
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-component reductive conjugate addition/aldol tandem reaction enabled by nickel/photoredox dual catalysis†
Hongping
Zhao
a and
Weiming
Yuan
 *abc
*abc
aKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Bioinorganic Chemistry and Materia Medica, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology (HUST), 1037 Luoyu Road, Wuhan 430074, PR China. E-mail: yuanwm@hust.edu.cn
bShenzhen Huazhong University of Science and Technology Research Institute, Shenzhen 518000, PR China
cGuangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, PR China
First published on 10th January 2023
Abstract
A three-component reductive cross-coupling of aryl halides, aldehydes, and alkenes by nickel/photoredox dual catalysis is disclosed. The key to success for this tandem transformation is to identify α-silylamine as a unique organic reductant, which releases silylium ions instead of protons to prevent unwanted protonation processes, and meanwhile serves as Lewis acid to activate aldehydes in situ. This dual catalytic protocol completes a traditional conjugate addition/aldol sequence that eliminates the requirement of organometallic reagents and metal-based reductants, thus providing a mild synthetic route to highly valuable β-hydroxyl carbonyl compounds with contiguous 1,2-stereocenters.
Introduction
Transition-metal-catalysed intermolecular three-component dicarbofunctionalization (DCF) of alkenes represents a powerful strategy to synthesize complex structures via simultaneously forging two vicinal carbon–carbon bonds in a single step operation.1 Compared to the traditional redox-neutral processes that generally involve the performance of organometallic reagents as nucleophilic coupling partners,2 nickel-catalysed reductive olefin DCF by harnessing two electrophiles across C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds has attracted increasing attention as such an alternative strategy bypasses the use of sensitive organometallics and features mild conditions and broad functional group compatibility.3 However, most of the reductive olefin DCFs rely on the coupling of two organohalide electrophiles, and thus, endeavouring to explore the reliability of other distinct and abundant electrophiles in olefin reductive DCF to broaden the structural and functional diversity of target molecules is highly desirable.
C bonds has attracted increasing attention as such an alternative strategy bypasses the use of sensitive organometallics and features mild conditions and broad functional group compatibility.3 However, most of the reductive olefin DCFs rely on the coupling of two organohalide electrophiles, and thus, endeavouring to explore the reliability of other distinct and abundant electrophiles in olefin reductive DCF to broaden the structural and functional diversity of target molecules is highly desirable.
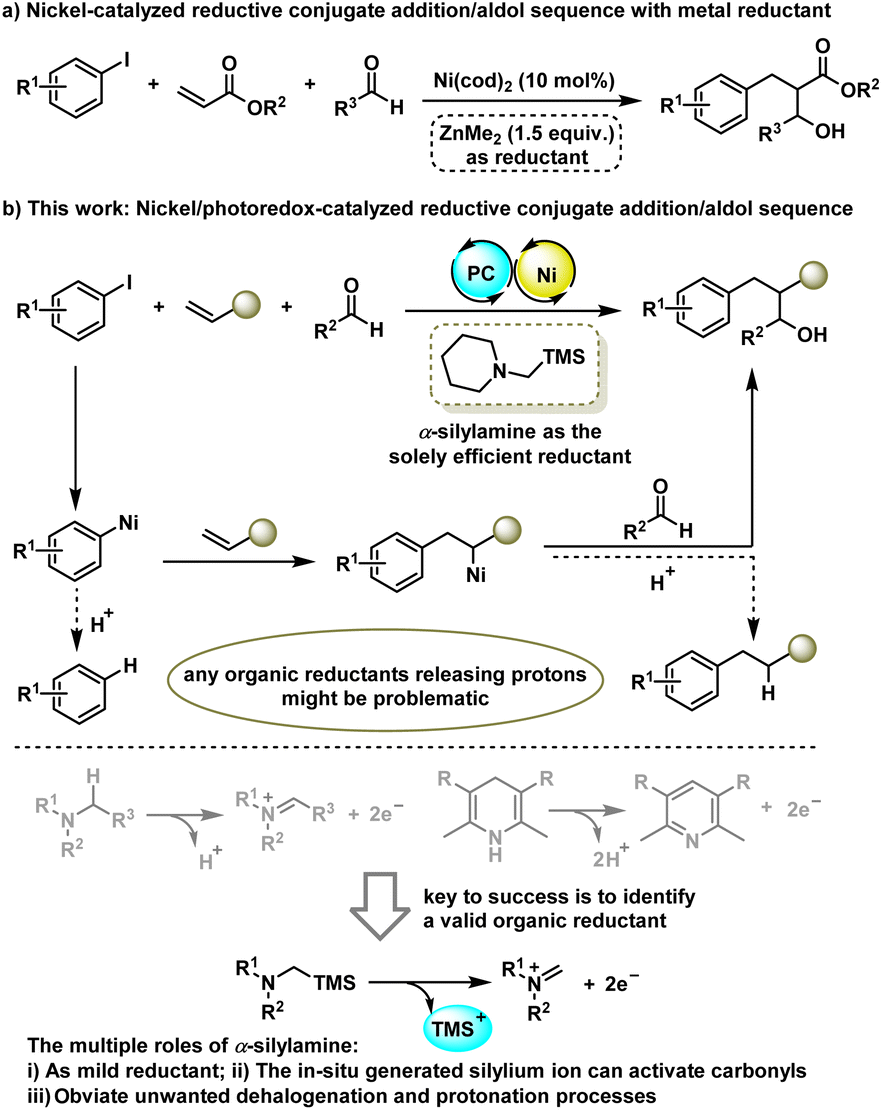
