
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble design of host and guest synergistically reinforces the Na-ion storage of sulfur cathodes†
Xiang Long
Huang
 *a,
Hong
Zhong
a,
Ce
Li
a,
Yaojie
Lei
c,
Shaohui
Zhang
d,
Yuhan
Wu
e,
Wenli
Zhang
f,
Hua Kun
Liu
bc,
Shi Xue
Dou
*bc and
Zhiming M.
Wang
*ag
*a,
Hong
Zhong
a,
Ce
Li
a,
Yaojie
Lei
c,
Shaohui
Zhang
d,
Yuhan
Wu
e,
Wenli
Zhang
f,
Hua Kun
Liu
bc,
Shi Xue
Dou
*bc and
Zhiming M.
Wang
*ag
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611137, China. E-mail: xlhuang_uestc@163.com; zhmwang@uestc.edu.cn
bInstitute of Energy Materials Science, University of Shanghai for Science and Technology, Shanghai 200093, China. E-mail: shi@usst.edu.cn
cInstitute for Superconducting and Electronic Materials, University of Wollongong, NSW 2500, Australia. E-mail: shi@uow.edu.au
dGuangdong Provincial Key Laboratory of Micro/Nano Optomechatronic Engineering, College of Mechatronics and Control Engineering, Shenzhen University, Shenzhen 518060, China
eSchool of Environmental and Chemical Engineering, Shenyang University of Technology, Shenyang 110870, China
fSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, 100 Waihuan Xi Road, Guangzhou 510006, China
gInstitute for Advanced Study, Chengdu University, Chengdu 610106, China
First published on 27th January 2023
Abstract
Development of room-temperature sodium–sulfur batteries is significantly hampered by the shuttle effect of soluble intermediates and intrinsically sluggish conversion kinetics. In this work, a double design host and guest strategy (i.e., implantation of a polar V2O3 adsorbent into a carbon substrate and selenium doping of a sulfur guest) is proposed to synergistically reinforce the electrochemical properties of sulfur electrodes in sodium ion storage. The V2O3 adsorbent efficiently immobilizes sulfur species via strong polar–polar interactions, while the selenium dopant improves the electronic conductivity of sulfur cathodes and accelerates the redox conversion of sulfur cathodes. The synergistic effect between the V2O3 adsorbent and selenium dopant is shown to inhibit the shuttle effect and improve the redox kinetics, thus realizing greatly enhanced Na-ion storage properties of sulfur cathodes. The as-designed sulfur cathode delivers a superior rate capability of 663 mA h g−1 at 2.0 A g−1 and demonstrates excellent cyclability of 405 mA h g−1 over 700 cycles at 1.0 A g−1.
Introduction
Room-temperature sodium–sulfur (RT Na–S) batteries are deemed to be an emerging and promising electrochemical energy storage technology and have drawn extensive research interest over the past decade due to integrating the advantages of both elemental Na and S.1,2 Their ubiquitous distribution in nature allows widespread and sustainable access to sodium resources and low-cost refinement of electrode materials. Moreover, the merits of high abundance, low cost, and nontoxicity make S an eco-friendly and large-scale commercial electrode material. Additionally, metallic Na and S demonstrate high theoretical specific capacities of 1166 and 1675 mA h g−1, respectively.3 Accordingly, RT Na–S batteries are cost-effective and high-energy density systems, which operate via a two-electron reaction pathway and show an impressive theoretical energy density of 1275 W h kg−1.4,5Although RT Na–S batteries can potentially meet all kinds of expectations for commercial applications, their development is hindered by challenges on the S cathode side. First, the low electronic conductivity of S and its discharge products leads to low utilization of active materials and large polarization.6,7 Second, electrochemically-induced solvated polysulfide moieties often give rise to instability of the S cathode and corrosion of the Na anode.8,9 Third, the dramatic volume variation of S cathodes is responsible for the microstructural deformation of materials and the pulverization of electrodes.10 Another concerning issue is that RT Na–S chemistry has to contend with the sluggish redox kinetics of S speciation and the low solid-state diffusivity of Na ions.11 These issues generally manifest in large voltage hysteresis in discharge–charge profiles, less well-defined and weak redox peaks in cyclic voltammetry (CV) curves, rapid capacity fading, and poor rate capabilities for RT Na–S batteries.
Engineering electrically conductive porous carbon hosts for dispersed encapsulation of S guest molecules has been widely shown to improve the electronic conductivity of S cathodes, confine the dissolution and shuttling of solvated polysulfides via the nanopore effect, and accommodate the volumetric fluctuation of active materials during the (de)sodiation process.12,13 Nevertheless, the substantial nonpolar nature of carbons renders them with too poor chemical affinity for polysulfides to inhibit the shuttle effect thoroughly. A useful design strategy is to implant polar adsorbents (e.g., heteroatoms and metal compounds) with strong bonding ability for polysulfides on/into conductive carbons, which can assist in capturing soluble polysulfides via surface polar sites to stabilize S cathodes.14–17 In this context, how to improve the redox kinetics of S guest molecules needs to be urgently considered.
In recent years, the effect of eutectic accelerators (i.e., Se doping for S) has been demonstrated as an effective strategy to regulate the electronic structure of S and boost the intrinsic kinetics of S cathodes, which exhibits some distinguished merits for redox conversions of S in comparison with the common design strategy of electrocatalysts.18,19 First, Se dopant can be easily dispersed amongst S molecules via S–Se bonding at a molecular level to regulate the reactivity of S molecules. Second, despite a low doping content, the Se dopant is able to function as electrochemically active sites to contribute extra capacity via redox reactions with Na ions. Third, the Se dopant is proved to favor improved Na ion diffusion in kinetic processes, thus inducing faster redox kinetics.20 Accordingly, such an approach has been attracting ever-increasing research interest, as the kinetic characteristics of S conversion can be regulated via the molecular design of guests instead of incorporating electrocatalysts into hosts.
In consideration of the significance of polar adsorption and Se doping for S conversion chemistry, we propose a double design host and guest strategy to inhibit the shuttle effect and improve the redox kinetics of RT Na–S batteries, that is, dispersing Se-doped S (SeS2) into polar V2O3 decorated porous carbon (denoted as SeS2/V2O3@C). Experimental and theoretical analysis shows that the V2O3 adsorbent is able to chemically capture sodium polysulfides/polyselenides via the formation of strong polar–polar interactions so as to stabilize the S cathode. The Se dopant induces higher capacity of the S electrode, due to its redox activity with Na ions, and more importantly, plays the role of accelerator in elevating the reaction kinetics of S cathodes. With the synergistic enhancement effect of host and guest, the as-designed SeS2/V2O3@C cathode demonstrates good cyclability (405 mA h g−1 after 700 cycles at 1.0 A g−1) and excellent rate capability (663 mA h g−1 at 2.0 A g−1). This strategy opens up a new pathway for designing high-performance S cathodes for Na-ion storage and sheds light on the concept that the design of guest molecules is equally as important as that of host design.
