
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeavy metalla vinyl-cations show metal–Lewis acid cooperativity in reaction with small molecules (NH3, N2H4, H2O, H2)†
Maximilian
Auer
a,
Janina
Bolten
a,
Klaus
Eichele
a,
Hartmut
Schubert
a,
Christian P.
Sindlinger
 *b and
Lars
Wesemann
*a
*b and
Lars
Wesemann
*a
aInstitut für Anorganische Chemie, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: lars.wesemann@uni-tuebingen.de
bInstitut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: christian.sindlinger@iac.uni-stuttgart.de
First published on 24th November 2022
Abstract
Halide abstraction from tetrylidene complexes [TbbE(Br)IrH(PMe3)3] [E = Ge (1), Sn (2)] and [Ar*E(Cl)IrH(PMe3)3] gives the salts [TbbEIrH(PMe3)3][BArF4] [E = Ge (3), Sn (4)] and [Ar*EIrH(PMe3)3][BArF4] [E = Ge (3′), E = Sn (4′)] (Tbb = 2,6-[CH(SiMe3)2]2-4-(t-Bu)C6H2, Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl). Bonding analysis suggests their most suitable description as metalla-tetrela vinyl cations with an Ir![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E double bond and a near linear coordination at the Ge/Sn atoms. Cationic complexes 3 and 4 oxidatively add NH3, N2H4, H2O, HCl, and H2 selectively to give: [TbbGe(NH2)IrH2(PMe3)3][BArF4] (5), [TbbE(NHNH2)IrH2(PMe3)3][BArF4] [E = Ge (7), Sn (8)], [TbbE(OH)IrH2(PMe3)3][BArF4] [E = Ge (9), Sn (10)], [TbbE(Cl)IrH2(PMe3)3][BArF4] [E = Ge (11a), Sn (12a)], [TbbGe(H)IrH2(PMe3)3][BArF4] (13), [TbbSn(μ-H3)Ir(PMe3)3][BArF4] (14), and [TbbSn(H)IrH2(PMe3)3][BArF4] (15). 14 isomerizes to give 15via an 1,2-H shift reaction. Hydride addition to cation 3 gives a mixture of products [TbbGeHIrH(PMe3)3] (16) and [TbbGeIrH2(PMe3)3] (17) and a reversible 1,2-H shift between 16 and 17 was studied. In the tin case 4 the dihydride [TbbSnIrH2(PMe3)3] (18) was isolated exclusively. The PMe3 and PEt3 derivatives, 18 and [TbbSnIrH2(PEt3)3] (19), respectively, could also be synthesized in reaction of [TbbSnH2]− with the respective chloride [(R3P)nIrCl] (R = Me, n = 4; R = Et, n = 3). Reaction of complex 19 with CO gives the substitution product [TbbSnIrH2(CO)(PEt3)2] (20). Further reaction with CO results in hydrogen transfer from the iridium to the tin atom to give [TbbSnH2Ir(CO)2(PEt3)2] (21). The reversibility of this ligand induced reductive elimination transferring 20 to 21 is shown.
E double bond and a near linear coordination at the Ge/Sn atoms. Cationic complexes 3 and 4 oxidatively add NH3, N2H4, H2O, HCl, and H2 selectively to give: [TbbGe(NH2)IrH2(PMe3)3][BArF4] (5), [TbbE(NHNH2)IrH2(PMe3)3][BArF4] [E = Ge (7), Sn (8)], [TbbE(OH)IrH2(PMe3)3][BArF4] [E = Ge (9), Sn (10)], [TbbE(Cl)IrH2(PMe3)3][BArF4] [E = Ge (11a), Sn (12a)], [TbbGe(H)IrH2(PMe3)3][BArF4] (13), [TbbSn(μ-H3)Ir(PMe3)3][BArF4] (14), and [TbbSn(H)IrH2(PMe3)3][BArF4] (15). 14 isomerizes to give 15via an 1,2-H shift reaction. Hydride addition to cation 3 gives a mixture of products [TbbGeHIrH(PMe3)3] (16) and [TbbGeIrH2(PMe3)3] (17) and a reversible 1,2-H shift between 16 and 17 was studied. In the tin case 4 the dihydride [TbbSnIrH2(PMe3)3] (18) was isolated exclusively. The PMe3 and PEt3 derivatives, 18 and [TbbSnIrH2(PEt3)3] (19), respectively, could also be synthesized in reaction of [TbbSnH2]− with the respective chloride [(R3P)nIrCl] (R = Me, n = 4; R = Et, n = 3). Reaction of complex 19 with CO gives the substitution product [TbbSnIrH2(CO)(PEt3)2] (20). Further reaction with CO results in hydrogen transfer from the iridium to the tin atom to give [TbbSnH2Ir(CO)2(PEt3)2] (21). The reversibility of this ligand induced reductive elimination transferring 20 to 21 is shown.
Introduction
Tetrylidyne complexes are heavier homologs of the parent carbyne complexes and exhibit a triple bond between a low-valent Group 14 element and a transition metal.1 With the synthesis of [Cp(CO)2Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) GeAr′] Power and co-workers have reported a pioneering example featuring a MoGe triple bond with a nearly linear C–GeMo unit.2,3 Filippou et al. developed the chemistry of the tetrylidyne coordination compounds and presented a broad variety of fascinating examples exhibiting a central triple bond unit ME with combinations between elements: M = Nb, Cr, Mo, W, Mn, Re, Fe, Ni, Pt and E = Si, Ge, Sn, Pb.4–19 The following synthetic procedures for tetrylidyne complexes were presented in the literature (Scheme 1): (1) starting with a metalloylidene complex, elimination of transition metal coordinated carbon monoxide yields the tetrylidyne complex;2,3,6,20 (2) addition of organotetrylene halides to transition metal complexes and elimination of nitrogen or phosphine ligands;5,8,9,12–19 (3) halide, hydride or NHC abstraction from ylidene complexes;4,7,10,11,21–24 (4) Tobita et al. reported dehydrogenation or stepwise proton and hydride abstraction;25–28 (5) metathetical exchange between metal–metal triple bonds;29 (6) hydrogen transfer in reaction of dihydride complexes with styrene.30
GeAr′] Power and co-workers have reported a pioneering example featuring a MoGe triple bond with a nearly linear C–GeMo unit.2,3 Filippou et al. developed the chemistry of the tetrylidyne coordination compounds and presented a broad variety of fascinating examples exhibiting a central triple bond unit ME with combinations between elements: M = Nb, Cr, Mo, W, Mn, Re, Fe, Ni, Pt and E = Si, Ge, Sn, Pb.4–19 The following synthetic procedures for tetrylidyne complexes were presented in the literature (Scheme 1): (1) starting with a metalloylidene complex, elimination of transition metal coordinated carbon monoxide yields the tetrylidyne complex;2,3,6,20 (2) addition of organotetrylene halides to transition metal complexes and elimination of nitrogen or phosphine ligands;5,8,9,12–19 (3) halide, hydride or NHC abstraction from ylidene complexes;4,7,10,11,21–24 (4) Tobita et al. reported dehydrogenation or stepwise proton and hydride abstraction;25–28 (5) metathetical exchange between metal–metal triple bonds;29 (6) hydrogen transfer in reaction of dihydride complexes with styrene.30
 | ||
| Scheme 1 Syntheses of tetrylidyne metal complexes (X = halide, hydride; L′ = N2, phosphine). | ||
