
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn artificial metallolyase with pliable 2-His-1-carboxylate facial triad for stereoselective Michael addition†
Ryusei
Matsumoto
a,
Saho
Yoshioka
b,
Miho
Yuasa
b,
Yoshitsugu
Morita
a,
Genji
Kurisu
 c and
Nobutaka
Fujieda
*a
c and
Nobutaka
Fujieda
*a
aDepartment of Applied Biological Chemistry, Graduate School of Agriculture, Osaka Metropolitan University, 1-1 Gakuen-cho, Naka-ku, Sakai-shi, Osaka 599-8531, Japan. E-mail: fujieda@omu.ac.jp
bDepartment of Applied Life Sciences, Graduate School of Life and Environmental Sciences, Osaka Prefecture University, 1-1 Gakuen-cho, Naka-ku, Sakai-shi, Osaka 599-8531, Japan
cInstitute for Protein Research, Osaka University, 3-2 Yamada-oka, Suita, Osaka 565-0871, Japan
First published on 13th March 2023
Abstract
We repurposed the metal-binding site of a cupin superfamily protein into the 2-His-1-carboxylate facial triad, which is one of the common motifs in natural non-heme enzymes, to construct artificial metalloenzymes that can catalyze new-to-nature reactions. The Cu2+-H52A/H58E variant catalyzed the stereoselective Michael addition reaction and was found to bear a flexible metal-binding site in the high-resolution crystal structure. Furthermore, the H52A/H58E/F104W mutant accommodated a water molecule, which was supported by Glu58 and Trp104 residues via hydrogen bonding, presumably leading to high stereoselectivity. Thus, the 2-His-1-carboxylate facial triad was confirmed to be a versatile and promising metal-binding motif for abiological and canonical biological reactions.
Introduction
Many natural enzymes accommodate metal ions for structural stability and to catalyze numerous challenging reactions.1 Mononuclear non-heme metalloenzymes catalyze the chemical transformation of substances, such as aromatic C–H bond hydroxylation and cis-dihydroxylation of arene double bonds.2,3 Based on this competent catalytic ability, various approaches to develop engineered metalloproteins with catalysis and artificial metalloenzymes (ArMs) have been developed. In ArMs, chemo-, regio-, and/or enantioselectivity is endowed with the bound synthetic metal complex by the matrix of the host protein. Additionally, the chemical stability and solubility in water are conferred to the protein-bound metal complexes. Therefore, ArMs are a powerful means of accelerating chemically difficult reactions under environmentally benign conditions. Several methods, such as covalent, supramolecular anchoring, and cofactor substitution, have been established for the site-directed anchoring of metal complexes to the protein cavity.4–10 An alternative strategy involves the direct linking of bare metal ions with coordinating endogenous amino acids via dative covalent bonds.11 This type of ArM is constructed by employing non-metalated natural or de novo proteins or by repurposing natural non-heme metalloenzymes, often aided by computational design.11–13 The construction of endogenous amino acid-ligated metal active centers within suitable protein cavities to achieve simultaneous selective substrate binding and catalytic activity is significantly more challenging.Recent crystal structures of mononuclear non-heme metalloenzymes have revealed the existence of a common metal-binding motif, which consists of two histidines and an amino acid with a carboxylic acid side chain, that is, glutamate or aspartate (His-His-Glu [HHE] or His-His-Asp [HHD]).2,14 This motif has been designated as a 2-His-1-carboxylate facial triad, where two nitrogen atoms and one oxygen atom coordinate to the central metal in fac fashion (Fig. 1).2 The fac-type coordination of three amino acid residues at the octahedral coordination center offers three vacant coordination sites on the opposite side, which provide important space for the substrates and coenzymes to bind.
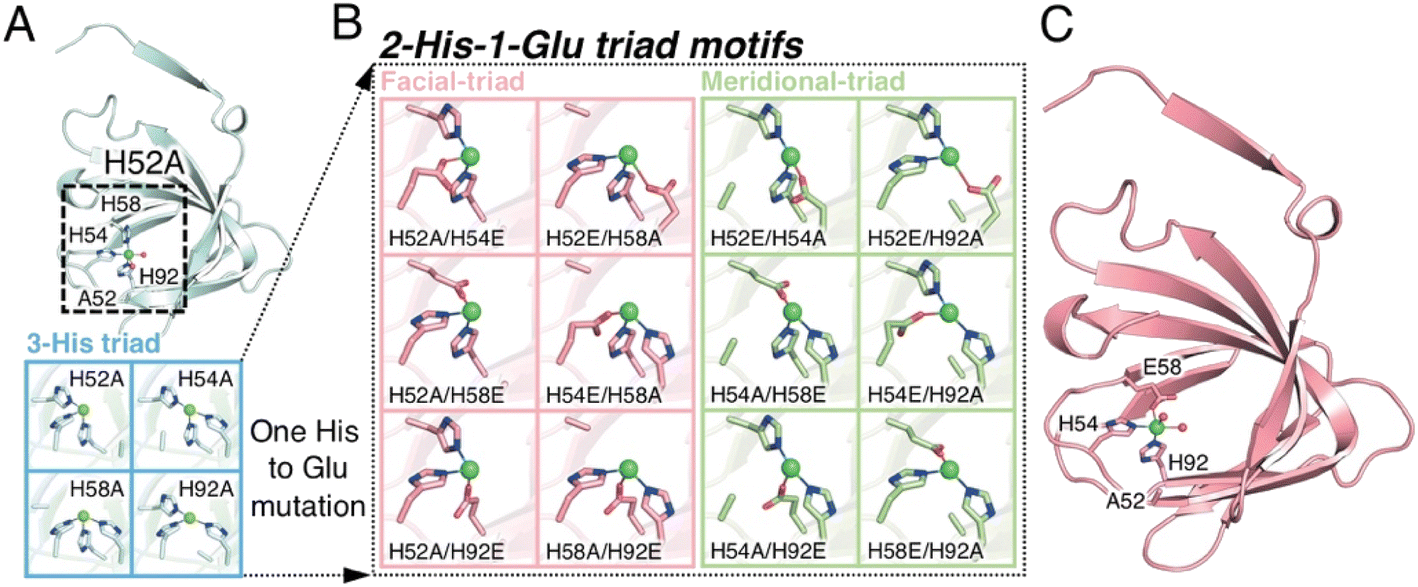 | ||
| Fig. 1 The extended metal ligand library with 2-His-1-carboxylate triad containing TM1459 cupin proteins for Michael addition reaction of methyl acetoacetate. (A) Subunit structure of Cu-bound H52A variant (subunit A, PDB code 6L2E) and model structures of 3-His triad motifs produced in silico. (B) Model structures of 2-His-1-Glu triad motifs manually produced in silico. (C) Subunit structure of Cu-bound H52A/H58E variant (subunit A, PDB code 8HJX, see Tables S1–S4 and Fig. S1, S2†). Protein main chain is displayed as a ribbon, key amino acid residues as sticks, Cu ions as green spheres, and water molecules as red spheres. | ||
