
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransient self-assembly of metal–organic complexes†
Jean-François
Ayme
 *a,
Bernd
Bruchmann
a,
Lydia
Karmazin
b and
Nathalie
Kyritsakas
b
*a,
Bernd
Bruchmann
a,
Lydia
Karmazin
b and
Nathalie
Kyritsakas
b
aBASF SE, Joint Research Network on Advanced Materials and Systems (JONAS), Carl-Bosch Str. 38, 67056 Ludwigshafen, Germany. E-mail: jeff.ayme@gmail.com
bService de Radiocristallographie, Fédération de chimie Le Bel FR2010, Université de Strasbourg, 1 rue Blaise Pascal, 67008 Strasbourg, France
First published on 9th January 2023
Abstract
Implementing transient processes in networks of dynamic molecules holds great promise for developing new functional behaviours. Here we report that trichloroacetic acid can be used to temporarily rearrange networks of dynamic imine-based metal complexes towards new equilibrium states, forcing them to express complexes otherwise unfavourable in their initial equilibrium states. Basic design principles were determined for the creation of such networks. Where a complex distribution of products was obtained in the initial equilibrium state of the system, the transient rearrangement temporarily yielded a simplified output, forcing a more structured distribution of products. Where a single complex was obtained in the initial equilibrium state of the system, the transient rearrangement temporarily modified the properties of this complex. By doing so, the mechanical properties of an helical macrocyclic complex could be temporarily altered by rearranging it into a [2]catenane.
Introduction
To rival the capabilities of biological systems, temporal control over chemical reactivity must be achieved in synthetic systems. Most synthetic self-assembly processes are designed to generate well-ordered structures with high thermodynamic or kinetic stabilities—these structures being at the global minimum of the energy landscape or trapped in local minima.1 By modifying the energy landscape using external stimuli (such as pH, light, or the addition of a chemical species) to create a new minimum, these structures could be forced to rearrange into new ones, yielding stimuli-responsive self-assembly processes.2 While this approach produces highly functional systems,3 it requires the repeated intervention of an operator to administer opposing stimuli at appropriate times to switch the system back and forth between its different functional states.To overcome this limitation and inspired by biological systems,1b,4 chemists have coupled self-assembly and energy-dissipating processes so that self-assembly processes could transiently express different structures via an influx of energy in the form of light, heat or chemicals.1b,5 These so-called “transient self-assemblies” require a constant input of energy to persist in time. If the energy supply is stopped, these structures dismantle, their components being recaptured by the initial state of the system (i.e., its state prior to the energy influx). Interest in chemically-induced transient self-assembly is rapidly growing encouraged by examples of their unique capabilities, including transient gel formation,6 operation of molecular machines,7 temporal control over host–guest systems,8 transient formation of supramolecular assemblies9 and non-equilibrium self-replication.10 In most current reports, however, chemically-induced transient processes are mostly used to modulate the behaviour of single component self-assemblies. Few examples exist in which the consumption of a chemical reactant regulates the organization of networks of dynamic molecules.10c,11 Using classical stimuli-responsive self-assembly, networks of dynamic molecules have enabled the emergence of complex functions inaccessible with simpler systems.3a,d,3g,12,13 Combining the capabilities of such networks with energy-dissipating processes would enable the development of new adaptive behaviours, furthering our understanding of complex systems (including biological ones).
To date, libraries of dynamic metal–organic complexes consisting of amine and 2-formylpyridine components reversibly condensed into imine ligands around labile metal-ions have yielded some of the most advanced artificial networks of dynamic molecules.3a,d,3g,12g,12k,13c–i
Here we demonstrate that a ‘transient acid’5h,7b–g,7i,8c,11f,12j can force libraries of imine-based metal complexes to temporarily rearrange towards a new equilibrium state, generating complexes otherwise unfavourable in the absence of the acid. The study provides basic design principles for creating networks of dynamic complexes with temporarily modifiable functions, showing that the properties of metal–organic complexes can be altered by their transient rearrangement.
Results and discussion
Rational and main design
Due to the differences of basicity between arylamine and alkylamine, we envisaged that trichloroacetic acid—an acid capable of temporarily acidifying a medium5h,7c,7d,i,11f,12j—could be used to induce the transient rearrangement of imine-based metal complexes.Here we will use the notation (a,b) to refer to the imine-based ligand obtained by the condensation of aldehyde a with amine b. The more basic ditopic amine 2 (Fig. 1A and B) should yield the most thermodynamically stable complex with Cu(I) and 2-formylpyridine 3 under neutral conditions, based on previous work.11f,13f,14 Complex [Cu(32,2)]+ should therefore be the unique complex observed when Cu(I) is added to an equimolar mixture of 1, 2 and 3. Upon addition of trichloroacetic acid, the initial acidification of the medium should displace 2 from the complex by protonation, allowing the incorporation of the less basic arylamine 1 (Fig. 2A and B). A new transient equilibrium state should quickly be achieved, expressing [Cu(3,1)2]+ as its most thermodynamically stable complex. The slow decarboxylation of trichloroacetate5h,7c,7d,i,11f,12j should then incrementally return the system of the metal complexes to its initial equilibrium state (i.e., prior to the addition of acid) via a quasi-static process continually maintaining the metal complexes at equilibrium (the rearrangement of the metal complexes being faster than the decarboxylation of trichloroacetate). The decarboxylation of trichloroacetate generates a strong base—trichloromethyl anions—that should restore neutral alkylamine 2, regenerating [Cu(32,2)]+ with chloroform and carbon dioxide as the only side products.