The metal-element triple bond usually consists of a σ-bond derived from the donation of a Group 14 element lone pair to the transition metal and two π-bonds, which result from transition metal d-orbital donation into the empty p-orbitals at the Group 14 element.19,24 In this continuum (Scheme 2) of π-backbonding interactions the electronic situation of the participating fragments play a key role and can give rise to potential fluctional internal frustration resulting in increased reactivity of the multiple bond.23
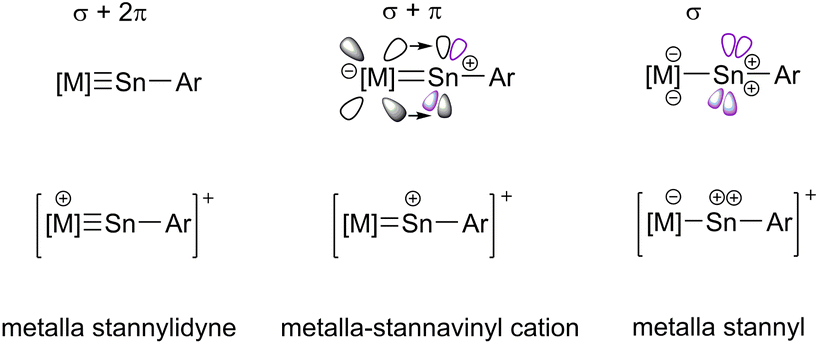 | ||
| Scheme 2 Bonding in stannylidyne complexes, continuum of π-backbonding. (Violet coloured orbital at Sn: without backdonation from transition metal). | ||
Results and discussion
Motivated by our recent progress in tetrylidyne rhodium chemistry we started a project to investigate multiple bonds between iridium and heavy Group 14 elements.30 In our initial efforts we reacted low valent organo element bromides of germanium [(TbbGeBr)2] and tin [TbbSnBr2][Li(thf)2] with the iridium hydride [(Me3P)4IrH] (Scheme 3 and Table 1).31,32,33 A phosphine was substituted against the bromoorgano tetrylene and the tetrylidene compounds [(TbbBrE)IrH(PMe3)3] (E = Ge: 1, E = Sn: 2, see Scheme 3 for depiction) were obtained in high yield (1: 96%, 2: 95%). In the case of the germanium derivative 1, determination of the molecular structure in the solid state was possible (see Table 2 and ESI† for details). The Ir–Ge distance in 1 of 2.2879(3) Å is a short distance between these elements (for comparison [{PhB(CH2PPh2)3}IrH2GeMes2] [Ge–Ir 2.339(1) Å],34–36 based on a CSD search, a shorter Ir–Ge interatomic distance was not reported) and the Ge atom shows a trigonal planar arrangement. Therefore, the bonding situation in the Ge–Ir complex was also investigated using DFT calculations in combination with NBO37 analysis, pointing toward a germylene acting as a σ-donor and π-acceptor ligand (see ESI†).38,39 The stannylene complex 2 exhibits a characteristic signal in the 119Sn-NMR spectrum at 566 ppm indicative for stannylene coordination.40,41 To synthesize the desired tetrylidyne iridium complexes, elimination of HBr was explored for both complexes 1 and 2. However, these experiments with strong bases like benzyl-potassium, KOtBu or Ph3CLi, were not successful. Instead, halide abstraction with Na[BArF4] was straightforward (Scheme 3) and the cationic iridium hydride complexes were isolated as [BArF4]-salts in high yield (3 Ge: 89%, 4 Sn: 88%). Examples of cationic transition metal hydride complexes featuring an M–E multiple bond with Group 14 elements are present in the literature (IrH–Si,42,43 MoH–Si,21 FeH–Sn,23 and OsH–Si22).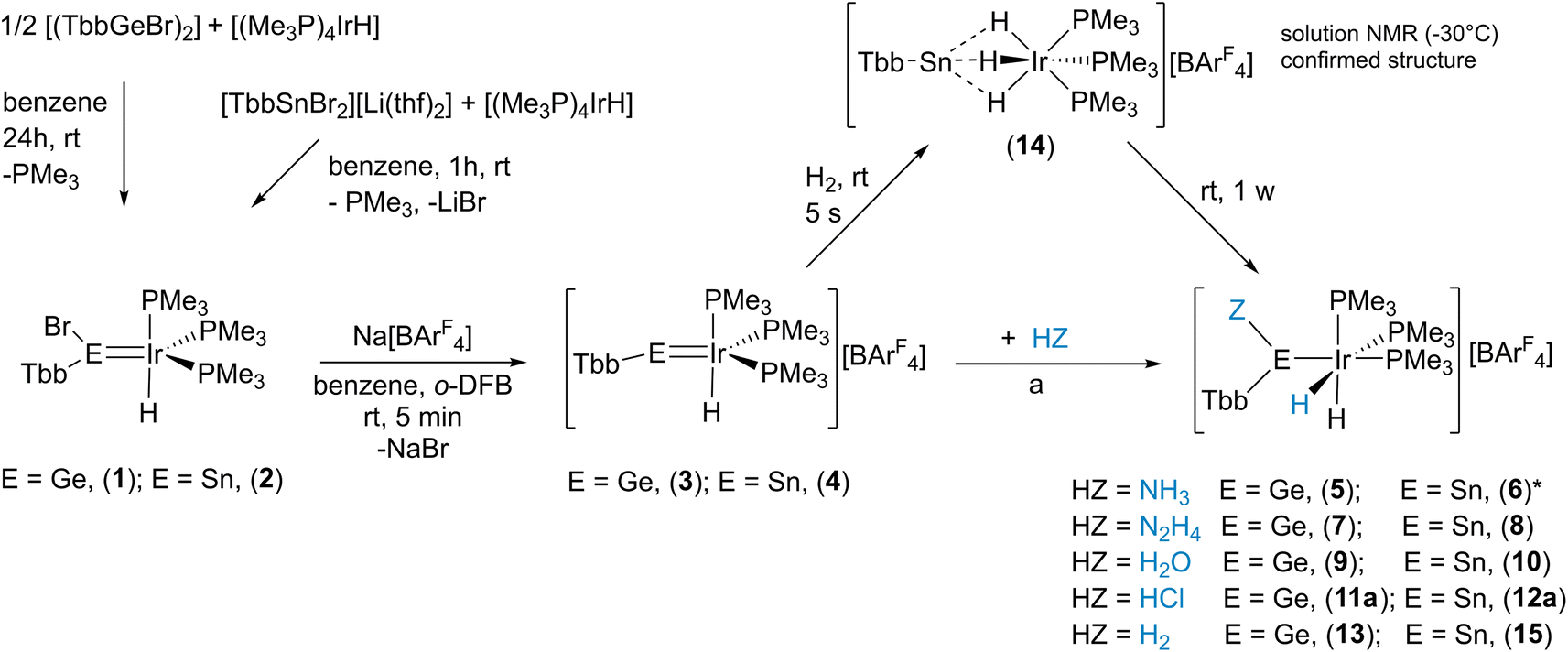 | ||
Scheme 3 Syntheses of 1, 2 and bromide abstraction to give tetrylidinium coordination compounds 3, 4. Reactivity studies of tetrylidinium–iridium cations (3, and 4). a: HZ = NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH3 (g), benzene/o-DFB 0.3/0.05 mL, rt, 5 s; HZ = N2H4:N2H4 (1 M thf), benzene/o-DFB 1:1, rt, 5 s; HZ = H2O:BaCl2·H2O, o-DFB, rt, 1 h; HZ = HCl:HCl·Et2O (1 M), o-DFB, rt, 5 s; HZ = H2:H2, rt 24 h (E = Ge). (6)*: 6 was not isolated, instead a reaction mixture between 4 and excess NH3 was characterized (vide infra). [o-DFB = 1,2-difluorobenzene, ArF = C6H3-3,5-(CF3)2]. :NH3 (g), benzene/o-DFB 0.3/0.05 mL, rt, 5 s; HZ = N2H4:N2H4 (1 M thf), benzene/o-DFB 1:1, rt, 5 s; HZ = H2O:BaCl2·H2O, o-DFB, rt, 1 h; HZ = HCl:HCl·Et2O (1 M), o-DFB, rt, 5 s; HZ = H2:H2, rt 24 h (E = Ge). (6)*: 6 was not isolated, instead a reaction mixture between 4 and excess NH3 was characterized (vide infra). [o-DFB = 1,2-difluorobenzene, ArF = C6H3-3,5-(CF3)2]. | ||
| 1H Ir–H δ | 2 J SnH | 2 J PH | 2 J HH | 1H E–Hδ | J SnH | 2 J PH | J HH | 31P δ | 2 J SnP | 119Sn δ | 2 J SnP | J SnH | δ calc. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a 119Sn DFT calculations of [TbbSn(NH2)IrH2(PMe3)3]+, calc. 709 ppm; [TbbSn(NH3)(NH2)IrH2(PMe3)3]+, calc. 88 ppm. | ||||||||||||||
| 1 | −11.82 | 20.8 | −45.6 | |||||||||||
| 2 | −12.65 | 219 | 18.6 | −43.7 | 598 | 566 | 603 | 218 | ||||||
| 3 | −11.87 | 8.8 | −43.9 | |||||||||||
| 4 | −11.30 | 119 | 10.3 | −38.5 | 347 | 1424, 1284 Solid | 347 | Broad | 1449 | |||||
| 5 | −12.80 | −62.3, −53.8 | ||||||||||||
| 4/NH3 | −13.67 | 127 | 11.4 | −60.9, −50.1 | 135, 1827 | −11 | 1831 | Broad | ||||||
| 7 | −13.03 | −62.3, −53.7 | ||||||||||||
| 8 | −13.47 | 251 | −60.1, −45.5 | 144, 1680 | 604 | 1682, 142 | 673.8 | |||||||
| 9 | −12.69 | −58.7, −51.9 | ||||||||||||
| 10 | −13.29 | −56.3, −43.8 | 124, 1714 | 648 | 1730, 128 | 778.4 | ||||||||
| 11a | −11.21 | −57.5, −50.9 | ||||||||||||
| 12a | −12.98 | 255 | −57.5, −42.6 | 119, 144 1714, 1777 | 772 | 1784, 126 | ||||||||
| 13 | −11.90 | 13.98 | 32.6, 7.6 | −54.0, −48.8 | ||||||||||
| 14 | −7.94 | 265 | −45.2 | 1592 | Broad | |||||||||
| 15 | −12.67 | 195 | 18.59 | 772 | 37.5, 4.7 | −54.9, −41.6 | 104, 1509 | 1013 | 1496 | 775 | ||||
| 16 | −10.79 | 12.04 | 13.6 | 3.5 | −42.1 Broad | |||||||||
| 17 | −11.07 | −58.8, −30.0 | ||||||||||||
| 18 | −12.10 | −60.9, 24.4 | 209, 320 | 3835 | 327 | |||||||||
| 19 | −13.26 | −26.4, 62.1 | 3827 | 458 | ||||||||||
| 20 | −10.49 | 21.3, 15.6 | 3.7 | −17.1, 42.9 | 336 | 3321 | 336 | |||||||
| −9.83 | 109, 17.1 | 3.7 | ||||||||||||
| 21 | −16.5 | 320, 305 | −388 | 320 | 1406 | |||||||||
In the crystalline state both products 3 and 4 exhibit a major disorder of the entire molecule and poor crystallinity. The TbbSn derivative 4 and the Ar*Ge complex 3′ (the terphenyl substituent Ar* was also employed to check for possible non-disordered structures of 3′ and 4′; Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl) are shown in Fig. 2. Syntheses of 3′ and 4′ were carried out using analogous starting materials and comparable procedures (see ESI† for details). However, in the case of 4′ we also developed a further synthesis employing the cation [Ar*Sn]+ reacting as a nucleophile substituting a phosphine ligand (Scheme 4). In the molecular structures of 3′ and 4 the absence of the bromide and the large C–E–Ir angle (>160°) are characteristic features for these cations. The found interatomic distances of Ge–Ir 2.225(2) Å and Sn–Ir 2.4135(5) Å are short bond lengths between these elements (Table 2).34,44–47 (The data of 3′ and 4 are results of disordered structures, 4: distance of the 90% component is shown).