Several ArMs containing His-His-Asp [HHD] fac-triad active sites have been reported based on this triad.15,16 An artificial Cu-Diels-Alderase was constructed by engineering an HHD fac-triad within the non-metalated TIM barrel protein, imidazole glycerol phosphate synthase (tHisF).17 The ArM possesses an engineered metal binding site consisting of Leu50 and Ile52, which was mutated to histidines, plus the native aspartic acid at position 11 and exhibited 73% conversion of 2-azachalcone compound 1 with good enantioselectivity (enantiomeric excess [ee] = 46%; endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo = 93:7) in the reaction with cyclopentadiene.17 Another artificial Cu-Diels-Alderase was generated by metal swapping the natural Fe2+ of HHD fac-triad active site in 1-aminocyclopropane carboxylic acid oxidase with the Cu2+ ion. In same reaction, it gave same product with high quantitative yield associated and excellent stereoselectivity (ee = 95%; endo:exo = 99:1).18 Artificial Fe proteins that simulate the HHD fac-triad active site in non-heme monooxygenases have been developed by combining metal complexes and streptavidin.19 On the other hand, artificial β-lactamases were built via the self-assembly of a monomeric redox protein that has His-His-Glu [HHE] triad zinc sites in its interfaces.20,21
:exo = 93:7) in the reaction with cyclopentadiene.17 Another artificial Cu-Diels-Alderase was generated by metal swapping the natural Fe2+ of HHD fac-triad active site in 1-aminocyclopropane carboxylic acid oxidase with the Cu2+ ion. In same reaction, it gave same product with high quantitative yield associated and excellent stereoselectivity (ee = 95%; endo:exo = 99:1).18 Artificial Fe proteins that simulate the HHD fac-triad active site in non-heme monooxygenases have been developed by combining metal complexes and streptavidin.19 On the other hand, artificial β-lactamases were built via the self-assembly of a monomeric redox protein that has His-His-Glu [HHE] triad zinc sites in its interfaces.20,21
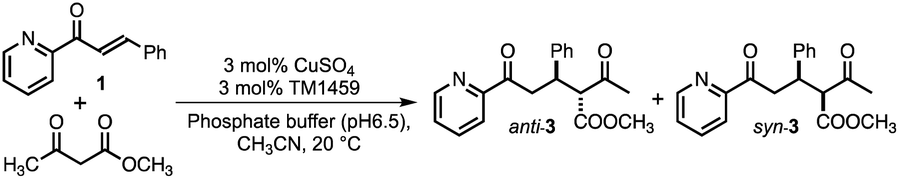
We constructed ArMs using the homodimeric protein from Thermotoga maritima, TM1459,22,23 which belongs to the cupin superfamily and has a small molecular mass of 12977 Da (residues 1–114). The protein possesses a metal-binding site consisting of four histidines, which is reminiscent of trispyridylamine (TPA) ligand, that can also be used as a macromolecular metal ligand.24 We repurposed Os-TM1459 into artificial peroxygenase to catalyze stereoselective cis-1,2-dihydroxylation reaction.24 Moreover, we reported artificial metallolyase based on the Cu-TM1459 cupin to accelerate stereoselective Michael addition reactions using nitroalkane and 2-azachalcone 1 as model substrates to simultaneously screen the primary and secondary coordination sphere with mini TM1459 variant library.25 Several TM1459 variants containing three histidine-bearing metal-binding site (3-His triad, Fig. 1A) and two histidine-bearing site (2-His dyad) are employed in the reaction screening.25 Michael addition reactions are very useful in the construction of carbon–carbon bonds that can be used in combination with various electrophiles and nucleophiles. Consequently, excellent enantioselectivities have been reported for Michael addition reactions of dimethylmalonate to unsaturated 2-acyl imidazoles or 2-azachalcone 1 with the catalytic system of ArMs based on DNA26–28 and small proteins.29 In particular, compounds obtained with chalcone derivatives as acceptors and malonate derivatives as donors can yield trans-cyclopropanes and oxetane derivatives, which are used as structural motifs for some pharmaceuticals and bioactive substances.30,31 Herein, we report our recent efforts to expand our approach by creating a 2-His-1-carboxylate triad (HHE, Fig. 1B and C, see Tables S1–S4 and Fig. S1, S2†), which can catalyze the Michael addition reaction of α,β-unsaturated ketone, 2-azachalcone 1, with representative active methylene compounds, such as dimethylmalonate and methyl acetoacetate.