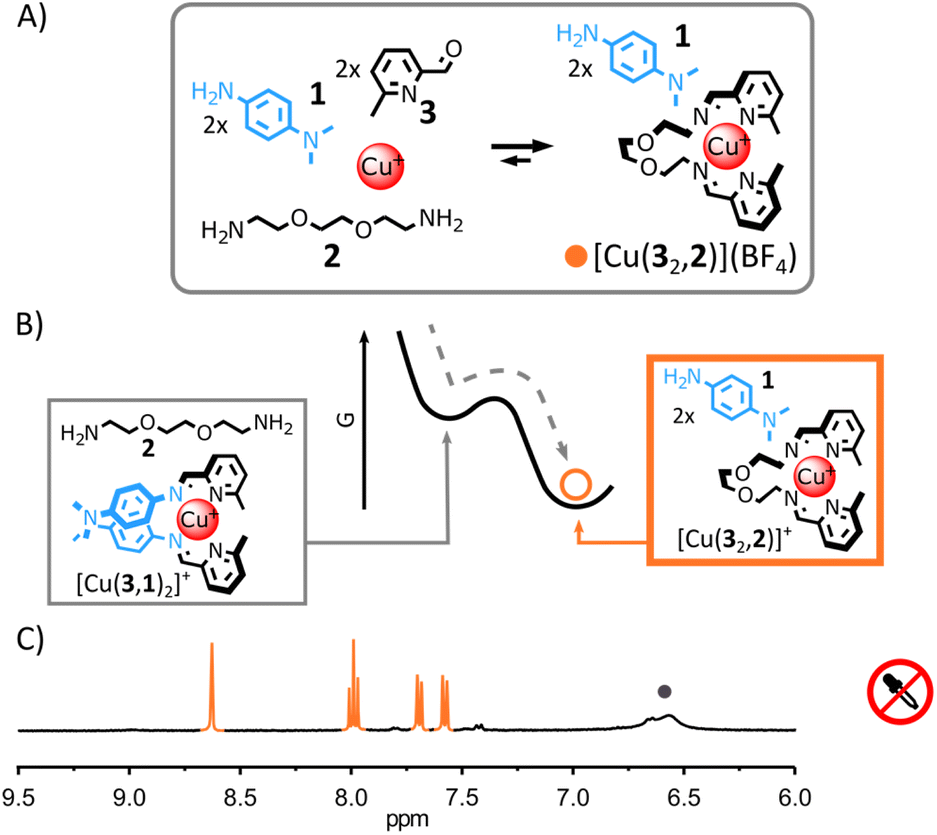 | ||
Fig. 1 (A) Self-assembly of [Cu(32,2)]+ under thermodynamic control. Reaction conditions: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:Cu(BF4) (2:1:2:1), CD3CN, 60 °C, 18 h. (B) Schematic representation of the Gibbs free energy landscape of the reaction; [Cu(3,1)2]+ only forms as a metastable intermediate during the self-assembly process. (C) Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 12 h at 60 °C; the diagnostic signals of [Cu(32,2)]+ are coloured in orange and two of the diagnostic signals of 1 are highlighted by a grey circle. :2:3:Cu(BF4) (2:1:2:1), CD3CN, 60 °C, 18 h. (B) Schematic representation of the Gibbs free energy landscape of the reaction; [Cu(3,1)2]+ only forms as a metastable intermediate during the self-assembly process. (C) Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 12 h at 60 °C; the diagnostic signals of [Cu(32,2)]+ are coloured in orange and two of the diagnostic signals of 1 are highlighted by a grey circle. | ||
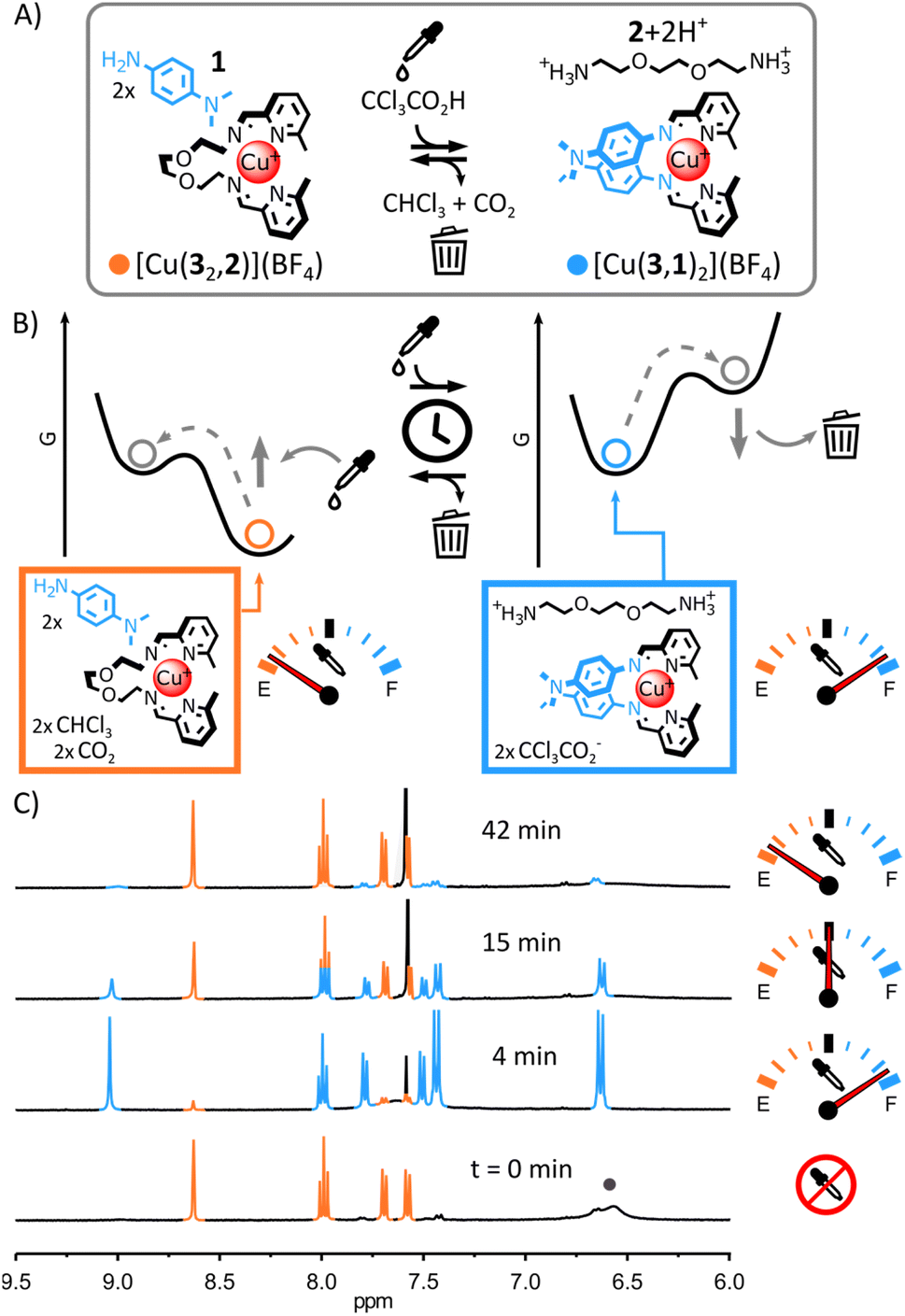 | ||
| Fig. 2 (A) Transient rearrangement of [Cu(32,2)]+ into [Cu(3,1)2]+. Conditions: CCl3COOH (2.5 eq.), CD3CN, r.t. (B) Schematic representation of the Gibbs free energy landscape of the rearrangement; [Cu(3,1)2]+ forms under thermodynamic control as part of a quasi-static process and [Cu(32,2)]+ forms under thermodynamic control in the default equilibrium state of the system. (C) Partial 1H NMR spectra (400 MHz, CD3CN, 297 K) showing the evolution of the crude reaction mixture upon addition of CCl3COOH; the diagnostic signals of [Cu(32,2)]+ and [Cu(3,1)2]+ are coloured in orange and blue, respectively, two of the diagnostic signals of 1 are highlighted by a grey circle. | ||
This rational was tested. Combining alkylamine 2, 2-formylpyridine 3 and Cu(I) in a 2:2:1 ratio in CD3CN led to the quantitative formation of [Cu(32,2)]+ after 12 h at 60 °C (see ESI† for details and full characterization data of new compounds). When arylamine 1 (2 eq.) was added to the solution, a new equilibrium state was quickly achieved.15 [Cu(32,2)]+ remained the only complex observable by 1H NMR spectroscopy (Fig. 1C and S17, ESI†) and electrospray ionization mass spectrometry (ESI-MS, ESI, Fig. S33†), hinting at its greater stability compared to [Cu(3,1)2]+. Addition of trichloroacetic acid (2.5 eq.) to this stable mixture of [Cu(32,2)]+ and 1 immediately caused the rearrangement of the system, yielding [Cu(3,1)2]+ and [2+2H+] as unique products observable by 1H NMR spectroscopy (Fig. 2C and S24–S26, ESI†). The formation of [Cu(3,1)2]+ was also confirmed by ESI-MS (ESI, Fig. S34†). After a few minutes, the signals of [Cu(3,1)2]+ and [2+2H+] started to diminish giving place to those of [Cu(32,2)]+ and 1—the decarboxylation of trichloroacetate slowly returning the system to its initial state (ESI, Fig. S24–S26†). After ca. 40 min, the 1H NMR spectra of the reaction mixture closely matched that prior to the addition of acid, differing only by the emergence of a strong signal at 7.58 ppm attributed to the side product CHCl3 (Fig. 2C).