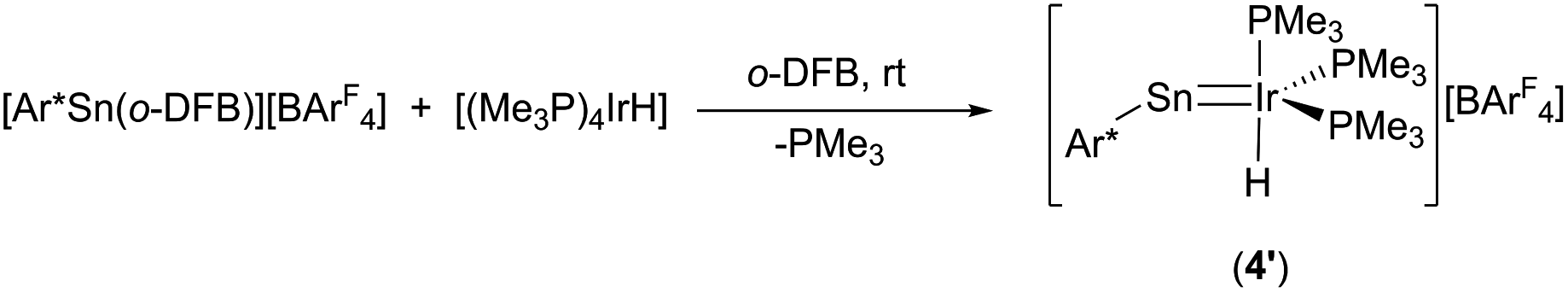 | ||
| Scheme 4 Synthesis of 4′via substitution. | ||
| Ir–E | E–X | E–C | Ir–E–C | Ir–P1 | Ir–P2 | Ir–P3 | Ir–C2 | C2–O1 | |
|---|---|---|---|---|---|---|---|---|---|
| a Molecular structures of 3′, 4, 5, 9, 10, 12b, 13, 19–21 show a disorder in the solid state. | |||||||||
| 1 E = Ge, X = Br | 2.2879(3) | 2.4562(4) | 1.982(3) | 140.8(1) | 2.2687(10) | 2.2791(9) | 2.3157(9) | ||
| 3′ E = Ge | 2.225(2) | 1.948(4) | 158.8(4) | 2.2906(12) | 2.2868(12) | 2.3166(14) | |||
| 4 E = Sn | 2.4135(5) | 2.133(5) | 159.2(1) | 2.2759(15) | 2.2832(15) | 2.3400(13) | |||
| 5 E = Ge, X = NH2 | 2.3538(4) | 1.778(2) | 1.944(2) | 130.9(1) | 2.3365(7) | 2.3092(7) | 2.3345(7) | ||
| 7 E = Ge, X = N2H3 | 2.3492(4) | 1.792(4) | 1.957(4) | 129.8(1) | 2.3543(12) | 2.3075(11) | 2.3372(10) | ||
| 9 E = Ge, X = OH | 2.3344(3) | 1.770(2) | 1.948(3) | 134.9(1) | 2.3421(8) | 2.3524(8) | 2.3052(8) | ||
| 10 E = Sn, X = OH | 2.5078(4) | 1.995(3) | 2.126(2) | 138.89(7) | 2.3389(10) | 2.2849(9) | 2.3508(9) | ||
| 12b E = Sn, X = Br | 2.5167(3) | 2.5387(6) | 2.130(4) | 137.36(11) | 2.3463(11) | 2.2955(12) | 2.3351(12) | ||
| 13 E = Ge, X = H | 2.3400(6) | 1.66(6) | 1.946(5) | 130.8(1) | 2.3394(15) | 2.3428(15) | 2.3151(15) | ||
| 19 E = Sn | 2.6353(2) | 2.229(3) | 115.0(1) | 2.3414(7) | 2.3249(6) | 2.3400(7) | |||
| 20 E = Sn | 2.5968(3) | 2.207(3) | 106.6(1) | 2.3209(9) | 2.4164(10) | 1.891(6) | 1.144(8) | ||
| 21 E = Sn, X = 2H | 2.6598(2) | 1.78(2) | 2.181(2) | 123.5(1) | 2.3434(6) | 2.3203(7) | 1.892(2) | 1.156(3) | |
In the 1H NMR spectrum the signal for the hydride was observed at low frequencies (Tables 1 and 3: q, −11.87 ppm, 2JP–H = 8.8 Hz; 4: q, −11.30 ppm + sat., IrH, 2JP–H = 10.3 Hz, 2JSn–H = 119 Hz) and in the 119Sn NMR spectrum of 4 (solution as well as solid state NMR data) a shift to high frequencies (1424 ppm) in comparison to starting material 2 was observed (Table 1). In compounds 1–4, the Ir atom is trigonally bipyramidally coordinated by three phosphine ligands, a hydride, and the Group 14 moiety. However, the hydride signal in the 1H NMR spectrum featuring a quartet due to coupling with the 31P nuclei and the 31P-NMR spectrum showing one resonance for the three phosphine ligands indicate time averaged high symmetry of compounds 1–4. To evaluate the presumed dynamic process in solution, temperature dependent NMR spectroscopy was carried out. Because the solubility of salts 3 and 4 was limited at low temperatures and compounds 1–4 exhibit the same ligand arrangement at iridium and comparable Ir–H hydride resonances and 31P NMR spectra low temperature experiments were carried out with a sample of 1.
Variable temperature 31P{1H} and 1H NMR studies of 1 at 11.75 T indeed demonstrate fluxional behavior (Fig. 1). At 299.7 K, this process makes the three phosphine ligands chemically and magnetically equivalent, resulting in a singlet in the 31P{1H} and a quartet in the 1H NMR spectra. However, cooling the sample to 190.0 K slows down the exchange sufficiently such that no phosphine ligand exchange is measurable and the 31P{1H} NMR spectrum corresponds to an AB2, the 1H NMR spectrum to an AB2X spin system. One rare conspicuous feature of the AB2X spin system is the asymmetry within the multiplet, partially reproduced by the simulation (Fig. 1b, bottom). It depends on the ratio of the coupling constants to the shift difference δAB, as well as on the relative signs of these parameters.48–50 Given that the sign of δAB is known from 31P NMR spectroscopy, the signs of the coupling constants are absolute. Additional spectra and simulations at intermediate temperatures are collected in the ESI.†
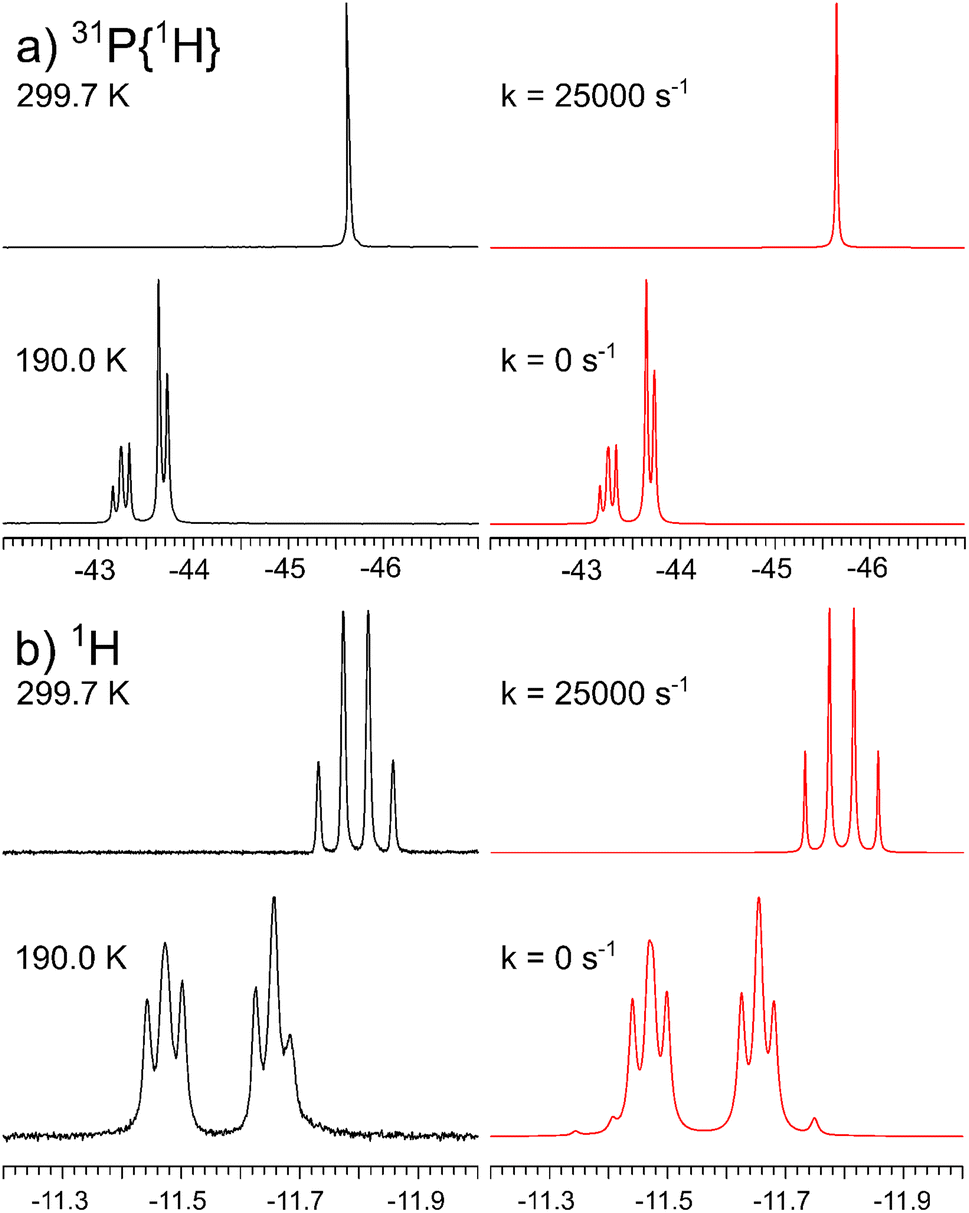 | ||
| Fig. 1 Experimental (left) and simulated (right) variable temperature (a) 202.46 MHz 31P{1H} and (b) 500.13 MHz 1H NMR spectra of 1. | ||
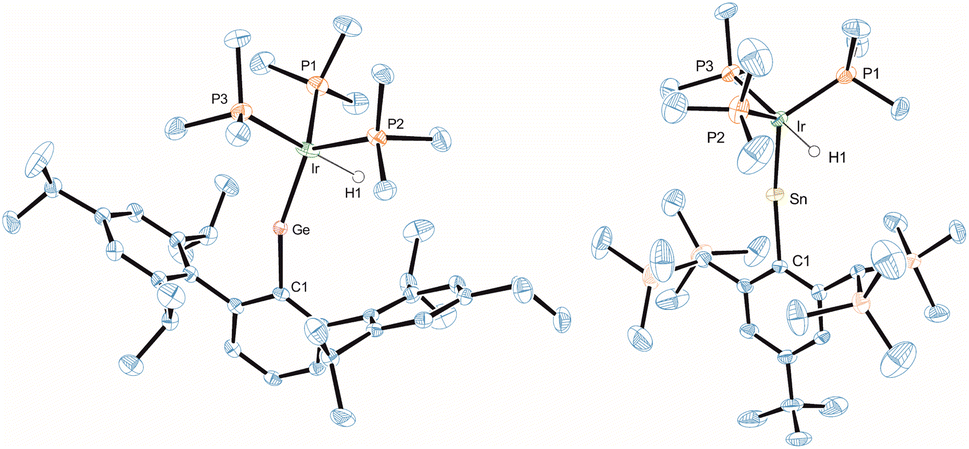 | ||
| Fig. 2 ORTEP of the molecular structure of the cation of 3′ and 4. Ellipsoids set at 50% probability. Hydrogen atoms except Ir–H are omitted for clarity. For selected interatomic distances and angles see Table 2. | ||
The short Ir–E bond length and particularly the crystallographically confirmed large Ir–E–C angle of ca. 160° are indicative for a tetrylidyne moiety and we were interested in addressing the bonding situation computationally by means of DFT. Interestingly, standard structure optimizations with a usually robust functional for organometallics such as BP86 (including dispersion corrections) fail completely to reproduce this large angle and predict (for example for tin compound 4) an Ir–Sn–C angle of 120°, along with a deviating T-shape geometry of the phosphine ligands at the Ir-atom. Such small angles would instead be more indicative of metallo-stannylene rather than a metallastannylidyne. Hybrid or range separated functionals such as M06-2X or ωB97X however optimized to local minima with angles around 160°. Compared to this experimental structure with a more linear coordination environment as also found by ωB97X, the more bent metallo-stannylene isomer structure (predicted by BP86 optimizations) was found the more stable structure by both model chemistries by respective single-point calculations. However, the extent of favourisation of the more bent-structure differed significantly between the two functionals (17.2 kcal mol−1 BP86; 2.7 kcal mol−1 ωB97X).
