
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo visualization of enantioselective targeting of amyloid and improvement of cognitive function by clickable chiral metallohelices†
Zhi
Du
a,
Chun
Liu
ab,
Zhenqi
Liu
ab,
Hualong
Song
 c,
Peter
Scott
c,
Xiubo
Du
d,
Jinsong
Ren
ab and
Xiaogang
Qu
*ab
c,
Peter
Scott
c,
Xiubo
Du
d,
Jinsong
Ren
ab and
Xiaogang
Qu
*ab
aLaboratory of Chemical Biology, State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: xqu@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, Anhui 230029, China
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
dCollege of Life Sciences and Oceanography, Shenzhen University, Shenzhen 518060, P. R. China
First published on 28th November 2022
Abstract
The pathogenesis of Alzheimer's disease (AD) is closely related to several contributing factors, especially amyloid-β (Aβ) aggregation. Bioorthogonal reactions provide a general, facile, and robust route for the localization and derivatization of Aβ-targeted agents. Herein, a pair of chiral alkyne-containing metallohelices (ΛA and ΔA) were demonstrated to enantioselectively target and modulate Aβ aggregation, which has been monitored in triple-transgenic AD model mice and proved to improve cognitive function. Compared with its enantiomer ΔA, ΛA performed better in blocking Aβ fibrillation, relieving Aβ-triggered toxicity, and recovering memory deficits in vivo. Moreover, clickable ΛA could act as a functional module for subsequent visualization and versatile modification of amyloid via bioorthogonal reaction. As a proof-of-concept, thioflavin T, tacrine, and magnetic nanoparticles were conjugated with ΛA to realize Aβ photo-oxygenation, acetylcholinesterase inhibition, and Aβ clearance, respectively. This proof-of-principle work provided new insights into the biolabeling and bioconjugation of multifunctional metallosupramolecules through click reactions for AD therapy.
Introduction
Alzheimer's disease (AD) is a stubborn neurodegenerative disorder that plagues millions of people worldwide.1,2 The accumulation and aggregation of amyloid-β (Aβ) peptides are implicated as the initial and prominent pathophysiology features of AD and further trigger a series of cascade reactions, such as oxidative stress, neurotransmitter deficiencies, and neuronal injury.3–5 Aβ-binding agents have been actively developed to modulate the amyloidogenic behavior and alleviate concomitant cytotoxicity.6 Tracking Aβ ligands in real-time is of great significance for precision medicine, which conduces to identify the distribution of the ligands and evaluate treatment effect. To tackle poly-pathological features in AD, the rational design of multifunctional agents by integrating two or more target-interacting moieties into one single molecule has gained particular attention.7–9As a quintessential bioorthogonal reaction, Cu(I)-mediated azide/alkyne cycloaddition (CuAAC) is characterized by high selectivity, mild conditions, and fast kinetics.10–12 Thus, it has been extensively investigated in the biochemical research, such as bioconjugation,13,14 biolabeling,15,16 and prodrug activation.17,18 From this point of view, bioorthogonal catalysis has a great potential to endow Aβ binders with additional capabilities for their localization and versatile modification to address the multifactorial nature of AD.
Owing to the distinctive physicochemical properties, various metal complexes display a fascinating affinity with Aβ and thus are exploited for the theranostics of AD.19–22 Given that Aβ aggregates carry the chirality at different-length scales, the incorporation of enantioselective recognition is a bonus for AD therapeutics.23–25 Chiral metallohelices, a type of three-dimensional metal complexes assembled by multidentate organic ligands wrapping around two or more metal centers, are analogous to α-helical peptides in terms of size, amphiphilicity, and stereochemistry.26–30 As potential non-peptide mimetics, a pair of alkyne-bearing metallohelices enantiomers (ΛA and ΔA, Scheme 1)31 are chosen, as an example to employ bioorthogonal activity for enantioselective targeting and visualization of amyloid without the need of de novo synthesis.
 | ||
| Scheme 1 The structures of Λ (left-handed twist) and Δ (right-handed twist) enantiomers of the chiral metallohelices. | ||
Results and discussion
Enantioselective modulation of Aβ aggregation by ΛA
