between the benzene rings of the TPE-based ligand and the aromatic rings of the EY (Fig. 2c and d). Moreover, hydrogen bonding interactions seemed to be present between the N–H of the amine group in the ligand and the oxygen atom of EY, with a N⋯O distance of around 3.10 Å. These multiple weak interactions are helpful in maintaining the stability of the host–guest complex.
However, the weak diffraction intensity limited the crystal structure analysis of Zn-1⊃SR101. Therefore, density functional theory (DFT) calculations were conducted to show the possible structure of the host–guest complex. The DFT result suggested that SR101 is encapsulated inside the cavity of Zn-1 (ESI, Fig. S12†). In the plausible structure of Zn-1⊃SR101, the average Zn–Zn distance is 17.40 Å, falling within the experimental range of 14.86–17.89 Å of Zn-1. The anionic SR101 was suggested to be located at the intersection angle between two adjacent TPE-based ligands, similar to the position of EY in the Zn-1 cavity. The C–H⋯π distance was shown to be approximately 3.71 Å between the inward pointing protons of the TPE-based ligand and the aromatic ring of the SR101 guest, and the C–H⋯O distance was around 3.36 Å between the oxygen atom of the TPE-based ligand and the hydrogen atom of SR101.
FRET process of the host–guest complexes
The UV-Vis absorption and emission spectra of ligand L and cage Zn-1 in DMF are shown in Fig. S16 in the ESI.† Compared with the free ligand L, the absorption spectrum of Zn-1 underwent a small red shift (from 320 to 355 nm). The free ligand showed a relatively weak and broad emission at 505 nm and the quantum efficiency fluorescence was 3.09% in DMF (ESI, Fig. S17d, Table S3†). The fluorescence spectra of Zn-1 showed different emission intensities in different solvent systems (ESI, Fig. S18†). Moreover, the emission of Zn-1 was dramatically enhanced at blue shift to 485 nm and the quantum efficiency fluorescence of Zn-1 reached 17.89% in DMF (ESI, Fig. S17a†), due to the motions of the ligand being restricted by coordination bonds, thus decreasing the nonradiative decay.The absorption of EY overlapped well with the emission of Zn-1 in DMF (Fig. 5a). The 3D fluorescence spectra of Zn-1, EY, and Zn-1⊃EY showed a series of excitation wavelengths with different intensity emissions in DMF (ESI, Fig. S19†). It was found that the optimized excitation wavelengths of Zn-1⊃EY were close to the excitation wavelength of Zn-1, indicating the possibility that the energy of Zn-1 was transferred to the guest EY. The FRET process was revealed by fluorescence titration of EY into a solution of Zn-1 (Fig. 5b). With the addition of EY, the broad emission peak of Zn-1 at 485 nm gradually decreased, while a new and sharp emission peak for Zn-1⊃EY appeared at 560 nm when excited at 375 nm, accompanied by a change in the color of the fluorescence from blue to yellow under UV light. The CIE chromaticity diagram also confirmed the fluorescence color change from blue to yellow following the titration of EY (ESI, Fig. S20a†). Alternatively, addition of Zn-1 to the solution of EY showed that the intensity of Zn-1 and EY gradually increased when excited at 375 nm (ESI, Fig. S21a†).