To finally corroborate the validity of the linear structure model as the dominant species beyond the single experimental data point from the X-ray geometry, we compared computationally predicted 119Sn-NMR chemical shifts and their anisotropic shielding tensors (Table 3) with the experimentally determined isotropic shifts from solution as well as from solid-state NMR-spectroscopy. The experimental isotropic shift of δexp(119Sn) = 1424 ppm (solution)/1284 ppm (solid state) is computationally (ADF2022, revPBE/TZ2P) very well reproduced for optimized structures in which the experimental angle (ca. 160°) was constrained (δcomp(119Sn) = 1449 ppm) as opposed to freely optimised structures with a metallo-stannylene-type angle (ca. 120°, δcomp(119Sn) = 2125 ppm). Moreover, the respective computational anisotropic magnetic shielding tensor (Table 3) matches the experimentally approximated tensor from 119Sn-solid-state NMR. This further experimental insight corroborates the anticipated relevance of the near-linear structure both in solid state and in solution.
| [C–Sn–Ir]° | δ iso [ppm] | δ 11 [ppm] | δ 22 [ppm] | δ 33 [ppm] |
|---|---|---|---|---|
| Exp. (4) 160 | 1284 | 2622 | 2622 | −978 |
| Calc. 160 | 1449 | 3108 | 2548 | −1308 |
| Calc. 120 | 2125 | 2467 | 2127 | 1780 |
Due to the ambiguity in the computational reproduction of the nature of the Ir–E interaction, we further probed the electronics by means of EDA-NOCV analysis and NBO analysis of a reduced model system [(Me3P)3(H)Ir–Sn–Me]+. Again, BP86 optimizes to a strongly bent structure (Ir–Sn–Me angle 107°) where ωB97X predicts an almost linear structure (172°). This probe with essentially non-bulky Me-substituents indicates that influences of dispersion or ligand repulsion are not key to the structural differences observed and that electronics must play a decisive role. NBO analysis (ωB97X-def2-SVP/TZVP(Sn,Ir)) of [(Me3P)3(H)Ir–Sn–Me]+ with an Ir–Sn–C angle constrained to the experimental value (160°) suggests a separation into trigonal-bipyramidal d8-[Ir(H)(PMe3)3] and [:Sn–Me]+ fragments with only very weak and very polarised bonding interactions of a σ-donating s-orbital based Sn-lone pair of electrons into a sd-hybrid orbital at the irdium atom, and only one inversely polarised π-backdonation from an Ir-d-orbital into one empty p-orbital at the Sn-atom, leaving an empty p-orbital at the tin atom (Fig. 3).
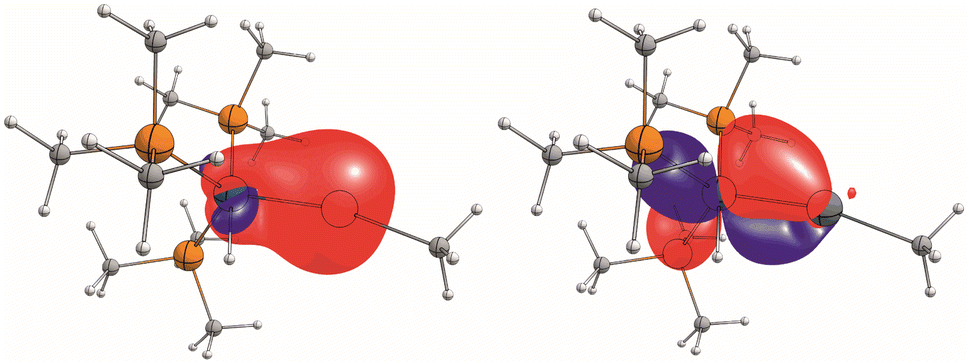 | ||
| Fig. 3 Depiction of NLMOs with significant contributions of Sn and Ir. Left: 82% Sn (s)/11% Ir (sd); right 83% Ir (d)/13% Sn(p); isovalues at. 0.05 a.u. | ||
EDA-NOCV analyses (ωB97X-def2-SVP/TZVP(Sn,Ir)) on the same structure with a respective fragment partitioning of a neutral [Ir(H)(PMe3)3] fragment and a cationic [:Sn–Me]+ gave an essentially identical picture with three leading interactions (see the ESI† for further details). The deformation densities of these interactions are shown in Fig. 4. Energetically, the π-backdonation contributes almost equally to the overall Sn–Ir bonding as the σ-donation contribution which is, for symmetry reasons, divided into two individual NOCV-contributions.
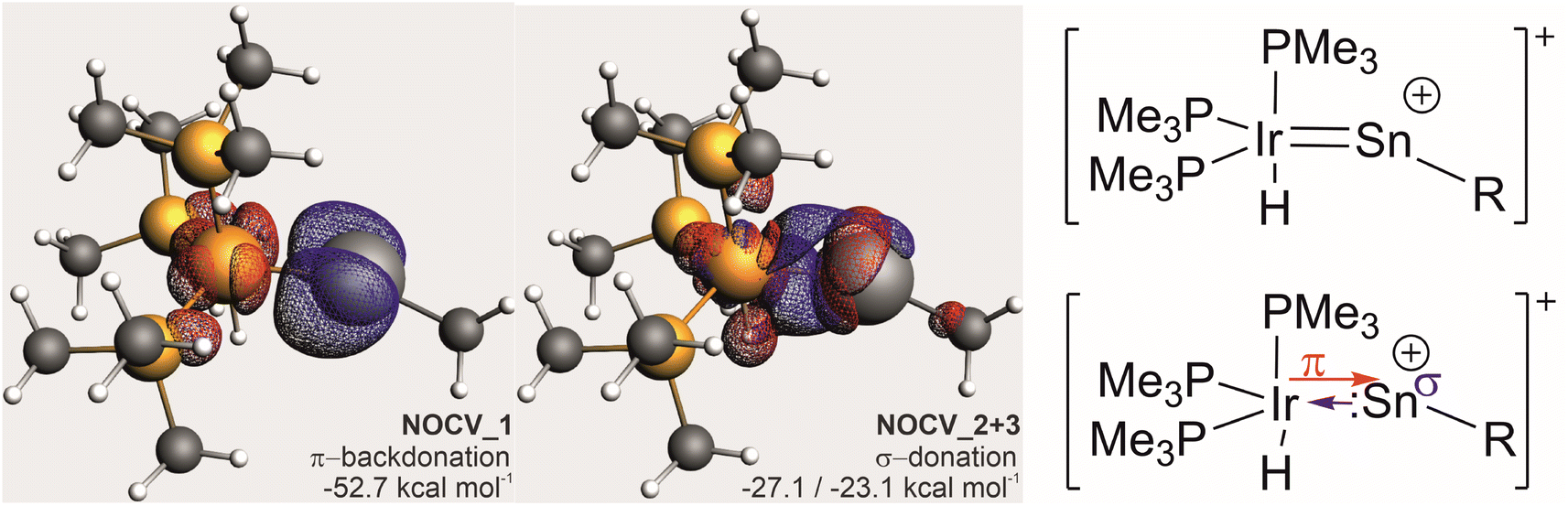 | ||
| Fig. 4 (Left): Depiction of deformation densities of leading interactions from EDA-NOCV analysis (isovalues at 0.004 a.u.; electron flow: red to blue). NOCV2 and 3 are combined in one depiction. (Right) Proposed leading bonding description for cation 4. | ||
From this computational insight, we conclude that there is no true triple-bonding between Ir and Sn but rather a double bond comprising of one σ- and one π-bond and an empty p-orbital at a near-linear coordinate Sn-atom with the cationic charge primarily residing at the Sn-atom. We would thus propose to see this moiety as a (metalla-)stannavinyl cation. This cationic nature of the Sn-site may also explain the electrophilicity and regioselective bond-additions in subsequent reactions (vide infra).
To further corroborate our assignment of the charges in the IrGe/Sn cation fragments of 3 and 4, NPA charges have been computed. In each case (3 Ge: +0.91/Ir: −0.5; 4 Sn: +1.18/Ir: −0.6) a polar distribution of charges is observed with a clearly negative Ir-atom and positive charge at the tetrel atom. This is in line with our rationale of the experimental observation involving a Lewis-acidic, positively charged tetrel atom as key center of reactiviy in these compounds. A slightly less pronounced distribution in the Ge-case may result from more effective π-overlap and thus increased Ir→Ge electron backdonation.
Because of the interesting bonding situation and electrophilicity of cations [Ge–Ir] 3 and [Sn–Ir] 4 we set up a systematic investigation of their chemical properties. Both cations have a good preparative accessibility [yield of 89% (3) and 88% (4)] enabling a study of their chemistry. The reactivity of 3 and 4 was explored in reactions with ammonia, hydrazine, water, hydrogen chloride, and hydrogen (Scheme 3). At ambient temperature with ammonia, hydrazine, water, or hydrogen chloride, both cations show a spontaneous heterolytic addition reaction across the E = Ir bond (Scheme 3). Upon heterolysis of the N–H bond, NH3 and N2H4 react with the electrophilic Group 14 element moiety to give an aryl amido/hydrazido tetrylene. The proton adds to the iridium fragment [IrH(PMe3)3] to result in a formal oxidation of Ir(I) to Ir(III). The reaction products ([Ge–NH2] 5, [Ge–N2H3] 7, [Sn–N2H3] 8) were characterized by NMR spectroscopy and the solid state molecular structures of 5 and 7 were determined by X-ray diffraction (Fig. 5). The Ir–Ge bond lengths found in 5 and 7 (Table 2) can be compared with interatomic Ge–Ir distances found for germylene–iridium coordination compounds.44,51 The Ge–N bond lengths of addition products [Ge–NH2] 5 and [Ge–N2H3] 7 exhibit Ge–N distances lying in the range [1.76(1) – 1.878(3) Å] of literature examples.52–58 The Ge atom in 5 and 7 is trigonal planar coordinated, and the iridium atom shows an octahedral ligand arrangement with the phosphine ligand in a facial coordination. DFT calculations using ORCA were performed to evaluate the coordination of the aryl amido- and hydrazido germylene at iridium. Based on the results of NBO analysis a partial delocalization of an electron pair of the NHR-substituent and Ir-fragment into the empty p-orbital at Ge was found (see ESI† for further details). The tin reaction product [Sn–N2H3] 8 was also characterized by 119Sn NMR spectroscopy (Table 1) and the chemical shift can be compared with the calculated isotropic shift (8: exp. 604 ppm, calc. 674 ppm). In the case of the reaction of 4 with NH3 a product structure analogous to the germanium NH3-addition product [Ge-NH2] 5 was assumed. However, the comparison between experimental 119Sn NMR data of the reaction mixture 4/excessive NH3 (exp. −11 ppm) and calculated NMR data based on the optimized structural model of 5 after replacement of the Ge atom against a Sn Atom ([TbbSn(NH2)IrH2(PMe3)3]+, calc. 709 ppm) exhibits a large difference. The low frequency signal of the mixture 4/NH3 at −11 ppm points toward an NH3 adduct [TbbSn(NH3)(NH2)IrH2(PMe3)3]+ to be present in NMR solutions with excess NH3. Therefore, this presumed NH3 adduct [TbbSn(NH3)(NH2)IrH2(PMe3)3]+ (structure optimized by DFT, 119Sn NMR by ADF: 88 ppm) was calculated by DFT methods and shows a calculated 119Sn NMR chemical shift (88 ppm) in good accordance to the experimental signal (−11 ppm). Furthermore, based on DFT calculations the NH3 adduct formation is an exergonic (ΔG = −3.94 kcal mol−1, BP86-D3BJ/def2SVP) reaction making the assumption of an adduct formation feasible. However, isolation of this adduct under an atmosphere of ammonia was not possible. The product of hydrolysis, compound 10 [Sn–OH], crystallized at −40 °C after several days.
