
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective rhodium-catalyzed addition of arylboronic acids to N-heteroaryl ketones: synthesis of α-hydroxy acids†
Jinbin
Zhu
 *a,
Zhenyue
Li
a,
Jiaqi
Li
a,
Duanshuai
Tian
b,
Ronghua
Xu
b,
Zhiyong
Tan
a,
Zhengwang
Chen
*a and
Wenjun
Tang
*b
*a,
Zhenyue
Li
a,
Jiaqi
Li
a,
Duanshuai
Tian
b,
Ronghua
Xu
b,
Zhiyong
Tan
a,
Zhengwang
Chen
*a and
Wenjun
Tang
*b
aKey Laboratory of Organo-Pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou 341000, China. E-mail: zhujinbin@gnnu.edu.cn; chenzwang2021@163.com
bState Key Laboratory of Bio-Organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling Ling Rd, Shanghai 200032, China. E-mail: tangwenjun@sioc.ac.cn
First published on 12th January 2023
Abstract
The enantioselective addition of arylboronic acids to N-heteroaryl ketones provides a convenient access to chiral α-heteroaryl tertiary alcohols, yet addition reactions of this type have been challenging due to catalyst deactivation. In this report, an efficient rhodium-catalyzed addition of arylboronic acids to N-heteroaryl ketones is established, affording a variety of valuable α-heteroaryl alcohols with excellent functional group compatibility. The employment of the WingPhos ligand containing two anthryl groups is crucial for this transformation. In particular, a range of chiral benzoxazolyl-substituted tertiary alcohols were formed with excellent ee values and yields by employing a Rh loading as low as 0.3 mol%, which can serve as a practical protocol to furnish a series of chiral α-hydroxy acids after hydrolysis.
Introduction
Chiral tertiary alcohol units exist widely in the structure of natural products, biologically active compounds, and functional molecules.1 In particular, chiral α-heteroaryl tertiary alcohols constitute important and privileged structural motifs in organic and medicinal chemistry. Among these molecules, Camptothecin is a leukemia and tumor inhibitor, and its modifications have been used to treat cancer.2 A triazole substituted tertiary alcohol has been identified as a 5-lipoxygenase inhibitor,3 while spiroborate is employed in asymmetric transformations as a ligand4 (Fig. 1a) and benzoxazolyl-substituted tertiary alcohol5 is explored as an antitumor compound. Prevalence and significance of chiral tertiary alcohols has attracted a great deal of attention and several common strategies have been developed for their asymmetric synthesis (Fig. 1b). For example, the past few decades have witnessed the development of kinetic resolution for optically active tertiary alcohols.6 However, the method needs synthesis of racemates in advance, but can only provide chiral products with 50% yield at most. Asymmetric addition of nucleophiles to ketones is one of the most straightforward approaches, and has gained enormous attention from the chemical community.7 Organometallic reagents such as