Results and discussion
We first screened TM1459 variants containing 3-His triads (H52A, H54A, H58A, and H92A, Fig. 1A) and 4-His tetrad (wild-type) as metal-binding motifs. When employing dimethylmalonate as a Michael donor (Tables S5–S7 and Fig. S1†), the wild-type TM1459 did not show enantioselectivity. On the other hand, H52A mutant could produce the (−)-isomer of compound 2 with excellent selectivity (enantiomeric excess (ee) = 86%; Table S7,† entry 3), whereas the H92A mutant generated the inverted stereoisomer (+)-compound 2 with moderate selectivity (ee = 58%; Table S7,† entry 6), demonstrating that the nucleophilic attack of active methylene species can be sterically controlled by the protein matrix of these Cu-TM1459 variants (Fig. S3†). The absolute configuration of (−)-compound 2 was determined to be (R)-compound 2 by comparison with the corresponding pyridine N-oxide adduct (N-oxygenated, following the treatment of (−)-compound 2 with H2O2, see ESI† Experimental section).The substrate scope of the reaction was then examined using methyl acetoacetate as the donor. In the case of methyl acetoacetate, the active methylene species attack α,β-unsaturated ketones, producing diastereomers because of the different substituents and acetyl and methyl ester groups. First, the catalytic reactions were conducted using the same series of TM1459 variants bearing the 3-His triad and wild-type containing one 4-His tetrad. Under the optimized conditions (Table S8†), the diastereomeric ratio (d.r.; Table 1, entry 1) was not observed without TM1459 variants. In contrast, TM1459 variants exhibited various d.r. and ee values (Table 1, entry 2–6). H52A and H92A, in particular, preferred the anti-form and showed very good enantioselectivity and very good yield, partly reflecting that the pKa value of α-proton of methyl acetoacetate (≅11) is lower than that of dimethylmalonate (≅13).32 Then, we chose the variant with 3-His triad as a scaffold, which was repurposed into 2-His-1-carboxylate triads, introduced by mutating one histidine to metal-coordinating glutamate. This mutation could slightly perturb the substrate-binding cavity and Cu-coordinating water molecule because of its smaller size than the imidazole and bidentate ability of the carboxyl group, while maintaining a coordination geometry similar to that of the 3-His triad. (Fig. 1B and C).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | TM1459 variant | Yieldb,c (%) | d.r. (anti:syn) |
eed (%) (anti) | Selectivitye | TON |
|
a Reaction conditions: TM1459 (0.3 mM), CuSO4 (0.3 mM), 1 (10 mM), methyl acetoacetate (60 mM), potassium phosphate buffer (pH 6.5)/CH3CN (9:1), 20 °C, 16 h.
b Yields, diastereomeric ratio (d.r.) and enantiomeric excesses (ee) were determined by chiral HPLC analysis.
c Yields were calculated based on the total amount of stereoisomers.
d (+) or (−) was determined by polarimeter on HPLC.
e Single isomer/total.
|
||||||
| 1 | — | 36 | 51:49 |
n.d. | n.d. | 12 |
| 2 | Wild type | 35 | 57:43 |
38(+) | 39% | 12 |
| 3 | H52A | 82 | 55:45 |
80(−) | 50% | 27 |
| 4 | H54A | 4 | n.d. | n.d. | n.d. | 1 |
| 5 | H58A | 31 | 46:54 |
16(+) | 27% | 10 |
| 6 | H92A | 72 | 70:30 |
86(+) | 65% | 24 |
| 7 | H52A/H54E | 33 | 61:39 |
24(−) | 38% | 11 |
| 8 | H52A/H58E | 64 | 52:48 |
96(−) | 52% | 21 |
| 9 | H52E/H54A | 37 | 60:40 |
36(−) | 41% | 12 |
| 10 | H54A/H58E | 42 | 56:44 |
4(+) | 29% | 14 |
| 11 | H52E/H58A | 70 | 56:44 |
12(−) | 31% | 23 |
| 12 | H54E/H58A | 76 | 59:41 |
4(−) | 31% | 25 |
| 13 | H52E/H92A | 37 | 55:45 |
20(−) | 33% | 12 |
| 14 | H52A/H58E/V19L | 93 | 57:43 |
88(−) | 54% | 31 |
| 15 | H52A/H58E/F41W | 97 | 62:38 |
94(−) | 60% | 32 |
| 16 | H52A/H58E/I49Q | 85 | 52:48 |
94(−) | 50% | 28 |
| 17 | H52A/H58E/F104W | 76 | 71:29 |
90(−) | 68% | 25 |
| 18 | H52A/H58Q | 10 | n.d. | n.d. | n.d. | 3 |
While the 3-His triad of H54A and H92A mutants exhibited meridional coordination geometry, those of H52A and H58A mutants presented facial coordination geometry in silico, although the 3-His facial triad structure was confirmed in the crystal structure of H52A (Fig. 1A). As a result of the mutation of a His residue to Glu, the corresponding mutants to H54A and H92A, and H52A and H58A displayed meridional and facial triads, respectively (Fig. 1B). The 7 corresponding mutants of 12 (Fig. S1 and S2†) were screened for the reaction with methyl acetoacetate (Table 1, entry 7–13). We found a drastic increase in enantioselectivity in the case of the H52A/H58E mutant, with good yield (Table 1, entry 8 and Fig. S4†). Typically, the 2-His-1-carboxylate facial triads in natural enzymes are considered beneficial structural motifs. In this experiment, we used mutants with meridional and facial triads. Screening revealed that the H52A/H58E mutant was obtained as a variant that exhibited high enantioselectivity and was postulated to have a facial triad, which is required for selective and efficient catalysis.