The influence of the chiral metallohelices ΛA and ΔA on the course of Aβ40 fibril formation was investigated using thioflavin T (ThT) assays, circular dichroism (CD), and transmission electron microscopy (TEM) (Fig. 1). ThT, whose fluorescence is augmented considerably upon binding to β-sheet-rich aggregates, is commonly exploited for monitoring the amyloid aggregation kinetics. ThT assays demonstrated that ΛA exhibited potent inhibition of Aβ40 aggregation while ΔA displayed a weak effect (Fig. 1b). The control experiment indicated the chiral metallohelices had a small impact on the binding of ThT to Aβ fibrils (Fig. S1b†). In addition, the free chiral ligands of ΛA and ΔA had very feeble regulation ability on Aβ40 aggregation (Fig. S1d†), indicating that stereoscopic structures of the chiral metallohelices played a vital role on Aβ discrimination.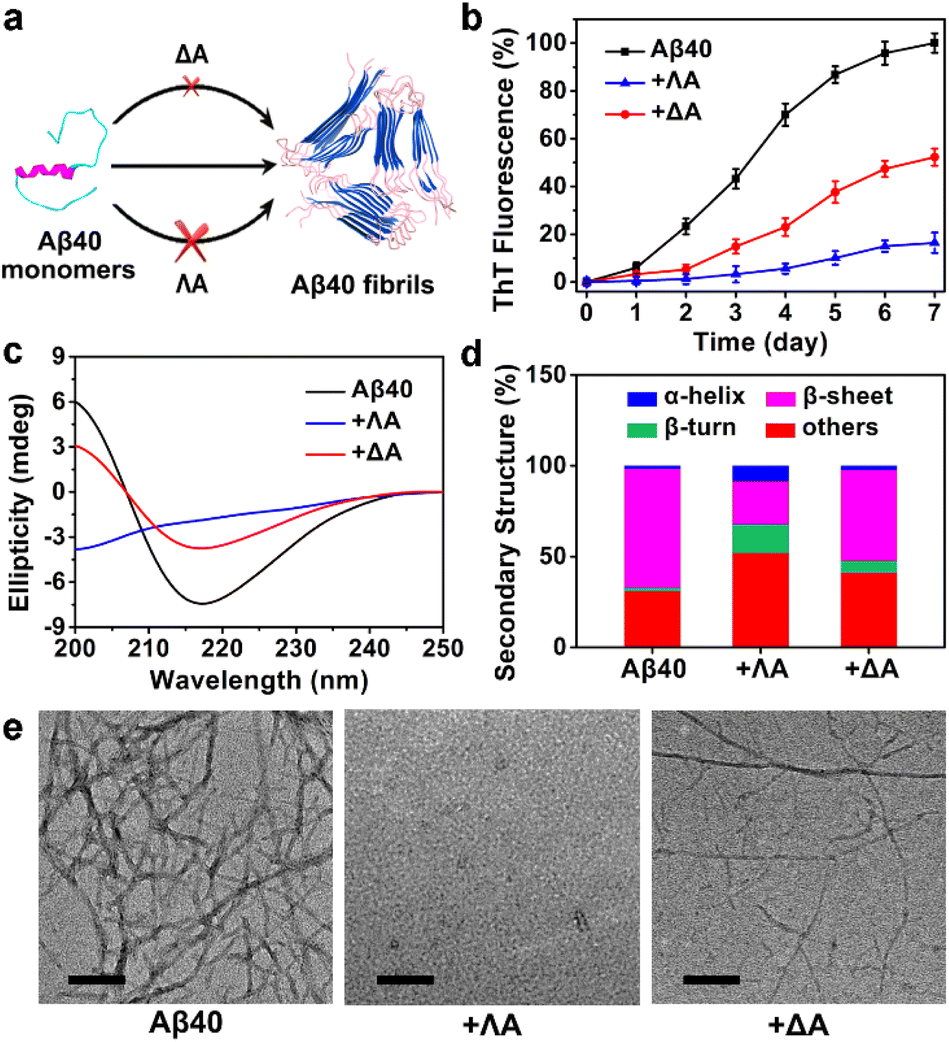 | ||
| Fig. 1 Enantioselective regulation on Aβ40 fibrillation with the chiral metallohelices. (a) Overview of interrupting Aβ40 aggregation with ΛA and ΔA. (b) ThT assays for monitoring Aβ40 fibrillogenesis. Error bars represented ±standard deviation (s.d.) of three independent experiments. (c) CD spectra of Aβ40. For the spectra of Aβ in the presence of metallohelices, the CD spectra of the metallohelices were subtracted to reduce interference. (d) Secondary structure content estimations. (e) TEM images of Aβ40 aggregates. Scale bars are 50 nm. | ||
CD spectra, secondary structure analysis, and TEM images further verified that ΛA prohibited Aβ40 from aggregating into β-sheet-rich fibrils more powerfully than its enantiomer (Fig. 1c–e). Besides, ΛA could also block Aβ42 fibrillation, as depicted in Fig. S2.† The performance of the chiral metallohelices on Aβ42 oligomerization was evaluated with a dot blotting immunoassay. As shown in Fig. S2d,† the sample treated with ΛA displayed a weaker reactivity to Aβ oligomer-specific antibody A11, indicating that ΛA decreased the formation of toxic oligomeric intermediates. UV-Vis absorption spectra and high-resolution mass spectra (HRMS) demonstrated the chiral metallohelices were stable in the phosphate-buffered saline (PBS) (Fig. S3†).
The complexation between ΛA and Aβ
Isothermal titration calorimetry (ITC), a distinguished method for evaluating molecular interactions, was employed to estimate the apparent binding constant (Ka), enthalpy change (ΔH), and binding stoichiometry (n) of Aβ40 with chiral metallohelices. According to Fig. 2a and b, an exothermic ITC isotherm was recorded and the binding stoichiometry was close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. ΛA showed a higher binding affinity to Aβ40 (Ka = 8.05 × 106 M−1) than the Δ enantiomer (Ka = 9.38 × 105 M−1), and the corresponding Gibbs free energy changes (ΔG) were −39.4 and −34.1 kJ mol−1, respectively. Competition dialysis experiment (Fig. S4†) further confirmed the higher affinity between Aβ40 and ΛA.
:1. ΛA showed a higher binding affinity to Aβ40 (Ka = 8.05 × 106 M−1) than the Δ enantiomer (Ka = 9.38 × 105 M−1), and the corresponding Gibbs free energy changes (ΔG) were −39.4 and −34.1 kJ mol−1, respectively. Competition dialysis experiment (Fig. S4†) further confirmed the higher affinity between Aβ40 and ΛA.
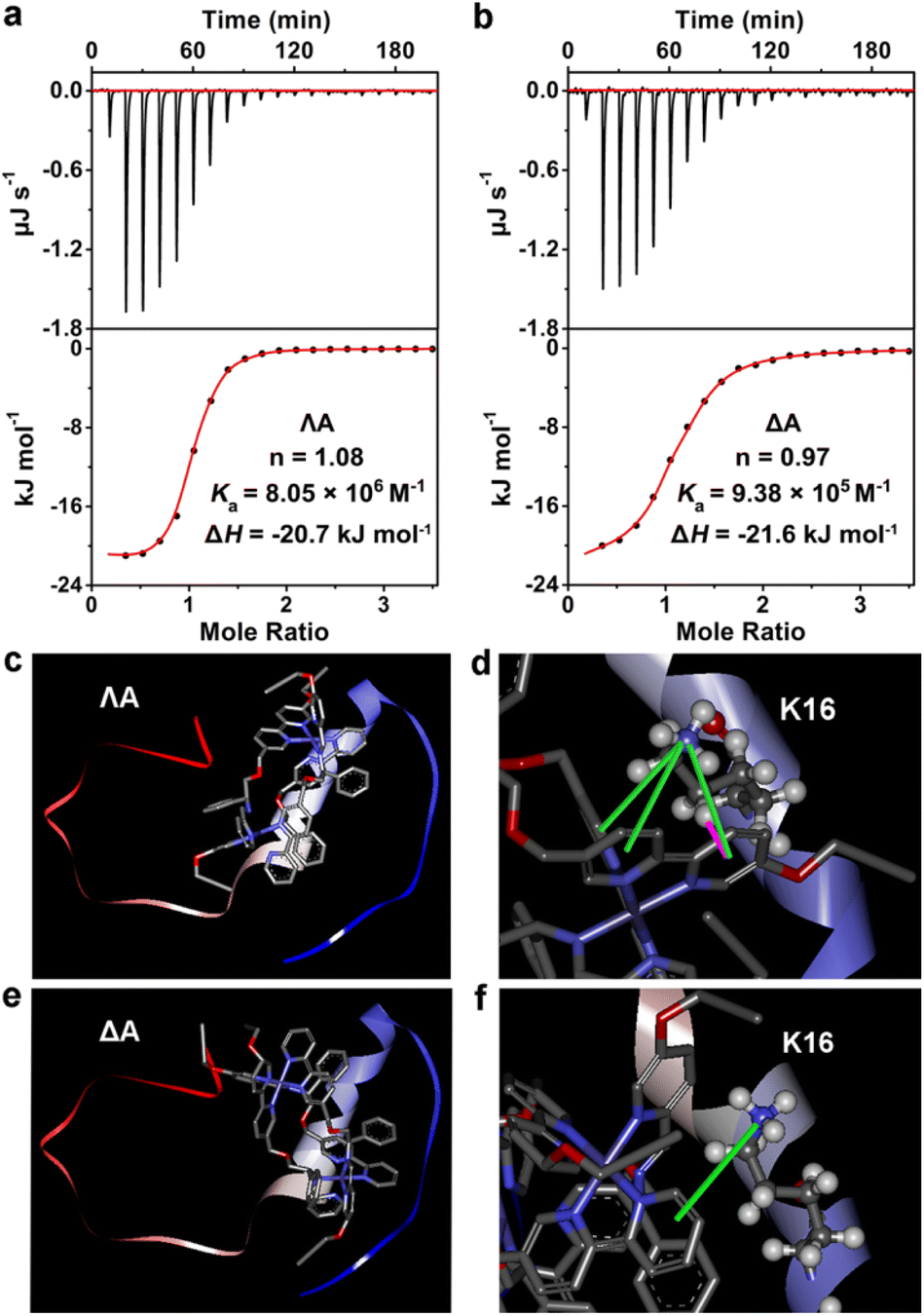 | ||
| Fig. 2 The interaction between Aβ40 and ΛA. (a) and (b) Presented ITC data of Aβ40 binding to ΛA and ΔA, respectively. The top panels are raw calorimetric data, and the bottom panels are fitting results. (c) and (d) Showed the lowest-energy conformations of Aβ40 with ΛA. (e) and (f) Displayed the lowest-energy conformations of Aβ40 with ΔA. The cation–π and σ–π interactions were indicated by green and magenta lines, respectively. | ||
The fragments 12–28 and 25–35, which are the critical regions responsible for Aβ aggregation,32 were utilized to explore the binding sites of Aβ to the chiral metallohelices. As shown in Fig. S5,†ΛA efficiently blocked the aggregation of Aβ12–28 while it displayed modest modulation ability to Aβ25–35, implying that ΛA preferentially binds to Aβ12–28 rather than Aβ25–35. Remarkably, Aβ15–20 is considered the self-recognition region and often exploited as an Aβ-target peptide.33 Table S1† revealed that ΛA possessed comparative regulation capability towards the fibrillation of Aβ15–20 and Aβ1–40. It is inferred from the above results that ΛA might bind to Aβ15–20 as the mechanism of altering Aβ aggregation.