organozinc, organoaluminum and Grignard agents have been employed in the asymmetric transformation but the protocol suffered from some drawbacks including poor functional group tolerance, moisture- and air-sensitive nature of reagents and operational tediousness.8 Therefore, asymmetric addition of organometallics to ketones is limited in synthesis of multifunctional group compounds or complex bioactive molecules. In contrast, organoboron reagents as nontoxic, stable and operationally simple species will be better nucleophiles for the addition reaction, in which many diverse functional groups can be tolerated (Fig. 1c).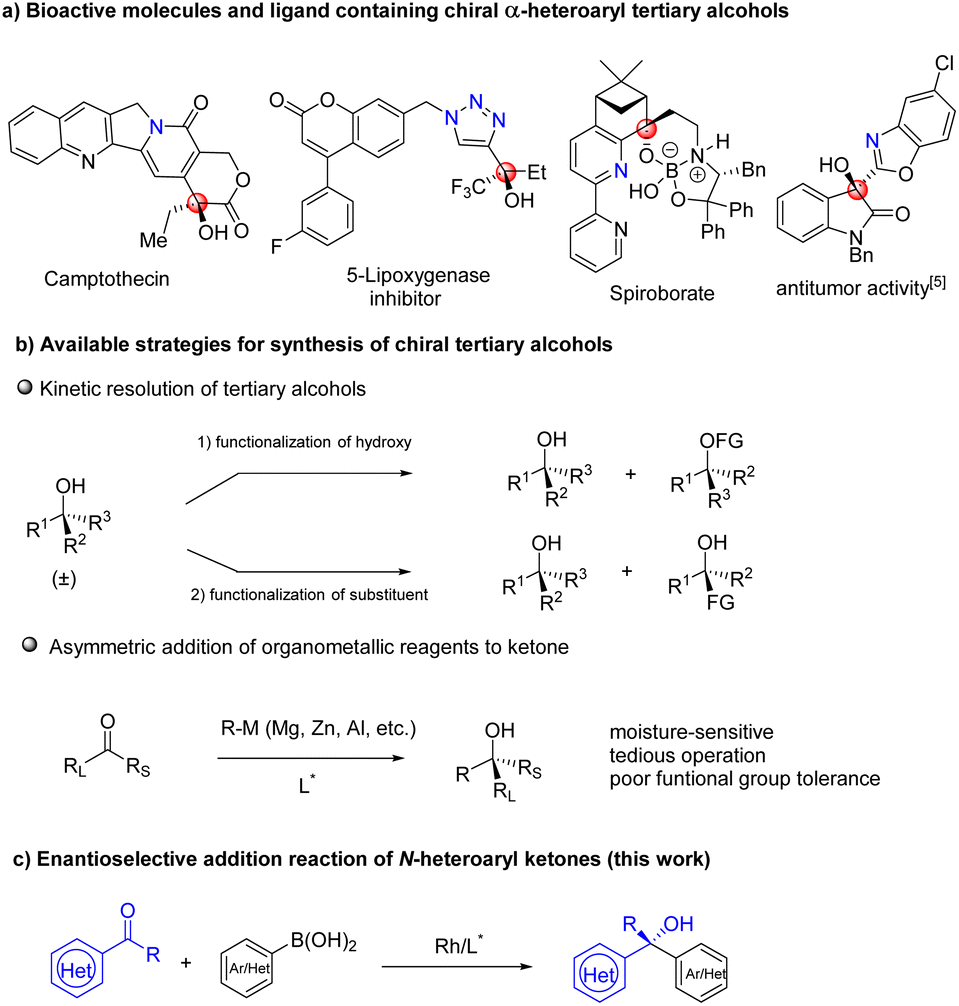 | ||
| Fig. 1 (a) Bioactive molecules and ligand containing chiral α-heteroaryl tertiary alcohols. (b) Strategies for synthesizing chiral tertiary alcohols. (c) Enantioselective addition of arylboronic acids to N-heteroaryl ketones. | ||
With the great efforts of chemists, many excellent results have been reported in the field. It is worth noting that studies mainly focused on asymmetric addition of arylboron reagents to aldehydes9 and activated ketones,10 as well as ketones in an intramolecular fashion.11 Low enantioselectivities were observed in challenging aryl ketone substrates.12 With progress in chiral ligand design, recently, our group reported enantioselective addition of arylboroxines to simple aryl ketones by using the Rh/WingPhos catalyst, where anthryl groups of the ligand were advantageous for high reactivity and enantioselectivity.13 Very recently, Shi and co-workers described enantioselective N-heterocyclic carbene/Ni-catalyzed addition of arylboronic esters to ketones.14 Nevertheless, the general addition reaction of arylboronic acids to N-heteroaryl ketones has not been realized, resulting in a lack of methods for catalytic asymmetric synthesis of α-heteroaryl tertiary alcohols. The key issue in the reaction is the intrinsic coordination ability of N-heteroaryl substrates to transition metals leading to catalyst deactivation.15 To address the challenge, we anticipated that WingPhos having two anthryl groups in its structure would promote reactivity by bringing two reaction partners into close proximity and avoiding heteroatoms of the substrate close to rhodium. At the same time, deep chiral pockets would control the enantioselectivity. Further, we hypothesized that employment of additives can attenuate catalyst deactivation when selected additive was suitable. Based on these guidelines, we herein established an enantioselective addition reaction of (hetero)aryl boronic acids to N-heteroaryl ketones with excellent tolerance of functional groups by using the WingPhos ligand having two anthryl groups. Furthermore, chiral tertiary alcohols derived from α-ketobenzoxazole provided a series of optically active α-hydroxy acids after direct hydrolysis. Herein, we report our results (Fig. 2).