To clarify the stereochemical mechanism that can account for the improvement in enantioselectivity, the crystal structure of the Cu-bound H52A/H58E mutant was determined with highly reliable crystallographic statistics at a resolution of 1.15 Å (Fig. 1C, 2A, S5A, S5B, and Tables S1–S3†). The coordination structure of the Cu center was shown to be a 2-His-1-carboxylate facial triad, completing the square pyramidal geometry of the N2O3 donor set, with two nitrogen atoms of histidines (His54 and His92), two water molecules in the equatorial position (O1 and O2), and an oxygen atom of glutamate (Glu58) in the axial position (Fig. 2A). In the reported crystal structure of H52A (PDB code 6L2E), Cu ions are supported by a 3-His triad with two water molecules. His54, His58, and two water molecules are located in the equatorial position and His92 sits in the axial position in chain A, whereas His58, His92, and two water molecules are located in the equatorial position and His54 is located in the axial position of chain B (O2′ and O3′, Fig. 2B). This change in the coordination structure implies that there are at least three types of binding modes between the Cu center and compound 1 as bidentate ligands, causing low stereoselectivity. In the case of H52A/H58E, the glutamate placed at 58 position supported the Cu ion by one carboxyl oxygen atom in a monodentate fashion, and the other carboxyl oxygen connected with Cu-coordinating water via hydrogen bonds (O1, Fig. 2A), likely preventing the ligand exchange of this water molecule with substrate compound 1. Therefore, compound 1 could access the Cu center only from the distal side of Glu58, fixing the binding mode to determine its chiral plane. In stark contrast to the Cu center of H52A, electron density due to minor Cu species was observed (Cu(B), Fig. 2A). Because of the rearrangement of coordination sphere, the ligand geometry of minor species is slightly different from major ones, where coordinate bond between Glu58 and Cu is slightly elongated from 2.20 ± 0.03 Å to 2.73 ± 0.04 Å, generating a sterically less hindered Cu center (Table S3†). This possibly makes it easy for the ketone oxygen and pyridine nitrogen of 1 to coordinate to the Cu ion from the positions trans to His92 and Glu58 (O2 and O3 positions, Fig. 2B), respectively, forming an octahedral intermediate containing the Jahn–Teller axis.
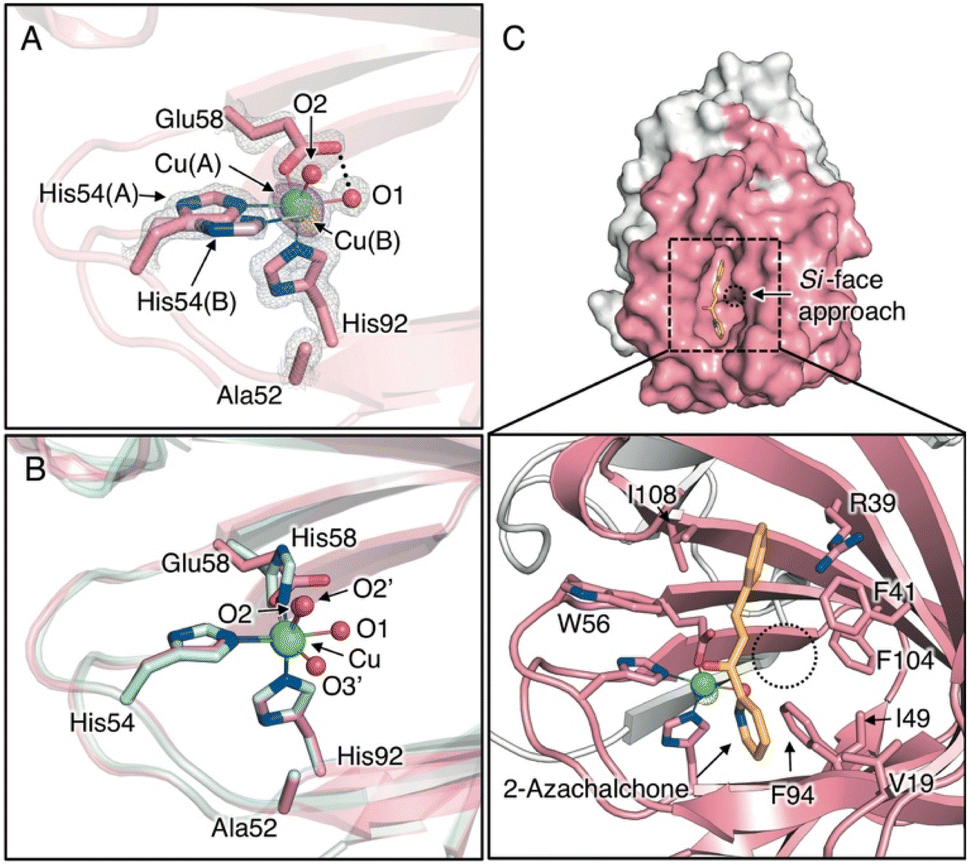 | ||
| Fig. 2 (A) Crystal structure of Cu-binding site of H52A/H58E (chain B, PDB code 8HJX). (B) Superimposed structures of the Cu-binding sites of H52A/H58E (pink) and H52A (blue, chain B, PDB code 6L2E) (C) Surface models and close-up views of Cu site of H52A/H58E of in silico-obtained azachalcone-TM1459 complex. 2Fo − Fc and anomalous maps contoured at 1.5 and 5.0σ are shown in gray and magenta mesh, respectively. | ||