The robustness of the system was tested by subjecting the same sample to five consecutive rearrangement cycles (ESI, Fig. S26–31†). The evolution of the system was monitored by 1H NMR spectroscopy, invariably showing behaviours identical to those observed during the first rearrangement cycle (ESI, Fig. S26–31†) and showing only minimal deterioration of the system even after five consecutive cycles (ESI, Fig. S32†). Having established that trichloroacetic acid could induce the transient rearrangement of a library of metal complexes—temporarily forming a complex unfavourable in the initial equilibrium state of the system—we sought to probe the tolerance of the system to modifications.
Structural tolerance of the design
The structural tolerance of the system shown in Fig. 2 was studied by systematic modification of the nature of (i) the alkylamine, (ii) the tetrahedral metal ion and (iii) the arylamine.(i) Transient self-assembly of [Cu(3,1)2](BF4) from a mixture of Cu(BF4), 4, 3 and 1. The substitution of ditopic alkylamine 1 for monotopic alkylamine 4 resulted in a distribution of homoleptic and heteroleptic complexes in the initial equilibrium state of the system (Fig. 3); however, the acid-induced rearrangement of the system remained unchanged. Upon addition of arylamine 1 (2 eq.) to [Cu(3,4)2]+ in CD3CN, 1H NMR signals corresponding to [Cu(3,1)2]+ and to the heteroleptic complex [Cu(3,1)(3,4)]+ appeared besides those of [Cu(3,4)2]+ (Fig. 3B and S18 and S42, ESI†). The lack of the self-sorting in the initial equilibrium state of the system15 could be attributed to the reduced entropic driving force for the formation of [Cu(3,4)2]+ compared to the formation of [Cu(32,2)]+ (loss of the chelate effect).14b,16 Adding trichloroacetic acid (2.5 eq.) to the system immediately simplified the reaction mixture, with only [Cu(3,1)2]+ and [4+2H+] being observable by 1H NMR spectroscopy (ESI, Fig. S35 and S36;† the formation of [Cu(3,1)2]+ was also confirmed by ESI-MS, ESI, Fig. S43†). This ‘simplified state’ persisted for a few minutes before slowly returning to the initial distribution of products (after ca. 40 min, ESI, Fig. S37†). This transient ‘simplified state’ could be recalled three times with little signs of fatigue (ESI, Fig. S27–41†). This system exemplifies how a network of dynamic complexes can be forced to temporarily express fewer products—a simplified output— by populating a different more structured equilibrium state via an energy-dissipating process.
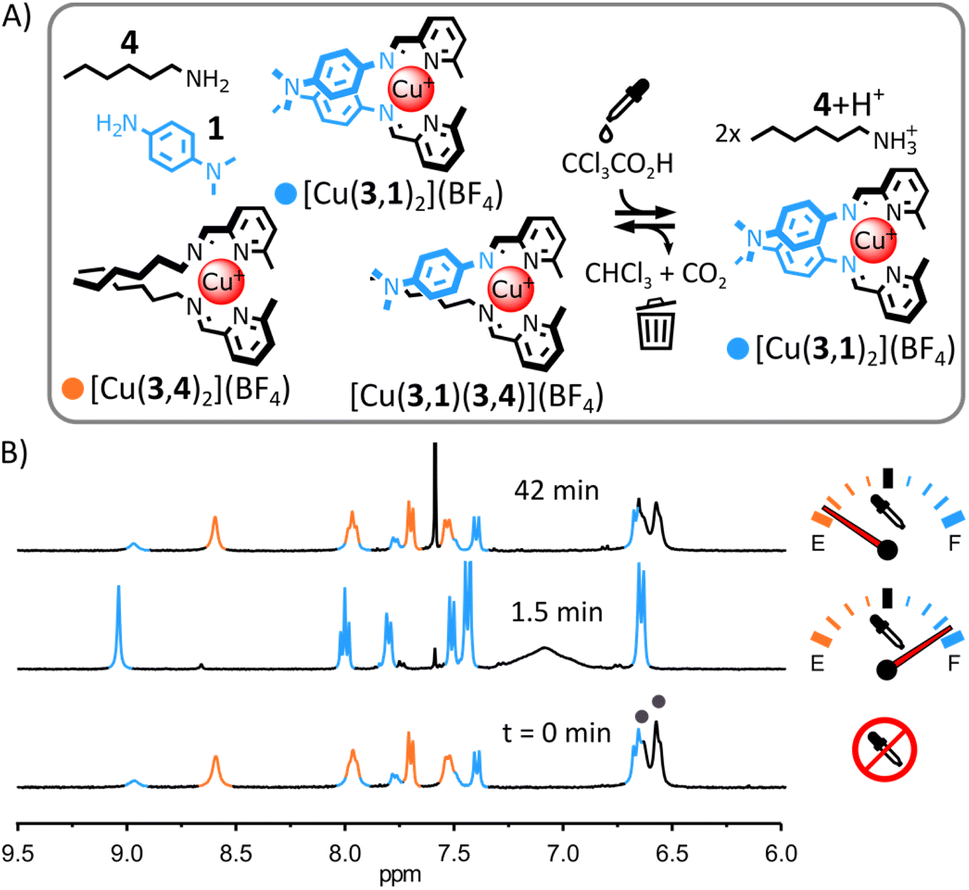 | ||
| Fig. 3 (A) Transient ‘simplification’ of a mixture of complexes into [Cu(3,1)2]+. Conditions: CCl3COOH (2.5 eq.), CD3CN, r. t. (B) Partial 1H NMR spectra (400 MHz, CD3CN, 297 K) showing the evolution of the crude reaction mixture upon addition of CCl3COOH; the diagnostic signals of [Cu(3,4)2]+ and [Cu(3,1)2]+ are coloured in orange and blue, respectively, two of the diagnostic signals of 1 are highlighted by a grey circles, the diagnostic signals of [Cu(3,1)(3,4)]+ overlap with those of [Cu(3,1)2]+ and [Cu(3,4)2]+. | ||
(ii) Transient self-assembly of [Ag(3,1)2](SbF6) from a mixture of Ag(SbF6), 3, 2 and 1. Upon addition of 1 to [Ag(32,2)]+ and equilibration at 60 °C for 18 h, the increased plasticity of the coordination geometry of Ag(I)17 yielded a distribution of complexes in the initial equilibrium state of the system (ESI, Fig. S19–21 and S49†), that immediately simplified upon addition of trichloroacetic acid. Following the addition of acid (2.5 eq.), [Ag(3,1)2]+ and [2+2H+] were the sole products observed in solution by 1H NMR spectroscopy (ESI, Fig. S44 and S45;† the formation of [Ag(3,1)2]+ was also confirmed by ESI-MS, ESI, Fig. S50†). The system gradually returned to its initial distribution of products over ca. 40 min (ESI, Fig. S46†). This process could be repeated with no visible deterioration (ESI, Fig. S46–48†), indicating the compatibility of Ag(I) with our transient rearrangement process. Our transient rearrangement process could also be operated from a non-equilibrated mixture of [Ag(32,2)](SbF6) and 1 (ESI, Fig. S51–S59†).