 | ||
| Fig. 2 (a) Reported examples of enantioselective addition reactions. (b) Our current study. | ||
Results and discussion
We chose 3-acetylpyridine as the model substrate to study the addition reaction with 4-methoxyphenylboroxine as the nucleophile. The reaction was performed in tert-butyl methyl ether (MTBE) under nitrogen for 16 hours in the presence of [{Rh(C2H4)2Cl}2] (1.5 mol%), (S,S,S,S)-WingPhos (3.6 mol%), MgBr2, and CsF as base, providing desired product 3aa in 53% yield and 99% ee (Table 1, entry 1). Without the additive, the reaction yielded chiral tertiary alcohol in similar yield and ee value under otherwise identical conditions (entry 2). Some representative commercially available ligands such as BINAP, QuinoxP*, Josiphos and Me-ferrocelane were tested and found to be inferior compared with WingPhos in terms of yield and enantioselectivity (entries 3–6). Actually, ligands based on the same dihydrobenzoxaphosphole skeleton gave lower yields (entries 7 and 8), although L6 afforded excellent enantioselectivity of 97% ee. The two anthryl groups in the structure of WingPhos were believed to promote reactivity by bringing reactants into close proximity. Further, we employed 4-methoxyphenylboronic acid to perform the rhodium-catalyzed addition, finding a better result with 72% yield and 99% ee (entry 9). The phenomenon indicated that there was difference between general aryl ketones and N-heteroaryl ketones in addition reactions, in contrast to our previous study.13 Screening of various inorganic and organic bases showed that cesium fluoride was the best one (entries 10–15). Study of various solvents failed to find a better solvent than MTBE (entries 16–19). To further improve the yield, the additive effect was proposed and investigated. Considering the coordination ability of the nitrogen atom, we tested several typical Lewis acids to attenuate catalyst deactivation. Gratifyingly, employment of B(OMe)3 delivered product 3aa in 90% isolated yield with 99% ee (entries 20–23), the positive effect was also found in the heteroaryl Suzuki–Miyaura cross-coupling reaction.16 Meanwhile, we found both lower and higher reaction temperatures afforded decreased yields (entries 24 and 25).|
|
|||||||
|---|---|---|---|---|---|---|---|
| No.a | L* | Base | Solvent | Additive | T [°C] | Yieldb [%] | eec [%] |
| a Unless otherwise specified, the reactions were performed under nitrogen for 16 h with 3-acetylpyridine (1a, 0.10 mmol), 4-methoxyphenylboronic acid (2, 0.30 mmol), base (0.20 mmol), additive (0.10 mmol) and solvent (3.5 mL) in the presence of [{Rh(C2H4)2Cl}2] (1.5 mol%) and L* (3.6 mol%). The R absolute configuration of 3aa was determined by comparing its analogue's optical rotation with reported data. b Yields were determined by 1H NMR with CH2Br2 as an internal standard. Isolated yield given within parentheses. c Determined by chiral HPLC using an AS column. d 4-Methoxyphenylboroxine (0.10 mmol, 0.30 mmol “B”) was employed. MTBE = methyl tert-butyl ether. | |||||||
| 1d | L7 | CsF | MTBE | MgBr2 | 80 | 53 | 99 |
| 2d | L7 | CsF | MTBE | — | 80 | 54 | 99 |
| 3d | L1 | CsF | MTBE | — | 80 | 13 | 3 |
| 4d | L2 | CsF | MTBE | — | 80 | 10 | 45 |
| 5d | L3 | CsF | MTBE | — | 80 | 29 | 33 |
| 6d | L4 | CsF | MTBE | — | 80 | 34 | 16 |
| 7d | L5 | CsF | MTBE | — | 80 | 18 | 32 |
| 8d | L6 | CsF | MTBE | — | 80 | 33 | 97 |
| 9 | L7 | CsF | MTBE | — | 80 | 72 | 99 |
| 10 | L7 | Cs2CO3 | MTBE | — | 80 | 44 | 99 |
| 11 | L7 | K3PO4 | MTBE | — | 80 | 54 | 99 |
| 12 | L7 | K2CO3 | MTBE | — | 80 | 34 | 99 |
| 13 | L7 | NaOH | MTBE | — | 80 | 30 | 99 |
| 14 | L7 | Me3SiOK | MTBE | — | 80 | 67 | 99 |
| 15 | L7 | DBU | MTBE | — | 80 | Trace | — |
| 16 | L7 | CsF | Toluene | — | 80 | 57 | 99 |
| 17 | L7 | CsF | THF | — | 80 | 15 | 99 |
| 18 | L7 | CsF | Dioxane | — | 80 | Trace | — |
| 19 | L7 | CsF | MeCN | — | 80 | Trace | — |
| 20 | L7 | CsF | MTBE | AlCl3 | 80 | 35 | 99 |
| 21 | L7 | CsF | MTBE | CuSO4 | 80 | 77 | 99 |
| 22 | L7 | CsF | MTBE | BF3OEt2 | 80 | 44 | 99 |
| 23 | L7 | CsF | MTBE | B(OMe)3 | 80 | 92(90) | 99 |
| 24 | L7 | CsF | MTBE | B(OMe)3 | 60 | 56 | 99 |
| 25 | L7 | CsF | MTBE | B(OMe)3 | 100 | 73 | 99 |

With the optimal conditions in hand (Table 1, entry 23), we then examined the substrate scope of the addition reaction. As shown in Table 2, heteroaryl ketones containing nitrogen atoms in different positions, as well as bearing various functional groups were well tolerated. Specifically, 4-acetylpyridine and 4-propionylpyridine with more steric hindrance provided corresponding products (3ba and 3ca) in excellent yields and ee values. 3-Acetylpyridine carrying electron-withdrawing groups such as fluorine, chlorine and bromine worked well to form desired tertiary alcohols in >94% yields and >98% ee (3da–3fa). Methoxyl-substituted heteroaryl ketones were also applicable under the conditions, whereas, the methyl-substituted substrate delivered 3ja with decreased yield (64%). Other interesting substrates including compound 3ka, 3-acetylquinoline 3la, and 5-acetylpyrimi-dine 3ma, afforded products in moderate to excellent yields and excellent enantioselectivities. For nucleophiles, we found that electron-rich arylboronic acids performed better than electron-deficient ones; halide arylboronic acids furnished alcohols in moderate yield when the temperature was raised to 100 °C (3ab–3af). Heteroarylboronic acid (3ag) successfully participated in the reaction with 79% yield. It is worth noting that brominated 2-acetylpyridine formed 3ga in 93% yield. In contrast, 2-acetylpyridine was not compatible in the reaction. The result indicated that the nitrogen atom combined with the adjacent carbonyl group strongly poisoned the catalyst. The method affords a straightforward access to a series of chiral α-heteroaryl tertiary alcohols which are cumbersome to prepare otherwise.