To scrutinize the binding mode of 1, a docking simulation with azachalcone was performed using this structural coordinate (PDB code 8HJX) and AutoDock Vina software. The lowest energy conformation of 1 is very similar to that of the H52A mutant in our previous study,15 and the ketone and pyridine moieties pointed to the Cu center (calculated free energy of binding [ΔGb]: −5.1 (kcal mol−1), Fig. 2C). Together with the crystal structure, it appears likely that the ketone oxygen and pyridine nitrogen bind to the Cu center from the positions trans to Glu58 and His92, respectively, to form the intermediate (Fig. 2C). In the azachalcone-docked structure, β-Si-face attack by methyl acetoacetate probably occurs at the position indicated by the dotted circle (Fig. 2C). This space is tightly packed by five hydrophobic amino acid residues, Val19, Phe41, Ile49, Phe94, and Phe104, while the opposite side is solvent-accessible (Fig. 2C).
To substantiate this assumption, we selected four amino acid residues (Val19, Phe41, Ile49, and Phe104) in the secondary coordination sphere as mutation targets for H52A/H58E based on the results of the docking simulation. To influence the interaction of this cavity with methyl acetoacetate, we replaced them with larger amino acid residues. The additional mutation of V19L and I49Q in H52A/H58E did not affect the diastereoselectivity, although the yields were enhanced to an excellent extent (Table 1; entries 14 and 16). On the other hand, H52A/H58E/F41W and H52A/H58E/F104W were found to give anti-preference up to 62:38 and 71:29 (d.r.), respectively, which were higher than those of H52A and H52A/H58E, retaining excellent enantioselectivity (ee = 94 and 90%, respectively; Table 1, entries 15, 17, and Fig. S4†). These results also partly support the hypothesis that methyl acetoacetate can access 1 from the Si-face of the double bond β-carbon of azachalcone in the docked structure.
To elucidate the substrate-binding mode in the H52A/H58E/F104W mutant, the crystal structure was determined at 1.18 Å resolution (Tables S1–S3†). The overall structure and coordination geometry of the Cu center were almost identical to those of the H52A/H58E mutant (Fig. 3A, B and S5A, S5C†). In this crystal structure, the major and minor copper species (Cu(A) and Cu(B), respectively, Fig. 3A) were observed, although Cu–Cu distance (0.83 ± 0.02 Å) was slightly larger than that in the H52A/H58E mutant (0.54 ± 0.02 Å, Fig. 3B and C; Tables S3†). Interestingly, the electron density of another water molecule was observed (O4, Fig. 3A), which was not observed in the H52A/H58E mutant because of the introduction of tryptophan at position 104 (Fig. 3B). This water molecule bridges the nitrogen atom of Trp104 and the carboxylic oxygen of Glu58 via hydrogen bonds, inducing the configuration of the hydrogen-bonding network starting from the Cu-coordinated water molecule to tryptophan. This water molecule sits near the aforementioned small cavity, which is surrounded by five amino acid residues, with methyl acetoacetate approaching. An additional F104W mutation improved the diastereoselectivity of the Michael addition reaction, perhaps by spatially fixing another water molecule between Glu58 and Trp104. This result suggests that the water molecule may have facilitated the acetyl group to intrude by the interaction with the ketone of the acetyl group via hydrogen bonding, and the bulky Trp104 residue prevented the slightly larger methoxycarbonyl side of the methyl acetoacetate molecule from intruding into this cavity rather than its acetyl group side (Fig. 3D). Reflecting such substrate binding mode, this mutant showed Michaelis–Menten saturation curve in the kinetic assay (Fig. S6†).