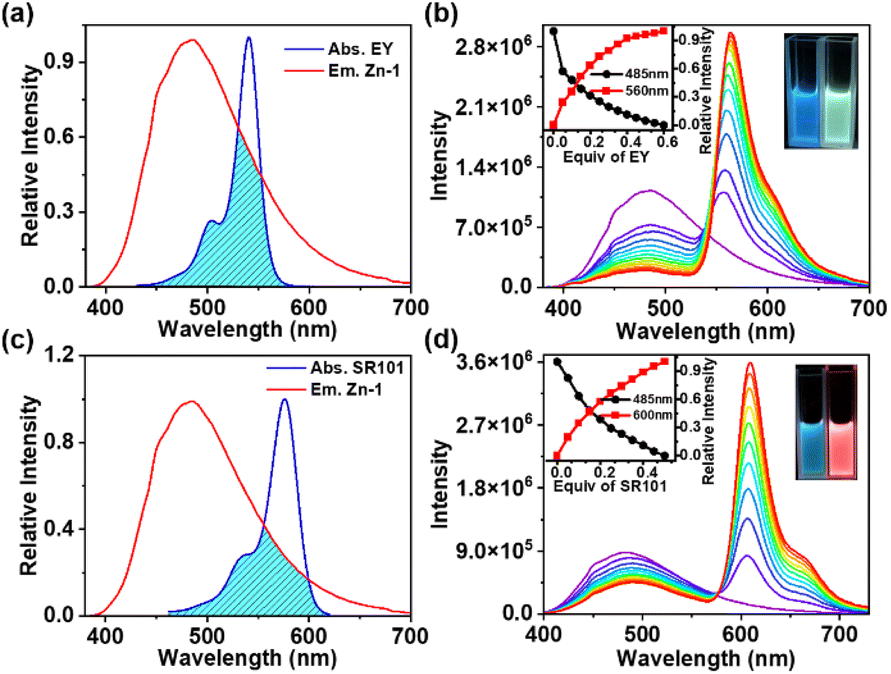 | ||
| Fig. 5 FRET process between Zn-1 and EY (SR101). (a) Normalized fluorescence spectrum of Zn-1 and absorbance spectrum of EY; (b) fluorescence spectra of Zn-1 ([Zn-1] = 1.0 × 10−5 M) in DMF with different concentrations of EY; (c) normalized fluorescence spectrum of Zn-1 and absorbance spectrum of SR101; (d) fluorescence spectra of Zn-1 ([Zn-1] = 1.0 × 10−5 M) in DMF with different concentrations of SR101. | ||
In addition, the FRET process was further evidenced by the decrease in the fluorescence lifetimes (τ) of the donor Zn-1 upon adding the acceptor EY (ESI, Fig. S22, Table S4†). The decay curve of the Zn-1 assembly was fitted to double exponential decay, which gave fluorescence lifetimes of τ1 = 1.68 ns and τ2 = 2.97 ns. Furthermore, the fluorescence lifetimes of the Zn-1⊃EY system decreased to τ1 = 1.28 ns and τ2 = 2.64 ns, suggesting that the energy was indeed transferred efficiently from Zn-1 to the EY acceptor. The process of FRET between Zn-1 and the guest was also studied from the increased fluorescence quantum yield (ΦF) (ESI, Fig. S17, Table S3†).19 The ΦF increased to 21.91% for Zn-1⊃EY, which was high compared to that of Zn-1(17.89%) alone in DMF.
It should be noted that the energy transfer efficiency (ΦET) was calculated as 82.4% (ESI, Table S5†) for Zn-1⊃EY according to the fluorescence intensity of Zn-1, which is quite high among those of reported supramolecular light-harvesting systems.13,15–19 This strong performance suggests that the cage-encapsulated host–guest complex is an excellent platform for light harvesting via the FRET process. The host–guest encapsulation drives the close through-space energy transfer between the donor and acceptor, without changing the luminescence properties arising from the introduction of covalent linking. In this supramolecular system, highly emissive Zn-1 has a large enough cavity into which the dye EY could be loaded to prevent its self-aggregation. Thus, the confined environment of Zn-1 sets a boundary for the EY guest, forcing the formation of a closely bound donor–acceptor pair in an intended ratio.