Aldehydes, in particular, are abundant and versatile building blocks in organic synthesis, while their application in catalytic reductive olefin DCF has been much less explored. An early example is from the Montgomery group who reported Ni(cod)2-catalyzed three-component reaction of acrylates, aryl iodides and aldehydes under net reductive conditions with a stoichiometric amount of ZnMe2 as the reductant (Scheme 1a).4 Later on, Gall and co-workers reported a similar transformation on using CoBr2 as the catalyst and Zn dust as the reductant.5 These reductive processes provide a complementary approach to a classical conjugate addition/aldol tandem sequence6 that requires sensitive arylmetallic reagents. However, the requirement of stoichiometric amounts of metal-based reductants inevitably produces a lot of metal salt waste and results in a certain degree of difficulty for synthetic scalability and practicability.
 | ||
| Scheme 1 Nickel-catalysed alkene dicarbofunctionalization with aldehydes as electrophiles. | ||
Taking advantage of the ability of the photoredox-assisted single-electron transfer (SET) process, researchers have recently applied Ni/photoredox dual catalysis7 to the area of cross-electrophile coupling reactions,8 enabling the coupling of organohalides in the absence of a stoichiometric metal reductant. However, most reported photo-redox-assisted reductive cross-couplings are limited to the two-component version9 with few exceptions have been extended to the intermolecular three-component reaction of aryl and alkyl halides across alkenes.10 Giving our longstanding interest in metallaphotoredox catalysis,9e,11 we reported here a Ni/photoredox-catalysed three-component conjunctive cross-electrophile coupling of aryl halides with aldehydes and alkenes (Scheme 1b). We envisioned that the key to success of this tandem transformation is to identify a suitable organic reductant as any organic reductants release protons might be problematic because they can protonate each tentative C–Ni bond species, thus producing unwanted outcomes such as a dehalogenation product and reductive Heck product. As such, the most commonly used organic reductants in literature including tertiary amines and Hantzsch esters (HEs) would be inferior to the reaction because they can release protons when using as reductant. To address this issue, we envisioned that α-silylamines might be a sort of valid organic reductant to furnish this tandem process as they release silylium ions instead of protons during the single electron oxidation stage. Moreover, the in situ generated silylium ion could act as a Lewis acid to activate aldehydes to promote the subsequent aldol reaction. α-Silylamines have been widely used as α-amino radical precursors in photocatalytic radical transformations,12 while their potential as mild organic reductants in reductive cross-couplings has not yet been explored. Here, we disclose the unique functions of α-silylamine in reductive conjugate addition/aldol tandem transformation.