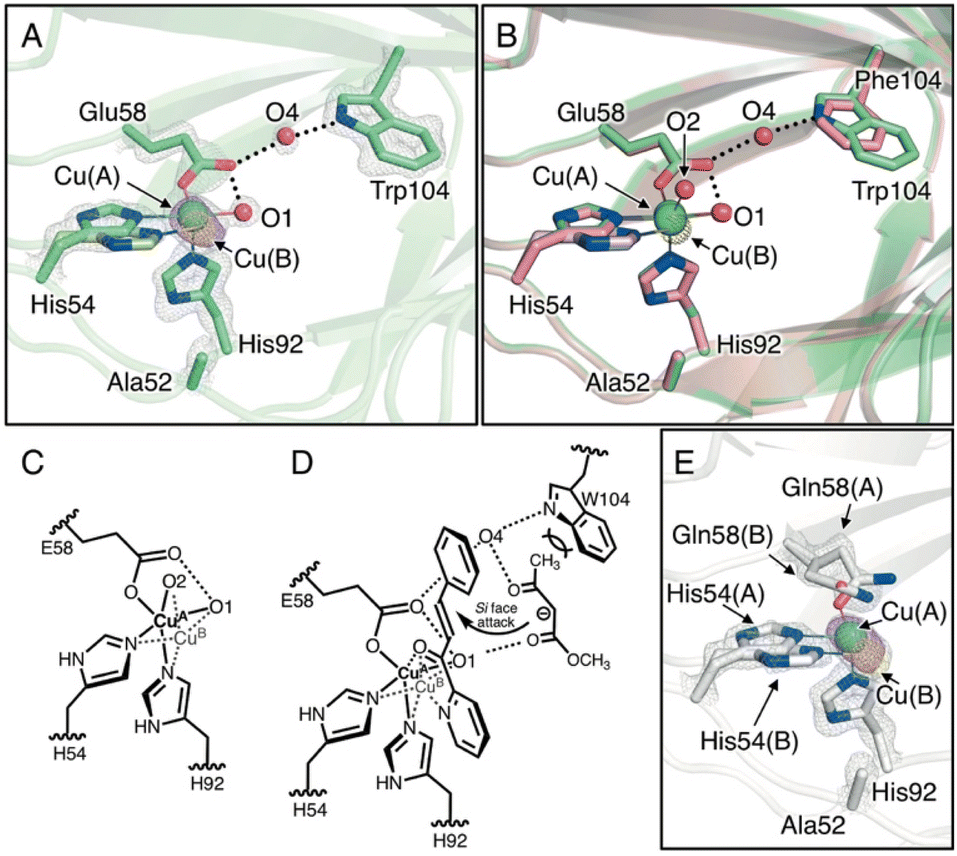 | ||
| Fig. 3 (A) Crystal structure of the Cu-binding site of H52A/H58E/F104W (chain B, PDB code 8HJY). (B) Superimposed structures of the Cu-binding sites of H52A/H58E/F104W(green) and H52A/H58E (pink). (C) Schematic representation of copper transition. (D) Plausible transition state of the quaternary complex of enzyme, Cu, azachalcone, and methyl acetoacetate. (E) Crystal structure of Cu-binding site of H52A/H58Q (chain B, PDB code: 8HJZ). 2Fo − Fc and anomalous maps contoured at 1.5 and 5.0σ are shown in gray and magenta mesh, respectively. | ||
To further investigate the pliable copper center, we also determined the crystal structure of Cu-bound H52A/H58Q mutants (1.22 Å resolution; Fig. S5A and S5D; Tables S1–S3†), the Cu center of which was supported by the fac-triad of His-His-Gln (HHQ). As in the H52A/H58E mutant, the nitrogen atom of the amide of the glutamine residue is hydrogen-bonded to the water molecule in the equatorial position. However, the dative bond length of Cu-amide oxygen of Gln58(A) (2.49 ± 0.11 Å) is longer than that of Cu-carboxyl oxygen of Glu58 in the H52A/H58E mutant (2.20 ± 0.03 Å), suggesting that the strength of the coordinate bond is weakened (Fig. 3E and Table S3†). In addition, we found the existence of not only minor Cu species but also another conformation of Gln58, indicating that Cu ions spatially fluctuate in the active center to a greater extent. In support of this notion, the minor Cu species was more significantly distant from the major Cu species (1.23 ± 0.02 Å) compared to that in the H52A/H58E mutant (0.54 ± 0.02 Å), breaking the coordination bond between Cu(B) and Gln58(B) residues and the hydrogen bond between Cu-coordinating water and Gln58(B) residue. This suggests that the binding affinity to Cu2+ ions for the H52A/H58Q mutant is relatively low. Actually, the H52A/H58Q mutant was found to have higher dissociation constant (Kd = 0.53 μM) than that of the H52A/H58E mutant (Kd = 0.11 μM, Fig. S7†). During the catalytic cycle, Cu ions can strip from the active center resulting in a decrease in reactivity, which leads to a drastic decrease in yields (Table 1; entry 18).
Cu active centers of three TM1459 variants (H52A, H52A/H58E, and H52A/H58Q) exhibited different degrees of flexibility, reflecting the pKa values of the amino acid residues at the 58 position (His, Glu, and Gln, respectively). In the H52A/H58E mutant, the appropriate flexibility of the Cu center appears to play a significant role in the binding of azachalcone 1, where the carboxyl group of Glu58 loosely restrains the location of the Cu ion via its moderate donating ability and somewhat immobilizes the Cu-coordinating water molecule by bridging the Cu ion and water molecule in a bidentate manner (Fig. 3C and D).