(iii) Transient self-assembly of [Cu(3,5)2](BF4) from a mixture of Cu(BF4), 5, 3 and 2. The use of the less electron-rich anisidine 5 in place of 4-(dimethylamino)aniline 1 was uninfluential on the initial self-assembly of the system (ESI, Fig. S22 and S64†) or its transient rearrangement (Fig. 4 and S60 and S61, ESI†). From 1H NMR spectra, [Cu(32,2)]+ and 5 were the only species present initially and [Cu(3,5)2]+ and [2+2H+] were the only species present upon addition of trichloroacetic acid (Fig. 4B and S62, ESI;† the formation of [Cu(3,5)2]+ was also confirmed by ESI-MS, ESI, Fig. S65†). However, in subsequent rearrangement cycles (Fig. 4B, top spectrum and ESI, Fig. S63†), 5 led to the rapid deterioration of the system via the trichloromethylation of imine (3,5) (ESI, Fig. S66†).18 This result indicates that in our original system (Fig. 2) the strong electron-donating N,N-dimethyl group of 1 must reduce the reactivity of imine (3,1) towards nucleophilic attack by the trichloromethyl anions generated by the decarboxylation of trichloroacetate.
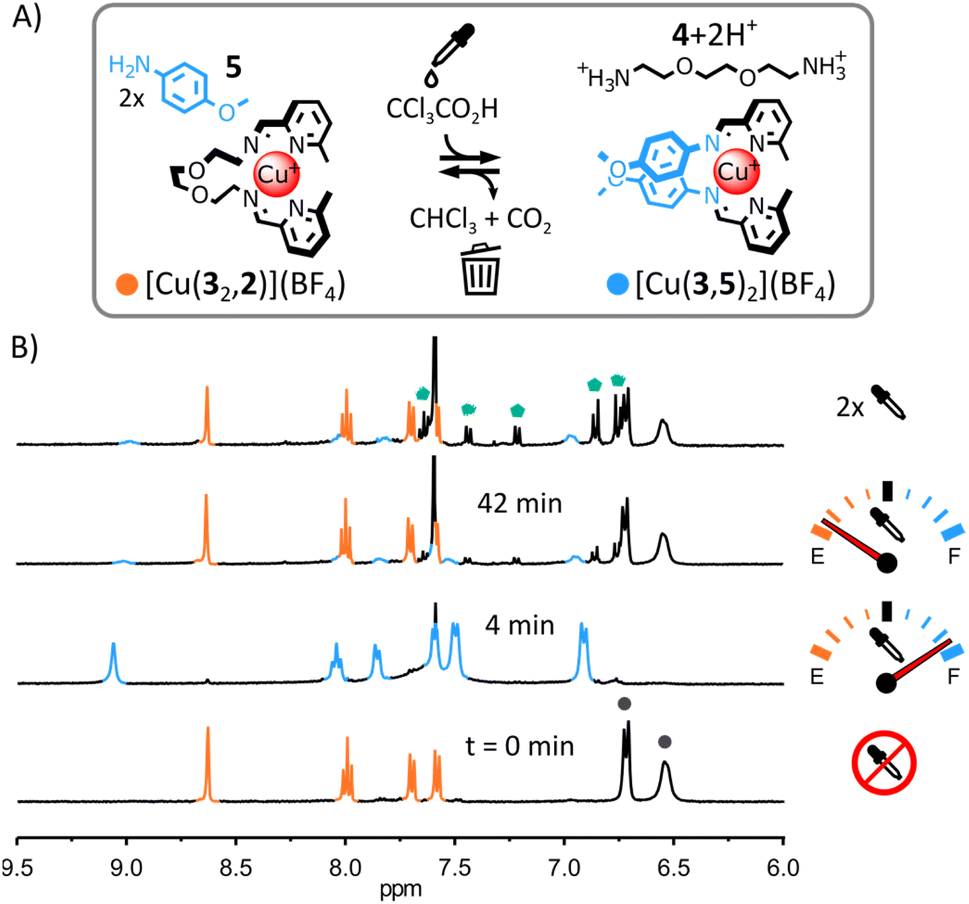 | ||
| Fig. 4 (A) Transient rearrangement of [Cu(32,2)]+ into [Cu(3,5)2]+. Conditions: CCl3COOH (2.5 eq.), CD3CN, r. t. (B) Partial 1H NMR spectra (400 MHz, CD3CN, 297 K) showing the evolution of the crude reaction mixture upon addition of CCl3COOH and after two rearrangement cycles; the diagnostic signals of [Cu(32,2)]+ and [Cu(3,5)2]+ are coloured in orange and blue, respectively, two of the diagnostic signals of 5 are highlighted by a grey circles, the diagnostic signals of trichloromethylated imine (3,5) are highlighted by green pentagons. | ||
Having established basic design principles, we envisaged that the transient rearrangement of metal–organic complexes described in Fig. 2 could be used to temporarily modify the properties of supramolecular architectures, enabling a macrocyclic complex to become a [2]catenane—two mechanically interlocked macrocycles.