Results and discussion
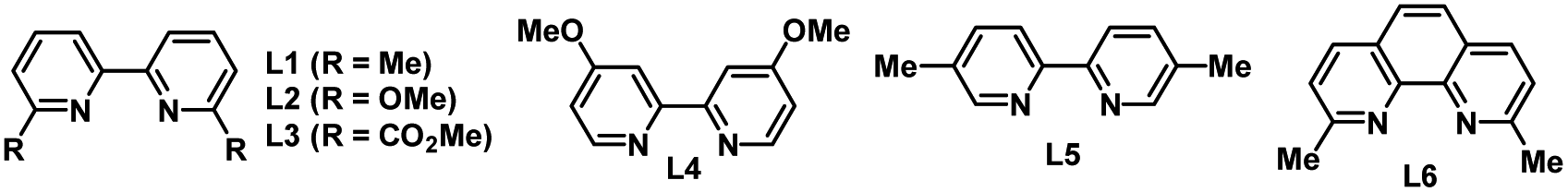
Our initial studies began with an investigation of various organic reductants. The three-component reductive conjugate addition/aldol tandem reaction with 4-iodo-1,1′-biphenyl (1a), methyl acrylate (2a), and benzaldehyde (3a) was performed in the presence of 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6, 10 mol% of NiBr2·DME, and 10 mol% of 6,6′-di(Me)bpy in DMAc (0.1 M) at room temperature under 1.5 W blue LED irradiation for 24 h. Various organic reductants including Hantzsch ester (HE), Et3N, iPr2NEt, and N,N,N′,N′-tetramethylethylenediamine (TMEDA) were examined with or without an extra base. Regrettably, all attempts failed to deliver any desired product. Instead, a huge amount of dehalogenation product 7 and/or reductive Heck product 5 was detected, suggesting that these proton-releasing organic reductants are not suitable for the transformation. Based on our previous work on nickel/photoredox-catalysed alkene dicarbofunctionalization,11a we tried to use an α-silyl substituted tertiary amine such as 1-((trimethylsilyl)methyl)piperidine as the organic reductant due to its easy preparation, low oxidation potential, and above all, because there are no protons released in the process. To our delight, the desired β-hydroxyl ester product 4 could be detected in 23% GC yield after the reaction with TBAF to remove the silyl group. Under this condition, only a trace amount of the dehalogenation product was detected. However, the reductive Heck product 5 and reductive carbonyl Heck product 6 were detected in 21% and 12% yield, respectively. After the initial result was obtained, we further examined other reaction parameters (Table 1). The screening of photocatalysts showed that Ir(ppy)3 is the most efficient photocatalyst (entries 1–4). Ortho-substituted bipyridines are prevailing ligands and 6,6′-di(OMe)bpy is proven to be the best (entries 5–9). NiBr2 and Ni(cod)2 could also promote the reaction and give a slightly lower yield (entries 10 and 11). The solvent has a dramatic influence on the reactivity (entries 12–14): DMF could only afford a trace amount of the product. Other less polar solvents such as MeCN or THF were all inferior to the reaction, which either results in a large decrease in yield or completely retards the reaction. The mole ratio of each component also affects the reaction and the highest yield of 78% could be obtained when using benzaldehyde as the limiting reagent (entry 15). Under the optimized conditions, reductive carbonyl Heck product 6 and dehalogenation product 7 were successfully inhibited. Not surprisingly, the yield of reductive Heck product 5 was increased due to the addition of an excess amount of 1a and 2a. The control experiments revealed that the photocatalyst, nickel and visible light are indispensable (entries 16–18). Interestingly, omitting a ligand could still afford 44% yield of the desired product (entry 19). Replacing aryl iodide with aryl bromide or aryl triflate gives a largely decreased yield for the desired outcome (entries 20 and 21).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entryb | Photocatalyst | [Ni] | Ligand | Solvent | GC yielda | ||
| 4 | 5 | 6 | |||||
a Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), NiBr2·DME (10 mol%), and 6,6′-di(Me)bpy (10 mol%). Yields are determined by GC with n-tridecane as the internal standard.
b Reaction conditions: photocatalyst (1 mol%), Ni catalyst (10 mol%), and ligand (10 mol%).
c The molar ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a:3a = 2:2:1.
d No light irradiation.
e 4-Bromo-1,1′-biphenyl instead of 1a.
f [1,1′-biphenyl]-4-yl trifluoromethanesulfonate instead of 1a. :2a:3a = 2:2:1.
d No light irradiation.
e 4-Bromo-1,1′-biphenyl instead of 1a.
f [1,1′-biphenyl]-4-yl trifluoromethanesulfonate instead of 1a.