Results and discussion
The design and fabrication process of the target material is schematically illustrated in Fig. 1a. The precursor was prepared as a template via a solvothermal method, the successful synthesis of which was confirmed from the X-ray diffraction (XRD) pattern and field-emission scanning electron microscopy (FESEM) image shown in Fig. S1 and S2,† in good accordance with the results of a previous study.21 The precursor is thermally treated to form porous carbon nanorods under an insert atmosphere (Fig. S3 and S4†). Pure carbon exhibits too weak a chemical affinity for soluble polysulfides and polyselenides to prevent their shuttle effect and hence, strong polar V2O3 nanoparticles are further grown onto the as-prepared carbon nanorods (V2O3@C), endowing them with improved capture ability for intermediates. Afterwards, the active SeS2 is composited with the V2O3@C to produce the SeS2/V2O3@C composite. | ||
| Fig. 1 (a) Schematic illustration of the synthetic procedure for the SeS2/V2O3@C; (b–d) TEM and HRTEM images of the V2O3@C and (e–g) the SeS2/V2O3@C; (h) STEM image and corresponding EDS mappings of the SeS2/V2O3@C. | ||
The V2O3@C nanocomposite inherits the nanorod-like shape, indicating that the growth process of V2O3 nanoparticles on porous carbon does not destroy the overall morphology and microstructure of the carbon substrate, as displayed in Fig. 1b. An interplanar spacing of 0.375 nm can be clearly witnessed from Fig. 2c, which actually corresponds to the (012) facet of V2O3 very well. In addition to that, it can be found that there is a tight coupling between polar V2O3 and the amorphous carbon structure, which is beneficial to the fast interfacial transport of electrons with improved kinetics. Internal nanocavities of the carbon nanorods can offer accommodation space for the inputted active materials, as clearly shown in Fig. 2d. The STEM image with corresponding EDS mappings in Fig. S5† reflect the uniform anchoring of V2O3 onto the carbon substrate. The content of V2O3 in the V2O3@C composite is about 26%, as shown in Fig. S6.† As SeS2 is infused into the V2O3@C through a typical melt-diffusion method, and the V2O3@C host still maintains its basic nanorod-like morphology (Fig. 1e). The carbon nanorod skeleton and the anchored V2O3 can still be clearly seen from the TEM image of the SeS2/V2O3@C (Fig. 1f), indicating their structural stability. The accodant color of the carbon nanorod region suggests the uniform dispersion of SeS2 into the internal space of the V2O3@C (Fig. 1f and g). The interplanar of SeS2 cannot be detected through high-resolution TEM, suggesting its amorphous nature. The STEM image and corresponding EDS mappings of the SeS2/V2O3@C (Fig. 1h) further prove the uniform distribution of elemental C, V, O, S, and Se, which was also verified from the linear elemental distributions of the composite shown in Fig. S7.†
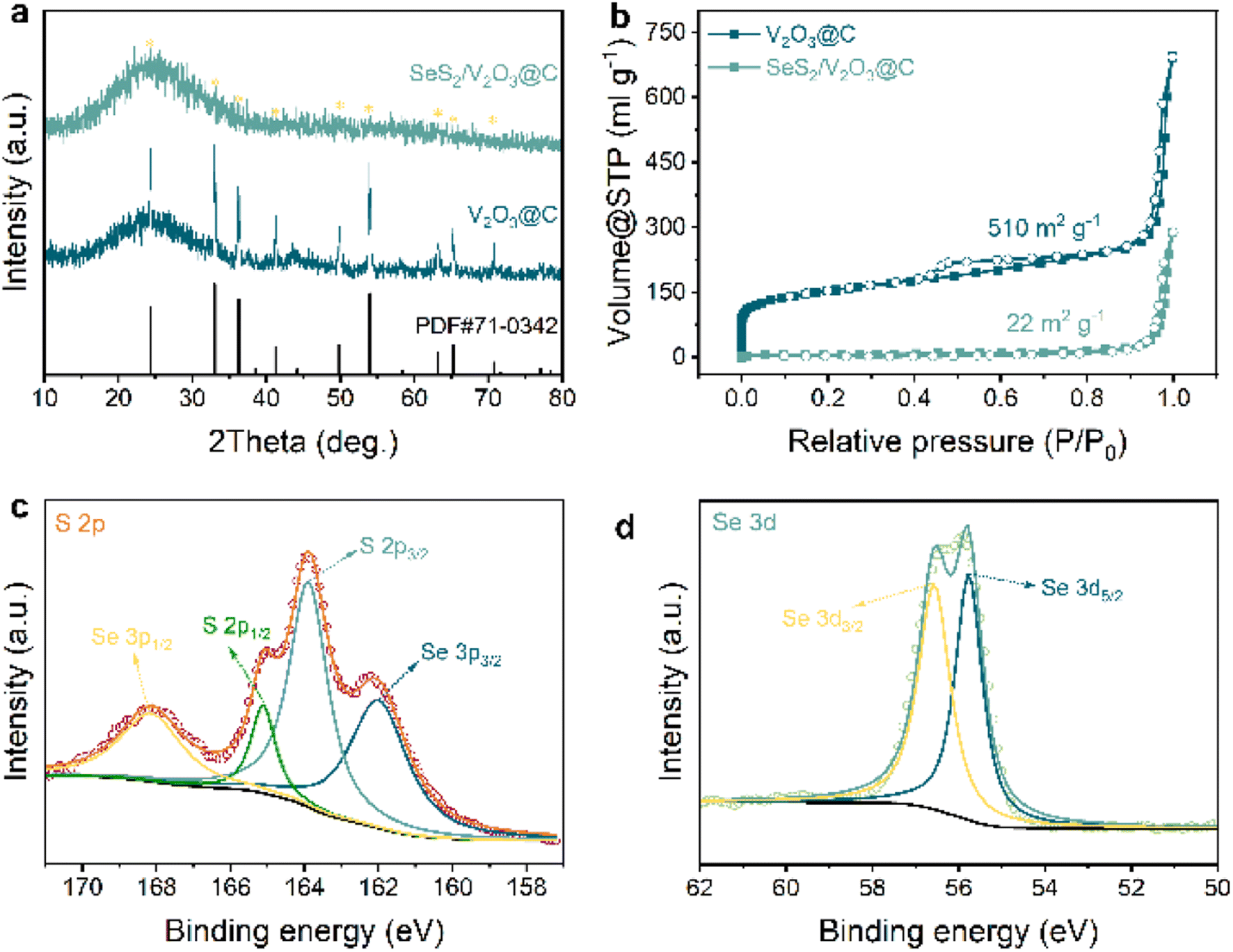 | ||
| Fig. 2 (a) XRD patterns of V2O3@C and SeS2/V2O3@C; (b) nitrogen; (c) XPS spectra of S 2p; (d) XPS spectra of Se 3d. | ||
The phase compositions and microstructures of the samples were further investigated via XRD and Raman spectroscopy, respectively. Fig. 2a displays the XRD pattern of the V2O3@C, which shows a series of well-defined and strong characteristic peaks in addition to peaks ascribed to carbon nanorods. These peaks are well matched with that of the standard card (PDF# 71-0342), confirming the successful preparation of the target host materials. When the encapsulation of SeS2 is completed, the XRD pattern of the SeS2/V2O3@C exhibits far weaker peak signals of V2O3 and the absence of characteristic peaks ascribed to SeS2. The absence of characteristic peaks ascribed to SeS2 indicates the perfect infusion of SeS2 within the internal nanopores of the host materials and its amorphous nature, as demonstrated in other similar literature.22–24 This result was verified by the Raman spectra of the samples shown in Fig. S8,† which is obtained within a range from 100 to 2000 cm−1, in which the Raman spectrum of SeS2/V2O3@C does not feature the peaks of SeS2 attributed to pure SeS2. The amorphous SeS2 can attain higher electrochemical reactivity to realize better electrochemical performance.25–28 Besides this, the intensity ratio of the two constant dominant peaks located at 1359 and 1594 cm−1 (D band and G band) indicates the graphitization degree of the carbon skeleton (i.e., ID/IG).29,30 The calculated ID/IG ratio value of 0.90 indicates the partially graphitized amorphous nature of the carbon skeleton, enabling fast electron transport in the electrochemical process.29
The porosity of the host materials usually plays a vital role in confining the dissolution of soluble species and alleviating the dramatic expansion of active materials. Here, the typical Brunauer–Emmett–Teller (BET) method was utilized to reveal the porous nature of all the samples, including their pore distribution, pore volume, and specific surface areas. As shown in Fig. 2b, the isotherm profile presents a dramatic rise in the low pressure region and an evident hysteresis in the high pressure region that suggest the existence of micropores and mesopores, respectively.31,32 The carbon substrate has a high specific surface area of 510 m2 g−1 and a pore volume of 0.993 cm3 g−1. The specific size of the nanopores was further analyzed from the pore size distribution curve obtained via density functional theory (DFT), as shown in Fig. S9.† Micropores, mesopores, and macropores can all be clearly observed. After the encapsulation of SeS2 into the carbon substrates, the plentiful nanopores disappear and the specific surface area is significantly decreased to 22 m2 g−1 with a much smaller pore volume of 0.373 cm3 g−1. This indicates that the infused SeS2 adequately occupies the internal nanocavities of the carbon substrate. The mass content of SeS2 was determined as 60% of the whole SeS2/V2O3@C nanocomposite, as shown in Fig. S10.† Notably, the temperature, at which the mass of the active materials was largely lost, was far higher than that of the preparation temperature (160 °C), further demonstrating that the SeS2 was completely confined within the carbon substrate.