Conclusions
To the best of our knowledge, this study is the first to construct His-His-Glu [HHE] facial-triad-coordinating copper-containing ArMs that can catalyze the abiological reactions, although there are several literature that reported ArMs containing His-His-Asp [HHD] facial-triad copper active sites.15–18 The 2-His-1-carboxylate facial triad motif is one of the most pivotal common structural motifs frequently observed in the active centers of natural metalloenzymes. In this system, three vacant sites located on the distal side of the facial triad play a significant role in the catalytic mechanisms, such as complex formation between cofactors and substrates. Small variations in the triad have also been discovered, such as the bidentate coordination of the carboxylato group in Rieske oxygenases, where the coordination geometry of the metal center by the three residues cannot be strictly referred to as the facial triad. In this context, the Cu active site near the heme moiety doesn't have the HHE triad motif in the artificial nitric oxide reductase.33 ACC oxidase mutant with the repurposed Cu center supported by the HHE triad did not show any catalytic activity, and concrete evidence is required for the clarification of its coordination geometry.34 In our study, the HHE motif, which was constructed to reduce the steric hindrance, allowed us to achieve high stereoselectivity in the Michael addition reaction, confirming the versatility of this fac-triad motif. Additionally, natural mononuclear copper metalloenzymes with the 2-His-1-carboxylate facial triad motif have not yet been reported.35 One of the variations, 3-His-1-Glu tetrad with Cu ion, was reported in quercetin 2,4-dioxygenase. This copper binding site forms two geometries in the resting state: a tetrahedral site of the 3-His motif plus water with Glu73 in an off-copper conformation (major form) and a distorted five-coordinate site in which Glu73 is an additional monodentate ligand (minor form).36,37 Gratifyingly, a similar equilibrium between the two geometries was observed in our ArMs, HHE fac-triad-coordinating Cu-TM1459 mutants. We envision that the use of the extended library of mutants of TM1459 for ArMs can help to expand the repertoire of artificial non-heme metalloenzymes. Moreover, investigating the detailed catalytic mechanisms of ArMs thus obtained in future studies will provide profound insights into natural enzymes and their catalytic activities.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
Ryusei Matsumoto: investigation, methodology, data curation, visualization, writing – original draft. Saho Yoshioka: investigation, data curation. Miho Yuasa: investigation. Yoshitsugu Morita: methodology, investigation. Genji Kurisu: methodology, writing – review and editing. Nobutaka Fujieda: conceptualization, methodology, visualization, writing – review and editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. F. received financial support/funding from JSPS, MEXT, Japan, (JSPS KAKENHI 18H04270 and 21H01954), the NOVARTIS Foundation (Japan) for the Promotion of Science, and the Sumitomo Foundation. This work was performed using a synchrotron beamline BL44XU at SPring-8 (Harima, Japan) under the Cooperative Research Program of the Institute for Protein Research, Osaka University (proposal numbers 2021A6500, 2021B6500, 2022B6500, and 2022B6500). We would also like to thank Dr E. Yamashita, Dr K. Sakurai, and Dr A. Nakagawa of SPring-8 BL44XU for their assistance with crystallographic data collection.Notes and references
- I. Bertini and A. Sigel, Handbook on Metalloproteins, CRC Press, 2001 Search PubMed.
- P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- G. D. Straganz and B. Nidetzky, ChemBioChem, 2006, 7, 1536–1548 CrossRef CAS PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS PubMed.
- J. C. Lewis, Acc. Chem. Res., 2019, 52, 576–584 CrossRef CAS PubMed.
- S. N. Natoli and J. F. Hartwig, Acc. Chem. Res., 2019, 52, 326–335 CrossRef CAS PubMed.
- W. J. Jeong, J. Yu and W. J. Song, Chem. Commun., 2020, 56, 9586–9599 RSC.
- A. G. Jarvis, Curr. Opin. Chem. Biol., 2020, 58, 63–71 CrossRef CAS PubMed.
- B. Maity, M. Taher, S. Mazumdar and T. Ueno, Coord. Chem. Rev., 2022, 469, 214593 CrossRef CAS.
- A. Pordea, Curr. Opin. Chem. Biol., 2015, 25, 124–132 CrossRef CAS PubMed.
- L. A. Churchfield and F. A. Tezcan, Acc. Chem. Res., 2019, 52, 345–355 CrossRef CAS PubMed.
- K. J. Koebke, T. B. J. Pinter, W. C. Pitts and V. L. Pecoraro, Chem. Rev., 2022, 122, 12046–12109 CrossRef CAS PubMed.
- S. Kal and L. Que, J. Biol. Inorg Chem., 2017, 22, 339–365 CrossRef CAS PubMed.
- B. Amrein, M. Schmid, G. Collet, P. Cuniasse, F. Gilardoni, F. P. Seebeck and T. R. Ward, Metallomics, 2012, 4, 379 CrossRef CAS PubMed.
- N. Fujieda, J. Schätti, E. Stuttfeld, K. Ohkubo, T. Maier, S. Fukuzumi and T. R. Ward, Chem. Sci., 2015, 6, 4060–4065 RSC.