|
|||||||
| 1 | Ir(ppy)3 | NiBr2·DME | L1 | DMAc | 34 | 23 | 21 |
| 2 | Ir(ppy)2bpyPF6 | NiBr2·DME | L1 | DMAc | 21 | 11 | 43 |
| 3 | Ru(bpy)3(PF6)2 | NiBr2·DME | L1 | DMAc | 32 | 16 | 5 |
| 4 | 4-CzIPN | NiBr2·DME | L1 | DMAc | 15 | 0 | 62 |
| 5 | Ir(ppy)3 | NiBr2·DME | L2 | DMAc | 62 | 20 | 5 |
| 6 | Ir(ppy)3 | NiBr2·DME | L3 | DMAc | 44 | 22 | 1 |
| 7 | Ir(ppy)3 | NiBr2·DME | L4 | DMAc | 19 | 9 | 3 |
| 8 | Ir(ppy)3 | NiBr2·DME | L5 | DMAc | 21 | 8 | 4 |
| 9 | Ir(ppy)3 | NiBr2·DME | L6 | DMAc | 24 | 16 | 51 |
| 10 | Ir(ppy)3 | NiBr2 | L2 | DMAc | 45 | 13 | 2 |
| 11 | Ir(ppy)3 | Ni(cod)2 | L2 | DMAc | 55 | 4 | 10 |
| 12 | Ir(ppy)3 | NiBr2·DME | L2 | DMF | 3 | 0 | 4 |
| 13 | Ir(ppy)3 | NiBr2·DME | L2 | MeCN | 24 | 43 | 2 |
| 14 | Ir(ppy)3 | NiBr2·DME | L2 | THF | 0 | 3 | 0 |
| 15 | Ir(ppy) 3 | NiBr 2 ·DME | L2 | DMAc | 78 | 43 | 0 |
| 16 | — | NiBr2·DME | L2 | DMAc | 0 | 0 | 0 |
| 17d | Ir(ppy)3 | NiBr2·DME | L2 | DMAc | 0 | 0 | 0 |
| 18 | Ir(ppy)3 | — | L2 | DMAc | 0 | 0 | 0 |
| 19 | Ir(ppy)3 | NiBr2·DME | — | DMAc | 44 | 13 | 0 |
| 20e | Ir(ppy)3 | NiBr2·DME | L2 | DMAc | 22 | 4 | 50 |
| 21f | Ir(ppy)3 | NiBr2·DME | L2 | DMAc | 15 | 6 | 8 |
|
|||||||
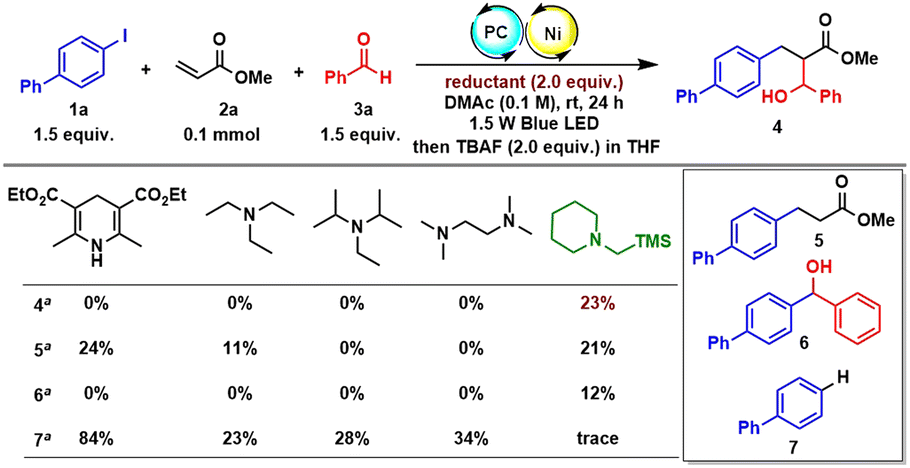
With the optimized reaction conditions in hand, we started to evaluate the scope generality of the three-component reaction (Table 2). First, a series of aryl iodides were examined. Aryl iodides with electron-donating and electron-withdrawing groups including phenyl (4), OMe (9), ester (11), ketone (12), SMe (13), and Cl (14) were well tolerated, delivering the corresponding products in moderate to good yields. A sterically hindered substrate such as ortho-methyl substituted iodobenzene (15) also reacted with a good yield. Heteroaryl iodide was tolerated as well (16). The structure of the trans-configuration of product 4 was determined by X-ray diffraction analysis. Regarding the scope of alkenes, except for different substituted acrylates (4, 17, and 18), various N-substituted acrylamides were investigated. Acyclic acrylamides (19, 20, 25, and 26) and cyclic acrylamides (21–24) all reacted smoothly and afforded a series of structurally abundant β-hydroxyl amide products. Particularly, α-methyl substituted methyl acrylate was also tolerated, enabling a product with an all-carbon quaternary stereocenter (27). Finally, the scope generality of aldehydes was evaluated. Again, not only electron-donating groups, but electron-withdrawing groups were also compatible (28–34). Sterically ortho-substituted benzaldehydes have no impact on the reactivity (35, 36). Of note, more reactive naphthaldehyde (37), furfural (38) and 2-thenaldehyde (39) successfully underwent conjugative coupling. Pleasingly, aliphatic aldehydes irrespective of containing an α-primary, or secondary, or tertiary alkyl substituent were smoothly incorporated (40–43). Both structures of trans- and syn-configuration of product 42 were determined by X-ray diffraction analysis. Regrettably, the reaction could only give a poor stereocontrol with diastereoisomeric ratios ranging from 1:1 to 2:1. The low level of diastereoselectivity might have contributed to the poor selectivity of the formed Z- vs. E-enolate in the conjugate addition step or the absence of a Zimmerman–Traxler-type transition state in the second aldol reaction step.13 We also added some metal-based Lewis acids such as ZnCl2 and MgCl2 to improve diastereoselection, but no further improvement was obtained. Anyway, both diastereoisomers can be separated by column chromatography on silica gel. The reaction could be scaled-up to 1 mmol (60%) and 3 mmol (48%) with an acceptable yield obtained.