To further dissect the structural features of the chiral metallohelices and Aβ40, we performed docking studies with AutoDock Vina. Aβ1–40 monomer (PDB ID 2LFM), in which the core hydrophobic region adopts a helical conformation,34 was used as a working model for molecular docking. Interestingly, ΛA was nearly parallel to the α-helix of the Aβ monomer while ΔA was sloping (Fig. 2c–f). As a result, there were four sets of cation–π and σ–π interactions together between the aromatic nuclei of ΛA and the K16 residue of Aβ (Fig. 2d); this is in accord with aforementioned observation from inhibiting aggregation assays of Aβ fragments. In contrast, only one set of cation–π interaction was found between ΔA and Aβ (Fig. 2f). This structural analysis provides a rationale for the stronger binding between ΛA and Aβ and more powerful modulation activity towards amyloid aggregation of ΛA.
Alleviation of Aβ-induced toxicity by ΛA in cells and the nematode model
Aβ-triggered oxidative stress is another feature in AD pathogenesis.35 It has been proven that quenching overproduced reactive oxygen species (ROS) plays an active role in delaying AD progression.36,37 A series of Fe complexes have been reported to mimic catalase (CAT) or superoxide dismutase (SOD) and display promising anti-oxidation features.38–40 Hence we speculated that the chiral metallohelices might also scavenge deleterious ROS. As expected, ΛA and ΔA efficiently decomposed H2O2 (Fig. 3a and b) and slowed down pyrogallol auto-oxidation (Fig. 3c and d), confirming their CAT-mimic and SOD-mimic activities.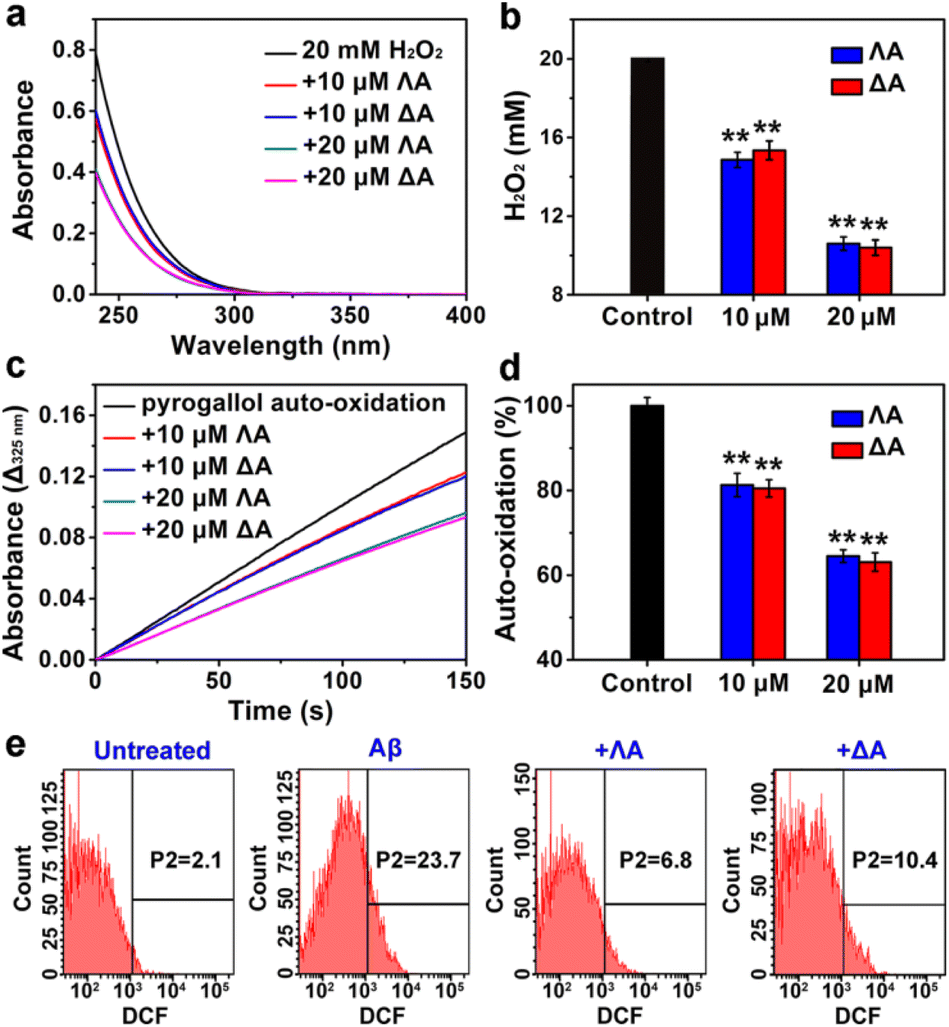 | ||
| Fig. 3 ROS-quenching properties of ΛA and ΔA. (a) CAT-mimic ability measured by UV-Vis absorption of H2O2. (b) The concentration of remained H2O2 after incubating with chiral metallohelices for 30 min. (c) SOD-like activity monitored by the kinetic study of pyrogallol auto-oxidation. The absorbance change at 325 nm (Δ325 nm) represented oxidated pyrogallol in 150 s. (d) Percent of pyrogallol auto-oxidation. Pyrogallol auto-oxidation without chiral metallohelices was set as the control. Each experiment was repeated three times. Error bars indicate ±s.d. Significance was determined by one-sided analysis of variance: **P < 0.01. (e) Flow cytometric analysis. PC12 cells were stained by ROS probe DCFH-DA. Untreated cells acted as the control. | ||
Based on the above encouraging results, we further investigated whether the metallohelices were able to protect PC12 cells from Aβ-induced toxicity. Intracellular ROS was monitored by the probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA), which can be oxidized to fluorescent dichlorofluorescein (DCF). Flow cytometric analysis revealed that ΛA-treatment significantly decreased Aβ-mediated oxidative stress, owing to the synergistic effect of regulating Aβ aggregation as well as CAT-mimic and SOD-mimic activities (Fig. 3e). Moreover, microscopic images presented the shrunken, rounded, and aggregated morphologies of Aβ-treated cells, while ΛA substantially restored these abnormalities (Fig. S6a†). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays further verified ΛA exhibited noticeable neuroprotection against Aβ-induced cytotoxicity (Fig. S6c†).