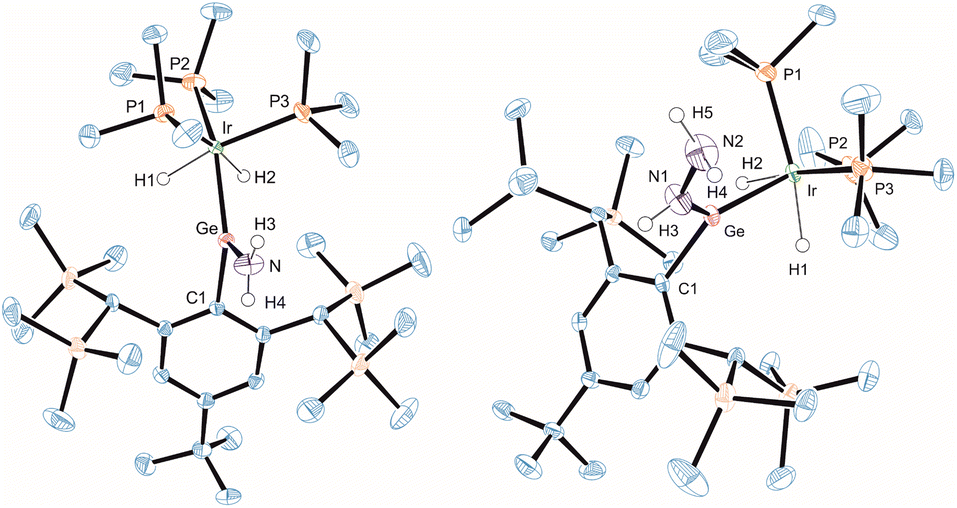 | ||
| Fig. 5 ORTEP of the molecular structures of 5 and 7. Ellipsoids set at 50% probability. Hydrogen atoms except N–H and Ir–H are omitted for clarity. For selected interatomic distances and angles see Table 2. | ||
Activation of ammonia and hydrazine is a very active field of research.59–64 Besides coordination induced bond weakening (Scheme 5e),59–61 oxidative addition at transition metal complexes65–69 or main group element compounds56,57,70–80 (Scheme 5a and b), cooperative reactions with metal–ligand systems81–85 and a N-heterocyclic germylene55 were reported (Scheme 5c and d). These literature examples of cooperative metal–ligand reactivity exhibit formation of a M–NH2 moiety (M = Ru, Rh, Ir, Ni; Ge) and transfer of a hydrogen atom to the ligand. Complexes 5, 7, 8 are the products of a cooperative heterolytic metal–ligand cleavage of a N–H bond and formation of an Ir–H and E-NHR moiety (R = H, NH2; E = Ge, Sn) was observed (Scheme 3).86,87 Obviously, the cations [Ge–Ir] 3 and [Sn–Ir] 4 exhibit metal–ligand cooperativity with the main group element ligand reacting as an electrophile (Schemes 3 and 5f).88 Metal–Lewis acid cooperativity was established for Lewis acidic triply coordinate aluminium or boron ligands and the coordination compounds were shown to activate hydrogen, water and C–H bonds.86,89–94
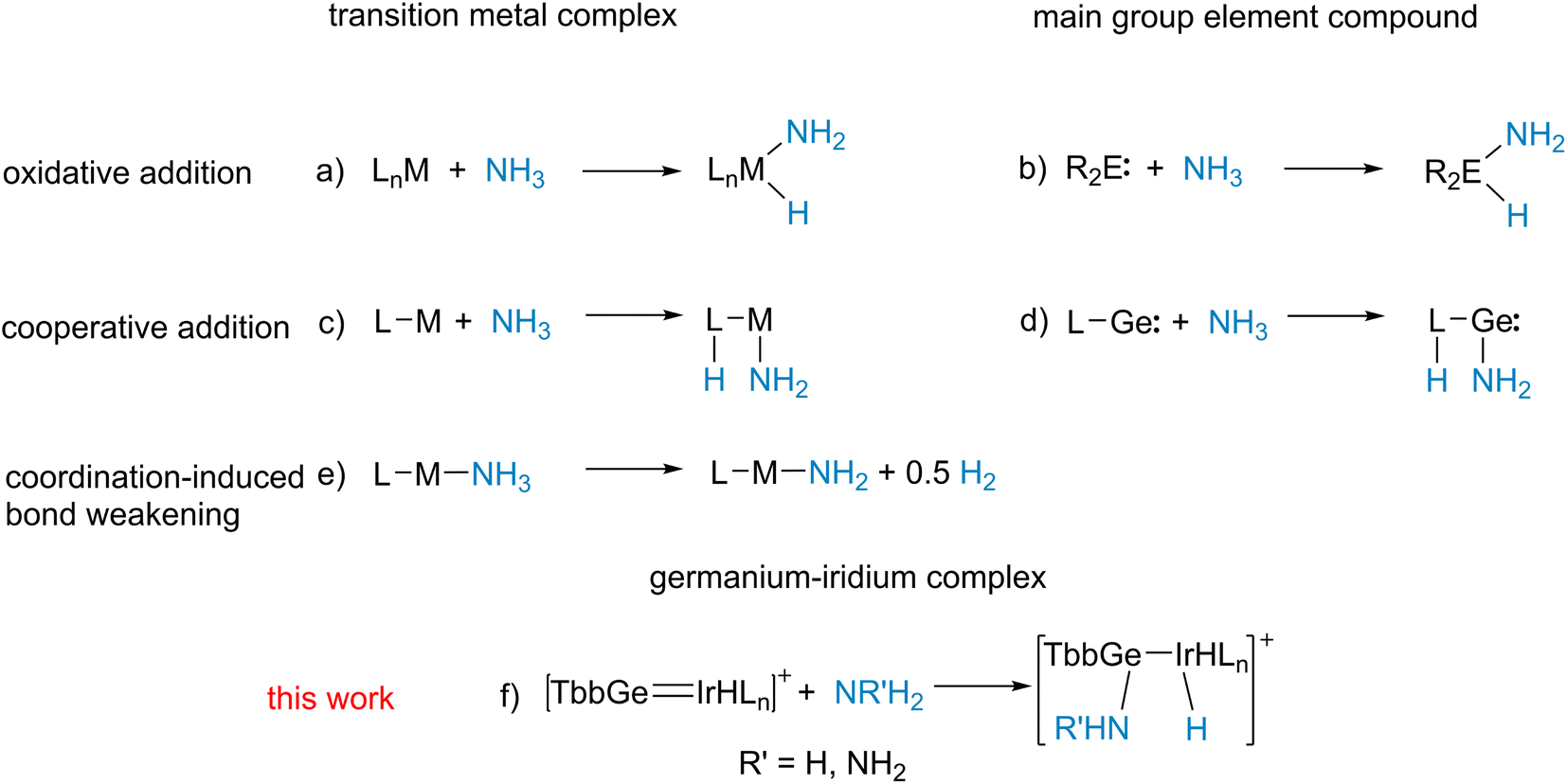 | ||
| Scheme 5 Reactions of ammonia with transition metal complexes and main group element compounds. | ||
Both cations [Ge–Ir] 3 and [Sn–Ir] 4 add water in reaction with two equivalents of adduct BaCl2·H2O in o-DFB. The BaCl2 adduct of water was used because the much higher molecular weight of the adduct allows a much more precise weighing (based on NMR spectroscopy, in the case of 3 with an excess of water the same selective product formation was found). The water molecule exhibits a comparable reactivity like NH3 and N2H4. In the product complexes [Ge–OH] 9 and [Sn–OH] 10 a hydroxide unit is bound at the Group 14 element and the proton is coordinated at the iridium atom. The molecular structure of the water addition at the germanium cation 9 is shown in Fig. 6 (structure of 10 see ESI†). The Ge–O distance found in complex 9 can be compared with a variety of organogermanium hydroxide structures.95–98 The found hydroxide coordination compounds are rare examples for Group 14 element hydroxides of coordination number three.97 Tetracoordinate low valent germanium and tin hydroxides, however, are well known in the literature.99–103 Products 9 and 10 exhibit in the 1H NMR spectrum a characteristic OH resonance (9: 5.29 ppm, d, 4JPH = 3.8 Hz; 10: 4.11 ppm, br. s). The signal in the 119Sn NMR spectrum was observed at 648 ppm, lying in the range of the calculated isotropic shift of 778 ppm (see ESI† for details). Water addition at transition metal complexes was studied intensively and in the case of cooperative transition metal/Lewis base bond activation, the hydroxide forms a bond with the transition metal.82,104–106 Figueroa et al. have presented an example for cooperative platinum/borane water activation. As found in the products 9 and 10, in this platinum/borane example, the hydroxide forms a bond to the electrophilic ligand.92
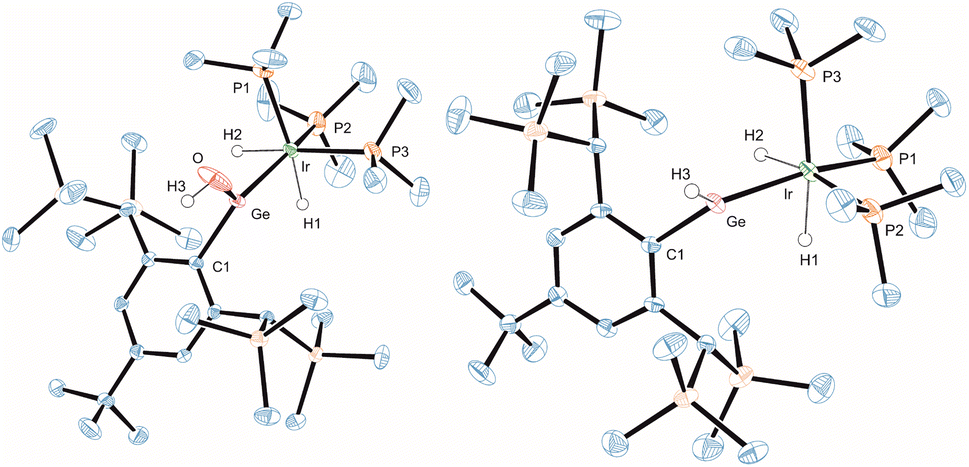 | ||
| Fig. 6 ORTEP of the molecular structures of 9 and 13. Ellipsoids set at 50% probability. Hydrogen atoms except OH, GeH, and Ir–H are omitted for clarity. For selected interatomic distances and angles see Table 2. | ||
Products of HCl addition 11a and 12a show in both cases protonation of the iridium atom and chloride addition to the Group 14 element (Scheme 3). The homologous bromide complex (12b) was synthesized by protonation of tetrylene coordination compound 2 with [H(OEt2)2][BArF4]. The molecular structure of the cation of bromide 12b is shown in the ESI.† In the 1H NMR spectrum for the IrH2 moiety, a signal at low frequencies was observed (Table 1) and in the case of the tin product a signal in the 119Sn NMR was found at 772 ppm (Table 1).