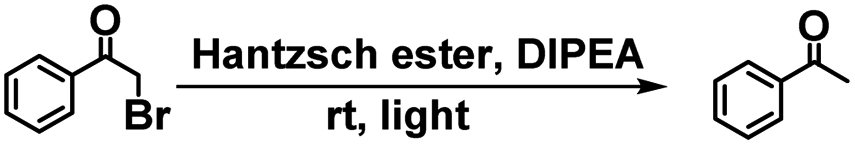
It has been reported that EY can be utilized as a photosensitizer to catalyze the dehalogenation of α-bromoacetophenone in the green light region due to its negligible absorption in the UV region.24 In this process, the cage host serves as an antenna to transfer energy from the UV radiation to EY owing to the strong absorption of Zn-1 in the UV region. Then, the UV light (365 nm) triggers a photocatalytic reaction. As shown in Table 1, in the presence of Zn-1⊃EY, the yield of acetophenone was 83.4% under 530 nm irradiation. A slightly low yield (76.1%) was obtained under 365 nm irradiation, indicating that the dehalogenation reaction not only uses green light but also exploits UV light through the absorption of Zn-1, expanding the range of the catalytic light source. A control experiment showed that only EY under 365 nm irradiation resulted in slight catalytic activity. A low yield (29.9%) was also obtained for free Zn-1. Moreover, almost no catalytic activity was observed in the dark for Zn-1⊃EY. These results revealed that light energy harvested by Zn-1 could be efficiently transferred to EY within the cage-encapsulated system (ESI, Fig. S25†).
|
|
|||
|---|---|---|---|
| Entry | Source light | Photocatalyst | Yield rate |
| a Reaction conditions: α-bromoacetophenone (20 μmol), Hantzsch ester (22 μmol), DIPEA (7 μl), DMF (1.2 mL), and catalyst (0.5 μmol), Ar, 8 h, rt. All yields were determined by GC analysis. | |||
| 1 | 530 nm | EY | 81.7% |
| 2 | 530 nm | Zn-1⊃EY | 83.4% |
| 3 | Dark | Zn-1⊃EY | n.d. |
| 4 | Dark | EY | 3.1% |
| 5 | 365 nm | EY | n.d. |
| 6 | 365 nm | Zn-1⊃EY | 76.1% |
| 7 | 365 nm | Zn-1 | 29.9% |
Energy transfer between Zn-1 and SR101 was also investigated by conducting fluorescence titration (Fig. 5c and d), fluorescence quantum yield (ESI, Fig. S17, Table S3†), fluorescence reverse titration (ESI, Fig. S21b†), and fluorescence decay (Fig. S22, Table S4†) measurements. The ΦET of Zn-1⊃SR101 was around 48.8% (ESI, Table S5†). Compared with that of EY, the absorption spectrum of SR101 showed less overlap with the emission of Zn-1 (Fig. 5c), and as a result, the ΦET of Zn-1⊃SR101 was slightly lower.
Encouraged by the emission color area of Zn-1 (485 nm, cyan color) and SR101 (600 nm, orange color) which are complementary for white emission, a white-light-emitting material was prepared, a type of material that has attracted much attention in recent years due to its potential uses in illumination and sensing.25 As expected, strong white-light emission could be attained at a concentration of 1.0 × 10−5 M (Zn-1) for a [Zn-1]/[SR101] molar ratio of 20:1 (Fig. 6). The color coordinates of the resulting Zn-1⊃SR101 system were calculated to be (0.32, 0.33), which are very close to those of a pure white system (0.33, 0.33). Compared with a single-molecule system, host–guest encapsulation is an ideal strategy for constructing a white-light emitting system as the photophysical properties can be easily tuned by altering the non-covalent interactions.