| a The reactions were performed with 1 (0.2 mmol), 2 (0.6 mmol, 3.0 equiv.), [Rh(C2H4)2Cl]2 (1.5 mol%), (S,S,S,S)-WingPhos (3.6 mol%), CsF (0.4 mmol) and B(OMe)3 (0.2 mmol, 1.0 equiv.) in MTBE at 80 °C for 16 h; isolated yields; ee values were determined by chiral HPLC. b Reaction temperature was 100 °C. |
|---|

|
Benzoxazole, an interesting N-heteroarene, is an important heterocycle moiety present in natural products and functional materials.17 The chiral tertiary alcohols containing the benzoxazole unit are of significant importance, and can serve as precursors of chiral α-hydroxy acids which are valuable building blocks in asymmetric synthesis. To our knowledge, no report is available on enantioselective arylation of α-ketobenzoxazoles. We used Rh/(S,S,S,S)-Wingphos as the catalyst to carry out addition of 2a to 1-(benzo[d]oxazol-2-yl)ethanone (4a). First, test of solvents showed that tert-butyl methyl ether and toluene provided almost equivalent yields and ee (Table 3, entries 1–4), while, a series of experiments confirmed that toluene was a more robust reaction solvent. Surprisingly, an equal yield was obtained in the absence of additives (entry 5). We believed that the greater steric hindrance due to the acetyl group and C4-H of benzoxazoles limited catalyst deactivation through coordination compared to the pyridine derivatives, in addition, the benzoxazole unit activated the carbonyl group. Therefore, α-ketobenzoxazoles exhibited excellent reactivity under the reaction conditions. Ligand examination revealed that lower yields and ees were acquired when anthryl groups of WingPhos were replaced by methoxyl or aryl groups (entries 6 and 7). Higher reaction temperature did not provide better results and decreasing the temperature to 50 °C afforded lower yield (entries 8 and 9).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| No.a | L* | Base | Solvent | Additive | T [°C] | Yieldb [%] | eec [%] |
| a Unless otherwise specified, the reactions were performed under nitrogen for 12 h with 1-(benzo[d]oxazol-2-yl)ethanone (4a, 0.20 mmol), 4-methoxyphenylboronic acid (2a, 0.50 mmol), base (0.40 mmol), additive (0.20 mmol) and solvent (3.5 mL) in the presence of [{Rh(C2H4)2Cl}2] (1.5 mol%) and L* (3.6 mol%). The R absolute configuration of 5aa was determined by comparing its analogue's optical rotation with reported data. b Isolated yields. c Determined by chiral HPLC using an AD column. | |||||||
| 1 | L7 | CsF | MTBE | B(OMe)3 | 60 | 93 | 97 |
| 2 | L7 | CsF | Toluene | B(OMe)3 | 60 | 94 | 97 |
| 3 | L7 | CsF | Dioxane | B(OMe)3 | 60 | 81 | 94 |
| 4 | L7 | CsF | THF | B(OMe)3 | 60 | 87 | 96 |
| 5 | L7 | CsF | Toluene | — | 60 | 94 | 97 |
| 6 | L5 | CsF | Toluene | — | 60 | 50 | 10 |
| 7 | L6 | CsF | Toluene | — | 60 | 58 | 78 |
| 8 | L7 | CsF | Toluene | — | 80 | 92 | 97 |
| 9 | L7 | CsF | Toluene | — | 50 | 85 | 97 |
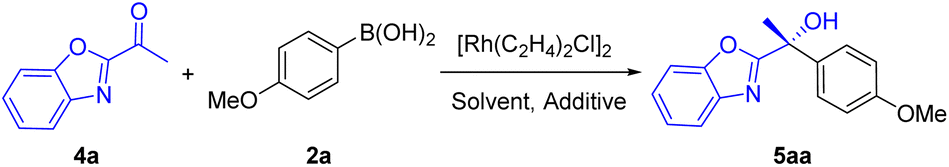
The substrate scope of enantioselective arylation of α-ketobenzoxazoles was investigated (Table 4). Thus, a wide range of chiral benzoxazolyl-substituted tertiary alcohols were prepared with excellent ee values and yields. Both electron-rich and electron-deficient α-ketobenzoxazoles with various substitution patterns on the aryl ring were all tolerated well (5ba–5ja). 1-(Naphtho[2,3-d]oxazol-2-yl)ethenone with a larger aromatic system was also applicable, furnishing product 5ka in 96% ee and 85% yield. Besides, the method afforded moderate to good yields and excellent enantioselectivities for ethyl and n-propyl α-ketobenzoxazoles (5la and 5ma). To our delight, α-ketobenzothiazole reacted smoothly to form 5na in 96% ee and 74% yield. Various aryl and heteroaryl boronic acids, including substituted phenyl, thienyl, furyl, pyridyl, and naphthyl groups, were compatible, offering desired products with satisfactory yields. In contrast, 3-chlorophenylboronic acid delivered chiral alcohol (5ae) in moderate yield. To demonstrate the practicality of this method, enantioselective addition of 4-methoxyphenylboronic acid to 4a was carried out in the presence of 0.15 mol% [{Rh(C2H4)2Cl}2] and 0.36 mol% WingPhos, providing 5aa in 92% yield and 97% ee. Electron-rich arylboronic acids furnished products in high yields (5ag and 5ah), while, electron-deficient aryl-boronic acids (5ad and 5ae) formed the corresponding products in relatively lower yields.