With wild-type N2 strain as the control, we further evaluated the therapeutic potential of chiral metallohelices in the transgenic AD model Caenorhabditis elegans (C. elegans) CL2006, which constitutively expressed Aβ.29 Thioflavin S (ThS) staining, ROS measurement, motility quantification, and survival curves demonstrated that Aβ aggregates led to paralysis phenotype, oxidative stress, behaviour defects, and shortened lifespan in the CL2006 model (Fig. 4 and S7†). Whereas the presence of ΛA reduced Aβ plaques, rescued Aβ-triggered paralysis, decreased the ROS level, and improved the motility of the CL2006 strain. Likewise, ΛA performed better than its enantiomer ΔA in relieving these Aβ-induced toxicities.
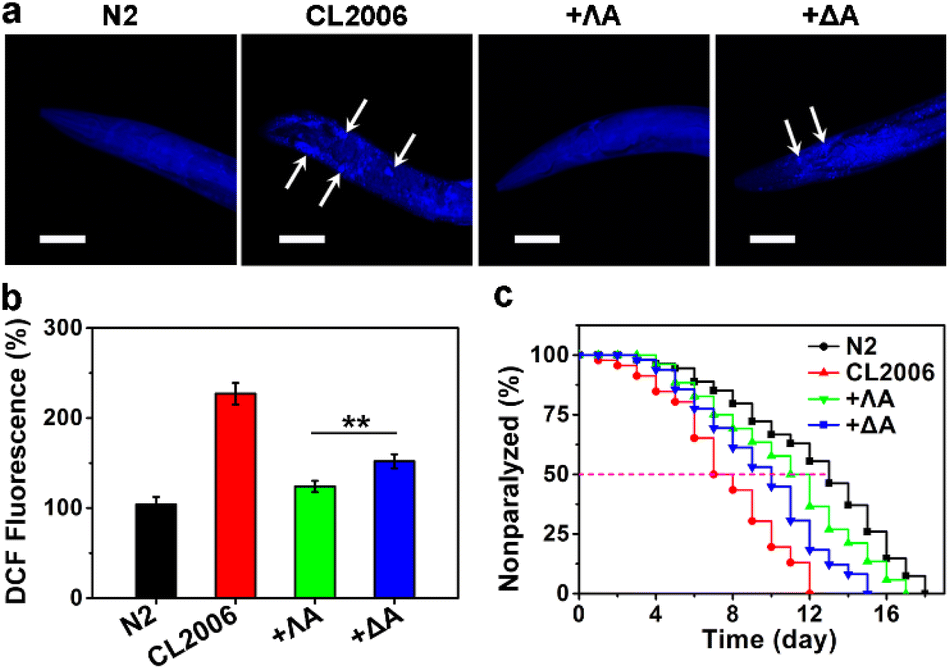 | ||
| Fig. 4 Rescuing Aβ-associated paralysis in CL2006 strain with ΛA. (a) ThS-staining Aβ plaques in CL2006 strain treated with ΛA or ΔA. Arrows indicate Aβ plaques. Scale bars: 50 μm. (b) ROS level in the worms monitored with DCFH-DA. All results were expressed as the mean ± s.d. from three independent experiments. Significance was determined by one-sided analysis of variance: **P < 0.01. (c) Survival curves of CL2006 strain treated with the chiral metallohelices. | ||
Improvement of cognitive function in AD model mice by ΛA
We next evaluated the performances of the metallohelices in the triple-transgenic mice model of AD (3×Tg-AD mice).41 The potential blood–brain barrier (BBB) permeability of the metallohelices was evaluated after the attachment of fluorescent Cyanine5 (Cy5). Fig. S8† indicated that ΛA successfully crossed the BBB and accumulated in the brain parenchyma. Besides, their potential toxicity was investigated through hematoxylin and eosin (H&E) staining and biochemistry analysis (Fig. S9 and S10†). Neither obvious tissue harm nor significant change in histopathology analysis was found, suggesting favorable biocompatibility of the metallohelices.The effects of the metallohelices on amyloid deposition were determined via immunoassay. As depicted in Fig. 5a, ΛA significantly decreased the levels of Aβ species in the brain compared to the treatment with ΔA, further confirming the enantioselectivity towards Aβ. Morris water maze (MWM) assay was conducted to assess the spatial learning behavior of 3×Tg-AD mice after administration. Four evaluation indicators were analyzed, including the escape latency, crossing frequency, time spent in the target quadrant, and swimming path of the mice (Fig. 5b–e). After the treatment of ΛA, 3×Tg-AD mice took shorter paths and less time to reach the target platform and displayed a spatially oriented swimming behavior. Taken together, these results indicate that ΛA could recover impaired learning and memory in 3×Tg-AD mice.