X-Ray photoelectron spectroscopy (XPS) was conducted on the SeS2/V2O3@C sample to characterize the chemical states of the relevant elements. The survey spectrum of the sample in Fig. S11† shows the co-existence of elemental C, V, O, Se, and S, in good accordance with the aforementioned EDS mappings. Next, the high-resolution XPS spectra of the three key elements V, Se, and S were further analyzed. In the spectra of V (Fig. S12†), there are two dominant peaks located at 517 and 524.6 eV, respectively, which can be ascribed to the spin–orbit splitting of V 2p3/2 and V 2p1/2 and indicates a chemical state of +3.33 Four clear peaks can be observed from the spectra of elemental S (Fig. 2c), which are positioned at 168.3, 165.1, 163.9, and 162.1 eV. The middle pair of peaks can be attributed to S–S bonds, while the two peaks at either side indicate the presence of Se–S bonds. Two peaks positioned at 55.8 and 56.6 eV reflect the elemental Se, as shown in Fig. 2d.
Owing to double design of host and guest, the as-obtained composite material is expected to realize excellent electrochemical performance. Accordingly, its sodium-ion storage capability was evaluated through assembling Na–SeS2 batteries (Fig. 3). In order to investigate the redox mechanism of the SeS2/V2O3@C composite during the (de)sodiation process, cyclic voltammogram (CV) curves were recorded at a scan rate of 0.1 mV s−1 (Fig. 3a) and compared with previous literature. At the first cathodic scan, there is a sharp peak around 1.0 V but no obvious peaks above 1.0 V. This shows the activation process of SeS2, where reduction reactions of SeS2 into Na2S/Na2Se and electrolyte-associated decomposition reactions are involved.19,34 At the corresponding anodic scan, Na2S/Na2Se is oxidized to S8/Se8 instead of pristine SeS2 and the three oxidation peaks positioned at 1.8, 1.9, and 2.2 V reveal the stepwise conversion process from Na2S/Na2Se to S8/Se8. This result is highly consistent with other reports and implies the distinguished redox behavior of the SeS2 cathode in different stages.19,24 At the second cycle, the reduction peak at 2.2 V can be ascribed to conversion from S8 to long-chain sodium polysulfides (Na2Sx), and the two strong peaks at 1.6 and 1.0 V reflect reduction reactions from long-chain polysulfides/polyselenides to short-chain polysulfides/polyselenides and finally to Na2S/Na2Se, respectively.35 The CV curves start to be stabilized from the third cycle, along with the appearance of three pairs of clear redox peaks, which suggest the reversible redox reactions of the cathode between S8/Se8 and Na2S/Na2Se and its electrochemical stability. As a whole, this mechanism is highly consistent with the pPAN/SeS2 cathode reported by Lou et al.19
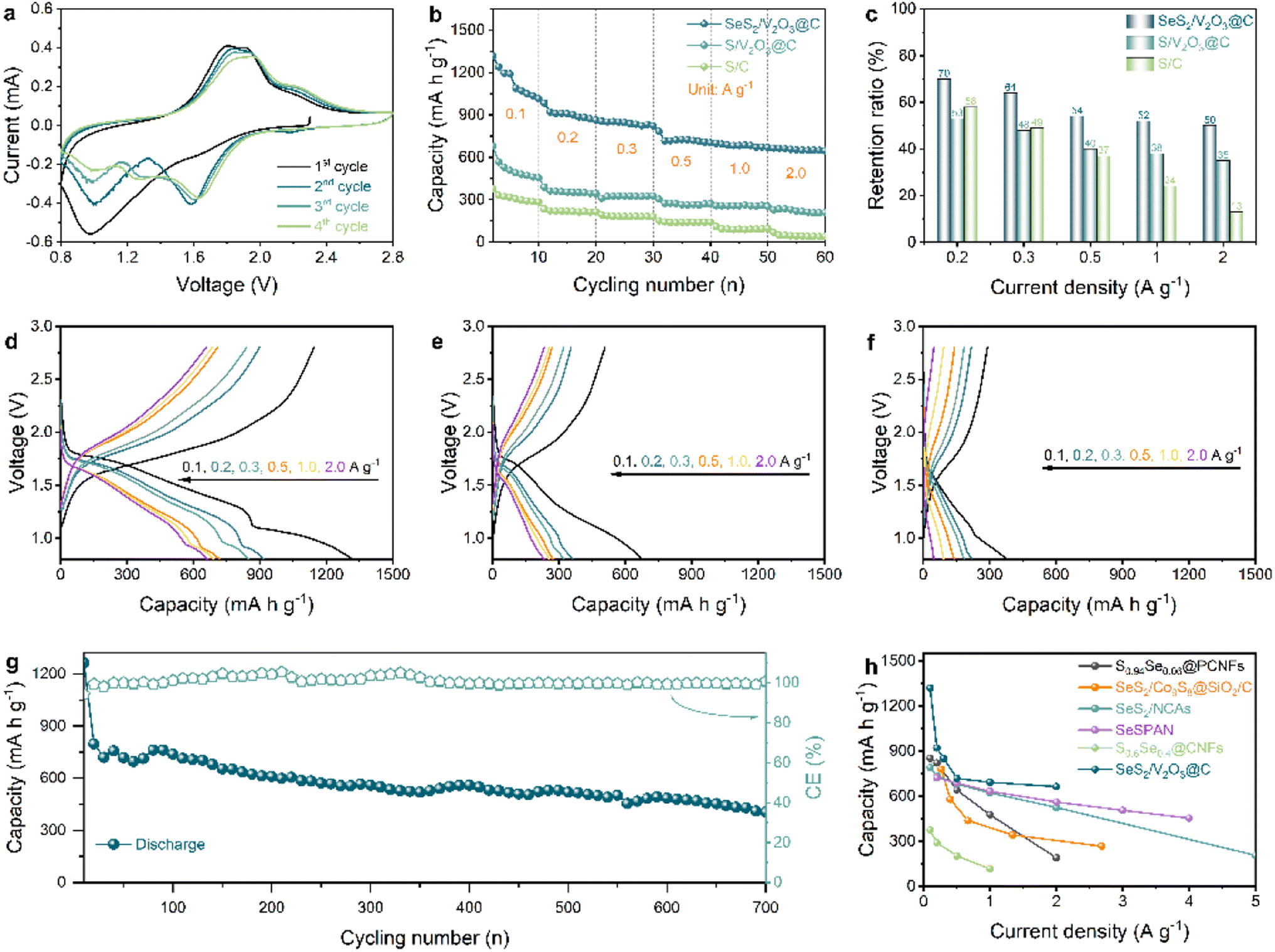 | ||
| Fig. 3 (a) CV curves at 0.1 mV s−1; (b) rate capabilities at different current densities; (c) capacity retention ratios based on 0.1 A g−1; (d–f) discharge–charge profiles of SeS2/V2O3@C, S/V2O3@C, and S/C at different current densities; (g) long-term cyclability of the SeS2/V2O3@C composite at 1.0 A g−1; (h) rate performance comparison between this work and other SexSy cathodes for Na-ion storage. | ||