Transient rearrangement of a macrocyclic complex into a [2]catenane
Dialdehyde 6 (Fig. 5A) is known to self-assemble with p-anisidine 5 and tetrahedral metal ions to produce dinuclear double-helical complexes (e.g., [Cu2(6,52)2]2+).13h Based on previous work by Nitschke,14b we anticipated that both a macrocyclic complex and a [2]catenane could be generated from this helical motif if appropriate diamines were used in place of 5 (Fig. 5A).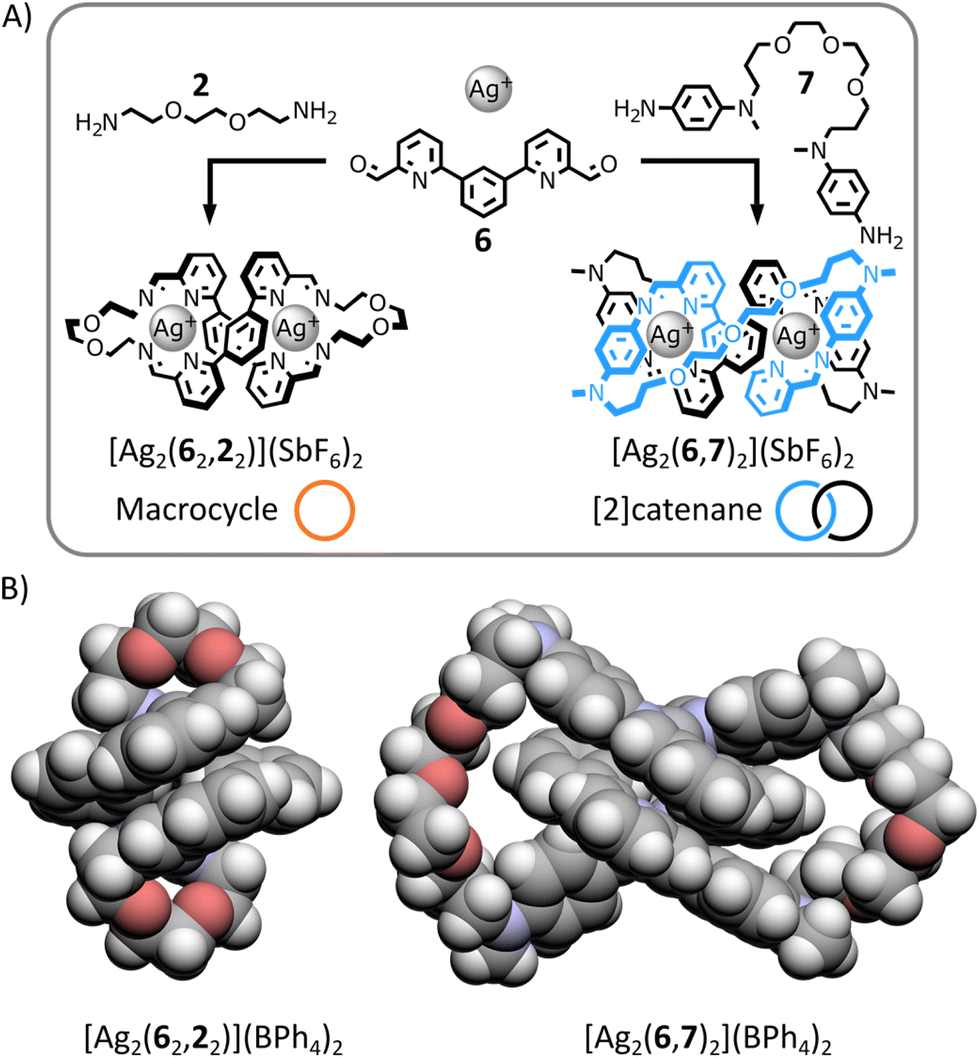 | ||
| Fig. 5 (A) Self-assembly of macrocycle [Ag2(62,22)]2+ and [2]catenane [Ag2(6,7)2]2+ under thermodynamic control. Reaction conditions: 6:2:Ag(SbF6) (1:1:1) or 6:7:Ag(SbF6) (1:1:1), CD3CN:CDCl3 3:1, 60 °C, 12 h. (B) Single crystal X-ray structures of macrocycle [Ag2(62,22)](BPh4)2 and of [2]catenane [Ag2(6,7)2](BPh4)2; solvent molecules and counterions have been omitted for clarity. | ||
In the presence of Ag(I) and aided by the gauche effect,19 dialkylamine 2 should have the right length and flexibility, to preferentially bridge two distinctive dialdehydes 6 while being incorporated in the helical complex, generating the dinuclear helical macrocycle [Ag2(62,22)]2+ (Fig. 5A). In contrast, the longer length and increased rigidity of diarylamine 7 should favour its condensation with the same dialdehyde 6 in the helical complex, yielding the dinuclear helical [2]catenane [Ag2(6,7)2]2+ (Fig. 5A).
To confirm their architectures, both complexes were prepared by heating an equimolar mixture of dialdehyde 6, diamine 2 or 7 and Ag(SbF6) in CD3CN:CDCl3 3:1 at 60 °C for 12 h. In both cases, a single product was obtained with NMR spectra and ESI-MS analyses consistent with the structures shown in Fig. 5 (ESI, Section 2.4.2 and 2.4.3†). X-ray-quality crystals of both architectures were grown by liquid–liquid diffusion (ESI, Section 5.1 and 5.2†). The solid-state structures of both complexes unambiguously confirmed their topologies (Fig. 5B and ESI, Section 5.1 and 5.2†).
The organic ligand of macrocycle [Ag2(62,22)]2+ forms a 42-atom-loop twisted in a double-helical shape by two Ag(I) ions each coordinated to two iminopyridine domains of the ligand in a flattened tetrahedral geometry. The architecture of [2]catenane [Ag2(6,7)2]2+ consists of two interlocked 36-membered rings entwined by an helical core created by two Ag(I) ions coordinating each to one of the iminopyridine domain of each macrocycles in an irregular four-coordinate geometry.
These two structures exemplify the importance of controlling crossing point connectivity while synthesizing interlocked architectures.20 Both complexes have the same helical core, providing the same initial three crossing points. The difference of chemical topology between macrocycle [Ag2(62,22)]2+ and [2]catenane [Ag2(6,7)2]2+ arises from the retention of a different number of these initial crossing points when connecting the end-groups of the helical core with diamine 2 or 7. By connecting the closest end-groups of the helical core, diamine 2 retains none of the initial crossing points, yielding a topologically trivial macrocycle. By connecting the end-groups of the same strand of the helical core, diamine 7 retains one of the initial crossing points, yielding a [2]catenane.
Having established the nature of both complexes, we attempted the transient rearrangement of [Ag2(62,22)]2+ and 7 into [Ag2(6,7)2]2+ and [2+2H+].
As shown in Fig. 6, when diarylamine 7 (1 eq.) was added to macrocycle [Ag2(62,22)]2+ in CD3CN:CDCl3 3:1, 1H NMR spectroscopy provided no evidence of reaction between these two species. This absence of reactivity combined with the ability of [Ag2(62,22)]2+ to selectively self-assemble from a mixture of Ag(SbF6), 7, 6 and 2 (ESI, Fig. S23†) hint at the greater stability of [Ag2(62,22)]2+ over [2]catenane [Ag2(6,7)2]2+ in the initial equilibrium state of the system.21 Addition of trichloroacetic acid (10 eq.) triggered the transient rearrangement of the system: macrocycle [Ag2(62,22)]2+ was gradually converted into [2]catenane [Ag2(6,7)2]2+ and dialkylamine 2 was trapped in its protonated form (Fig. 6B, S67 and S68, ESI†). The concentration of [Ag2(6,7)2]2+ peaked at ca. 240 min before starting to decrease—slowly at first, but accelerating with depletion of the acid (ESI, Fig. S71 and S73†). After ca. 870 min, macrocycle [Ag2(62,22)]2+ was again the only complex visible by 1H NMR spectroscopy.