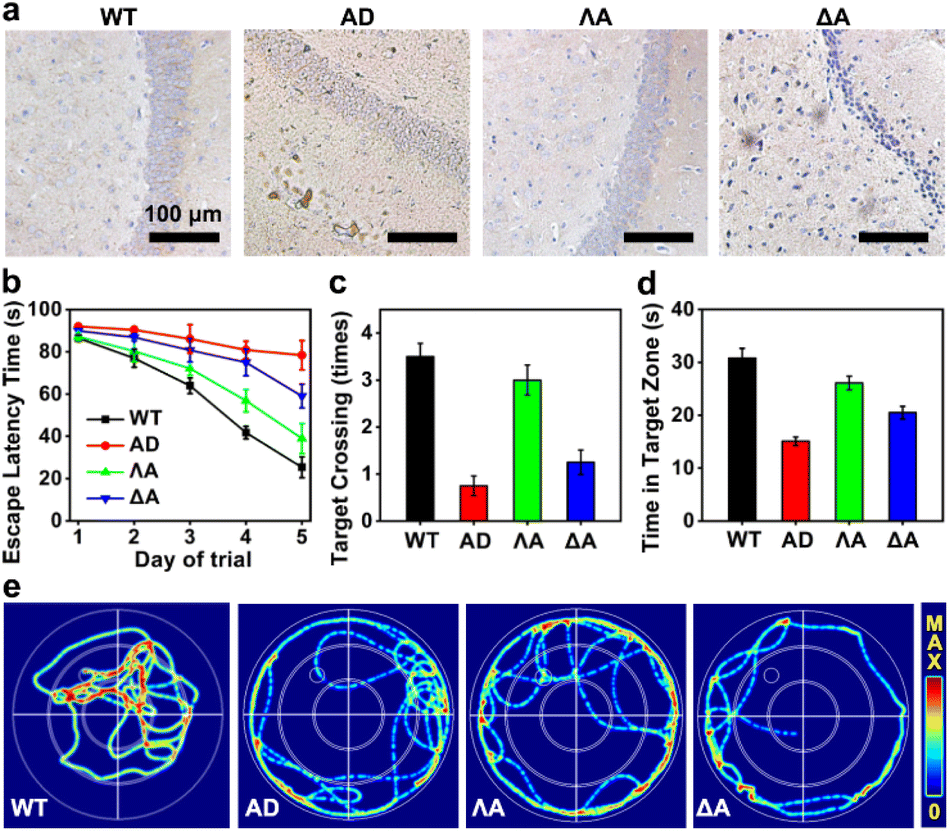 | ||
| Fig. 5 Improvement of cognitive function in AD model mice with ΛA. (a) Representative images of Aβ staining in the hippocampus. (b) Escape latency time in the training trial. The escape latency of all groups was gradually shortened after successive training. (c) The frequency to cross the target platform. (d) Time spent in the target quadrant. (e) Representative swimming paths of mice to seek the target platform (the small circular region in the left upper corner). | ||
Bioorthogonal PAM of ΛA to extend its biochemical applications
Inspired by protein post-translational modification (PTM), post-assembly modification (PAM) has been developed as a powerful tool to optimize metallosupramolecular performance by covalently introducing new functional groups, in which case the cautious pre-assembly ligand design is avoided while retaining the desired framework and properties.42,43 In this context, we investigated the chemoselective conjugation of bioactive moieties to the alkyne-bearing metallohelix ΛAvia CuAAC reaction to broaden its biochemical applications. A commercial fluorescent dye Cyanine5–azide (Cy5–N3) was used for labelling the metallohelices in both PC12 cells and C. elegans CL2006 strain (Fig. 6). Colocalization quantitative analysis was evaluated via Manders' colocalization coefficients (MCC), conducted using ImageJ software with the JACoP plugin.44 Encouragingly, most of ΛA (about 90%) was found to be co-localized with Aβ antibodies in PC12 cells (Fig. 6a), and the MCC value of ΛA with Aβ was more significant than that of ΔA (Fig. 6c and d). This is well consistent with the above observation that ΛA dramatically outperforms ΔA in terms of specificity on targeting Aβ, inhibition of amyloid aggregation, and attenuation of Aβ-mediated toxicity. Fig. 6b and e further displayed the potential of click reaction for metallohelix visualization in the worms.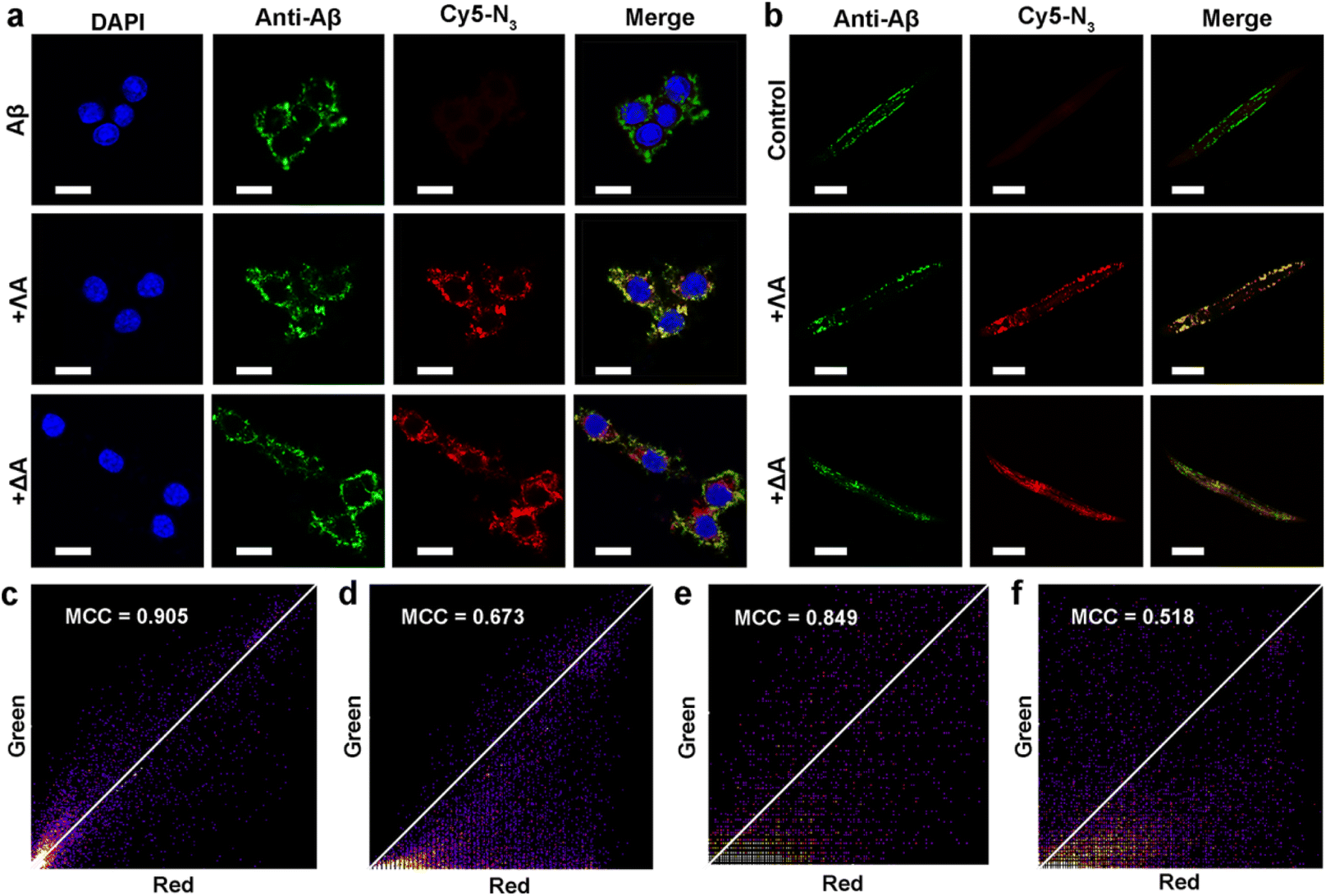 | ||
| Fig. 6 Co-localization of ΛA and Aβ in the PC12 cells and CL2006 strain. (a) Immunofluorescence staining of PC12 cells treated with Aβ and the chiral metallohelices. Aβ was labeled with Aβ-specific antibody B4 (green channel), and the alkyne-bearing metallohelices ΛA and ΔA were visualized by Cy5–N3 (red channel) via CuAAC reaction. The nucleus was dyed with DAPI. Scale bars: 10 μm. (b) Fluorescence images of the CL2006 strain, which were treated with the chiral metallohelices and then labeled with Cy5–N3 and Aβ antibody. Scale bars: 100 μm, (c) and (d) were the MCC values of Aβ with Cy5-labelled metallohelices in the PC12 cells, respectively, (e) and (f) were the MCC values of Aβ with Cy5-labelled metallohelices in the CL2006 strain, respectively. | ||