Fig. 3b displays the rate performance of the SeS2/V2O3@C and two control samples. The SeS2/V2O3@C shows high discharge capacities of 1319, 920, 850, 718, 693, and 663 mA h g−1 at 0.1, 0.2, 0.3, 0.5, 1.0, and 2.0 A g−1, respectively. In contrast, the S/V2O3@C and S/C composites show far lower discharge capacities of 362/221, 322/186, 274/142, 257/92, and 234/48 mA h g−1 at the corresponding rates. For acquiring deeper insight into the rate capability of these samples, the rate capacity retention ratio based on 0.1 A g−1 was calculated, as exhibited in Fig. 3c. The SeS2/V2O3@C composite displayed capacity retention ratios of 70, 64, 54, 52, and 50% at 0.2, 0.3, 0.5, 1.0, and 2.0 A g−1, respectively. For the S/V2O3@C composite, capacity retention ratios of 53, 48, 40, 38, and 35% were obtained. As for the S/C composite, its capacity retention ratios were 58, 49, 37, 24, and 13%. It is worth mentioning that the SeS2/V2O3@C and S/V2O3@C composites both show higher capacity retention ratios than that of the S/C composite at ultrahigh rates (1.0 and 2.0 A g−1), suggesting that the incorporation of polar V2O3 plays a vital role in promoting rate capability. Next, we moved to the analysis of the discharge–charge profiles of these samples. The discharge–charge profiles of the SeS2/V2O3@C in Fig. 3d show clear and overlapped voltage plateaus and low voltage polarization, which correspond very well to the aforementioned redox peaks, further indicating the highly reversible electrochemical reactions of the active materials. Conversely, the S/V2O3@C and S/C composites present shorter voltage plateaus, more inclined slopes, and far greater polarization (Fig. 3e and f). The better specific capacities, higher capacity retention ratios, and well-defined plateaus jointly demonstrate the enhanced rate performance of the SeS2/V2O3@C composite in comparison with the S/V2O3@C and S/C composites. Besides this, the SeS2/C composite shows negligible capacity improvement in comparison with the S/C composite, as shown in Fig. S13.† This proves that the performance improvement should be attributed to a synergistic effect between Se doping and the polar adsorbent instead of a single added component.
The long-term cyclability of the SeS2/V2O3@C composite was also evaluated at a high current density of 1.0 A g−1, as illustrated in Fig. 3g. For full activation of the active materials, the SeS2/V2O3@C composite cathode was pre-cycled for 20 cycles at 0.1 A g−1. Afterwards, a discharge specific capacity of 798 mA h g−1 was observed and a high discharge specific capacity of 405 mA h g−1 was still retained after 700 cycles. In addition to that, an ultrahigh coulombic efficiency of close to 100% is observed, suggesting high energy conversion efficiency. Fig. S14† shows the cycling performance of the V2O3@C composite under the same test conditions, which can be ignored due to the low contribution and suggests that the capacity of the whole cathode is from the reversible conversions of the active materials. The long cyclability of the electrode is superior to the reports of most studies regarding Na–SexSy batteries through comprehensively considering multiple factors such as current density, lifespan, and capacity retention.19,22–24,35–38Table 1 summarizes the long-term cycling stability of currently reported SexSy cathodes for Na-based batteries. Furthermore, the rate capabilities are compared with those of studies on Na–SexSy batteries. It can be clearly seen from Fig. 3h that the rate capabilities of the as-designed SeS2/V2O3@C composite are obviously higher than most of the values reported in the literature.22,24,35,37,38 To explore the influence of active material loading content on the performance of the cathodes, the loading content of SeS2 in the composite was elevated to 70%. As shown in Fig. S15†, the SeS2 cathode with a higher content shows the worst cycling performance. This enlightens that more work needs to be done to realize high-loading electrodes with excellent performance in the future.