| a The reactions were performed at 60 °C for 12 h with α-ketobenzoxazoles (4, 0.20 mmol), arylboronic acid (2, 0.50 mmol), CsF (0.40 mmol), toluene (3.5 mL) in the presence of [{Rh(C2H4)2Cl}2] (1.5 mol%) and (S,S,S,S)-WingPhos (3.6 mol%). b Isolated yields shown within parentheses were obtained when reactions were performed in the presence of [{Rh(C2H4)2Cl}2] (0.15 mol%) and (S,S,S,S)-WingPhos (0.36 mol%). c 50 °C. d Using 4.0 equiv. 4-MeOC6H4B(OH)2, 4 mL toluene, 14 h. e Using 3.0 equiv. 3-thiopheneboronic acid, 50 °C, 14 h. f 3.5 equiv. 2-furanboronic acid, 4 mL toluene. |
|---|

|
In order to illustrate the high efficiency of the Rh/WingPhos catalyst for enantioselective arylation, the mechanism for this asymmetric reaction of N-heteroaryl ketones is proposed in Fig. 3. First, transmetallation of the arylboronic acid with the [Rh(Cl)/{(S,S,S,S)-WingPhos}] species formed the aryl-Rh species I. Next, coordination of an N-heteroaryl ketone provided the species II, in which a π–π effect between the anthryl group and substrate was proposed based on the ligand and substrate test results.18 Migratory insertion of the aryl-Rh in N-heteroaryl ketones would adopt more favorable conformation to generate chiral tertiary alcohol species III, in which the catalyst keenly discriminated the re- and si- face of ketones with a deep chiral pocket. Transmetallation between III and another arylboronic acid provided the product and regenerated I.
 | ||
| Fig. 3 Proposed mechanism. | ||
Optically active α-hydroxy acids present significant importance because of their prevalence among natural products and biologically active molecules, and as building blocks for asymmetric synthesis.19 Chiral tertiary α-hydroxy acids having quaternary carbon centers, have gained enormous attention from synthetic chemists. Catalytic asymmetric addition of organometallics to α-ketoesters provided a straightforward approach for synthesizing precursors of tertiary α-hydroxy acids. However, high catalyst loadings were needed.20 We prepared chiral benzoxazolyl-substituted tertiary alcohols with a Rh loading as low as 0.3 mol%, which were suitable precursors for synthesis of α-hydroxy acids. When basic reaction conditions were employed, KOH in ethylene glycol, tertiary alcohols cleanly converted into the corresponding α-hydroxy acids (Scheme 1). A series of arylmethyl α-hydroxy acids were obtained in good yields with no loss in ees, albeit a low yield was obtained with ethyl-substituted alcohol (6 h).
 | ||
| Scheme 1 Conversion of chiral α-heteroaryl tertiary alcohols into the corresponding α-hydroxy acids. | ||
The chiral α-hydroxy acids can be applied for synthesis of valuable chiral compounds, according to the reported literature. For example, 6c reacted with (rac)-2-bromobutanoyl bromide to form chiral 1,4-dioxane-2,5-diones 7, which served as a precursor for polylactides and a promising new chiral template.21 Reduction of 6c provided chiral diol 8 in 91% yield,22a,b which was a useful intermediate in synthetic chemistry. Further, oxirane 9 can be obtained through tosylation and ring closure.22b,c Similarly, the diol furnished an interesting azidoalcohol 10 by treatment with tosyl chloride and sodium azide (Scheme 2).22b
 | ||
| Scheme 2 Transformations of 6c as building blocks. | ||
Conclusions
We have realized a highly enantioselective addition of arylboronic acids to N-heteroaryl ketones for the synthesis of chiral α-heteroaryl tertiary alcohols. The method features mild reaction and excellent functional group compatibility. The use of the WingPhos ligand having two anthryl groups is crucial for this transformation due to promotion of reactivity by bringing two reaction partners into close proximity and excellent enantioselectivity control. Besides, trimethyl borate weakens the coordination of N-heteroaryl substrates to rhodium. In particular, enantioselective addition of α-ketobenzoxazoles furnishes various chiral benzoxazolyl-substituted tertiary alcohols with excellent ee values and yields by employing rhodium loading as low as 0.3 mol%, which can serve as a practical protocol to deliver valuable chiral tertiary α-hydroxy acids after hydrolysis.Data availability
The data supporting this study are available within the main text and the associated ESI.Author contributions
J. Z. and W. T. directed this project. Z. L., J. Z., J. L., D. T., R. X., and Z. T. performed the experiments. J. Z., W. T. and Z. C. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to NSFC-22002019 and the Jiangxi Natural Science Foundation (20212ACB213002) for financial support. We thank Dr Kang Du and Dr He Yang for valuable discussion.Notes and references
- For books, see: (a) Quaternary Stereocenters, challenges and solutions for organic synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed.
