
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox and guest tunable spin-crossover properties in a polymeric polyoxometalate†
Mario
Palacios-Corella
 a,
Víctor
García-López
a,
Joao Carlos
Waerenborgh
b,
Bruno J. C.
Vieira
b,
Guillermo
Mínguez Espallargas
a,
Miguel
Clemente-León
*a and
Eugenio
Coronado
*a
a,
Víctor
García-López
a,
Joao Carlos
Waerenborgh
b,
Bruno J. C.
Vieira
b,
Guillermo
Mínguez Espallargas
a,
Miguel
Clemente-León
*a and
Eugenio
Coronado
*a
aInstituto de Ciencia Molecular (ICMol), Universitat de València, C/Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: miguel.clemente@uv.es; eugenio.coronado@uv.es; Fax: +34 963543273; Tel: +34 963544419
bCentro de Ciências e Tecnologias Nucleares, DECN, Instituto Superior Técnico, Universidade de Lisboa, 2695-066 Bobadela LRS, Portugal
First published on 22nd February 2023
Abstract
A bifunctionalized polyoxometalate (POM), [V6O19(C16H15N6O)2]2−, which contains a redox active hexavanadate moiety covalently linked to two tridentate 2,6-bis(pyrazol-1-yl)pyridine (1-bpp) ligands, has been prepared and characterized. Reaction of this hybrid molecule with Fe(II) or Zn(II) ions produces crystalline neutral 1D networks of formula Fe[V6O19(C16H15N6O)2]·solv (2) and Zn[V6O19(C16H15N6O)2]·solv (3) (solv = solvent molecules). Magnetic properties of 2 show an abrupt spin-crossover (SCO) with the temperature, which can be induced by light irradiation at 10 K (Light-Induced Excited Spin-State Trapping, LIESST effect). Interestingly, this porous and flexible structure enables reversible exchange of solvents in 2, which allows tuning the temperature of the thermal SCO. In 2 and 3, the hexavanadate unit can be reduced by electrochemical or chemical means in a reversible way. Chemical reduction and reoxidation by a postsynthetic method is accompanied by the insertion in the structure of the reductant and oxidant molecules (cobaltocene and tribromide, respectively), which provokes drastic changes in the spin state of Fe(II). This leads to an elegant switching multifunctional material in which SCO properties of the Fe(II) complexes coexist with the redox properties of the POM and can be tuned by a variety of stimuli such as temperature, light, solvent exchange or electron transfer. During the reduction process, 3 undergoes a single-crystal-to-single-crystal one-electron reduction, which confirms the presence of cobaltocenium cations by single crystal X-ray diffraction.
Introduction
Spin crossover (SCO) complexes undergo an entropy-driven spin state change, which can be triggered by a variety of external stimuli (temperature, pressure, analytes or electric fields).1,2 The majority of SCO research has been dedicated to the study of octahedral Fe(II) complexes (3d6), which switch between the diamagnetic low-spin state (LS, S = 0) and the paramagnetic high-spin state (HS, S = 2). Since this phenomenon induces reversible changes of several physical properties, SCO materials are attracting considerable interest due to their potential applications as chemical switches, sensors or memory devices.2Bis-chelated Fe(II) complexes based on tridentate ligands of the type 2,6-bis(pyrazol-1-yl)pyridine (1-bpp)3 are an archetypical family of SCO complexes. They usually present abrupt thermal spin transitions and a Light-Induced Excited Spin State Trapping (LIESST) effect4 at relatively high temperatures.5 In addition, the spin state of these systems is known to be very sensitive to intermolecular interactions with solvent molecules and counteranions through the molecular network.6–9 Consequently, they could act as chemo-responsive materials. Most of the previous results with this ligand and derivatives are based on salts of mononuclear complexes, which very often lose crystallinity or dissolve in contact with organic solvents. Therefore, the organization of this type of complexes into coordination polymers (CPs) could be a suitable strategy to optimize the SCO properties of these systems by increasing the stability and adding new functionalities such as porosity, magnetism (magnetic centers coupled magnetically to the Fe(II) SCO center or with single-molecule magnet properties) or redox activity. Previous attempts to reach this goal were based on ditopic “back-to-back” neutral 1-bpp ligands.10,11 However, the resultant materials were often poorly crystalline, with incomplete and very gradual SCO transitions.12