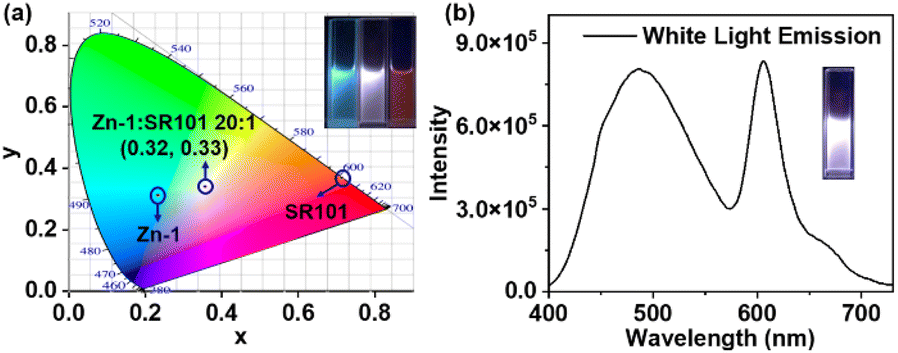 | ||
| Fig. 6 (a) CIE chromaticity coordinates of Zn-1, SR101, and Zn-1⊃SR101; (b) fluorescence spectrum of the white-light emission coordinate (0.32, 0.33), inset: fluorescence image of the white-light emitting solution, [Zn-1] = 1.0 × 10−5 M, [SR101] = 5 × 10−7 M. | ||
The effect of the host–guest interaction on the FRET processes was also identified by fluorescence titration of EY or SR101 with a solution of ligand L. Even though the absorption of EY (SR101) overlapped well with the emission of L in DMF, the broad emission peak of ligand L at 505 nm was almost unchanged, with the gradual addition of EY (SR101) to a solution of ligand L (ESI, Fig. S26†).
The FRET process within the cage can be inhibited by binding a competing guest to the enzyme-like host.26 Anthraquinone-2,7-disulfonic acid disodium salt (2,7-ADA), a negatively charged aromatic anion similar to the dyes EY and SR101, was employed as a competitive guest and introduced into the host–guest system (ESI, Fig. S27†). With the gradual addition of 2,7-ADA to a solution of Zn-1⊃EY (Zn-1⊃SR101), the emission peaks of EY (SR101) and Zn-1 gradually decreased in intensity (ESI, Fig. S28†), showing that 2,7-ADA acts as an inhibitor, competing with EY and SR101 to be encapsulated in the cage and reducing the FRET process.
Conclusions
In summary, a new emissive TPE-based metal–organic cage Zn-1 was constructed as an energy donor as well as host to encapsulate the guest acceptors EY or SR101. The energy acceptors EY and SR101 were matched to generate highly efficient FRET through close space distances forced by the host–guest interactions within the cage. The host–guest complex Zn-1⊃EY was shown to serve as a light-harvesting system for the photocatalytic dehalogenation of α-bromoacetophenone. Furthermore, by adjusting the molar ratio of Zn-1 and SR101, bright white-light emission was achieved with the CIE coordinate (0.32, 0.33). Overall, this work was devoted to developing an ideal approach for the creation of a host–guest system via cage encapsulation to enhance the efficiency of the FRET process, serving as a versatile platform for mimicking natural light-harvesting systems.Experimental
Preparation of Zn-1
A mixture of L (101 mg, 0.09 mmol) and zinc tetrafluoroborate (28.6 mg, 0.12 mmol) was dissolved in 30 mL DMF. The resulting reaction mixture was then heated at 80 °C for 12 hours. This solution was thereafter added to diethyl ether and the product was obtained after centrifugation and drying. The desired product was obtained as a light brown solid in 61% yield (based on the solid dried under vacuum). 1H NMR (400 MHz, DMSO-d6): δ = 10.77 (s, 1H), 9.04–8.65 (m, 3H), 8.52–8.00 (m, 3H), 7.77–7.48 (m, 3H), 7.05 (dd, J = 23.4, 15.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ = 166.69, 148.50, 147.54, 146.16, 145.51, 143.54, 142.02, 139.83, 139.30, 139.01, 136.85, 132.74, 130.78, 127.77, 127.64, 127.20, 123.94, 122.92. 19F NMR (400 MHz, DMSO-d6): δ = −148.22.General procedure for the cage-based catalysis