| Material | Content | Voltage | Rate | Lifespan | Retention |
|---|---|---|---|---|---|
| This work | 60% | 0.8–2.8 V | 1 A g − 1 | 700 | 405.0 mA h g − 1 |
| SeS2–C36 | 50.0% | 0.8–2.8 V | 0.05 A g−1 | 30 | 228.0 mA h g−1 |
| S0·6Se0.4@CNFs22 | 60.0% | 0.8–3.0 V | 0.1 A g−1 | 100 | 375.0 mA h g−1 |
| pPAN/SeS2 (ref. 19) | 69.0% | 0.8–2.8 V | 1 A g−1 | 400 | 800 mA h g−1 |
| S0·96Se0.04@PCNFs24 | 72.0% | 0.8–3.0 V | 0.5 A g−1 | 200 | 456.0 mA h g−1 |
| SeS2/Co9S8@SiO2/C35 | 40.0% | 0.5–2.8 V | 1C | 300 | 191.0 mA h g−1 |
| CCN/SeS2 (ref. 23) | 70.0% | 0.7–2.8 V | 0.5 A g−1 | 470 | 556.0 mA h g−1 |
| SeS2@NCAs37 | 54.0% | 1.0–3.0 V | 0.5 A g−1 | 1000 | 536.0 mA h g−1 |
| SeSPAN38 | 60.0% | 0.8–2.8 V | 0.5 A g−1 | 150 | 632.0 mA h g−1 |
The aforementioned excellent electrochemical performance should be attributed to the double design of the SeS2/V2O3@C composite in terms of chemical compositions of both the host and guest materials, which can potentially help to stabilize the cathode and improve the reaction kinetics. The immobilization effect of V2O3 for soluble intermediates was explored via theoretical and experimental evidence. DFT calculations were conducted on graphene and V2O3 to gain insight into molecular configurations and adsorption energy, as displayed in Fig. 4 and S16.† Various polysulfides/polyselenides cannot chemically bond with pure graphene. This suggests that pure carbon substrate is unable to interact with these soluble polysulfides/polyselenides via strong chemical bonds instead of weak van der Waals forces. In contrast, polysulfides interact with the surface atoms of V2O3via Na–O and V–S bonds. Likewise, a series of polyselenides tend to couple with V2O3 to generate Na–O and V–Se bonds. The chemical affinity of V2O3 for polysulfides/polyselenides can be quantificationally determined from their adsorption energies. The adsorption energies of polysulfides/polyselenides with different substrates are shown in Fig. 4b and c. The adsorption energies of Na2Sn (n = 8, 6, 4, 2, and 1) on graphene were −0.06, −0.05, −0.07, −0.44, and −0.53 eV, respectively, while the adsorption energies on the V2O3 surface were −2.99, −3.09, −3.37, −4.66, and −4.07 eV, respectively. The adsorption energies of Na2Sen (n = 8, 6, 4, 2, and 1) on the graphene were −0.08, −0.06, −0.05, −0.39, and −0.63 eV, respectively, while the adsorption energies on the V2O3 surface were −2.93, −4.47, −3.18, −4.16, and −3.87 eV, respectively. This fully demonstrates the great chemical affinity of V2O3 for various polysulfides/polyselenides. Besides this, it is also worth mentioning that with an increase in the chain length of polysulfides and polyselenides, the adsorption energy is relatively lowered (weakened chemical bonds), suggesting that polysulfides/polyselenides easily dissociate into shorter-chain sulfides on the surfaces of V2O3.
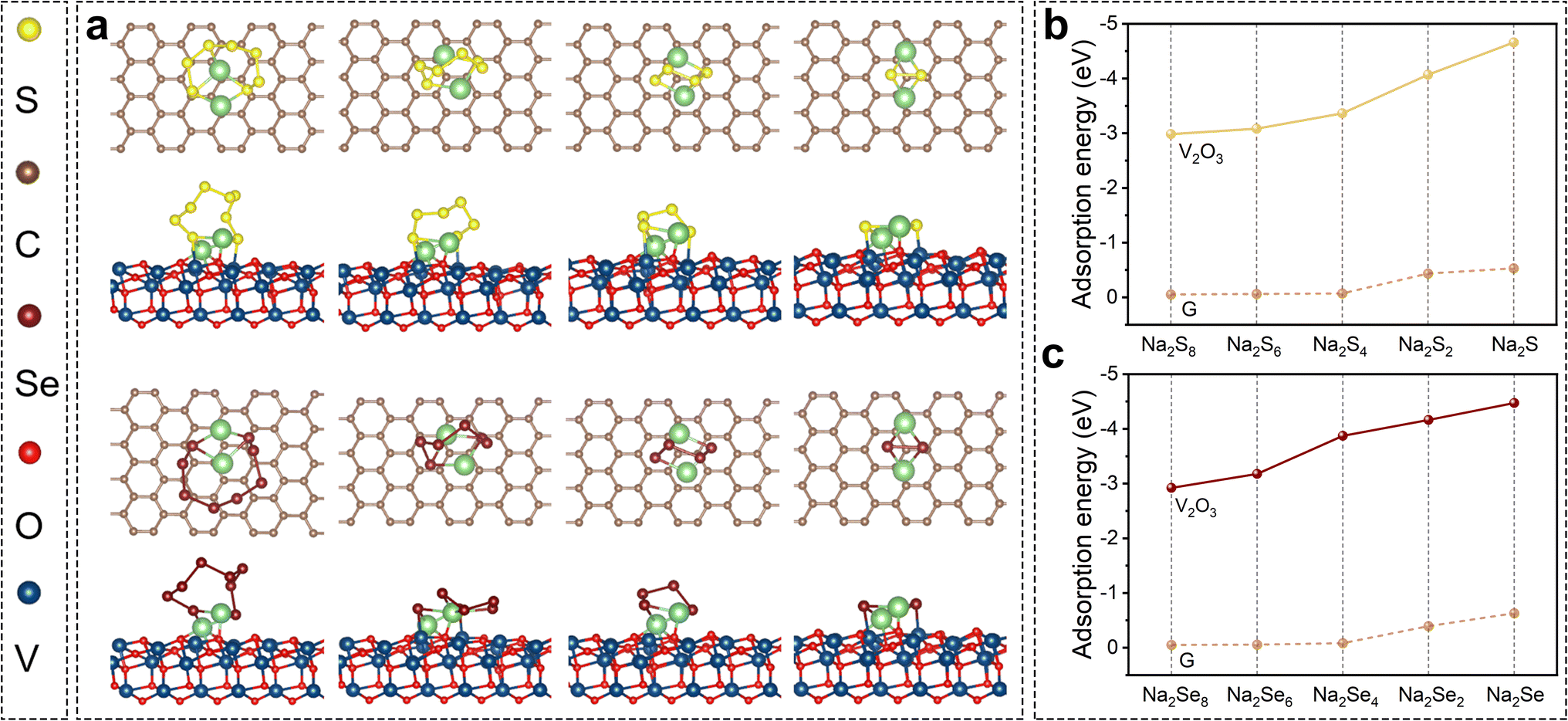 | ||
| Fig. 4 (a) Molecular configurations between polysulfides/polyselenides and different substrates (graphene and V2O3); adsorption energy between graphene/V2O3 and (b) polysulfides or (c) polyselenides. | ||
These results are further supported by optical observations, UV-vis spectroscopy of the polysulfide solutions, and XPS spectra of the sample before and after discharging. As shown in Fig. S17a,† Na2S6 solution was prepared as a representative of polysulfides/polyselenides to be soaked with different samples and further observe its color change after standing for 12 h. The solution soaked with the pure carbon substrate remained the same yellow color as the blank solution, while the solution soaked with V2O3@C became colorless and transparent. At the same time, the absorbance intensity of the solution soaked with the V2O3@C was significantly reduced (Fig. S17b†), suggesting complete adsorption of the polysulfide molecules. This contrast effectively proves the strong chemical affinity of the V2O3 for polysulfides. Moreover, the XPS spectra of elemental V from the SeS2/V2O3@C composite cathode undergoes an evident shift after discharging to 2.0 V (Fig. S17c†), indicating chemical bonding between S/Se-based products and V2O3@C in the electrochemical process.