In this work, an alternative strategy has been attempted: instead of using a neutral and electrically innocent organic spacer between the two 1-bpp units of the ditopic ligand, a charged ligand containing a polyoxometalate (POM) moiety has been prepared. POMs are polyanionic molecular-metal oxides of groups 5 or 6 transition metals in their highest oxidation states. POMs can accept various numbers of electrons, while keeping their structure almost intact, leading to mixed-valence systems in which the extra electrons are extensively delocalized over the whole POM framework.13 Redox-active ligands have been the focus of recent research in SCO as they enable controlling the SCO properties with electrical stimuli. Most of the SCO systems of this type reported in the literature are based on 1D and 3D CPs containing tetrathiafulvalene (TTF).14–19 The use of a POM could be an interesting alternative for obtaining redox-active SCO materials thanks to their robustness and redox tunability.20,21 To obtain a 1-bpp functionalized POM, we have used tris-alkoxo-amide tripods as linking units. This is a well-known method to design POM-based hybrid structures,22,23 and robust POM frameworks linked through metal ions,24,25 exhibiting multifunctional properties.26–29
To our knowledge, there are very few reports of POMs showing SCO behavior.30,31 In a previous work, we explored this topic through the incorporation of 1-bpp into a MnIII Anderson POM.32 We first prepared the 1-bpp bifunctionalized Anderson POM and subsequently, by reaction with Fe2+, the amorphous polymeric compound (C16H36N)[Fe(MnMo6O24(C16H15N6O)2)]·(H2O)4. This compound showed a LIESST effect with a small but significant photoconversion (∼8%) combined with a field-induced slow relaxation of magnetization due to the presence of isolated Mn(III) anisotropic magnetic ions.
Herein an optimization of this strategy has been achieved by taking advantage of a different POM unit, the Lindqvist hexavanadate. We have chosen this POM because it can be reversibly reduced. In addition to this, the 2− charge of the functionalized hexavanadate POM matches with the 2+ charge of the iron center. This has enabled us to obtain a crystalline neutral polymer with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Fe:POM stoichiometry, which contrasts with the amorphous charged polymers obtained by reacting the 3− charged Anderson POM with Fe2+ mentioned above and those so far obtained with 1-bpp ditopic ligands with neutral organic spacers.
:1 Fe:POM stoichiometry, which contrasts with the amorphous charged polymers obtained by reacting the 3− charged Anderson POM with Fe2+ mentioned above and those so far obtained with 1-bpp ditopic ligands with neutral organic spacers.
Results and discussion
Synthesis and structure
In a first step we report the preparation of the molecular hexavanadate POM bifunctionalized with 1-bpp linkers (compound (C16H36N)2[V6O19(C16H15N6O)2]·(C4H9NO)2, (1)). The formation of this molecular building block is confirmed by several techniques (see Fig. S1–S6 and Scheme S1 and associated text in the ESI†). Single crystal X-ray diffraction structure shows that this POM presents the common Lindqvist hexavanadate structure in which the six V(V) atoms from six octahedral edge-sharing VO6 form an octahedron. The two 1-bpp ligands, linked through a tris-alkoxo-amide tripod, occupy opposite faces of that octahedron (see Fig. 1). | ||
| Fig. 1 View of the structure of the functionalized-POM in compound 1. (V (pink), C (black), N (blue), O (red)). Hydrogen atoms have been omitted for clarity. | ||
In a second step, 1 is reacted with one equivalent of Fe2+ or Zn2+ by slow diffusion in DMF/MeCN mixtures to afford the crystalline materials Fe[V6O19(C16H15N6O)2]·solv (2) and Zn[V6O19(C16H15N6O)2]·solv (3) (solv = solvent). The composition of 2 and 3, studied by energy dispersive X-ray spectroscopy (EDX) measurements, is consistent with the expected 1:6 Fe:V and Zn:V ratios. In addition, elemental analysis, 1H NMR and thermogravimetric analysis (TGA) have been used to characterize the composition of these materials (see Experimental section in the ESI†). It is important to note that these solids are not stable in absence of solvent (see below) and therefore need to be characterized and stored in the mother liquor (DMF/MeCN mixture in as-synthesized samples 2·DMF and 3·DMF) or in MeCN (samples 2·MeCN and 3·MeCN). Powder X-ray diffraction (PXRD) measurements of 2·MeCN and 3·MeCN in contact with the solvent confirmed that Fe and Zn derivatives are isostructural (see Experimental section, Table S2 and Fig. S7 in the ESI†).