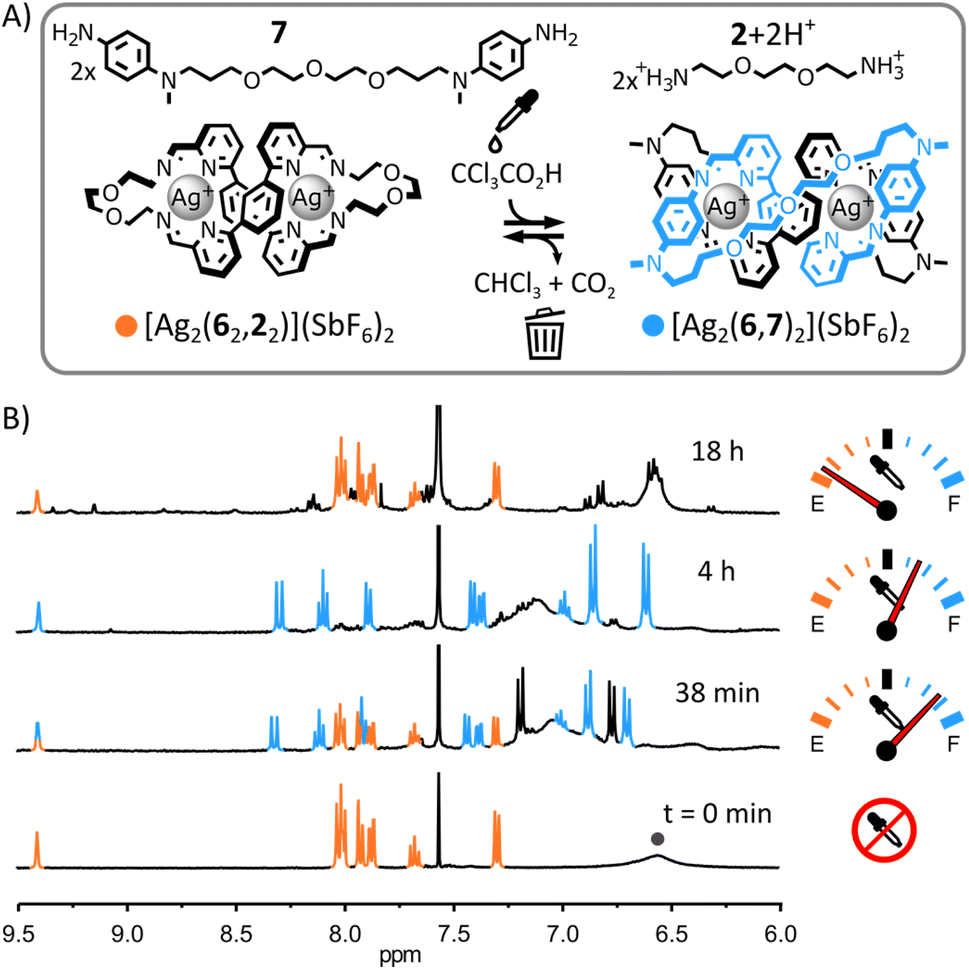 | ||
| Fig. 6 (A) Transient rearrangement of macrocycle [Ag2(62,22)]2+ into [2]catenane [Ag2(6,7)2]2+. Conditions: CCl3COOH (10 eq.), CD3CN:CDCl3 3:1, r. t. (B) Partial 1H NMR spectra (400 MHz, CD3CN:CDCl3 3:1, 297 K) showing the evolution of the crude reaction mixture upon addition of CCl3COOH; the diagnostic signals of [Ag2(62,22)]2+ and [Ag2(6,7)2]2+ are coloured in orange and blue, respectively, two of the diagnostic signals of 7 are highlighted by a grey circle. | ||
Compared to the systems of mononuclear complexes in Fig. 2–4, the rate of rearrangement of this system was considerably lower ([Ag2(6,7)2]2+ peaked after ca. 240 min whereas [Ag(3,1)2]+ peaked after ca. 2 min), reflecting the increased complexity of this rearrangement process. Metal–organic complexes involving dialdehyde and diamine components are known to self-assemble via the initial formation of metastable ill-defined oligomers and polymers slowly rearranging into (kinetically or thermodynamically) more stable structures.19b,22 A similar scenario is likely when the present system is rearranging towards [2]catenane [Ag2(6,7)2]2+ following the addition of trichloroacetic acid. Indications of the formation of ill-defined oligomeric and polymeric species were found by monitoring the rearrangement of the system by 1H NMR spectroscopy. Despite [Ag2(62,22)]2+ and [Ag2(6,7)2]2+ being the only species visible by 1H NMR spectroscopy throughout the rearrangement process (Fig. 6B and S71, ESI†), their combined populations remained under ca. 80% of the initial population of [Ag2(62,22)]2+ (i.e., prior to the addition of trichloroacetic acid), the missing material being presumably trapped in ill-defined oligomers and polymers undetectable by 1H NMR spectroscopy. After the concentration of [Ag2(6,7)2]2+ peaked, the baseline of the 1H NMR spectra of the reaction became broad and uneven in the aromatic region (Fig. 6B and S71, ESI†), indicating an increased formation of oligomeric and polymeric species. This observation is consistent with the progressive deprotonation of [2+2H+] during the decomposition process of trichloroacetate into CHCl3 and CO2, making both diamines 2 and 7 available to form oligomeric and polymeric species as the system returns to its initial equilibrium state.
Compared to the systems of mononuclear complexes in Fig. 2–4, a large excess of trichloroacetic acid was required to maximize the formation of [2]catenane [Ag2(6,7)2]2+ (10 eq. of trichloroacetic acid were needed in this case vs. 2.5 eq. in the case of the simpler systems). The amount of [Ag2(6,7)2]2+ generated during the transient process and its lifetime were both found to be roughly proportional to the amount of acid added (ESI, Fig. S69–S71 and S73†). When the amount of acid added was increased from 7.5 to 10 eq., the peak population of [Ag2(6,7)2]2+ increased from ca. 35 to 70% of the initial population of [Ag2(62,22)]2+ and the amount of time required for the system to return to its initial equilibrium state increased from ca. 450 to 810 min. This high requirement in acid could suggest that the rearrangement of the system is slow compared to the rate of decarboxylation of trichloroacetate. An excess of acid would therefore be needed to reach a steady state in the system, maximizing the transient formation of catenane [Ag2(6,7)2]2+ under thermodynamic control.
Higher amounts of acid (up to 12 eq., ESI, Fig. S72 and S73†) did not increase the peak population of [Ag2(6,7)2]2+ further, possibly indicating that some of the oligomeric and polymeric intermediates formed during the rearrangement of the system may be too kinetically stable to rearrange in the time scale of the transient process (in the conditions used).
While all the initial macrocycle [Ag2(62,22)]2+ could be recovered at the end of the rearrangement using 7.5 eq. of trichloroacetic acid (ESI, Fig. S69 and S73†), the amount of macrocycle recovered at the end of the rearrangement cycle diminished when more acid was used (90 and 70% of the initial macrocycle were recovered when 8.5 or 12 eq. of trichloroacetic acid were used, respectively, ESI, Fig. S70–S73†). Performing a second rearrangement cycle led to significant deterioration of the system (ESI, Fig. S75†).
Conclusions
Trichloroacetic acid has been used to control in time the composition of imine-based metal complexes via energy-dissipation. By exploiting the base-promoted decarboxylation of trichloroacetic acid, it was possible to cycle between the preferential incorporation of an arylamine or an alkylamine component in a metal complex, enabling the transient expression of complexes otherwise unfavourable in the absence of trichloroacetic acid.The electronic properties of the arylamine component appeared to be crucial to the robustness of the transient rearrangement process, whereas the nature of the alkylamine component and tetrahedral metal ions had little influence. Transient processes can temporarily simplify the output of a self-assembly process by populating a different, more structured, equilibrium state. The properties of metal complexes can be controlled in time via the transient modification of their compositions. We showed that the mechanical properties of a macrocyclic complex could be temporarily altered by rearranging it into a topologically more complex [2]catenane.