In order to gain insight into the reaction mechanism, some control experiments were conducted. The reaction was completely inhibited by adding radical scavenger TEMPO [(2,2,6,6-tetramethylpiperidin-1-yl)oxyl] (Scheme 2(1)). Besides, all reactions failed to deliver any desired product when various different fluoride sources added (Scheme 2(2)). We speculated that the in situ generated silylium ion species (see the mechanistic discussion) was trapped by a fluoride source to form the useless TMSF. These interesting results indicate the significant importance of silyl groups for this reaction. We deduced that the TMS group plays at least two roles: (1) may serve as a Lewis acid to activate a carbonyl group for aldol reaction; (2) re-generate the active nickel catalyst via transmetallation of the O–Ni bond to the stronger O–Si bond. A control experiment without aldehyde was performed where no three-component conjunctive addition product was detected, and only a large amount of reductive Heck product 5 was afforded (Scheme 2(3)), suggesting that an α-amino radical might not be generated11a under this condition. The intermediate 5 may possibly react with aldehyde in the presence of a base under thermal conditions to get the final product. However, the reaction cannot produce 4, thus excluding this possibility (Scheme 2(4)). To better understand the behavior of the photoexcited step, Stern–Volmer fluorescence quenching experiments14 were performed (see the ESI† for details). The results revealed that the oxidative quenching of the excited state of the photocatalyst by the nickel(II) catalyst is much more