Apart from efficient immobilization of the active materials, the fast conversion of the cathodes with high kinetics can also contribute to improving the electrochemical performance to a large extent. The above discussion highlighted the adsorption effect of V2O3 for soluble intermediates, so the improvement effect of the Se doping on conversion kinetics will be discussed next. As shown in Fig. 5a, SeS2/V2O3@C exhibits much lower voltage polarization than that of the S/V2O3@C, suggesting that the Se dopant promotes the kinetical conversion of S guests. Fig. 5b displays the XRD patterns of the cycled SeS2/V2O3@C and S/V2O3@C electrodes. The latter suffers from more obvious electrochemical deposition of Na2Sx and Na2S in comparison to the former. This indicates that the Se doping is able to facilitate the reversible conversion of the active S molecules to lower the accumulation of inactive species and deliver higher capacity. The SeS2/V2O3@C shows a much lower electron transfer impedance than that of S/V2O3@C, which is conducive to promoting a faster electrochemical reaction rate (Fig. 5c), suggesting the improved electronic conductivity of the whole electrode is induced by the Se dopant. The CV curves at various scan rates were recorded to analyze the Na-ion diffusion behavior (Fig. S17†). It can be observed that despite increased current density, the CV curve still presents clear redox peaks and a strong current response, which are indicators of the electrochemical stability of electrodes. Furthermore, the peaks can be linearly fitted to attain the b value and determine the electrochemical behavior of Na ions based on their scan rates and current response. As is known, Na-ion transfer is controlled by diffusion if the b value is close to 0.5, while it exhibits capacitance with faster kinetics if the b value is close to 1.0.39,40 As shown in Fig. S19,† the reduction and oxidation process both show b values of 0.79 and 0.80, respectively, which suggest the redox reactions of the SeS2/V2O3@C composite cathode exhibit fast conversion kinetics.
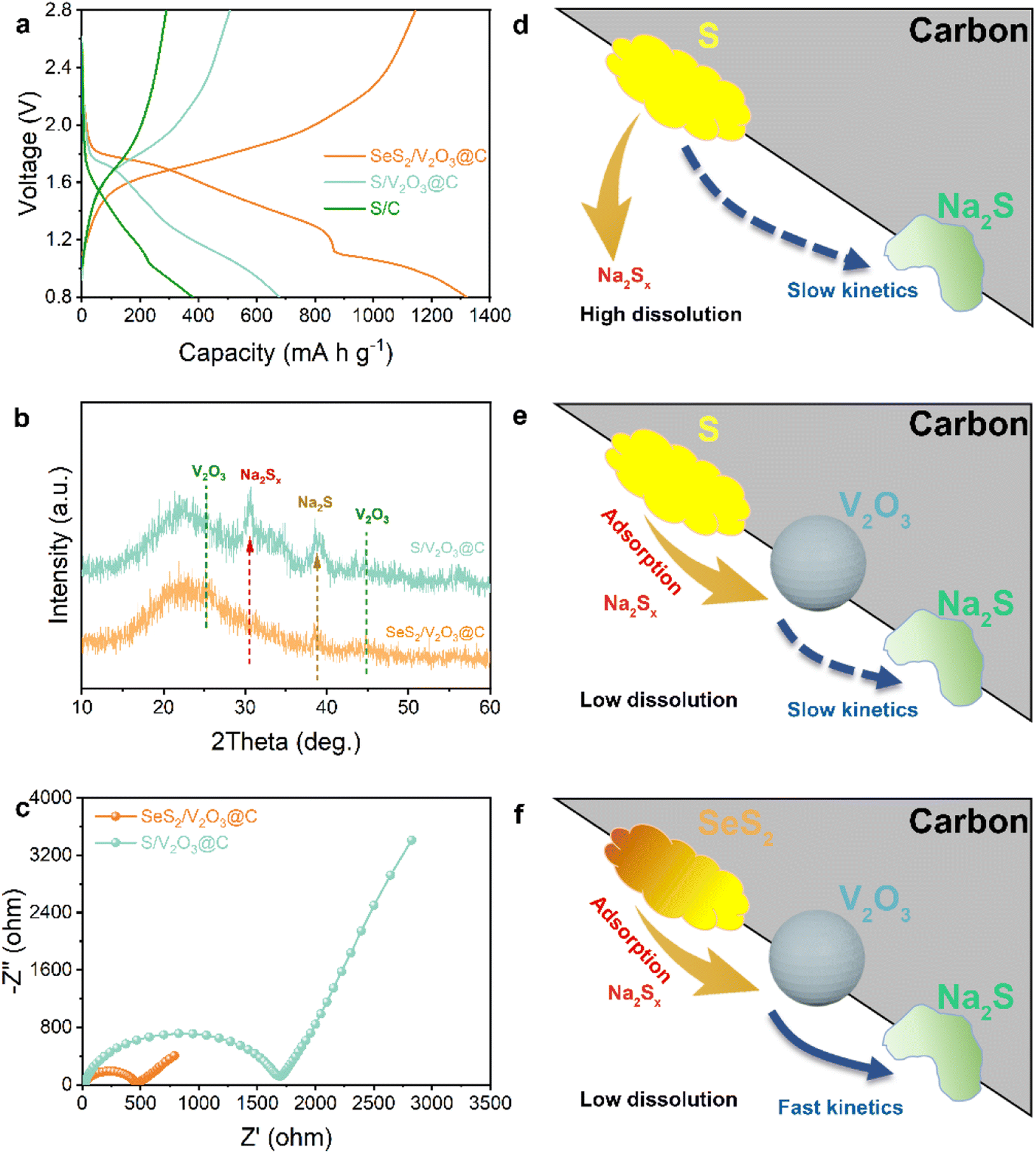 | ||
| Fig. 5 (a) Discharge–charge profiles of three samples at the same rate; (b) XRD patterns of the SeS2/V2O3@C and S/V2O3@C composite after cycling; (c) EIS spectra of the SeS2/V2O3@C and S/V2O3@C; (d–f) schematic illustrations of the electrochemical behavior of different samples. | ||
Specially, post-analyses are carried out through disassembling the batteries after cycling to further characterize the stability of the as-designed cathode. Fig. S20† shows the separators from batteries assembled with different cathodes. The separator from that with the S/C cathode is obviously yellow, suggesting the dissolution and shuttling of the S species. The separator from that with S/V2O3@C is slightly yellow, suggesting that the V2O3 effectively helps to inhibit the shuttle effect of polysulfides to a large extent. In contrast, the separator from that with SeS2/V2O3@C remains white, suggesting the completely controlled shuttle effect via the joint action of V2O3 implantation and Se doping. Besides this, the microscopic morphology of the cycled electrode was observed via the TEM (Fig. S21†) imaging of multiple regions, where the materials still maintains its nanorod shape. This confirms the structural stability of the substrate materials, which can assist in alleviating the mechanical stress from the volume change of the active materials and consequently induces a prolonged cycling lifespan. The STEM with corresponding EDS mappings in Fig. S22† reveals the uniform distribution of all elements after the cycling process. The observable elemental F and Cl are products of the decomposition reactions of electrolyte to construct the cathode interface layer, as the sample is fully cleaned before testing.
As a whole, the excellent electrochemical properties of the as-designed SeS2/V2O3@C composite result from the synergistic effect of polar V2O3 and doped Se, in which the V2O3 plays a significant role in adsorbing soluble polysulfides/polyselenides to stabilize the whole cathode without loss of the active materials and the Se dopant is responsible for improving the electronic conductivity of the S molecules and accelerating the conversion kinetics. The carbon substrate offers a conductive network for fast electron transport and accommodates the volume effect of the active materials during the electrochemical process. The synergistic mechanism is schematically illustrated in Fig. 5d–f.