Cations [Ge–Ir] 3 and [Sn–Ir] 4 were also reacted with hydrogen at ambient temperatures and diverging reactivity with this unpolar substrate was found. The [Ge–Ir] 3 cation splits the hydrogen molecule to add a hydride at the germanium atom and a proton to the [IrH(PMe3)3] moiety (Scheme 3, 13). This type of cooperative transition metal/Lewis acid hydrogen activation was found only for transition metal–borane complexes so far.92,107–110 In the tin case, we found a stepwise reaction: first addition of H2 at iridium was observed (Scheme 3, 14), which can be compared with hydrogenation of a rhodium–tin triple bond in the stannylidyne complex [(Me3P)2(Ph3P)RhSnAr*] [see Scheme 1 reaction (6)] adding one equivalent of hydrogen at rhodium to give [(Me3P)2(Ph3P)RhH2–SnAr*].30 The iridium trihydride fragment [Ir(H)3(PMe3)3] of 14 (see Scheme 3 for depiction of 14) shows high symmetry in solution even at −30 °C (because of the low solubility of 14, NMR experiments below −30 °C were not possible): only one signal in the 1H NMR spectrum for the IrH3 unit at −7.94 ppm with tin satellites (1JSn–H = 265 Hz) and one signal in the 31P NMR spectrum (Table 1) were observed. The signal in the 119Sn NMR spectrum was found at 1592 ppm. We interpret these spectroscopic findings, which can be compared with the hydride bridged Rh-complex [Ar*Sn(μ-H2)Rh(PPh3)2] [1H: −4.13 ppm (JSn–H 220 Hz), 119Sn 1728 ppm],30 as an indicator for a triply hydride-bridged structure of 14 [TbbSn(μ-H3)Ir(PMe3)3]+. This type of [M(μ-H3)Ir(PEt3)3]+ structural motif is already known for a rhodium fragment [M = (dppe)Rh].111 Remarkably, after one week, cation 14 undergoes a slow hydrogen shift of one hydrogen atom from the bridging position [Sn(μ-H)Ir] to the tin substituent to give 15 (Scheme 3). Obviously, the hydridostannylene coordination at iridium (15) is energetically favoured over the hydrido-bridged structure (14).112–114 Complexes 13 and 15 are examples for hydridotetrylene coordination featuring a characteristic signal in the 1H NMR spectrum for the coordinated TbbE-H moiety at high frequencies (Table 1, 13: GeH 13.98 ppm; 15: SnH 18.59 ppm).23,42,113–117 The molecular structure of Ge-hydride 13 is shown in Fig. 6.
Finally, we also studied the addition of hydride (delivered by K[HBEt3]) to the iridium–tetrylidinium cations 3 [Ge–Ir] and 4 [Sn–Ir] (Scheme 6). In the germanium case a mixture of two products was isolated: the hydridogermylene complex (16, 50%) and the hydridoirido-germylene (17, 50%). Both products can be reversibly converted via an 1,2-H shift into each other (Scheme 6, see ESI† for NMR spectra of the transfer). Hydridogermylene 16, which is the more stable isomer, was converted under the influence of light (460 nm) into the irido-germylene 17 almost quantitatively. After 5 days gently heating at 40 °C the hydrogen atom is transferred back to the germanium to give 16 (at rt a 1:1 mixture with 17 was isolated). This type of reversible 1,2-H shift was also observed in the case of a tungsten–germylene complex.118 Chemistry of hydrido(hydridotetrylene)coordination compounds was intensively studied by Tobita and Tilley et al. for a variety of systems MH-EH (Mo–Ge,25 W–Si,116 W–Ge,27,119 Ru–Si,120 Os–Sn,114 Ir–Si42).
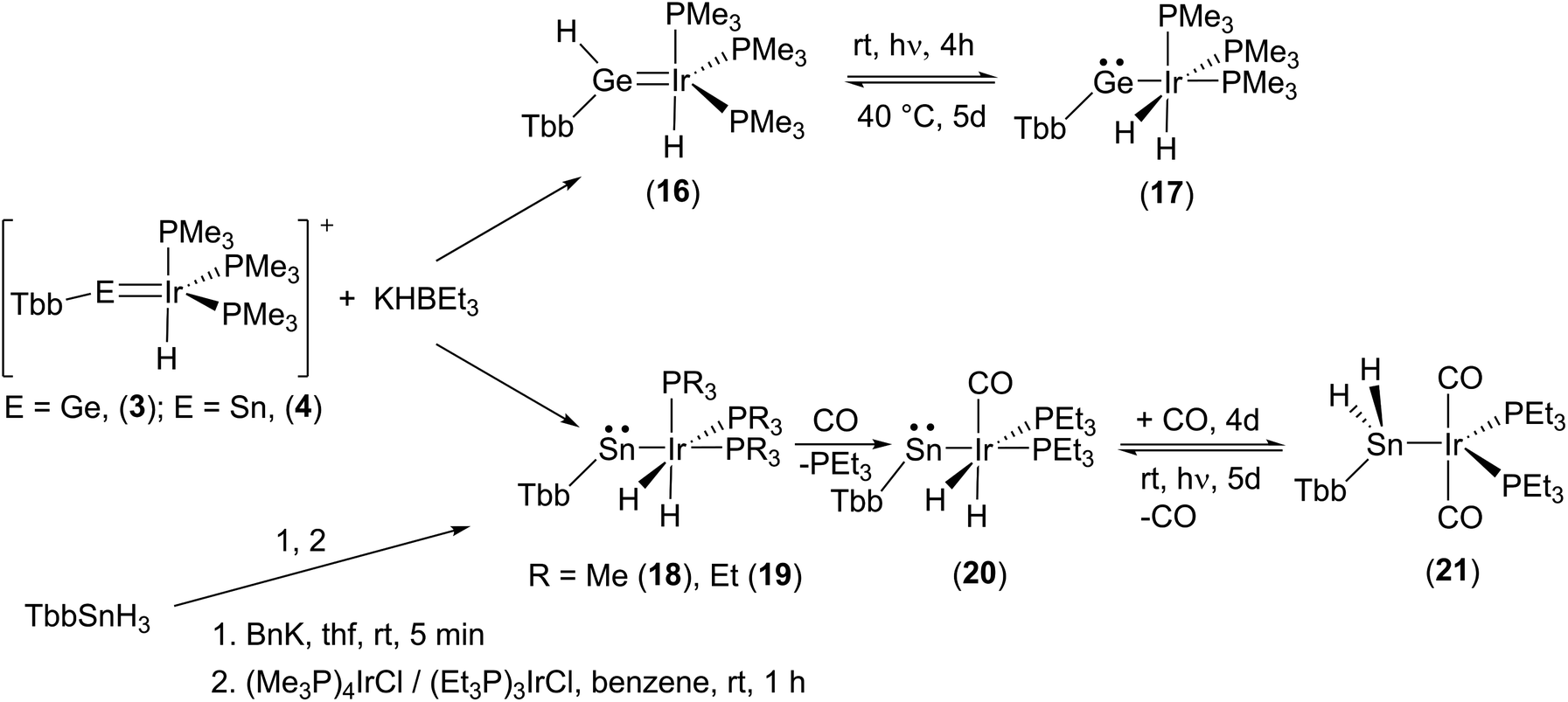 | ||
| Scheme 6 Hydride addition at cations 3 and 4. Reversible 1,2-H-shift reactions 16–17, hν = 460 nm. Ligand induced hydrogen transfer 20–21, hν = UV-lamp. | ||
To our surprise, the homologous tin cation 4 [Sn–Ir] adds the hydride almost exclusively at the iridium atom and only a slight amount of addition at the tin atom was observed by NMR spectroscopy in the course of the low temperature synthesis. An irido-stannylene (18) is formed, which was also synthesized with varying phosphines (18-PMe3, 19-PEt3) by an alternative approach starting from the trihydride TbbSnH3 (Scheme 6).33 Deprotonation using benzyl-potassium yields [TbbSnH2K] and is followed by reaction with the electrophile [(R3P)nIrCl] (R = Me n = 4, R = Et n = 3).121,122 During this synthesis of complexes 18 and 19, two hydrogen atoms show a 1,2-H shift from tin to iridium. Tilley et al. reported a stepwise 1,2-H shift of two hydrogen atoms from tin to iron, ruthenium, and osmium, respectively.23,113,114
The stability of hydridogermylene (16) and hydrido-metallostannylene (18) isomers goes along with findings from Tilley and co-workers.112–114,123,124
To investigate a possible transfer of the hydrogen atoms back to the tin, we started to exchange the phosphine ligands against carbonyl ligands. The aim was to destabilize the oxidation state three of iridium in complex 19 with the coordination of strong π-accepting ligands. Substitution by a second carbonyl ligand finally resulted in transfer of the hydrogen atoms back to the tin atom to yield a stannyl complex 21. Remarkably, this ligand induced hydrogen transfer is a reversible reaction. Under the influence of light (UV-lamp) for 5 days the carbonyl is released and both hydrogen atoms are transferred back to the iridium atom to give metallostannylene 20 in a mixture with 21 (80:20) (Scheme 6, see ESI†). To the best of our knowledge this is a rare case for such a reversible ligand-induced transfer. Examples for the ligand-induced reductive elimination reactions were reported in the literature for alkane elimination from Zr(VI) or Pd(II) complexes,125–127 hydrogen from dinuclear platinum compounds,128 and arene from a tungsten complex.129,130 Complexes 18–21 were characterized by NMR spectroscopy (Table 1) and molecular structures of 19–21 are shown in Fig. 7 (Table 2).