DMF (1.2 ml), α-bromoacetophenone (4.0 mg, 0.02 mmol), diethyl-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (Hantzsch ester) (5.6 mg, 2.2 × 10−2 mmol), N,N-diisopropylethylamine (DIPEA) (7.0 μl, 0.07 mmol), and catalyst (Zn-1 4.32 mg, 5 × 10−4 mmol and EY 0.35 mg, 5 × 10−4 mmol) were added to a 5 mL flask. The flask was sealed with a septum, and then degassed by bubbling with argon gas for 15 min under atmospheric pressure at room temperature. After that, the mixture was irradiated using a light source at room temperature for 8 h. The product was monitored by gas chromatography relative to the internal standard 1,3,5-trimethoxybenzene.Crystallography
Crystals of Zn-1 and the host–guest complex Zn-1⊃EY suitable for X-ray diffraction measurements were obtained via the slow diffusion of diethyl ether into a mixed DMF and CH3CN solution of the complex over a few days. The acquired crystals were found to be hypersensitive to the loss of solvent. In spite of rapid handling times and low-temperature data collection, the quality of the data was less than ideal.Crystal data of Zn-1: C422H299B11.50F47N72O27.75 Zn8, M = 8362.59, monoclinic, space group P21/c, white block, a = 27.515(2), b = 48.566(3), c = 55.058(3) Å, β = 91.649(1), V = 73544(7) Å3, Z = 4, Dc = 0.755 g cm−3, μ(MoKα) = 0.309 mm−1, T = 120(2) K. 129439 unique reflections [Rint = 0.1120]. Final R1 [with I > 2σ(I)] = 0.1153, wR2 (all data) = 0.3155 for the data collected. CCDC number 2195613.†
Crystal data of Zn-1⊃EY: C453.50H351.50B7.50BrF30N77.50O62Zn8, M = 9132.55, triclinic, space group P, orange block, a = 27.217(7), b = 33.119(7), c = 38.817(2) Å, α = 88.598(2), β = 88.357(1), γ = 83.075(2), V = 34712(3) Å3, Z = 2, Dc = 0.874 g cm−3, μ(Mo-Kα) = 0.71073 mm−1, T = 180(2) K. 122093 unique reflections [Rint = 0.1005]. Final R1 [with I > 2σ(I)] = 0.1497, wR2 (all data) = 0.450 for the data collected. CCDC number 2195617.†
Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
C. He and D. Li designed the research and co-wrote the manuscript. D. Li, H. Li, and G. Guo performed all experiments. X. Liu carried out DFT calculation. L. Yang and X. Li assisted in analyzing the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22171033, 21701019, and 21573034) and the Fundamental Research Funds for the Central Universities [DUT17RC(3)094].Notes and references
- (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (c) D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS; (d) D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed; (e) M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed; (f) Y. Tamura, H. Takezawa and M. Fujita, J. Am. Chem. Soc., 2020, 142, 5504–5508 CrossRef CAS PubMed.
- (a) M. D. Ludden, C. G. P. Taylor and M. D. Ward, Chem. Sci., 2021, 12, 12640–12650 RSC; (b) T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562–5566 CrossRef CAS PubMed; (c) X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed; (d) M. Ueda, N. Kishida, L. Catti and M. Yoshizawa, Chem. Sci., 2022, 13, 8642–8648 RSC.
- (a) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (b) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed; (c) R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed; (d) Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC; (e) W.-X. Gao, H.-N. Zhang and G.-X. Jin, Coord. Chem. Rev., 2019, 386, 69–84 CrossRef CAS; (f) L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS PubMed; (g) X.-X. Gou, T. Liu, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 16683–16689 CrossRef CAS PubMed; (h) L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS PubMed; (i) V. MartÍ-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed.