Next, we integrated various functional moieties with ΛAvia bioorthogonal PAM to modulate multiple facets of AD, as illustrated in Fig. 7a. Previous studies have demonstrated that photoactivated ThT and its analogs can oxygenate Aβ and thereby decrease the aggregation property and pathogenicity of Aβ.45,46 The azide derivative of ThT (ThT–N3) was grafted onto ΛA employing the CuAAC reaction. The obtained ΛA–ThT hybrid thus bore three copies of ThT moiety (Fig. S11a†). UV-Vis absorption and CD spectra indicated that PAM did not disrupt the metallohelix architecture (Fig. S11b and c†). Interestingly, photoactivated ΛA–ThT could selectively oxygenate Aβ (Fig. 7b, c and S11e†), presumably because of the transient lifetime and restricted diffusion range of highly reactive singlet oxygen (1O2) in biological systems.47,48 8-Anilinonaphthalene-1-sulfonate (ANS), which displays enhanced fluorescence upon binding to protein hydrophobic residues, was used for monitoring Aβ fibrillation. Our observations manifested that photoactivated ΛA–ThT reduced Aβ hydrophobicity (Fig. S11f†) and prohibited Aβ aggregation (Fig. S11g†) through a synergistic mechanism.
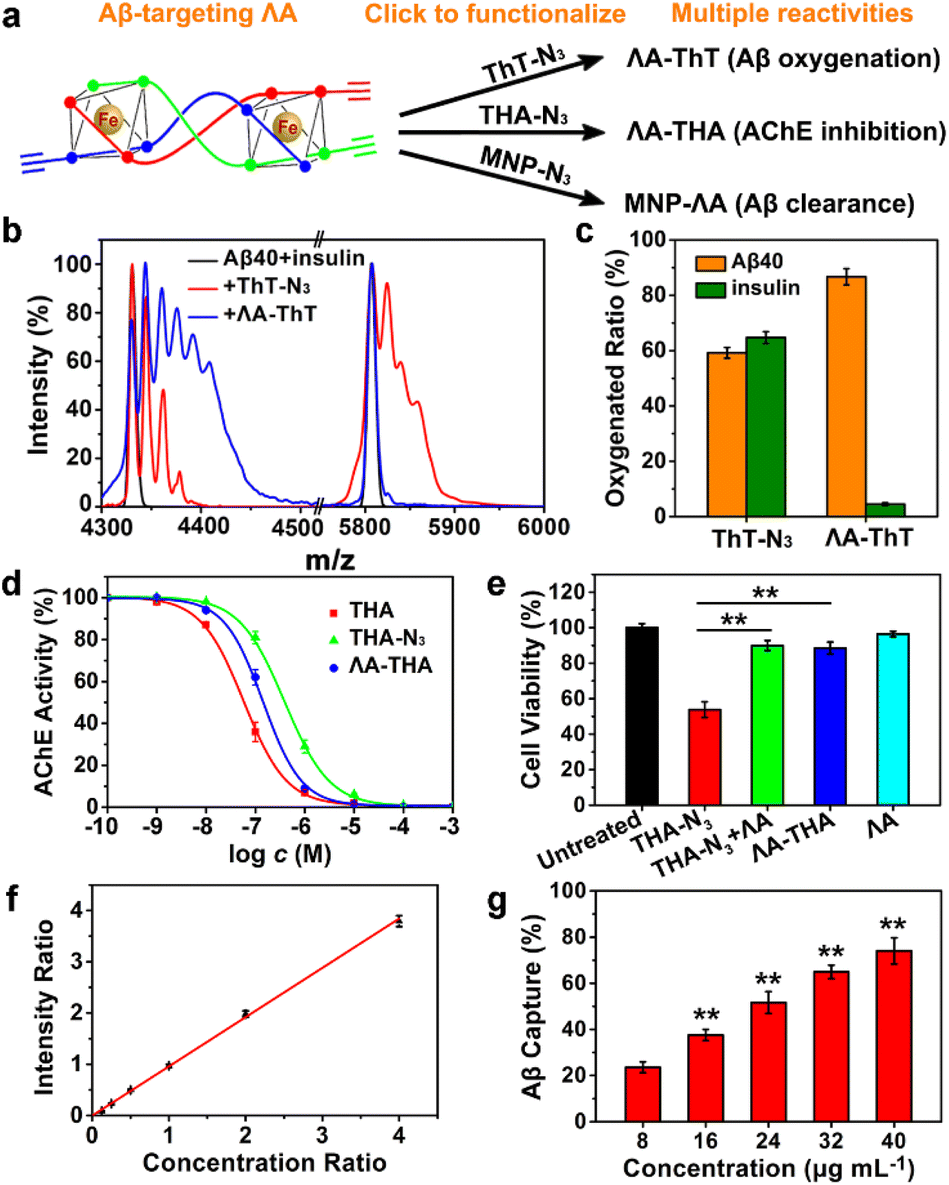 | ||
| Fig. 7 Bioorthogonal PAM of ΛA to introduce multiple reactivities. (a) Scheme of PAM of clickable ΛA. (b) The mass spectra of Aβ and insulin oxidized by photo-activated ThT–N3 or ΛA–ThT. Insulin, which could form amyloid fibrils under certain conditions, was used as the control to verify Aβ-specific oxygenation with ΛA–ThT. (c) The oxygenated ratio of Aβ40 and insulin. Error bars represented ±s.d. of three independent experiments. (d) AChE inhibition study. (e) In vitro hepatotoxicity evaluated with HepG2 cells. The cells were treated with THA–N3 (120 μM), ΛA–THA (40 μM), or ΛA (40 μM) for 24 h. (f) MS intensity ratio of Aβ40/Aβ42 versus the concentration ratio. (g) The concentration-dependent Aβ40 capture with MNP–ΛA. Each experiment was repeated three times. Error bars indicate ±s.d. Significance was determined by one-sided analysis of variance: **P < 0.01. | ||
Tacrine (1,2,3,4-tetrahydroacridine-9-amine, THA), the first clinically-used acetylcholinesterase (AChE) inhibitor for AD treatment, is hindered by severe adverse reactions, especially hepatic toxicity, which perhaps results from oxidative stress.49,50 Due to reliable anti-AChE potency, classical pharmacophore, and synthetic accessibility, THA-based scaffolds are still at the forefront of developing safer AChE inhibitors.51–53 Recent reports revealed that antioxidants could repress THA-triggered hepatotoxicity by scavenging overproduced ROS during administration.54,55 In these contexts, the azide derivative of THA (THA–N3) was also integrated with ΛA through click reaction to obtain ΛA–THA (Fig. S12†). The AChE inhibitory activity of ΛA–THA was evaluated by modifying Ellman's method.51 As shown in Fig. 7d and e and Table S2,† the hybrid metallohelix ΛA–THA exhibited a modest anti-AChE activity and diminished hepatotoxicity, which might be ascribed to a ROS-quenching feature inherited from ΛA (Fig. S12d–f†). In addition, ΛA–THA retained the capacity to intervene Aβ aggregation (Fig. S12g†).