efficient over other components (Scheme 2(5)), suggesting that a single-electron-transfer (SET) event occurred between the nickel(II) catalyst and *Ir(ppy)3 that initiates the photocatalytic process. To exclude the possibility of the nickel complex affecting the emission intensity of the photocatalyst, the absorption spectra of each component were recorded and the results showed that all components have no absorption in this region (Scheme 2(6)).
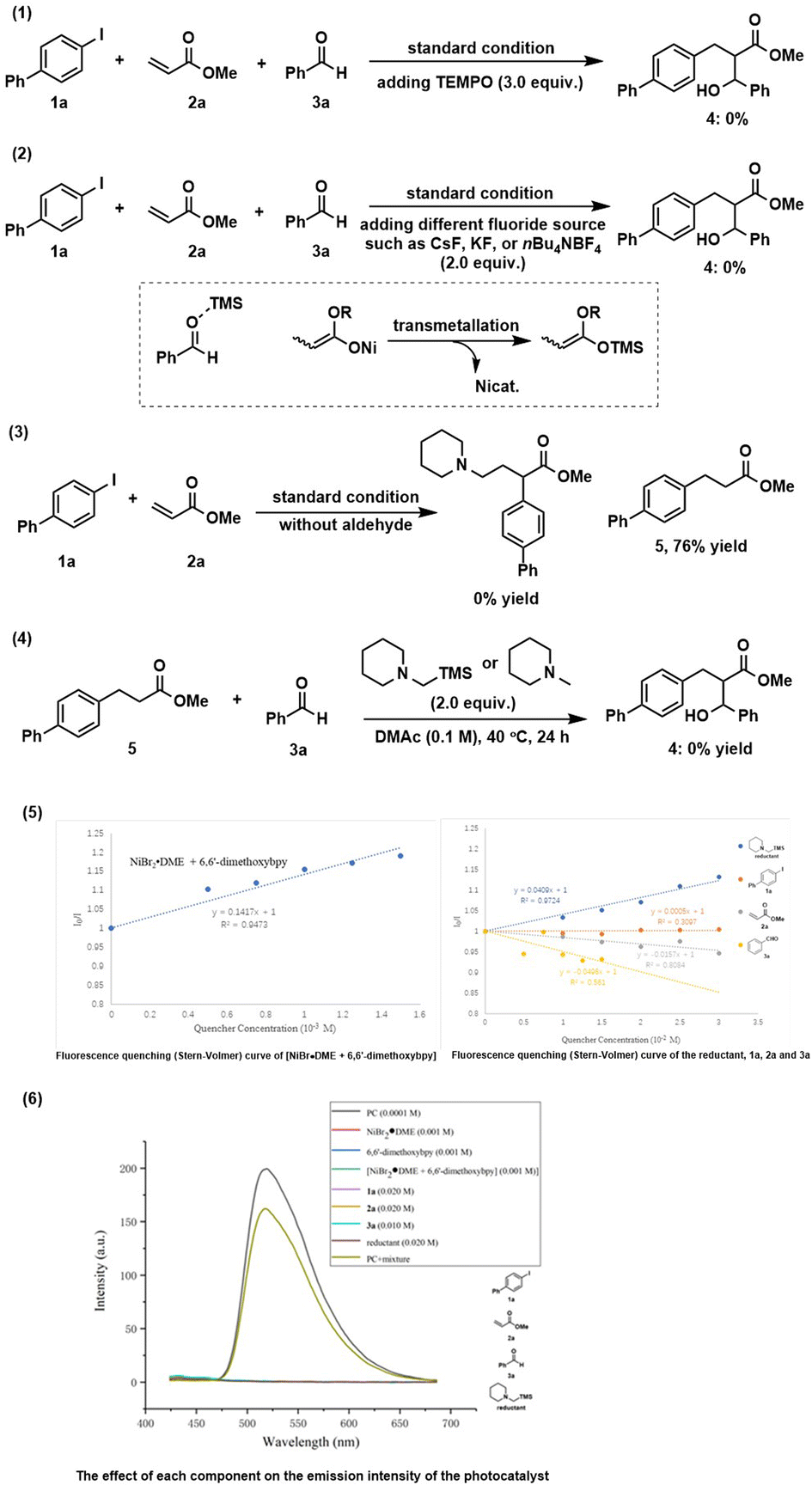 | ||
| Scheme 2 Control experiments and spectroscopic investigations. | ||
Based on the above mechanistic studies and our previous reports,11 a plausible mechanism was proposed for this reductive conjugate addition/aldol tandem sequence (Scheme 3). First, successive SET process between the photoexcited *IrIII (EIV/*III1/2 = −1.73 V vs. SCE)15 and NiII [E1/2(NiII/Ni0) = −1.2 V vs. SCE]16 generates Ni0 and IrIV. IrIV possessing a strong reductive potential (EIV/III1/2 = +0.77 V vs. SCE)15 can oxidize α-silylamine (Eox1/2 = +0.71 V vs. SCE)17,18 to get iminium ion by successive SET. Meanwhile, IrIV is reduced back to the ground state of IrIII. On the other hand, Ni0 species undergoes oxidative addition with ArX to afford ArNiIIX, followed by migration insertion into CC bond to get α-carbonyl-NiII intermediate (IV). An equilibration between nickel O-enolate (V) and C-enolate (IV) tautomer is possible,19 which could be further transformed into a more stable silyl ketene acetal (VI) via a Ni/Si transmetallation. Final aldol reaction promoted by Lewis acidic silylium ion delivers products and regenerate NiII catalyst.