The structure of the Zn derivative (3) was solved by single crystal X-ray diffraction at 120 K in a crystal of 3·DMF at 120 K. It confirms the formation of a polymeric structure where the 1-bpp bifunctionalized POMs are connected to each other through the metallic center (Fig. 2). It crystallizes in the monoclinic space group P21/c showing a single molecule of [V6O19(C16H15N6O)2]2− POM, one Zn atom, one MeCN, one DMF and disordered solvent molecules in the asymmetric unit. 3 forms a one-dimensional (1D) neutral coordination polymer of formula {Zn[V6O19(C16H15N6O)2]}n running along the [10−1] direction (see Fig. 2). The Zn atom is six-coordinated to two tridentate 1-bpp ligands from two [V6O19(C16H15N6O)2]2− units, which at the same time are coordinated to two Zn(II) ions through their two terminal 1-bpp ligands.
 | ||
| Fig. 2 View of the structure of the chains of compound 3 with hydrogen-bonded solvent molecules (VO6 octahedra (yellow), C (black), N (blue), O (red), Zn (gray)). Dashed lines represent hydrogen bonds. Hydrogen atoms have been omitted for clarity. | ||
These 1D coordination polymers are symmetry related by an inversion center placed at [0, 0, 0] and connected to each other by weak intermolecular interactions leading to double layers in the ac plane and channels filled with solvent molecules parallel to the chain direction (see Fig. 3). Thus, one of the two NH groups from the amide groups of [V6O19(C16H15N6O)2]2− form hydrogen bonds with a MeCN molecule (see Fig. 2). Among these interactions, the closest contacts between the coordinated ligands of Zn involve CO groups with CH groups or C atoms from pyrazolyl groups (Fig. S8 in the ESI†). Each Zn(II) complex interacts through this type of interactions with four complexes from neighboring chains belonging to the same double layer (Fig. S8 in the ESI†). This leads to a minimal interchain distance between Zn ions of 10.47 Å, which is much shorter than the shortest intrachain Zn–Zn distance of 25.2 Å. These two distances are longer than those found in other salts of Fe(II) complexes of 1-bpp with similar triol substituents (8.702 Å in [Fe(1-bpp-triolH3)2](ClO4)2·MeCN (triolH3 = C(O)NHC(CH2OH)3)).33 Therefore, the insertion of these 1-bpp complexes into the POM network provides a better isolation than that reported for simple salts of 1-bpp complexes with similar substituents. However, since DMF and MeCN solvent molecules occupy the channels and present numerous short contacts with 1-bpp ligands, important structural changes are expected if they are removed or replaced by other solvents.
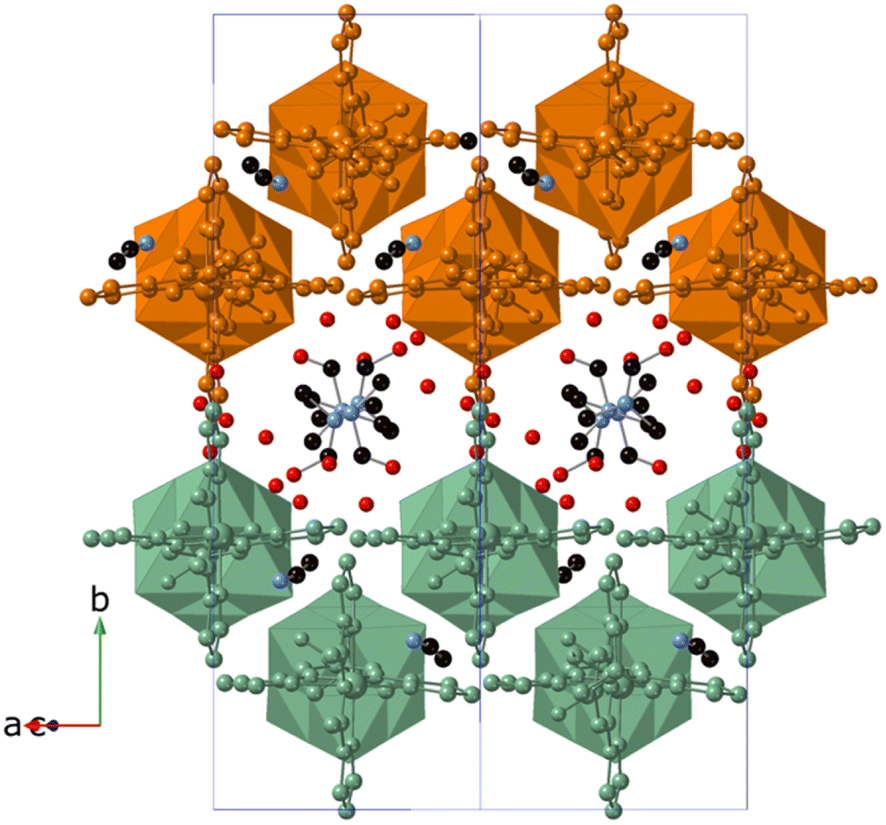 | ||
| Fig. 3 Crystal structure of 3·DMF viewed along the [−101] direction (C (black), N (blue), O (red)). Atoms of the Zn[V6O19(C16H15N6O)2] chains belonging to the same double layer are colored orange or green. Hydrogen atoms have been omitted for clarity. | ||
Exchange of solvents