To further assess the universality of bioorthogonal chemistry, ΛA was also conjugated with azide functionalized nanoparticles. Magnetic nanoparticles (MNPs) have been widely exploited as versatile nano-platforms for the theranostics of neurodegenerative diseases, because of their excellent biocompatibility, BBB permeability, and attractive magnetic properties.56–59 Recently, Hyeon's group constructed multifunctional nano-assemblies (magnetite and ceria nanocomposites) for Aβ clearance through selective binding and magnetic separation.60 Inspired by that, ΛA was attached to the MNPs to form MNP–ΛA through the same click reaction (Fig. S13 and S14†). As a proof of concept, MNP–ΛA was harnessed for Aβ40 capture, and the adsorbed Aβ40 was measured by MS quantitative analysis with Aβ42 as the internal standard. The amino acid sequences of Aβ1–40 and Aβ1–42 differ by only two amino acids, so Aβ42 is an ideal internal standard for Aβ40 in MS analysis (Fig. 7f). Fig. 7g showed that MNP–ΛA was able to remove Aβ40 from the mice serum in a concentration-dependent manner. Taken together, these results indicate that, through bioorthogonal PAM, ΛA can act as a universal scaffold to integrate various bioactive moieties, thus conferring the superiority of multidirectional treatment for AD.
Conclusion
In summary, chiral metallohelix ΛA displayed enantioselectivity on targeting and regulating Aβ aggregation. Meanwhile, it mimicked CAT and SOD to quench overproduced ROS and maintain redox homeostasis. Such synergism enabled the impressive ability of ΛA to attenuate Aβ-mediated toxicity in cells and the C. elegans CL2006 strain, while the Δ enantiomer was far less efficient. Moreover, ΛA efficiently reduced the Aβ burden in the brain of 3×Tg-AD mice and recovered their impaired learning and memory. Beyond the inherent bioactivity, the alkyne-bearing ΛA served as a building block that allowed subsequent chemoselective conjugation via click reaction for extended functionalities, including biolabeling, bioconjugation, and nanoplatform construction. Through fluorescent tagging, clickable ΛA was found to be well co-localized with Aβ in vivo. As representatives, several types of multi-target-directed hybrid agents against the interrelated pathological features in AD were constructed, where ΛA–ThT reduced Aβ hydrophobicity and aggregation tendency; ΛA–THA showed modest anti-AChE potential and attenuated hepatotoxicity; and MNP–ΛA conveniently captured and cleared Aβ. More importantly, incorporating new functional units did not interfere with the core metallohelix framework, and the final hybrid simultaneously inherited the biological activities from diverse parent frameworks. Hence, bioorthogonal chemistry promises a fresh and unique perspective for customized supramolecules with desirable functionality against multifaceted AD.Data availability
We have included all data in the ESI Section.†Author contributions
X. Q. and J. R. conceived the project. Z. D. performed the experimental study and drafted the manuscript. H. S. and P. S. synthesized the chiral triplex metallohelices, and P. S. assisted in revising the manuscript. C. L., and Z. L. carried out the experiments on the AD mice and analyzed the data. X. D. helped to analyze the data. All authors approved the final version. Z. D., C. L., and Z. L. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFA0709202), NSFC (21820102009, 21871249, and 22237006) and Key Program of Frontier of Sciences (CAS QYZDJ-SSW-SLH052).References
- J. Gaugler, B. James, T. Johnson, J. Reimer, M. Solis, J. Weuve, R. F. Buckley and T. J. Hohman, Alzheimer’s Dementia, 2022, 18, 700–789 CrossRef.
- P. C. Ke, M. A. Sani, F. Ding, A. Kakinen, I. Javed, F. Separovic, T. P. Davis and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 6492–6531 RSC.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- Z. Du, M. Li, J. Ren and X. Qu, Acc. Chem. Res., 2021, 54, 2172–2184 CrossRef CAS PubMed.
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299–310 CrossRef CAS PubMed.
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. Wee Yong, S. Tsutsui, P. K. Stys and T. Storr, Chem. Sci., 2017, 8, 5636–5643 RSC.
- T. Yang, Z. Zhu, E. Yin, Y. Wang, C. Zhang, H. Yuan, H. Zhang, S. Jin, Z. Guo and X. Wang, Chem. Sci., 2019, 10, 10149–10158 RSC.
- L. Taiariol, C. Chaix, C. Farre and E. Moreau, Chem. Rev., 2022, 122, 340–384 CrossRef CAS.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231–1232 RSC.
- J. Chen, K. Li, S. E. Bonson and S. C. Zimmerman, J. Am. Chem. Soc., 2020, 142, 13966–13973 CrossRef CAS PubMed.
- Y. Wang, J. Weng, J. Lin, D. Ye and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 2787–2794 CrossRef CAS PubMed.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- Y. You, Q. Deng, Y. Wang, Y. Sang, G. Li, F. Pu, J. Ren and X. Qu, Nat. Commun., 2022, 13, 1459 CrossRef CAS PubMed.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS.
- H. Liu, Y. Qu and X. Wang, Future Med. Chem., 2018, 10, 679–701 CrossRef CAS PubMed.
- J. M. Suh, G. Kim, J. Kang and M. H. Lim, Inorg. Chem., 2019, 58, 8–17 CrossRef CAS PubMed.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- L. M. F. Gomes, J. C. Bataglioli and T. Storr, Coord. Chem. Rev., 2020, 412, 213255 CrossRef CAS.