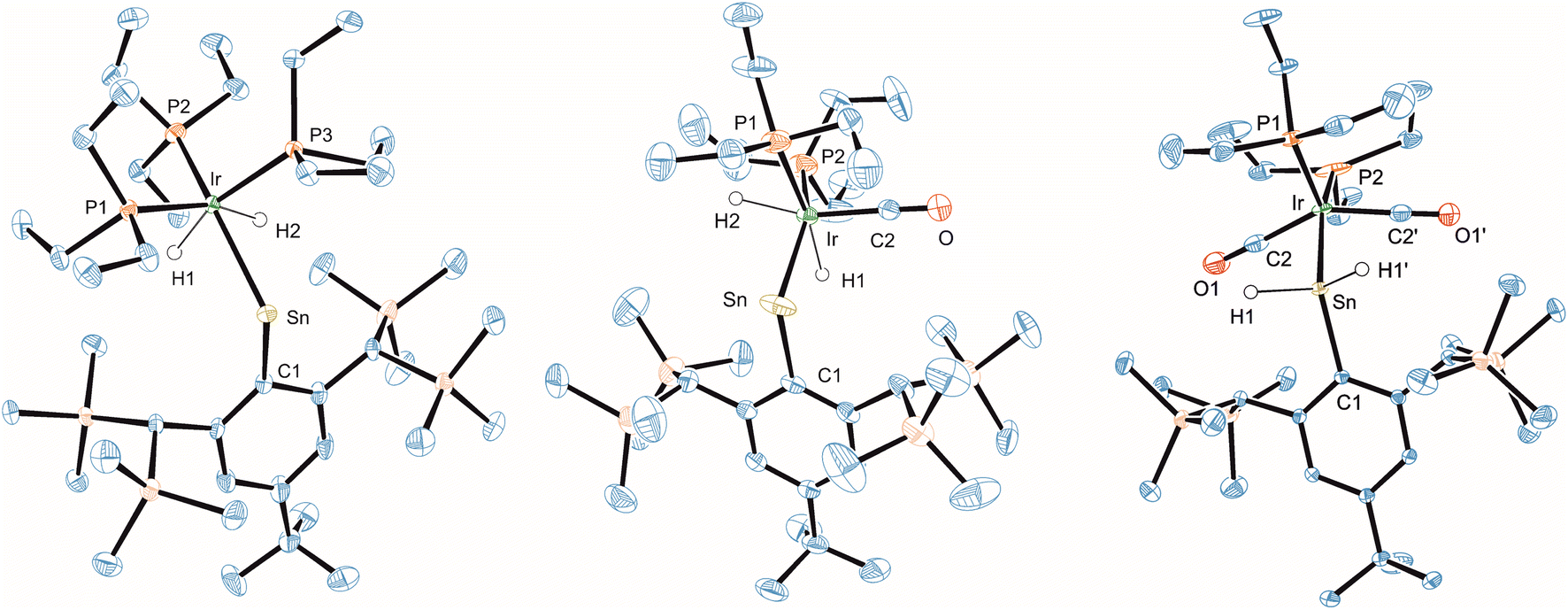 | ||
| Fig. 7 ORTEP of the molecular structures of 19, 20 and 21. Ellipsoids set at 50% probability. Hydrogen atoms except SnH and Ir–H are omitted for clarity. For selected interatomic distances and angles see Table 2. | ||
Conclusions
Tetrylidinium–iridium cations [ArGeIrHL3]+ and [ArSnIrHL3]+ show a vinyl-cation type bonding situation with a double bond between tetrylene and iridium atoms and an empty p-orbital at the Group 14 element. These tetrylidinium cations are highly reactive electrophiles acting as a cooperative metal–ligand Lewis acid. Selective addition of polar as well as nonpolar Z–H bonds (Z = NH2, N2H3, OH, Cl, H) at the cationic moiety [E = Ir]+ (E = Ge, Sn) was characterized.
The germylidinium cation adds hydride at the germanium atom and the product shows a reversible 1,2-H shift between the germanium [GeH–IrH] and iridium atom [Ge–IrH2]. The tin cation adds a hydride exclusively at the iridium atom and the products of a reversible ligand induced transfer of two hydrogen atoms between tin [Sn–IrH2] and iridium [SnH2–Ir] were isolated after PEt3/CO substitution reactions.
These findings stimulate for a more general study of cationic low valent main group element coordination at transition metals.
Data availability
Full experimental and computational details are provided as part of the ESI.†Author contributions
Investigations, writing, original draft preparation, review M. A.; preparation of 1, 5, 9, and 13 J. B.; special NMR experiments K. E.; discussion of X-ray measurements H. S.; DFT calculation, manuscript review C. P. S.; supervision, funding acquisition, DFT calculation, manuscript writing and review L. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. thanks the Fonds der Chemischen Industrie for a scholarship. We acknowledge support of the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (Justus 2 Cluster).Notes and references
- H. Hashimoto and K. Nagata, Chem. Lett., 2021, 50, 778–787 CrossRef.
- R. S. Simons and P. P. Power, J. Am. Chem. Soc., 1996, 118, 11966–11967 CrossRef.
- L. Pu, B. Twamley, S. T. Haubrich, M. M. Olmstead, B. V. Mork, R. S. Simons and P. P. Power, J. Am. Chem. Soc., 2000, 122, 650–656 CrossRef.
- P. Ghana, M. I. Arz, U. Chakraborty, G. Schnakenburg and A. C. Filippou, J. Am. Chem. Soc., 2018, 140, 7187–7198 CrossRef PubMed.
- P. Ghana, M. I. Arz, G. Schnakenburg, M. Straßmann and A. C. Filippou, Organometallics, 2018, 37, 772–780 CrossRef.
- A. C. Filippou, D. Hoffmann and G. Schnakenburg, Chem. Sci., 2017, 8, 6290–6299 RSC.
- A. C. Filippou, P. Ghana, U. Chakraborty and G. Schnakenburg, J. Am. Chem. Soc., 2013, 135, 11525–11528 CrossRef CAS PubMed.
- A. C. Filippou, U. Chakraborty and G. Schnakenburg, Chem.–Eur. J., 2013, 19, 5676–5686 CrossRef CAS PubMed.
- A. C. Filippou, A. Barandov, G. Schnakenburg, B. Lewall, M. van Gastel and A. Marchanka, Angew. Chem., Int. Ed., 2012, 51, 789–793 CrossRef CAS PubMed.
- A. C. Filippou, O. Chernov and G. Schnakenburg, Chem.–Eur. J., 2011, 17, 13574–13583 CrossRef CAS PubMed.
- A. C. Filippou, O. Chernov, K. W. Stumpf and G. Schnakenburg, Angew. Chem., Int. Ed., 2010, 49, 3296–3300 CrossRef CAS PubMed.
- A. C. Filippou, N. Weidemann and G. Schnakenburg, Angew. Chem., Int. Ed., 2008, 47, 5799–5802 CrossRef CAS PubMed.
- A. C. Filippou, N. Weidemann, A. I. Philippopoulos and G. Schnakenburg, Angew. Chem., 2006, 118, 6133–6137 CrossRef.
- A. C. Filippou, N. Weidemann, G. Schnakenburg, H. Rohde and A. I. Philippopoulos, Angew. Chem., Int. Ed., 2004, 43, 6512–6516 CrossRef CAS PubMed.
- A. C. Filippou, H. Rohde and G. Schnakenburg, Angew. Chem., 2004, 116, 2293–2297 CrossRef.
- A. C. Filippou, P. Portius, A. I. Philippopoulos and H. Rohde, Angew. Chem., Int. Ed., 2003, 42, 445–447 CrossRef CAS PubMed.
- A. C. Filippou, A. I. Philippopoulos and G. Schnakenburg, Organometallics, 2003, 22, 3339–3341 CrossRef CAS.
- A. C. Filippou, P. Portius and A. I. Philippopoulos, Organometallics, 2002, 21, 653–661 CrossRef CAS.
- A. C. Filippou, A. I. Philippopoulos, P. Portius and D. U. Neumann, Angew. Chem., Int. Ed., 2000, 39, 2778–2781 CrossRef CAS PubMed.
- J. Hicks, T. J. Hadlington, C. Schenk, J. Li and C. Jones, Organometallics, 2013, 32, 323–329 CrossRef CAS.
- B. V. Mork and T. D. Tilley, Angew. Chem., Int. Ed., 2003, 42, 357–360 CrossRef CAS.
- P. G. Hayes, Z. Xu, C. Beddie, J. M. Keith, M. B. Hall and T. D. Tilley, J. Am. Chem. Soc., 2013, 135, 11780–11783 CrossRef CAS.
- R. C. Handford, M. A. Nesbit, P. W. Smith, R. D. Britt and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 358–367 CrossRef CAS PubMed.
- P. M. Keil and T. J. Hadlington, Chem. Commun., 2022, 58, 3011–3014 RSC.
- T. P. Dhungana, H. Hashimoto, M. Ray and H. Tobita, Organometallics, 2020, 39, 4350–4361 CrossRef.
- T. Fukuda, T. Yoshimoto, H. Hashimoto and H. Tobita, Organometallics, 2016, 35, 921–924 CrossRef.
- H. Hashimoto, T. Fukuda, H. Tobita, M. Ray and S. Sakaki, Angew. Chem., Int. Ed., 2012, 51, 2930–2933 CrossRef PubMed.
- T. Fukuda, H. Hashimoto and H. Tobita, J. Organomet. Chem., 2017, 848, 89–94 CrossRef.
- J. D. Queen, A. C. Phung, C. A. Caputo, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2020, 142, 2233–2237 CrossRef.
- M. Widemann, K. Eichele, H. Schubert, C. P. Sindlinger, S. Klenner, R. Pöttgen and L. Wesemann, Angew. Chem., Int. Ed., 2021, 60, 5882–5889 CrossRef CAS PubMed.
- D. L. Thorn and T. H. Tulip, Organometallics, 1982, 1, 1580–1586 CrossRef CAS.
- T. Sasamori, T. Sugahara, T. Agou, J.-D. Guo, S. Nagase, R. Streubel and N. Tokitoh, Organometallics, 2015, 34, 2106–2109 CrossRef CAS.
- M. Auer, F. Diab, K. Eichele, H. Schubert and L. Wesemann, Dalton Trans., 2022, 51, 5950–5961 RSC.
- J. D. Feldman, J. C. Peters and T. D. Tilley, Organometallics, 2002, 21, 4065–4075 CrossRef CAS.
- N. A. Bell, F. Glockling, M. L. Schneider, H. M. M. Shearer and M. D. Wilbey, Acta Crystallogr., Sect. C: Struct. Chem., 1984, 40, 625–628 CrossRef.