- (a) M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1996, 88, 3888–3890 CrossRef; (b) W. D. Kingsbury, J. C. Boehm, D. R. Jakas, K. G. Holden, S. M. Hecht, G. Gallagher, M. J. Caranfa, F. L. Mccabe, L. F. Faucette, R. K. Johnson and R. P. Hertzberg, J. Med. Chem., 1991, 34, 98–107 CrossRef CAS PubMed; (c) H.-J. Ban, I.-J. Oh, K.-S. Kim, J.-Y. Ju, Y.-S. Kwon, Y.-I. Kim, S.-C. Lim and Y.-C. Kim, Tuberc. Respir. Dis., 2009, 66, 93–97 CrossRef; (d) Y. Kawato, M. Aonuma, Y. Hirota, H. Kuga and K. Sato, Cancer Res., 1991, 51, 4187–4191 CAS.
- S. G. Ouellet, D. Gauvreau, M. Cameron, S. Dolman, L.-C. Campeau, G. Hughes, P. D. O'Shea and I. W. Davies, Org. Process Res. Dev., 2012, 16, 214–219 CrossRef CAS.
- X.-R. Huang, C. Chen, G.-H. Lee and S.-M. Peng, Adv. Synth. Catal., 2009, 351, 3089–3095 CrossRef CAS.
- (a) P. Hewawasam, N. A. Meanwell, V. K. Gribkoff, S. I. Dworetzky and C. G. Boissard, Bioorg. Med. Chem. Lett., 1997, 7, 1255–1260 CrossRef CAS; (b) G.-W. Wang, A.-X. Zhou, J.-J. Wang, R.-B. Hu and S.-D. Yang, Org. Lett., 2013, 15, 5270–5273 CrossRef CAS PubMed.
- For review, see: (a) Y. Chen, W. Liu and X. Yang, Chin. J. Org. Chem., 2022, 42, 679–697 CrossRef , for selected examples, see: ; (b) E. R. Jarvo, C. A. Evans, G. T. Copeland and S. J. Miller, J. Org. Chem., 2001, 66, 5522–5527 CrossRef CAS PubMed; (c) I. Čorić, S. Müller and B. List, J. Am. Chem. Soc., 2010, 132, 17370–17373 CrossRef PubMed; (d) S. Qu, S. M. Smith, V. Laina-Martín, R. M. Neyyappadath, M. D. Greenhalgh and A. D. Smith, Angew. Chem. Int. Ed., 2020, 59, 16572–16578 ( Angew. Chem. , 2020 , 132 , 16715–16721 ) CrossRef CAS PubMed.
- (a) R. Noyori and M. Kitamura, Angew. Chem. Int. Ed., 1991, 30, 49–69 ( Angew. Chem. , 1991 , 103 , 34–55 ) CrossRef; (b) L. Pu and H.-B. Yu, Chem. Rev., 2001, 101, 757–824 CrossRef CAS PubMed; (c) J. F. Collados, R. Solà, S. R. Harutyunyan and B. Maciá, ACS Catal., 2016, 6, 1952–1970 CrossRef CAS.
- For selected examples, see: Zn: (a) P. I. Dosa and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 445–446 CrossRef CAS; (b) D. J. Ramón and M. Yus, Angew. Chem. Int. Ed., 2004, 43, 284–287 ( Angew. Chem. , 2004 , 116 , 286–289 ) CrossRef PubMed; (c) H. Li, C. García and P. J. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5425–5427 CrossRef CAS PubMed , Al: ; (d) C.-A. Chen, K.-H. Wu and H.-M. Gau, Angew. Chem., Int. Ed., 2007, 46, 5373–5376 ( Angew. Chem. , 2007 , 119 , 5469–5472 ) CrossRef CAS PubMed , Ti: ; (e) K.-H. Wu, Y.-Y. Kuo, C.-A. Chen, Y.-L. Huang and H.-M. Gau, Adv. Synth. Catal., 2013, 355, 1001–1008 CrossRef CAS , Mg: ; (f) A. V. R. Madduri, S. R. Harutyunyan and A. J. Minnaard, Angew. Chem. Int. Ed., 2012, 51, 3164–3167 ( Angew. Chem. , 2012 , 124 , 3218–3221 ) CrossRef CAS PubMed; (g) E. Fernández-Mateos, B. Maciá and M. Yus, Eur. J. Org. Chem., 2014, 6519–6526 CrossRef.