- J. Podtetenieff, A. Taglieber, E. Bill, E. J. Reijerse and M. T. Reetz, Angew. Chem., Int. Ed., 2010, 122, 5277–5281 CrossRef.
- W. Ghattas, V. Dubosclard, S. Tachon, M. Beaumet, R. Guillot, M. Reglier, A. J. Simaan and J. Mahy, Angew. Chem., Int. Ed., 2019, 58, 14605–14609 CrossRef CAS PubMed.
- K. R. Miller, J. D. Paretsky, A. H. Follmer, T. Heinisch, K. Mittra, S. Gul, I.-S. Kim, F. D. Fuller, A. Batyuk, K. D. Sutherlin, A. S. Brewster, A. Bhowmick, N. K. Sauter, J. Kern, J. Yano, M. T. Green, T. R. Ward and A. S. Borovik, Inorg. Chem., 2020, 59, 6000–6009 CrossRef CAS PubMed.
- W. J. Song and F. A. Tezcan, Science, 2014, 346, 1525–1528 CrossRef CAS PubMed.
- W. J. Song, J. Yu and F. A. Tezcan, J. Am. Chem. Soc., 2017, 139, 16772–16779 CrossRef CAS PubMed.
- L. Jaroszewski, R. Schwarzenbacher, F. von Delft, D. McMullan, L. S. Brinen, J. M. Canaves, X. Dai, A. M. Deacon, M. DiDonato, M.-A. Elsliger, S. Eshagi, R. Floyd, A. Godzik, C. Grittini, S. K. Grzechnik, E. Hampton, I. Levin, C. Karlak, H. E. Klock, E. Koesema, J. S. Kovarik, A. Kreusch, P. Kuhn, S. A. Lesley, T. M. McPhillips, M. D. Miller, A. Morse, K. Moy, J. Ouyang, R. Page, K. Quijano, R. Reyes, F. Rezezadeh, A. Robb, E. Sims, G. Spraggon, R. C. Stevens, H. van den Bedem, J. Velasquez, J. Vincent, X. Wang, B. West, G. Wolf, Q. Xu, K. O. Hodgson, J. Wooley and I. A. Wilson, Proteins: Struct., Funct., Bioinf., 2004, 56, 611–614 CrossRef CAS PubMed.
- I. Hajnal, K. Faber, H. Schwab, M. Hall and K. Steiner, Adv. Synth. Catal., 2015, 357, 3309–3316 CrossRef CAS.
- N. Fujieda, T. Nakano, Y. Taniguchi, H. Ichihashi, H. Sugimoto, Y. Morimoto, Y. Nishikawa, G. Kurisu and S. Itoh, J. Am. Chem. Soc., 2017, 139, 5149–5155 CrossRef CAS PubMed.
- N. Fujieda, H. Ichihashi, M. Yuasa, Y. Nishikawa, G. Kurisu and S. Itoh, Angew. Chem., Int. Ed., 2020, 59, 7717–7720 CrossRef CAS PubMed.
- D. Coquière, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2007, 46, 9308–9311 CrossRef PubMed.
- J. Wang, E. Benedetti, L. Bethge, S. Vonhoff, S. Klussmann, J.-J. Vasseur, J. Cossy, M. Smietana and S. Arseniyadis, Angew. Chem., Int. Ed., 2013, 52, 11546–11549 CrossRef CAS PubMed.
- P. M. Punt, M. D. Langenberg, O. Altan and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 3555–3561 CrossRef CAS PubMed.
- D. Coquière, J. Bos, J. Beld and G. Roelfes, Angew. Chem., Int. Ed., 2009, 48, 5159–5162 CrossRef PubMed.
- Y. Ye, C. Zheng and R. Fan, Org. Lett., 2009, 11, 3156–3159 CrossRef CAS PubMed.
- W.-C. Gao, F. Hu, J. Tian, X. Li, W.-L. Wei and H.-H. Chang, Chem. Commun., 2016, 52, 13097–13100 RSC.
- J. A. C. Oliveira, G. Kiala, F. Siopa, A. Bernard, G. Gontard, J. Oble, C. A. M. Afonso and G. Poli, Tetrahedron, 2020, 76, 131182 CrossRef CAS.
- Y.-W. Lin, N. Yeung, Y.-G. Gao, K. D. Miner, L. Lei, H. Robinson and Y. Lu, J. Am. Chem. Soc., 2010, 132, 9970–9972 CrossRef CAS PubMed.
- N. El Bakkali-Tahéri, S. Tachon, M. Orio, S. Bertaina, M. Martinho, V. Robert, M. Réglier, T. Tron, P. Dorlet and A. J. Simaan, Arch. Biochem. Biophys., 2017, 623–624, 31–41 CrossRef PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- F. Fusetti, K. H. Schröter, R. A. Steiner, P. I. van Noort, T. Pijning, H. J. Rozeboom, K. H. Kalk, M. R. Egmond and B. W. Dijkstra, Structure, 2002, 10, 259–268 CrossRef CAS PubMed.
- I. M. Kooter, R. A. Steiner, B. W. Dijkstra, P. I. van Noort, M. R. Egmond and M. Huber, Eur. J. Biochem., 2002, 269, 2971–2979 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06809e |
| This journal is © The Royal Society of Chemistry 2023 |