- S. M. Hawkins, P. B. Hitchcock, M. F. Lappert and A. K. Rai, Chem. Commun., 1986, 1689–1690 RSC.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- J. Henoch, A. Auch, F. Diab, K. Eichele, H. Schubert, P. Sirsch, T. Block, R. Pöttgen and L. Wesemann, Inorg. Chem., 2018, 57, 4135–4145 CrossRef CAS.
- K. M. Krebs, S. Freitag, H. Schubert, B. Gerke, R. Pöttgen and L. Wesemann, Chem.–Eur. J., 2015, 21, 4628–4638 CrossRef CAS.
- R. S. Simons, J. C. Gallucci, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2002, 654, 224–228 CrossRef CAS.
- E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 9226–9227 CrossRef CAS PubMed.
- J. A. Cabeza, J. M. Fernández-Colinas, P. García-Álvarez, L. González-Álvarez and E. Pérez-Carreño, Dalton Trans., 2019, 48, 10996–11003 RSC.
- T. J. Malosh, J. R. Shapley, R. J. Lawson, D. N. T. Hay and T. N. Rohrabaugh, J. Organomet. Chem., 2013, 745–746, 98–105 CrossRef CAS.
- R. D. Adams, Y. Kan and Q. Zhang, Organometallics, 2011, 30, 328–333 CrossRef CAS.
- R. D. Adams, M. Chen, E. Trufan and Q. Zhang, Organometallics, 2011, 30, 661–664 CrossRef CAS.
- T. W. Dingle and K. R. Dixon, Inorg. Chem., 1974, 13, 846–851 CrossRef CAS.
- P. Diehl and J. A. Pople, Mol. Phys., 1960, 3, 557–561 CrossRef CAS.
- R. J. Abraham, E. O. Bishop and R. E. Richards, Mol. Phys., 1960, 3, 485–494 CrossRef CAS.
- K. Bakthavachalam, S. Dutta, C. Arivazhagan, B. Raghavendra, A. Haridas, S. S. Sen, D. Koley and S. Ghosh, Dalton Trans., 2018, 47, 15835–15844 RSC.
- A. Jana, H. W. Roesky, C. Schulzke, P. P. Samuel and A. Döring, Inorg. Chem., 2010, 49, 5554–5559 CrossRef CAS.
- Z. D. Brown, J.-D. Guo, S. Nagase and P. P. Power, Organometallics, 2012, 31, 3768–3772 CrossRef CAS.
- A. Hinz, Chem.–Eur. J., 2019, 25, 7843–7846 CrossRef CAS PubMed.
- A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2009, 48, 798–800 CrossRef CAS PubMed.
- M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem.–Eur. J., 2016, 22, 11685–11698 CrossRef CAS.
- Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS PubMed.
- A. Jana, S. S. Sen, H. W. Roesky, C. Schulzke, S. Dutta and S. K. Pati, Angew. Chem., Int. Ed., 2009, 48, 4246–4248 CrossRef CAS PubMed.
- L. Nurdin, Y. Yang, P. G. N. Neate, W. E. Piers, L. Maron, M. L. Neidig, J.-B. Lin and B. S. Gelfand, Chem. Sci., 2021, 12, 2231–2241 RSC.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef PubMed.
- G. W. Margulieux, M. J. Bezdek, Z. R. Turner and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 6110–6113 CrossRef PubMed.
- J. I. van der Vlugt, Chem. Soc. Rev., 2010, 39, 2302–2322 RSC.
- M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Würtele, B. de Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290–9302 CrossRef PubMed.
- G. P. Connor, D. Delony, J. E. Weber, B. Q. Mercado, J. B. Curley, S. Schneider, J. M. Mayer and P. L. Holland, Chem. Sci., 2022, 13, 4010–4018 RSC.
- A. L. Casalnuovo, J. C. Calabrese and D. Milstein, Inorg. Chem., 1987, 26, 971–973 CrossRef CAS.
- J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed.
- E. Morgan, D. F. MacLean, R. McDonald and L. Turculet, J. Am. Chem. Soc., 2009, 131, 14234–14236 CrossRef CAS PubMed.
- C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318–10319 CrossRef CAS PubMed.
- M. P. Betoré, M. A. Casado, P. García-Orduña, F. J. Lahoz, V. Polo and L. A. Oro, Organometallics, 2016, 35, 720–731 CrossRef.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef PubMed.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef PubMed.
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef PubMed.
- Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031–2034 CrossRef PubMed.
- A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef PubMed.
- A. Meltzer, S. Inoue, C. Präsang and M. Driess, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef.
- S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef.
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef PubMed.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef PubMed.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef PubMed.
- D. C. H. Do, A. V. Protchenko, M. Á. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684–4687 RSC.
- E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef PubMed.
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef.
- Y.-H. Chang, Y. Nakajima, H. Tanaka, K. Yoshizawa and F. Ozawa, J. Am. Chem. Soc., 2013, 135, 11791–11794 CrossRef.
- R. M. Brown, J. Borau Garcia, J. Valjus, C. J. Roberts, H. M. Tuononen, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2015, 54, 6274–6277 CrossRef.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- O. Chernov, Dissertation, Rheinische Friedrich-Wilhelms-University of Bonn, 2012.
- R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 2021, 3488–3498 CrossRef CAS PubMed.
- G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef.
- N. Gorgas, A. J. P. White and M. R. Crimmin, J. Am. Chem. Soc., 2022, 144, 8770–8777 CrossRef PubMed.
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262–10265 CrossRef.
- D. You and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 6843–6846 CrossRef.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef PubMed.
- L. Pu, N. J. Hardman and P. P. Power, Organometallics, 2001, 20, 5105–5109 CrossRef CAS.
- L.-C. Pop, N. Kurokawa, H. Ebata, K. Tomizawa, T. Tajima, M. Ikeda, M. Yoshioka, M. Biesemans, R. Willem, M. Minoura and M. Saito, Can. J. Chem., 2014, 92, 542–548 CrossRef CAS.
- W.-P. Leung, K.-W. Kan, Y.-C. Chan and T. C. W. Mak, Inorg. Chem., 2013, 52, 4571–4577 CrossRef CAS.
- A. C. Filippou, K. W. Stumpf, O. Chernov and G. Schnakenburg, Organometallics, 2012, 31, 748–755 CrossRef CAS.
- P. Nie, Y. Li, Q. Yu, B. Li, H. Zhu and T.-B. Wen, Eur. J. Inorg. Chem., 2017, 2017, 3892–3899 CrossRef CAS.
- C. Bibal, S. Mazières, H. Gornitzka and C. Couret, Organometallics, 2002, 21, 2940–2943 CrossRef CAS.
- R. Jambor, B. Kašná, S. G. Koller, C. Strohmann, M. Schürmann and K. Jurkschat, Eur. J. Inorg. Chem., 2010, 2010, 902–908 CrossRef.
- L. W. Pineda, V. Jancik, J. F. Colunga-Valladares, H. W. Roesky, A. Hofmeister and J. Magull, Organometallics, 2006, 25, 2381–2383 CrossRef CAS.
- A. Jana, S. P. Sarish, H. W. Roesky, C. Schulzke and P. P. Samuel, Chem. Commun., 2010, 46, 707–709 RSC.
- D. Morales-Morales, D. W. Lee, Z. Wang and C. M. Jensen, Organometallics, 2001, 20, 1144–1147 CrossRef CAS.
- P. Kläring, S. Pahl, T. Braun and A. Penner, Dalton Trans., 2011, 40, 6785–6791 RSC.
- O. V. Ozerov, Chem. Soc. Rev., 2009, 38, 83–88 RSC.
- N. Tsoureas, Y.-Y. Kuo, M. F. Haddow and G. R. Owen, Chem. Commun., 2011, 47, 484–486 RSC.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS.
- H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053–3062 CrossRef CAS.
- B. E. Cowie and D. J. H. Emslie, Chem.–Eur. J., 2014, 20, 16899–16912 CrossRef CAS PubMed.
- A. Albinati, A. Musco, R. Naegeli and L. M. Venanzi, Angew. Chem., Int. Ed., 1981, 20, 958–959 CrossRef.
- P. W. Smith, R. C. Handford and T. D. Tilley, Organometallics, 2019, 38, 4060–4065 CrossRef.
- H.-J. Liu, J. Guihaumé, T. Davin, C. Raynaud, O. Eisenstein and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 13991–13994 CrossRef PubMed.
- P. G. Hayes, C. W. Gribble, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 4606–4607 CrossRef PubMed.
- M. E. Fasulo and T. D. Tilley, Chem. Commun., 2012, 48, 7690–7692 RSC.
- T. Watanabe, H. Hashimoto and H. Tobita, Angew. Chem., Int. Ed., 2004, 43, 218–221 CrossRef.
- Q. Zhu, J. C. Fettinger and P. P. Power, Dalton Trans., 2021, 50, 12555–12562 RSC.
- M. Widemann, S. Jeggle, M. Auer, K. Eichele, H. Schubert, C. P. Sindlinger and L. Wesemann, Chem. Sci., 2022, 13, 3999–4009 RSC.
- H. Hashimoto, T. Tsubota, T. Fukuda and H. Tobita, Chem. Lett., 2009, 38, 1196–1197 CrossRef.
- M. Ochiai, H. Hashimoto and H. Tobita, Angew. Chem., Int. Ed., 2007, 46, 8192–8194 CrossRef PubMed.
- R. S. Simons, J. C. Gallucci, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2002, 654(1), 224–228 CrossRef.
- T. Herskovitz, C. Kampe, H. D. Kaesz and W. M. Seidel, Inorg. Synth., 1982, 99–103 Search PubMed.
- H.-J. Liu, C. Landis, C. Raynaud, O. Eisenstein and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 9186–9194 CrossRef PubMed.
- P. G. Hayes, R. Waterman, P. B. Glaser and T. D. Tilley, Organometallics, 2009, 28, 5082–5089 CrossRef.
- K. I. Gell and J. Schwartz, J. Am. Chem. Soc., 1981, 103, 2687–2695 CrossRef.
- M. Tanabe, N. Ishikawa and K. Osakada, Organometallics, 2006, 25, 796–798 CrossRef.
- S. Otsuka, T. Yoshida, M. Matsumoto and K. Nakatsu, J. Am. Chem. Soc., 1976, 98, 5850–5858 CrossRef.
- R. H. Hill and R. J. Puddephatt, J. Am. Chem. Soc., 1983, 105, 5797–5804 CrossRef.
- D. J. Burkey, J. D. Debad and P. Legzdins, J. Am. Chem. Soc., 1997, 119, 1139–1140 CrossRef.
- D. M. Crumpton-Bregel and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 9442–9456 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2208289–2208301. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05620h |
| This journal is © The Royal Society of Chemistry 2023 |