- (a) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed; (b) J. Dong, Y. Liu and Y. Cui, Acc. Chem. Res., 2021, 54, 194–206 CrossRef CAS PubMed; (c) J.-S. Wang, K. Wu, C. Yin, K. Li, Y. Huang, J. Ruan, X. Feng, P. Hu and C.-Y. Su, Nat. Commun., 2020, 11, 4675 CrossRef PubMed; (d) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Duerr, I. Ivanovic-Burmazovic and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- (a) L. Zhao, X. Jing, X.-Z. Li, X.-Y. Guo, L. Zeng, C. He and C.-Y. Duan, Coord. Chem. Rev., 2019, 378, 151–187 CrossRef CAS; (b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed; (c) W. Cullen, H. Takezawa and M. Fujita, Angew. Chem., Int. Ed., 2019, 58, 9171–9173 CrossRef CAS PubMed; (d) H. Sunohara, K. Koyamada, H. Takezawa and M. Fujita, Chem. Commun., 2021, 57, 9300–9930 RSC; (e) D. M. Daiton, S. R. Ellis, E. M. Nichols, R. A. Mathies, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 10128–10131 CrossRef PubMed; (f) A. J. Musser, P. P. Neelakandan, J. M. Richter, H. Mori, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 12050–12059 CrossRef CAS PubMed.
- (a) M. Hao, G. Sun, M. Zuo, Z. Xu, Y. Chen, X. Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 10095–10100 CrossRef CAS PubMed; (b) D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 143, 1313–1317 CrossRef CAS PubMed; (c) Z. Zhang, Z. Zhao, Y. Hou, H. Wang, X. Li, G. He and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 8862–8866 CrossRef CAS PubMed.
- (a) X.-M. Chen, Q. Cao, H. K. Bisoyi, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 10493–10497 CrossRef CAS PubMed; (b) X.-F. Hou, S. Zhang, X. Chen, H. K. Bisoyi, T. Xu, J. Liu, D. Chen, X.-M. Chen and Q. Li, ACS Appl. Mater. Interfaces, 2022, 14, 22443–22453 CrossRef CAS PubMed.
- (a) N. Melnychuk, S. Egloff, A. Runser, A. Reisch and A. S. Klymchenko, Angew. Chem., Int. Ed., 2020, 59, 6811–6818 CrossRef CAS PubMed; (b) T. Xiao, C. Bao, L. Zhang, K. Diao, D. Ren, C. Wei, Z.-Y. Li and X.-Q. Sun, J. Mater. Chem. A, 2022, 10, 8528–8534 RSC.
- (a) Y.-X. Yuan, J.-H. Jia, Y.-P. Song, F.-Y. Ye, Y.-S. Zheng and S.-Q. Zang, J. Am. Chem. Soc., 2022, 144, 5389–5399 CrossRef CAS PubMed; (b) H. Liu, Z. Zhang, C. Mu, L. Ma, H. Yuan, S. Ling, H. Wang, X. Li and M. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207289 CAS.
- Y. Jiang and J. McNeill, Chem. Rev., 2017, 117, 838–859 CrossRef CAS.
- (a) K. Wang, K. Velmurugan, B. Li and X.-Y. Hu, Chem. Commun., 2021, 57, 13641–13654 RSC; (b) J.-J. Li, H.-Y. Zhang, X.-Y. Dai, Z.-X. Liu and Y. Liu, Chem. Commun., 2020, 56, 5949–5952 RSC; (c) K. Diao, D. J. Whitaker, Z. Huang, H. Qian, D. Ren, L. Zhang, Z.-Y. Li, X.-Q. Sun, T. Xiao and L. Wang, Chem. Commun., 2022, 58, 2343–2346 RSC.
- (a) J. Yang, M.-C. Yoon, H. Yoo, P. Kim and D. Kim, Chem. Soc. Rev., 2012, 41, 4808–4826 RSC; (b) Y.-H. Jeong, M. Son, H. Yoon, P. Kim, D.-H. Lee, D. Kim and W.-D. Jang, Angew. Chem., Int. Ed., 2014, 53, 6925–6928 CrossRef CAS PubMed; (c) P. Parkinson, C. E. I. Knappke, N. Kamonsutthipaijit, K. Sirithip, J. D. Matichak, H. L. Anderson and L. M. Herz, J. Am. Chem. Soc., 2014, 136, 8217–8220 CrossRef CAS PubMed.