By providing a generalizable method to transiently access different equilibrium states in systems of metal–organic complexes (or architectures), the information gleaned from our investigation will facilitate the development of new adaptive behaviours in these systems; behaviours exploiting both the capabilities of the default equilibrium (or kinetically trapped) state of the system and the capabilities of transient equilibrium states accessible via energy-dissipation.
Data availability
The synthetic procedures, characterization, and spectral data supporting this article have been uploaded as part of the ESI.† CCDC 2162663 and CCDC 2162664 contain the supplementary crystallographic data for this paper.Author contributions
J. F. A. conceived and conducted the experiments. L. K. and N. K. solved and refined the X-ray structures. J. F. A. and B. B. supervised the project. The manuscript was written by J. F. A. All authors reviewed the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge BASF SE (via its JONAS program) for the funding of this research. JFA gratefully acknowledges Prof. Jean-Marie Lehn and Dr Dean Thomas for useful discussions, Hava Aksoy and Cyril Antheaume for performing the ESI-HRMS measurements, Dr Artem Osypenko for repeating the synthesis of [Cu(32,2)]+ and [Cu(3,4)2]+, Corinne Bailly for her assistance regarding crystallography, Prof. Leroy Cronin and the University of Glasgow for permitting the generous usage of their facilities.Notes and references
- For selected resources on self-assembly: (a) G. M. Whitesides and B. A. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed; (b) K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- For selected general resources on stimuli-responsive self-assembly: (a) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, John Wiley & Sons, Ltd, 1995 CrossRef; (b) Intelligent Stimuli-Responsive Materials, ed. Q. Li, John Wiley & Sons, Ltd, 2013 Search PubMed; (c) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, 3rd edn, 2022 Search PubMed; (d) H. Ivan and L. Jean-Marie, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef PubMed; (e) O. Sijbren, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef PubMed.
- For recent review articles covering applications of stimuli-responsive self-assembly: (a) J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (c) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC; (d) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (e) A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC; (f) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed; (g) J.-F. Ayme and J.-M. Lehn, in Advances in Inorganic Chemistry, ed. R. van Eldik and R. Puchta, Academic Press, 2018, vol. 71, pp. 3–78 Search PubMed; (h) M. Grzelczak, L. M. Liz-Marzán and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC; (i) A. Goswami, S. Saha, P. K. Biswas and M. Schmittel, Chem. Rev., 2020, 120, 125–199 CrossRef CAS PubMed; (j) S. M. Jansze and K. Severin, J. Am. Chem. Soc., 2019, 141, 815–819 CrossRef CAS PubMed; (k) S. M. Jansze, G. Cecot and K. Severin, Chem. Sci., 2018, 9, 4253–4257 RSC.
- For selected general resources on energy-consumption in biological processes: (a) B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, W.W. Norton & Company, 2015 Search PubMed; (b) S. Mann, Angew. Chem., Int. Ed., 2008, 47, 5306–5320 CrossRef CAS PubMed.
- For selected general resources on man-made dissipative self-assembly: (a) M. Fialkowski, K. J. M. Bishop, R. Klajn, S. K. Smoukov, C. J. Campbell and B. A. Grzybowski, J. Phys. Chem. B, 2006, 110, 2482–2496 CrossRef CAS PubMed; (b) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC; (c) S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC; (d) M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC; (e) G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed; (f) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed; (g) N. Singh, G. J. M. Formon, S. de Piccoli and T. M. Hermans, Adv. Mater., 2020, 32, 1906834 CrossRef CAS PubMed; (h) C. Biagini and S. Di Stefano, Angew. Chem., Int. Ed., 2020, 59, 8344–8354 CrossRef CAS PubMed; (i) B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552–578 CrossRef; (j) S. Amano, S. Borsley, D. A. Leigh and Z. Sun, Nat. Nanotechnol., 2021, 16, 1057–1067 CrossRef CAS PubMed; (k) L. S. Kariyawasam, M. M. Hossain and C. S. Hartley, Angew. Chem., Int. Ed., 2021, 60, 12648–12658 CrossRef CAS PubMed; (l) H. Lee, J. Tessarolo, D. Langbehn, A. Baksi, R. Herges and G. H. Clever, J. Am. Chem. Soc., 2022, 144, 3099–3105 CrossRef CAS PubMed; (m) R. G. DiNardi, A. O. Douglas, R. Tian, J. R. Price, M. Tajik, W. A. Donald and J. E. Beves, Angew. Chem. Int. Ed., 2022, 61, e202205701 CrossRef CAS PubMed; (n) V. W. L. Gunawardana, T. J. Finnegan, C. E. Ward, C. E. Moore and J. D. Badjić, Angew. Chem., Int. Ed., 2022, 61, e202207418 Search PubMed.
- (a) J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed; (b) J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef CAS PubMed; (c) N. Singh, B. Lainer, G. J. M. Formon, S. de Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS PubMed.
- (a) M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235–240 CrossRef CAS PubMed; (b) J. A. Berrocal, C. Biagini, L. Mandolini and S. Di Stefano, Angew. Chem., Int. Ed., 2016, 55, 6997–7001 CrossRef CAS PubMed; (c) E.-C. Sundus, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef PubMed; (d) C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 CrossRef CAS PubMed; (e) D. Mariottini, D. Del Giudice, G. Ercolani, S. Di Stefano and F. Ricci, Chem. Sci., 2021, 12, 11735–11739 RSC; (f) A. Goswami, S. Saha, E. Elramadi, A. Ghosh and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 14926–14935 CrossRef CAS PubMed; (g) A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 5319–5323 CrossRef CAS PubMed; (h) S. Amano, S. D. P. Fielden and D. A. Leigh, Nature, 2021, 594, 529–534 CrossRef CAS PubMed; (i) D. Thomas, D. J. Tetlow, Y. Ren, S. Kassem, U. Karaca and D. A. Leigh, Nat. Nanotechnol., 2022, 17, 701–707 CrossRef CAS PubMed; (j) S. Borsley, E. Kreidt, D. A. Leigh and B. M. W. Roberts, Nature, 2022, 604, 80–85 CrossRef CAS PubMed.
- (a) C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504–509 CrossRef CAS PubMed; (b) L. S. Kariyawasam and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 11949–11955 CrossRef CAS PubMed; (c) F. Rispoli, E. Spatola, D. Del Giudice, R. Cacciapaglia, A. Casnati, L. Baldini and S. Di Stefano, J. Org. Chem., 2022, 87, 3623–3629 CrossRef CAS PubMed.
- (a) S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed; (b) B. G. P. van Ravensteijn, W. E. Hendriksen, R. Eelkema, J. H. van Esch and W. K. Kegel, J. Am. Chem. Soc., 2017, 139, 9763–9766 CrossRef CAS PubMed; (c) R. K. Grötsch, C. Wanzke, M. Speckbacher, A. Angı, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2019, 141, 9872–9878 CrossRef PubMed.