Conclusion
In summary, we presented a double design strategy of a carbon host and S guest to fabricate a SeS2/V2O3@C composite for preparing S cathodes with enhanced electrochemical properties. Theoretical and experimental evidence has shown that the polar V2O3 nanoparticles implanted into carbon nanorods are able to chemically adsorb the soluble sodium polysulfides/polyselenides via strong chemical affinity and the Se dopant regulates the electronic structure of the S molecules, improving their electronic conductivity and promoting the redox conversion of S species. The synergistic effect between Se doping and polar V2O3 inhibits the shuttle effect and induces better reaction kinetics, eventually leading to outstanding electrochemical performance. The work sheds light on the significance of the co-design of host and guest as well as provides a new pathway to improve the overall properties of Na–S batteries.Experimental section
Preparation of the precursor
First, 1.20 g of 2,5-dihydroxyterephtalic acid and 2.016 g of zinc acetate dihydrate were separately dissolved in mixed solutions of deionized water and dimethylformamide. Afterwards, these two solutions were mixed and stirred for 30 min. Then, the uniform solution obtained was transferred into a Teflon-lined autoclave and heated for 24 h at 100 °C. After that, the resulting solid precipitate was centrifuged, washed with ethanol, and dried overnight at 80 °C to produce the precursor.Preparation of carbon and V2O3@C
The precursor was placed into a tube furnace and annealed for about 4.5 h at 900 °C under an Ar atmosphere. After cooling, the black powder was carbon nanorods. 500 mg of the carbon nanorods and 400 mg of vanadium chloride were dispersed in 20 mL of ethanol via stirring for 12 h. After this time, the solution was dried for 8 h at 80 °C. The as-obtained black powder was further annealed for 5 h at 900 °C under an Ar atmosphere at a heating rate of 5 °C min−1 to produce V2O3@C.Preparation of SeS2/V2O3@C
SeS2 powder and V2O3@C powder were mixed, ground, and heated for 12 h at 160 °C to produce the SeS2/V2O3@C composite. The control sample S/V2O3@C was fabricated using the same method, except that a temperature of 155 °C was used.Materials characterization
The morphologies of the samples were observed via FESEM (ZEISS, GeminiSEM 300) and TEM (Tecnai G2 F20 S-TWIN). The elemental distributions were acquired by energy-dispersive X-ray spectroscopy (EDS). The crystal structures were analyzed using an X-ray diffractometer (XRD) equipped with a Cu Kα radiation source. X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALAB 250Xi) was used to characterize the chemical states of the elements. Raman spectra were examined using an InviaRefl (Renishaw, UK) equipped with a 532 nm laser. The porosity was tested using the Brunauer–Emmett–Teller method (BSD PS2, China). Thermal analysis was evaluated by thermogravimetric analysis (TG 209, Germany).Electrochemical measurements
The target materials, Ketjen black, and PVDF were ground in a mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 to form a black slurry. The resulting slurry was then coated on carbon-coated Al foil and further dried at 80 °C. The areal mass loading content was 1.2–1.5 mg cm−2. The amount of electrolyte in every coin cell was 50 μL. The cathode slice, Na foil, and glass fiber were assembled into 2032-type coin cells in an argon-filled glove box with an oxygen/water content of <0.01 ppm. The electrolyte was 1 M NaClO4 salt dissolved in a EC-PC (1:1) mixed solvent with 5 wt% FEC additive. An electrochemical workstation (Chenhua Instrument, CHI 660E) was harnessed to measure CV curves over a voltage window of 0.8–2.8 V and EIS spectra were recorded over a frequency range from 0.01 Hz to 100 kHz. The galvanostatic discharge–charge properties were measured on a LAND instrument testing system.
:1:1 to form a black slurry. The resulting slurry was then coated on carbon-coated Al foil and further dried at 80 °C. The areal mass loading content was 1.2–1.5 mg cm−2. The amount of electrolyte in every coin cell was 50 μL. The cathode slice, Na foil, and glass fiber were assembled into 2032-type coin cells in an argon-filled glove box with an oxygen/water content of <0.01 ppm. The electrolyte was 1 M NaClO4 salt dissolved in a EC-PC (1:1) mixed solvent with 5 wt% FEC additive. An electrochemical workstation (Chenhua Instrument, CHI 660E) was harnessed to measure CV curves over a voltage window of 0.8–2.8 V and EIS spectra were recorded over a frequency range from 0.01 Hz to 100 kHz. The galvanostatic discharge–charge properties were measured on a LAND instrument testing system.
Calculation details
First-principle calculations were implemented in the VASP program based on density functional theory. The generalized gradient approximation of Perdew–Burke–Ernzerhof was employed for investigating the electronic exchange and correlation. The plane wave pseudopotential with a kinetic cutoff energy of 450 eV within the projector augmented wave (PAW) method was utilized. van der Waals dispersion-corrected DFT (DFT-D3) was also performed. The self-consistent total energy convergence criteria were less than 10−4 eV. The geometry optimization was finalized once the forces on all atoms were smaller than 0.02 eV Å−1. The K-point was produced using a 3 × 3 × 1 grid employing the Monkhorst–Pack method. The cell parameters (a × b × c) were 4 × 4 × 1 for graphene and 2 × 1 × 1 for V2O3 (012), respectively. All surface models were constructed under vacuum conditions of ∼15 Å along the c-axis. The absorbed energies between slab materials (graphene and V2O3) and different Na2Sn/Na2Sen molecules were calculated according to the equation: Ead = E(slab+molecules) − Eslab − Emolecules.Author contributions
X. L. H. conceived the project, carried out the experiments, and wrote the manuscript. H. Z., C. L., Y. L., and S. Z. performed the materials characterization. Y. W., W. Z., and H. K. L. read and edited the manuscript. S. X. D. and Z. W. had advisory roles in the project and completing the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Authors acknowledge financial support from the National Key Research and Development Program of China (2019YFB2203400), the “111 Project” (B20030), and ARC DP210102215.References
- Y. Qi, Q.-J. Li, Y. Wu, S.-j. Bao, C. Li, Y. Chen, G. Wang and M. Xu, Nat. Commun., 2021, 12, 6347 CrossRef CAS PubMed.
- X. L. Huang, Y.-X. Wang, S.-L. Chou, S. X. Dou and Z. M. Wang, Energy Environ. Sci., 2021, 14, 3757–3795 RSC.
- Y. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. Wang, M. Armand, T. Rojo and G. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 RSC.
- X. L. Huang, S. X. Dou and Z. M. Wang, Mater. Horiz., 2021, 8, 2870–2885 RSC.
- X. L. Huang, X. Zhang, L. Zhou, Z. Guo, H. K. Liu, S. X. Dou and Z. Wang, Adv. Sci., 2022, 2206558 CrossRef PubMed.
- X. Xu, D. Zhou, X. Qin, K. Lin, F. Kang, B. Li, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Nat. Commun., 2018, 9, 3870 CrossRef PubMed.
- H. Liu, W. Pei, W.-H. Lai, Z. Yan, H. Yang, Y. Lei, Y.-X. Wang, Q. Gu, S. Zhou, S. Chou, H. K. Liu and S. X. Dou, ACS Nano, 2020, 14, 7259–7268 CrossRef CAS PubMed.
- X. Ye, J. Ruan, Y. Pang, J. Yang, Y. Liu, Y. Huang and S. Zheng, ACS Nano, 2021, 15, 5639–5648 CrossRef CAS PubMed.