- (a) Y.-X. Hu, W.-J. Li, P.-P. Jia, X.-Q. Wang, L. Xu and H.-B. Yang, Adv. Opt. Mater., 2020, 8, 2000265 CrossRef CAS; (b) Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2021, 143, 382–389 CrossRef CAS; (c) T. Xiao, L. Zhang, H. Wu, H. Qian, D. Ren, Z.-Y. Li and X. Sun, Chem. Commun., 2021, 57, 5782–5785 RSC.
- A. Melis, Energy Environ. Sci., 2012, 5, 5531–5539 RSC.
- (a) K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed; (b) Y. Li, S. S. Rajasree, G. Y. Lee, J. Yu, J.-H. Tang, R. Ni, G. Li, K. N. Houk, P. Deria and P. J. Stang, J. Am. Chem. Soc., 2021, 143, 2908–2919 CrossRef CAS PubMed; (c) Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed; (d) W.-J. Li, X.-Q. Wang, D.-Y. Zhang, Y.-X. Hu, W.-T. Xu, L. Xu, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2021, 60, 18761–18768 CrossRef CAS PubMed.
- (a) Y.-N. Jing, S.-S. Li, M. Su, H. Bao and W.-M. Wan, J. Am. Chem. Soc., 2019, 141, 16839–16848 CrossRef CAS PubMed; (b) L. Xu, Z. Wang, R. Wang, L. Wang, X. He, H. Jiang, H. Tang, D. Cao and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9908–9913 CrossRef CAS PubMed.
- (a) M. Zeng, A. Ren, W. Wu, Y. Zhao, C. Zhan and J. Yao, Chem. Sci., 2020, 11, 9154–9161 RSC; (b) Q. Mu, J. Liu, W. Chen, X. Song, X. Liu, X. Zhang, Z. Chang and L. Chen, Chem.–Eur. J., 2019, 25, 1901–1905 CrossRef CAS PubMed.
- C. Li, J. Zhang, S. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 1643–1647 CrossRef CAS PubMed.
- (a) K. Acharyya, S. Bhattacharyya, S. Lu, Y. Sun, P. S. Mukherjee and P. J. Stang, Angew. Chem., Int. Ed., 2022, 61, e202200715 CrossRef CAS PubMed; (b) J. R. Piper, L. Cletheroe, C. G. P. Taylor, A. J. Metherell, J. A. Weinstein, I. V. Sazanovich and M. D. Ward, Chem. Commun., 2017, 53, 408 RSC.
- D. R. Martir, A. Pizzolante, D. Escudero, D. Jacquemin, S. L. Warriner and E. Zysman-Colman, ACS Appl. Energy Mater., 2018, 1, 2971–2978 CrossRef CAS.
- A. Kumar, R. Saha and P. S. Mukherjee, Chem. Sci., 2021, 12, 5319–5329 RSC.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- (a) X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed; (b) Z. Guo, G. Li, H. Wang, J. Zhao, Y. Liu, H. Tan, X. Li, P. J. Stang and X. Yan, J. Am. Chem. Soc., 2021, 143, 9215–9221 CrossRef CAS PubMed; (c) X. Yan, M. Wang, T. R. Cook, M. Zhang, M. L. Saha, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580–4588 CrossRef CAS PubMed; (d) V. M. Suresh, A. De and T. K. Maji, Chem. Commun., 2015, 51, 14678–14681 RSC; (e) P. Sutar, V. M. Suresh, K. Jayaramulu, A. Hazra and T. K. Maji, Nat. Commun., 2018, 9, 3587–3598 CrossRef PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- (a) H.-T. Feng, X. Zheng, X. Gu, M. Chen, J. W. Y. Lam, X. Huang and B. Z. Tang, Chem. Mater., 2018, 30, 1285–1290 CrossRef CAS; (b) Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2021, 143, 382–389 CrossRef CAS PubMed.
- J. Wei, L. Zhao, C. He, S. Zheng, J. N. H. Reek and C. Duan, J. Am. Chem. Soc., 2019, 141, 12707–12716 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures and additional spectroscopic data. CCDC 2195613 and 2195617. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06022a |
| This journal is © The Royal Society of Chemistry 2023 |