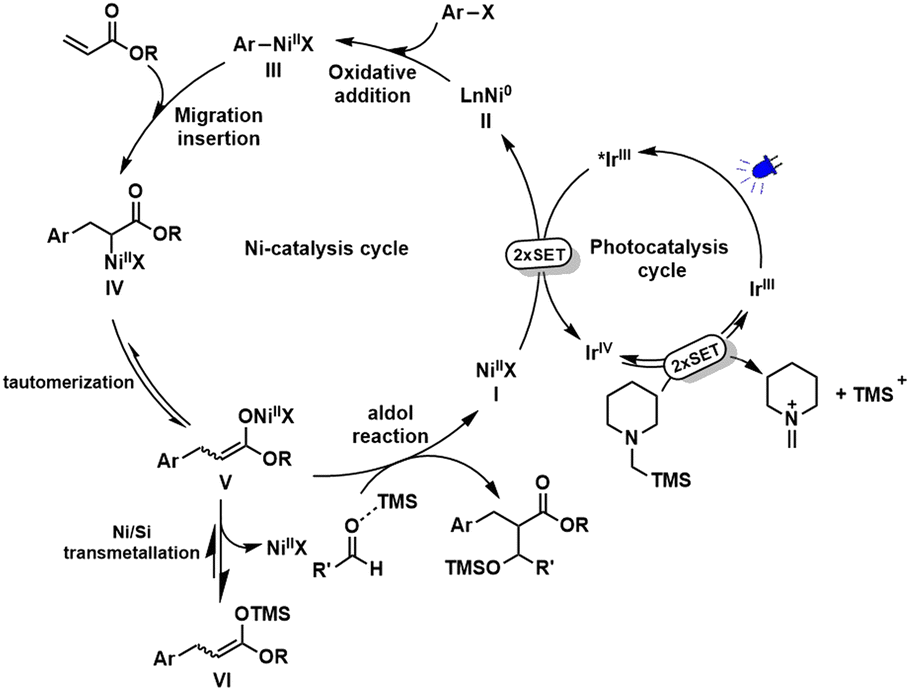 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we report here a nickel/photoredox dual catalytic platform for enabling a reductive conjugate addition/aldol reaction with aryl halides, electron-deficient alkenes, and aldehydes. α-Silylamine is identified as an efficient organic reductant due to its unique properties. Our strategy provides a complementary route to previous procedures that bypassing the use of organometallic reagents or metal reductants for this three-component transformation. Further studies on designing asymmetric reaction and gaining a deep insight into the mechanism are currently ongoing in the laboratory.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound 4 and 42 have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2217184, 2217195, and 2217198.Author contributions
H. Z. performed all the experiments and prepared the ESI.† W. Y. supervised the research and wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22201087), Guangdong Basic and Applied Basic Research Foundation (2019A1515110788, 2022A1515012507), Guangdong Provincial Key Laboratory of Catalysis (2020B121201002), and the Innovation and Talent Recruitment Base of New Energy Chemistry and Device (B21003) of Huazhong University of Science and Technology (HUST) for the financial support.Notes and references
- For selected reviews on transition-metal-catalysed 1,2-dicarbofunctionalization of alkenes, see: (a) R. K. Dhungana, S. Kc, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314 CrossRef CAS PubMed; (b) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287 RSC; (c) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542 CrossRef CAS PubMed; (d) S. Zhu, X. Zhao, H. Li and L. Chu, Chem. Soc. Rev., 2021, 50, 10836 RSC.
- For selected examples of nickel-catalysed 1,2-dicarbofunctionalization of alkenes involving organometallics, see: (a) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657 CrossRef CAS PubMed; (b) B. Shrestha, P. Basnet, R. K. Dhungana, S. Kc, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653 CrossRef CAS PubMed; (c) M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 14245 CrossRef CAS PubMed; (d) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC; (e) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (f) J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270 CrossRef CAS PubMed; (g) P. Gao, L.-A. Chen and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 10653 CrossRef CAS PubMed; (h) H. Wang, C.-F. Liu, R. T. Martin, O. Gutierrez and M. J. Koh, Nat. Chem., 2022, 14, 188 CrossRef CAS PubMed; (i) O. Apolinar, T. Kang, T. M. Alturaifi, P. G. Bedekar, C. Z. Rubel, J. Derosa, B. B. Sanchez, Q. N. Wong, E. J. Sturgell, J. S. Chen, S. R. Wisniewski, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2022, 144, 19337 CrossRef CAS PubMed; (j) C.-F. Liu, Z.-C. Wang, X. Luo, J. Lu, C. H. M. Ko, S.-L. Shi and M. J. Koh, Nat. Catal., 2022, 5, 934 CrossRef CAS.