- H. Liu, W.-H. Lai, Y. Liang, X. Liang, Z.-C. Yan, H.-L. Yang, Y.-J. Lei, P. Wei, S. Zhou, Q.-F. Gu, S.-L. Chou, H. K. Liu, S. X. Dou and Y.-X. Wang, J. Mater. Chem. A, 2021, 9, 566–574 RSC.
- X. L. Huang, S. X. Dou and Z. M. Wang, Energy Storage Mater., 2022, 45, 265–280 CrossRef.
- Z. Yan, J. Xiao, W. Lai, L. Wang, F. Gebert, Y. Wang, Q. Gu, H. Liu, S.-L. Chou, H. Liu and S.-X. Dou, Nat. Commun., 2019, 10, 4793 CrossRef PubMed.
- C. Wu, Y. Lei, L. Simonelli, D. Tonti, A. Black, X. Lu, W.-H. Lai, X. Cai, Y.-X. Wang, Q. Gu, S.-L. Chou, H.-K. Liu, G. Wang and S.-X. Dou, Adv. Mater., 2022, 34, 2108363 CrossRef CAS PubMed.
- Y.-X. Wang, J. Yang, W. Lai, S.-L. Chou, Q.-F. Gu, H. K. Liu, D. Zhao and S. X. Dou, J. Am. Chem. Soc., 2016, 138, 16576–16579 CrossRef CAS PubMed.
- A. Y. S. Eng, Y. Wang, D.-T. Nguyen, S. Y. Tee, C. Y. J. Lim, X. Y. Tan, M.-F. Ng, J. Xu and Z. W. Seh, Nano Lett., 2021, 21, 5401–5408 CrossRef CAS PubMed.
- F. Xiao, X. Yang, H. Wang, J. Xu, Y. Liu, D. Y. W. Yu and A. L. Rogach, Adv. Energy Mater., 2020, 10, 2000931 CrossRef CAS.
- J. Zhou, Y. Yang, Y. Zhang, S. Duan, X. Zhou, W. Sun and S. Xu, Angew. Chem., Int. Ed., 2021, 60, 10129–10136 CrossRef CAS PubMed.
- A. Kumar, A. Ghosh, A. Roy, M. R. Panda, M. Forsyth, D. R. MacFarlane and S. Mitra, Energy Storage Mater., 2019, 20, 196–202 CrossRef.
- S. Li, Z. Zeng, J. Yang, Z. Han, W. Hu, L. Wang, J. Ma, B. Shan and J. Xie, ACS Appl. Energy Mater., 2019, 2, 2956–2964 CrossRef CAS.
- Z. Li, J. Zhang, Y. Lu and X. W. Lou, Sci. Adv., 2018, 4, eaat1687 CrossRef PubMed.
- L. Wang, X. Chen, S. Li, J. Yang, Y. Sun, L. Peng, B. Shan and J. Xie, J. Mater. Chem. A, 2019, 7, 12732–12739 RSC.
- Y. Sun, J. Liao, J. Sun, L. Duan, Y. Du, J. Bao and X. Zhou, ACS Appl. Energy Mater., 2022, 5, 13023–13030 CrossRef CAS.
- Y. Yao, L. Zeng, S. Hu, Y. Jiang, B. Yuan and Y. Yu, Small, 2017, 13, 1603513 CrossRef PubMed.
- J. Ma, L. Gao, S. Li, Z. Zeng, L. Zhang and J. Xie, Batteries Supercaps, 2020, 3, 165–173 CrossRef CAS.
- L. Zeng, Y. Yao, J. Shi, Y. Jiang, W. Li, L. Gu and Y. Yu, Energy Storage Mater., 2016, 5, 50–57 CrossRef.
- C. Feng, X. L. Huang, Y. Li, Y. Wang, C. Li, W. Qiu, S. Zhang, H. Liu, Y. Zhang, H. K. Liu, S. X. Dou and Z. Wang, Chem. Eng. J., 2022, 442, 136189 CrossRef CAS.
- C. Liu, X. Huang, J. Wang, H. Song, Y. Yang, Y. Liu, J. Li, L. Wang and C. Yu, Adv. Funct. Mater., 2018, 28, 1705253 CrossRef.
- H. Zhang, L. Zhou, X. Huang, H. Song and C. Yu, Nano Res., 2016, 9, 3725–3734 CrossRef CAS.
- C. Shen, T. Wang, X. Xu and X. Tian, Electrochim. Acta, 2020, 349, 136331 CrossRef CAS.
- X. L. Huang, X. Zhang, M. Yi, Y. Wang, S. Zhang, S. Chong, H. K. Liu, S. X. Dou and Z. Wang, Chem. Sci., 2022, 13, 11585–11593 RSC.
- X. L. Huang, P. Xiang, H. Liu, C. Feng, S. Zhang, Z. Tian, H. K. Liu, S. X. Dou and Z. Wang, Inorg. Chem. Front., 2022, 9, 5486–5494 RSC.
- X. Huang, J. Deng, Y. Qi, D. Liu, Y. Wu, W. Gao, W. Zhong, F. Zhang, S. Bao and M. Xu, Inorg. Chem. Front., 2020, 7, 1182–1189 RSC.
- L. Zhang, B. Zhang, Y. Dou, Y. Wang, M. Al-Mamun, X. Hu and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 20422–20428 CrossRef CAS PubMed.
- X. Liu, Z. Wang, Y. Niu, C. Liu, H. Chen, X. Ren, Z. Liu, W.-M. Lau and D. Zhou, ACS Appl. Energy Mater., 2022, 5, 3525–3535 CrossRef CAS.
- Y. Lei, C. Wu, X. Lu, W. Hua, S. Li, Y. Liang, H. Liu, W.-H. Lai, Q. Gu, X. Cai, N. Wang, Y.-X. Wang, S.-L. Chou, H.-K. Liu, G. Wang and S.-X. Dou, Angew. Chem., Int. Ed., 2022, 61, e202200384 CAS.
- T. Yang, Y. Qi, W. Zhong, M. Tao, B. Guo, Y. Wu, S.-J. Bao and M. Xu, Adv. Funct. Mater., 2021, 31, 2001952 CrossRef CAS.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- Y. Deng, L. Gong, H. Ahmed, Y. Pan, X. Cheng, S. Zhu and H. Zhang, Nano Res., 2020, 13, 967–974 CrossRef CAS.
- V. H. Pham, J. A. Boscoboinik, D. J. Stacchiola, E. C. Self, P. Manikandan, S. Nagarajan, Y. Wang, V. G. Pol, J. Nanda, E. Paek and D. Mitlin, Energy Storage Mater., 2019, 20, 71–79 CrossRef.
- X. Zhou, Z. Yu, Y. Yao, Y. Jiang, X. Rui, J. Liu and Y. Yu, Adv. Mater., 2022, 34, 2200479 CrossRef CAS PubMed.
- H. Hao, Y. Wang, N. Katyal, G. Yang, H. Dong, P. Liu, S. Hwang, J. Mantha, G. Henkelman, Y. Xu, J. A. Boscoboinik, J. Nanda and D. Mitlin, Adv. Mater., 2022, 34, 2106572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06831a |
| This journal is © The Royal Society of Chemistry 2023 |