- (a) S. M. Morrow, I. Colomer and S. P. Fletcher, Nat. Commun., 2019, 10, 1011 CrossRef PubMed; (b) A. H. J. Engwerda, J. Southworth, M. A. Lebedeva, R. J. H. Scanes, P. Kukura and S. P. Fletcher, Angew. Chem., Int. Ed., 2020, 59, 20361–20366 CrossRef CAS PubMed; (c) S. Yang, G. Schaeffer, E. Mattia, O. Markovitch, K. Liu, A. S. Hussain, J. Ottelé, A. Sood and S. Otto, Angew. Chem., Int. Ed., 2021, 60, 11344–11349 CrossRef CAS PubMed; (d) A. H. J. Engwerda, J. Southworth, M. A. Lebedeva, R. J. H. Scanes, P. Kukura and S. P. Fletcher, Angew. Chem., Int. Ed., 2020, 59, 20361–20366 CrossRef CAS PubMed.
- (a) M. Tena-Solsona, C. Wanzke, B. Riess, A. R. Bausch and J. Boekhoven, Nat. Commun., 2018, 9, 2044 CrossRef PubMed; (b) M. M. Hossain, J. L. Atkinson and C. S. Hartley, Angew. Chem., Int. Ed., 2020, 59, 13807–13813 CrossRef CAS PubMed; (c) C. M. E. Kriebisch, A. M. Bergmann and J. Boekhoven, J. Am. Chem. Soc., 2021, 143, 7719–7725 CrossRef CAS PubMed; (d) M. G. Howlett, R. J. H. Scanes and S. P. Fletcher, JACS Au, 2021, 1, 1355–1361 CrossRef CAS PubMed; (e) J. Deng and A. Walther, Nat. Commun., 2020, 11, 3658 CrossRef CAS PubMed; (f) D. Del Giudice, M. Valentini, G. Melchiorre, E. Spatola and S. Di Stefano, Chem. – Eur. J., 2022, e202200685 CAS; (g) P. S. Schwarz, M. Tena-Solsona, K. Dai and J. Boekhoven, Chem. Commun., 2022, 58, 1284–1297 RSC.
- For selected review article covering complex functions in systems of dynamic molecules: (a) F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed; (b) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC; (c) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC; (d) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed; (e) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC; (f) N. Roy, B. Bruchmann and J.-M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC; (g) D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC; (h) F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed; (i) P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed; (j) D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS; (k) I. Jahović, Y.-Q. Zou, S. Adorinni, J. R. Nitschke and S. Marchesan, Matter, 2021, 4, 2123–2140 CrossRef; (l) N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- For selected research article on complex functions in systems of dynamic molecules: (a) H. Ivan and L. Jean-Marie, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef PubMed; (b) O. Sijbren, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef PubMed; (c) A. D. W. Kennedy, N. de Haas, H. Iranmanesh, E. T. Luis, C. Shen, P. Wang, J. R. Price, W. A. Donald, J. Andréasson, F. Huang and J. E. Beves, Chem.–Eur. J., 2019, 25, 5708–5718 CrossRef CAS PubMed; (d) M. He and J.-M. Lehn, J. Am. Chem. Soc., 2019, 141, 18560–18569 CrossRef CAS PubMed; (e) A. Osypenko, S. Dhers and J.-M. Lehn, J. Am. Chem. Soc., 2019, 141, 12724–12737 CrossRef CAS PubMed; (f) J.-F. Ayme and J.-M. Lehn, Chem. Sci., 2020, 11, 1114–1121 RSC; (g) J.-F. Ayme, S. Dhers and J.-M. Lehn, Angew. Chem., Int. Ed., 2020, 59, 12484–12492 CrossRef CAS PubMed; (h) J.-F. Ayme, J.-M. Lehn, C. Bailly and L. Karmazin, J. Am. Chem. Soc., 2020, 142, 5819–5824 CrossRef CAS PubMed; (i) B.-N. T. Nguyen, J. D. Thoburn, A. B. Grommet, D. J. Howe, T. K. Ronson, H. P. Ryan, J. L. Bolliger and J. R. Nitschke, J. Am. Chem. Soc., 2021, 143, 12175–12180 CrossRef CAS PubMed; (j) O. Borodin, Y. Shchukin, C. C. Robertson, S. Richter and M. von Delius, J. Am. Chem. Soc., 2021, 143, 16448–16457 CrossRef CAS PubMed; (k) R. Gu and J.-M. Lehn, J. Am. Chem. Soc., 2021, 143, 14136–14146 CrossRef CAS PubMed; (l) N. F. König, D. Mutruc and S. Hecht, J. Am. Chem. Soc., 2021, 143, 9162–9168 CrossRef PubMed.
- (a) D. Schultz and J. R. Nitschke, J. Am. Chem. Soc., 2006, 128, 9887–9892 CrossRef CAS PubMed; (b) M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem. - Eur. J., 2006, 12, 4069–4076 CrossRef CAS PubMed.
- The distribution of products observed by 1H NMR spectroscopy minutes after the addition of the arylamine matched that obtained when the same system of components was left to equilibrate for 18 h at 60 °C from an identical or a different starting point (see ESI† Section 3), indicating that the system has reached equilibrium—or is close to equilibrium—in this short time span.
- (a) G. Schwarzenbach, Helv. Chim. Acta, 1952, 35, 2344–2359 CrossRef CAS; (b) D. Schultz and J. R. Nitschke, Angew. Chem., Int. Ed., 2006, 45, 2453–2456 CrossRef CAS PubMed.
- (a) E. C. Constable, S. M. Elder, M. J. Hannon, A. Martin, P. R. Raithby and D. A. Tocher, J. Chem. Soc., Dalton Trans., 1996, 2423–2433 RSC; (b) E. C. Constable, A. J. Edwards, G. R. Haire, M. J. Hannon and P. R. Raithby, Polyhedron, 1998, 17, 243–253 CrossRef CAS; (c) J. R. Price, Y. Lan and S. Brooker, Dalton Trans., 2007, 1807–1820 RSC.
- (a) E. J. Corey, J. O. Link and Y. Shao, Tetrahedron Lett., 1992, 33, 3435–3438 CrossRef; (b) Y. Li, T. Zheng, W. Wang, W. Xu, Y. Ma, S. Zhang, H. Wang and Z. Sun, Adv. Synth. Catal., 2012, 354, 308–312 CrossRef CAS.
- (a) M. A. Murcko and R. A. DiPaola, J. Am. Chem. Soc., 1992, 114, 10010–10018 CrossRef CAS; (b) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed.
- (a) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700–1712 RSC; (b) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed.
- The distribution of products observed by 1H NMR spectroscopy minutes after the addition of diarylamine 7 to [Ag2(62,22)]2+ matched that obtained during the selective self-assembly of [Ag2(62,22)]2+ from a mixture of Ag(SbF6), 7, 6 and 2 at 60 °C for 18 h (see ESI† Section 3.2), indicating that the system has reached equilibrium—or is close to equilibrium—in this short time span.
- J. P. Carpenter, C. T. McTernan, J. L. Greenfield, R. Lavendomme, T. K. Ronson and J. R. Nitschke, Chem, 2021, 7, 1534–1543 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2162663 and 2162664. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06374c |
| This journal is © The Royal Society of Chemistry 2023 |