- For selected examples of rhodium catalysts, see: (a) M. Sakai, M. Ueda and N. Miyaura, Angew. Chem. Int. Ed., 1998, 37, 3279–3281 ( Angew. Chem. , 1998 , 110 , 3475–3477 ) CrossRef CAS; (b) T. Focken, J. Rudolph and C. Bolm, Synthesis, 2005, 429–436 CAS; (c) K. Suzuki, S. Ishii, K. Kondo and T. Aoyama, Synlett, 2006, 648–650 CAS; (d) H.-F. Duan, J.-H. Xie, W.-J. Shi, Q. Zhang and Q.-L. Zhou, Org. Lett., 2006, 8, 1479–1481 CrossRef CAS PubMed; (e) S. Morikawa, K. Michigami and H. Amii, Org. Lett., 2010, 12, 2520–2523 CrossRef CAS PubMed; (f) T. Nishimura, H. Kumamoto, M. Nagaosa and T. Hayashi, Chem. Commun., 2010, 46, 3010–3012 RSC , for ruthenium catalysts, see: ; (g) Y. Yamamoto, K. Kurihara and N. Miyaura, Angew. Chem. Int. Ed., 2009, 48, 4414–4416 ( Angew. Chem. , 2009 , 121 , 4478–4480 ) CrossRef CAS PubMed; (h) K. Li, N. Hu, R. Luo, W. Yuan and W. Tang, J. Org. Chem., 2013, 78, 6350–6355 CrossRef CAS PubMed , for copper catalysts, see: ; (i) D. Tomita, M. Kanai and M. Shibasaki, Chem.–Asian J., 2006, 1, 161–166 CrossRef CAS PubMed , for nickel catalysts, see: ; (j) T. Arao, K. Kondo and T. Aoyama, Tetrahedron Lett., 2007, 48, 4115–4117 CrossRef CAS; (k) K. Yamamoto, K. Tsurumi, F. Sakurai, K. Kondo and T. Aoyama, Synthesis, 2008, 3585–3590 CAS , for cobalt catalysts, see: ; (l) J. Karthikeyan, M. Jeganmohan and C.-H. Cheng, Chem.–Eur. J., 2010, 16, 8989–8992 CrossRef CAS PubMed , for amino- or iminoalcohol-catalyzed asymmetric arylation in combination with an arylboronic acid and diethylzinc, see: ; (m) C. Bolm and J. Rudolph, J. Am. Chem. Soc., 2002, 124, 14850–14851 CrossRef CAS PubMed.
- For additions to α-keto esters, see: (a) H.-F. Duan, J.-H. Xie, X.-C. Qiao, L.-X. Wang and Q.-L. Zhou, Angew. Chem. Int. Ed., 2008, 47, 4351–4353 ( Angew. Chem. , 2008 , 120 , 4423–4425 ) CrossRef CAS PubMed; (b) Y. Yamamoto, T. Shirai, M. Watanabe, K. Kurihara and N. Miyaura, Molecules, 2011, 16, 5020–5034 CrossRef CAS PubMed; (c) F. Cai, X. Pu, X. Qi, V. Lynch, A. Radha and J. M. Ready, J. Am. Chem. Soc., 2011, 133, 18066–18069 CrossRef CAS PubMed; (d) T.-S. Zhu, S.-S. Jin and M.-H. Xu, Angew. Chem. Int. Ed., 2012, 51, 780–783 ( Angew. Chem. , 2012 , 124 , 804–807 ) CrossRef CAS PubMed , for additions to isatins, see: ; (e) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem. Int. Ed., 2006, 45, 3353–3356 ( Angew. Chem. , 2006 , 118 , 3431–3434 ) CrossRef CAS PubMed; (f) P. Y. Toullec, R. B. C. Jagt, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Org. Lett., 2006, 8, 2715–2718 CrossRef CAS PubMed; (g) H. Lai, Z. Huang, Q. Wu and Y. Qin, J. Org. Chem., 2009, 74, 283–288 CrossRef CAS PubMed; (h) Z. Liu, P. Gu, M. Shi, P. McDowell and G. Li, Org. Lett., 2011, 13, 2314–2317 CrossRef CAS PubMed , for additions to trifluoromethyl ketones, see: ; (i) S. L. X. Martina, R. B. C. Jagt, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Chem. Commun., 2006, 4093–4095 RSC; (j) R. Luo, K. Li, Y. Hu and W. Tang, Adv, Synth. Catal., 2013, 355, 1297–1302 CrossRef CAS.