Exchange of solvents was tested to study the robustness and flexibility of the CP. We first studied the stability of 2 and 3 in absence of solvent. The PXRD pattern of a filtered sample of 2·MeCN reveals drastic changes with respect to that measured in contact with the mother liquor. Thus, it presents fewer and broader peaks than that of the solvated sample (see Fig. S9 in the ESI† and associated text). This indicates a loss of crystallinity and a different packing of the M[V6O19(C16H15N6O)2] chains upon desolvation, as a result of the decrease of the space between chains (see Fig. S10 in the ESI† and associated text). It is possible to recover partially the initial crystalline pattern by soaking these dry crystals again in acetonitrile (see Fig. S9 in the ESI†) but with poorer crystallinity. This is an indication of a certain degree of reversibility in the desolvation/resolvation process.Interestingly, reversible solvent exchange of the initial DMF/MeCN solvent mixture could be achieved preserving the crystallinity if the crystals were always kept in contact with the mother liquor or clean solvent. Thus, different clean MeCN, MeOH, EtOH, nitromethane (MeNO2) or benzonitrile (PhCN) solvents can be exchanged by soaking the as-synthesized crystals of 2·DMF or 3·DMF, covered with a small amount of the mother liquor, in the fresh solvent for 3 days and storing in the same solvent (see Experimental section in the ESI†). This gives rise to 2·MeCN, 2·MeOH, 2·EtOH, 2·MeNO2, 2·PhCN and 3·MeCN exchanged samples where the ·solvent notation refers to the solvent used in the exchange process, which in some cases does not enter in the final structure. Thus, characterization of the composition of these exchanged samples performed by 1H NMR of freshly filtered crystals dissolved in dimethyl sulfoxide-d6, elemental analyses and thermogravimetric analyses (TGA) of the filtered samples indicate that the organic solvent molecules found in the structure of 3·DMF (DMF and MeCN) can be replaced by the other solvents or water molecules present in air or in the solvents (see ESI†).
PXRD of all these samples in contact with the mother liquor was performed to understand the structural changes originated by the exchange of solvents (see Fig. 4 and S14 in the ESI†). PXRD patterns of the solvent-exchanged samples 2·PhCN, 2·DMF, 2·MeNO2, 2MeCN and 2·EtOH suggest a similar structure to that solved by single crystal X-ray diffraction for 3·DMF (see Fig. 4). However, close inspection of the powder patterns reveals some differences in the first diffraction peak, corresponding to the (020) planes, which is directly related to the interlayer distance. Specifically, this peak is shifted to lower 2θ values (i.e. larger d spacing) for the bulkiest solvent (2·PhCN), with a value of 6.2° (d = 14.2 Å) while that containing only water molecules (2·EtOH) shows the highest 2θ value of this peak at 6.5° (d = 13.6 Å). These data support that the size of the solvents inserted between the layers could tune the distance between the double layers of chains found in the structure of 3·DMF in the ac plane. A similar trend is observed for the other samples, with lower 2θ values of this peak for the samples with bulkier solvents (2·DMF and 2·MeNO2) (see ESI† for a more detailed discussion).