- N. Rubin, E. Perugia, M. Goldschmidt, M. Fridkin and L. Addadi, J. Am. Chem. Soc., 2008, 130, 4602–4603 CrossRef CAS PubMed.
- J. A. Raskatov, A. R. Foley, J. M. Louis, W. M. Yau and R. Tycko, J. Am. Chem. Soc., 2021, 143, 13299–13313 CrossRef CAS.
- M. Tornquist and S. Linse, Angew. Chem., Int. Ed., 2021, 60, 24008–24011 CrossRef CAS PubMed.
- H. Song, M. Postings, P. Scott and N. J. Rogers, Chem. Sci., 2021, 12, 1620–1631 RSC.
- A. D. Faulkner, R. A. Kaner, Q. M. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797–803 CrossRef CAS PubMed.
- S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2011, 4, 31–36 CrossRef PubMed.
- Y. Guan, Z. Du, N. Gao, Y. Cao, X. Wang, P. Scott, H. Song, J. Ren and X. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS PubMed.
- H. Song, N. J. Rogers, S. J. Allison, V. Brabec, H. Bridgewater, H. Kostrhunova, L. Markova, R. M. Phillips, E. C. Pinder, S. L. Shepherd, L. S. Young, J. Zajac and P. Scott, Chem. Sci., 2019, 10, 8547–8557 RSC.
- J. Wang, C. Zhao, A. Zhao, M. Li, J. Ren and X. Qu, J. Am. Chem. Soc., 2015, 137, 1213–1219 CrossRef CAS.
- V. Armiento, A. Spanopoulou and A. Kapurniotu, Angew. Chem., Int. Ed., 2020, 59, 3372–3384 CrossRef CAS PubMed.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Commun., 2011, 411, 312–316 CrossRef CAS PubMed.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed.
- H. J. Kwon, M. Y. Cha, D. Kim, D. K. Kim, M. Soh, K. Shin, T. Hyeon and I. Mook-Jung, ACS Nano, 2016, 10, 2860–2870 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang, X. Li, L. Wang, M. Yin, L. Wang, N. Chen, C. Fan and H. Song, Adv. Mater., 2016, 28, 1387–1393 CrossRef CAS.
- L. M. F. Gomes, A. Mahammed, K. E. Prosser, J. R. Smith, M. A. Silverman, C. J. Walsby, Z. Gross and T. Storr, Chem. Sci., 2019, 10, 1634–1643 RSC.
- X. M. Zhang, J. Tang, L. N. Wang, D. Yao, Q. Yu, F. P. Huang and H. D. Bian, Polyhedron, 2017, 133, 433–440 CrossRef CAS.
- T. P. Ribeiro, F. L. Fonseca, M. D. de Carvalho, R. M. Godinho, F. P. de Almeida, T. D. Saint'Pierre, N. A. Rey, C. Fernandes, A. Horn, Jr. and M. D. Pereira, Biochem. J., 2017, 474, 301–315 CrossRef CAS PubMed.
- M. Ma, Z. Liu, N. Gao, Z. Pi, X. Du, J. Ren and X. Qu, J. Am. Chem. Soc., 2020, 142, 21702–21711 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC.
- M. M. Gan, J. Q. Liu, L. Zhang, Y. Y. Wang, F. E. Hahn and Y. F. Han, Chem. Rev., 2018, 118, 9587–9641 CrossRef CAS.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723–C742 CrossRef CAS.
- Z. Du, D. Yu, X. Du, P. Scott, J. Ren and X. Qu, Chem. Sci., 2019, 10, 10343–10350 RSC.
- A. Taniguchi, Y. Shimizu, K. Oisaki, Y. Sohma and M. Kanai, Nat. Chem., 2016, 8, 974–982 CrossRef CAS.
- P. Rajaputra, M. Bio, G. Nkepang, P. Thapa, S. Woo and Y. You, Bioorg. Med. Chem., 2016, 24, 1540–1549 CrossRef CAS PubMed.
- J. L. Pan and J. Shi, Adv. Funct. Mater., 2014, 24, 7318–7327 CrossRef.
- B. Sameem, M. Saeedi, M. Mahdavi and A. Shafiee, Eur. J. Med. Chem., 2017, 128, 332–345 CrossRef CAS PubMed.
- D. Galimberti and E. Scarpini, Expert Opin. Invest. Drugs, 2016, 25, 1181–1187 CrossRef CAS.
- A. Kochi, T. J. Eckroat, K. D. Green, A. S. Mayhoub, M. H. Lim and S. Garneau-Tsodikova, Chem. Sci., 2013, 4, 4137–4145 RSC.
- W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CrossRef CAS PubMed.
- M. Ouberai, K. Brannstrom, M. Vestling, A. Olofsson, P. Dumy, S. Chierici and J. Garcia, Org. Biomol. Chem., 2011, 9, 1140–1147 RSC.
- M. Benchekroun, A. Romero, J. Egea, R. Leon, P. Michalska, I. Buendia, M. L. Jimeno, D. Jun, J. Janockova, V. Sepsova, O. Soukup, O. M. Bautista-Aguilera, B. Refouvelet, O. Ouari, J. Marco-Contelles and L. Ismaili, J. Med. Chem., 2016, 59, 9967–9973 CrossRef CAS.
- E. Nepovimova, J. Korabecny, R. Dolezal, K. Babkova, A. Ondrejicek, D. Jun, V. Sepsova, A. Horova, M. Hrabinova, O. Soukup, N. Bukum, P. Jost, L. Muckova, J. Kassa, D. Malinak, M. Andrs and K. Kuca, J. Med. Chem., 2015, 58, 8985–9003 CrossRef CAS.
- Y. Li, Y. Li, W. Ji, Z. Lu, L. Liu, Y. Shi, G. Ma and X. Zhang, J. Am. Chem. Soc., 2018, 140, 4164–4171 CrossRef CAS PubMed.
- K. L. Viola, J. Sbarboro, R. Sureka, M. De, M. A. Bicca, J. Wang, S. Vasavada, S. Satpathy, S. Wu, H. Joshi, P. T. Velasco, K. MacRenaris, E. A. Waters, C. Lu, J. Phan, P. Lacor, P. Prasad, V. P. Dravid and W. L. Klein, Nat. Nanotechnol., 2015, 10, 91–98 CrossRef CAS PubMed.
- C. Wang, X. Wang, H. N. Chan, G. Liu, Z. Wang, H. W. Li and M. S. Wong, Adv. Funct. Mater., 2020, 30, 1909529 CrossRef CAS.
- J. Jang and C. B. Park, Sci. Adv., 2022, 8, eabn1675 CrossRef CAS PubMed.
- D. Kim, H. J. Kwon and T. Hyeon, Adv. Mater., 2019, 31, e1807965 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05897a |
| This journal is © The Royal Society of Chemistry 2023 |