- (a) G. Liu and X. Lu, J. Am. Chem. Soc., 2006, 128, 16504–16505 CrossRef CAS PubMed; (b) G. Liu and X. Lu, Tetrahedron, 2008, 64, 7324–7330 CrossRef CAS; (c) D. W. Low, G. Pattison, M. D. Wieczysty, G. H. Churchill and H. W. Lam, Org. Lett., 2012, 14, 2548–2551 CrossRef CAS PubMed; (d) G. L. Galllego and R. Sarpong, Chem. Sci., 2012, 3, 1338–1342 RSC; (e) C. Ni, J. Gao and X. Fang, Chem. Commun., 2020, 56, 2654–2657 RSC.
- (a) J. Bouffard and K. Itami, Org. Lett., 2009, 11, 4410–4413 CrossRef CAS PubMed; (b) T. Korenaga, A. Ko, K. Uotani, Y. Tanaka and T. Sakai, Angew. Chem. Int. Ed., 2011, 50, 10703–10707 ( Angew. Chem. , 2011 , 123 , 10891–10895 ) CrossRef CAS PubMed; (c) Y.-X. Liao, C.-H. Xing and Q.-S. Hu, Org. Lett., 2012, 14, 1544–1547 CrossRef CAS PubMed.
- L. Huang, J. Zhu, G. Jiao, Z. Wang, X. Yu, W.-P. Deng and W. Tang, Angew. Chem. Int. Ed., 2016, 55, 4527–4531 ( Angew. Chem. , 2016 , 128 , 4603–4607 ) CrossRef CAS PubMed.
- Y. Cai, L.-X. Ruan, A. Rahman and S.-L. Shi, Angew. Chem. Int. Ed., 2021, 60, 5262–5267 ( Angew. Chem. , 2021 , 133 , 5322–5327 ) CrossRef CAS PubMed.
- C. G. Watson and V. K. Aggarwal, Org. Lett., 2013, 15, 1346–1349 CrossRef CAS PubMed.
- V. M. Kassel, C. M. Hanneman, C. P. Delaney and S. E. Denmark, J. Am. Chem. Soc., 2021, 143, 13845–13853 CrossRef CAS PubMed.
- (a) A. D. Rodríguez, C. Ramírez, I. I. Rodríguez and E. González, Org. Lett., 1999, 1, 527–530 CrossRef PubMed; (b) J. P. Davidson and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 13486–13489 CrossRef CAS PubMed; (c) S. M. Johnson, S. Connelly, I. A. Wilson and J. W. Kelly, J. Med. Chem., 2008, 51, 260–270 CrossRef CAS PubMed; (d) H. Razavi, S. K. Palaninathan, E. T. Powers, R. L. Wiseman, H. E. Purkey, N. N. Mohamedmohaideen, S. Deechongkit, J. C. Sacchettini and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 2758–2761 CrossRef CAS PubMed; (e) M. Gjorgjieva, T. Tomašič, M. Barančokova, S. Katsamakas, J. Ilaš, P. Tammela, L. P. Mašič and D. Kikelj, J. Med. Chem., 2016, 59, 8941–8954 CrossRef CAS PubMed; (f) S. Park, S. Kim, J. Seo and S. Y. Park, Macromolecules, 2005, 38, 4557–4559 CrossRef CAS.
- In the enantioselective addition reaction, WingPhos provided better yield and ee than L5 and L6. Substrates (3c, 5l, and 5m) with greater steric hindrance in alkyl were applicable to furnish alcohols with excellent ees; a π–π effect between the anthryl group and substrate was proposed to promote reactivity and enantioselectivity.
- G. M. Coppola and H. F. Schuster, α-Hydroxy Acids in Enantioselective Synthesis, VCH, Weinheim, 1997 Search PubMed.
- For selected examples, see: (a) E. F. DiMauro and M. C. Kozlowski, Org. Lett., 2002, 4, 3781–3784 CrossRef CAS PubMed; (b) L. C. Wieland, H. Deng, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 15453–15456 CrossRef CAS PubMed; (c) G. Blay, I. Fernández, A. Marco-Aleixandre and J. R. Pedro, Org. Lett., 2006, 8, 1287–1290 CrossRef CAS PubMed; (d) H.-L. Wu, P.-Y. Wu, Y.-Y. Shen and B.-J. Uang, J. Org. Chem., 2008, 73, 6445–6447 CrossRef CAS PubMed.
- R. Nagase, Y. Iida, M. Sugi, T. Misaki and Y. Tanabe, Synthesis, 2008, 3670–3674 CAS.
- (a) S. Qu, S. M. Smith, V. Laina-Martin, R. M. Neyyappadath, M. D. Greenhalgh and A. D. Smith, Angew. Chem. Int. Ed., 2020, 59, 16572–16578 ( Angew. Chem. , 2020 , 132 , 16715–16721 ) CrossRef CAS PubMed; (b) R. Infante, J. Nieto and C. Andres, Chem.–Eur. J., 2012, 18, 4375–4379 CrossRef CAS PubMed; (c) X.-Y. Han, H. Liu, C.-H. Liu, B. Wu, L.-F. Chen, B.-H. Zhong and K.-L. Liu, Bioorg. Med. Chem. Lett., 2005, 15, 1979–1982 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05907j |
| This journal is © The Royal Society of Chemistry 2023 |