- For recent progress of Ni-catalysed reductive conjunctive cross-coupling of alkenes, see: (a) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed; (b) W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812 CrossRef CAS PubMed; (c) H.-Y. Tu, F. Wang, L. Huo, Y. Li, S. Zhu, X. Zhao, H. Li, F.-L. Qing and L. Chu, J. Am. Chem. Soc., 2020, 142, 9604 CrossRef CAS PubMed; (d) D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198 CrossRef CAS PubMed; (e) X.-X. Wang, X. Lu, S.-J. He and Y. Fu, Chem. Sci., 2020, 11, 7950 RSC; (f) T. Yang, Y. Jiang, Y. Luo, J. J. H. Lim, Y. Lan and M. J. Koh, J. Am. Chem. Soc., 2020, 142, 21410 CrossRef CAS PubMed; (g) X. Xi, Y. Chen and W. Yuan, Org. Lett., 2022, 24, 3938 CrossRef CAS PubMed; (h) J.-X. Zhang and W. Shu, Org. Lett., 2022, 24, 3844 CrossRef CAS PubMed; (i) T. Yang, X. Chen, W. Rao and M. J. Koh, Chem, 2020, 6, 738 CrossRef CAS.
- K. Subburaj and J. Montgomery, J. Am. Chem. Soc., 2003, 125, 11210 CrossRef CAS PubMed.
- J. Paul, M. Presset, F. Cantagrel, E. L. Gall and E. Léonel, Chem.–Eur. J., 2017, 23, 402 CrossRef CAS PubMed.
- For selected reviews, see: (a) H.-C. Guo and J.-A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354 CrossRef CAS PubMed; (b) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039 RSC.
- For selected reviews on TM/photoredox catalysis, see: (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (b) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429 CrossRef CAS PubMed; (c) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (b) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC.
- For recent reports on cross-electrophile reactions under Ni-photoredox catalysis, see: (a) Z. Duan, W. Li and A. Lei, Org. Lett., 2016, 18, 4012 CrossRef CAS PubMed; (b) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (c) J. Yi, S. O. Badir, L. M. Kammer, M. Ribagorda and G. A. Molander, Org. Lett., 2019, 21, 3346 CrossRef CAS PubMed; (d) M. Parasram, B. J. Shields, O. Ahmad, T. Knauber and A. G. Doyle, ACS Catal., 2020, 10, 5821 CrossRef CAS PubMed; (e) X. Xi, Y. Luo, W. Li, M. Xu, H. Zhao, Y. Chen, S. Zheng, X. Qi and W. Yuan, Angew. Chem., Int. Ed., 2022, e202114731 CAS; (f) H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172 CrossRef CAS PubMed.
- (a) S.-Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370 CrossRef CAS PubMed; (b) P. Qian, H. Guan, Y.-E. Wang, Q. Lu, F. Zhang, D. Xiong, P. J. Walsh and J. Mao, Nat. Commun., 2021, 12, 6613 CrossRef CAS PubMed; (c) A. Luridiana, D. Mazzarella, L. Capaldo, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, M. Nuño, W. J. Buma and T. Noël, ACS Catal., 2022, 12, 11216 CrossRef CAS PubMed.
- (a) S. Zheng, Z. Chen, Y. Hu, X. Xi, Z. Liao, W. Li and W. Yuan, Angew. Chem., Int. Ed., 2020, 59, 17910 CrossRef CAS PubMed; (b) S. Zheng, W. Wang and W. Yuan, J. Am. Chem. Soc., 2022, 144, 17776 CrossRef CAS PubMed.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946 CrossRef CAS PubMed.
- H. E. Zimmerman and M. D. Traxler, J. Am. Chem. Soc., 1957, 79, 1920 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, CA, 1978 Search PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- C. Remeur, C. B. Kelly, N. R. Patel and G. A. Molander, ACS Catal., 2017, 7, 6065 CrossRef CAS PubMed.
- Given the fact that α-amino radical (Eox1/2 ≈ −1.0 V vs. SCE) is more readily oxidized than the starting material α-silylamine (Eox1/2 = +0.4–0.8 V vs. SCE), it can undergo a successive single-electron oxidation process to get iminium ion species and release silylium ions. For data of the redox-potentials, see: (a) J. Yoshida, T. Maekawa, T. Murata, S. Matsunaga and S. Isoe, J. Am. Chem. Soc., 1990, 112, 1962 CrossRef CAS; (b) D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189 CrossRef CAS.
- (a) J. Cámpora, C. M. Maya, P. Palma, E. Carmona, E. Gutiérrez, C. Ruiz, C. Graiff and A. Tiripicchio, Chem.–Eur. J., 2005, 11, 6889 CrossRef PubMed; (b) E. R. Burkhardt, R. G. Bergman and C. H. Heathcock, Organometallics, 1990, 9, 30 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copy of NMR spectra. CCDC 2217184, 2217195 and 2217198. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06303d |
| This journal is © The Royal Society of Chemistry 2023 |