 | ||
| Fig. 4 PXRD patterns of 3·MeCN, 2·PhCN, 2·DMF, 2·MeNO2, 2·MeCN and 2·EtOH. | ||
Finally, to test the reversibility of the process, we performed additional solvent exchanges in the sample with the bulkiest solvent (2·PhCN), which was subsequently soaked for 3 days in MeCN (2·PhCN·MeCN), and then again in PhCN (2·PhCN·MeCN·PhCN). 1H NMR and elemental analyses confirm that this multiple exchange of solvents is reversible (see Experimental section in the ESI†). PXRD patterns of 2·PhCN·MeCN and 2·PhCN·MeCN·PhCN are very similar to those of 2·MeCN and 2·PhCN, respectively, confirming that it is possible to exchange more than one solvent with the same sample in a reversible way, while retaining the structure of each solvate (see Fig. S13d and S15 in the ESI†). The crucial point to achieve solvent exchange is to maintain always a small amount of solvent covering the crystals.
Magnetic properties of the exchanged samples
The temperature dependence of the product χMT (χM is the molar paramagnetic susceptibility) of the exchanged samples in contact with the solvent was measured to study the influence of the exchanged solvents in the spin transition (see Fig. 5 and S16a in the ESI†). The most complete and cooperative SCO is observed in 2·MeCN, 2·MeNO2 and 2·EtOH (see Fig. 5). They present abrupt decreases of χMT from 3.5 cm3 K mol−1, typical of 100% of Fe(II) in the HS state, at 300 K to 0.9 cm3 K mol−1 (2·MeCN and 2·EtOH) and 0.5 cm3 K mol−1 (2·MeNO2) below 100 K. This corresponds to almost complete spin transitions with ∼25% and ∼15% of residual HS Fe(II) ions at low temperatures, respectively. In addition, thermal hystereses are observed in 2·MeCN (8 K), 2·MeNO2 (3 K) and 2·EtOH (3 K) at a scan rate 2 K min−1. To our knowledge, these are the first POM compounds showing a complete and cooperative SCO, in spite of the long distances found between the metal centers in the structure. This cooperativity could be induced by the intermolecular interactions between [Fe(1-bpp)2]2+ complexes of neighboring chains. To our knowledge, there is only one example of a polymeric compound containing 1-bpp displaying a similar cooperative SCO behavior. This is the poorly crystalline compound {[FeL]n(BF4)2n}·solv (L = ditopic back-to-back ligand with two 1-bpp coupled through 1,4-phenylene), which shows an hysteresis loop of 10 K.10a Another interesting feature is that the exchange of solvents enables tuning the temperature of the SCO, with T1/2 values (T1/2 = temperature of 50% HS to LS conversion) ranging from 140 K in 2·MeCN to 260 K in 2·EtOH and intermediate value in 2·MeNO2 (220 K). In contrast to these three samples, 2·PhCN and the as-synthesized 2·DMF crystals present gradual and incomplete spin transitions (see Fig. S16a in the ESI†). Remarkably, temperature dependence of χMT of 2·PhCN·MeCN and 2·PhCN·MeCN·PhCN are very similar to those of 2·MeCN and 2·PhCN, respectively, (see Fig. S16b in the ESI†) confirming that the structure and spin state of Fe(II) in the same crystal can be tuned in a reversible way by successive soaking in different solvents. | ||
| Fig. 5 Thermal variation of χMT of solvent exchanged samples of 2 measured in contact with the solvent. 2·MeCN; red circles: 2·MeNO2; pink circles: 2·EtOH; orange circles. | ||
To rationalize these results, we have to take into account that the spin state of solvates of the same salt of [FeII(1-bpp)2]2+ is very sensitive to different factors such as intermolecular interactions with solvent molecules and counteranions or pressure effects due to different solvent filling. Thus, different magnetic behaviors for solvates of the same complex are quite usual.3,8,9 In our case, there are two very simple magneto-structural trends. On one hand, the gradual SCO found on the samples with the bulkiest solvents in the structure (2·DMF and 2·PhCN) suggest that the intercalation of bulky solvent molecules between the [FeII(1-bpp)2]2+ centers decreases the cooperativity of the SCO, which is in agreement with the presence of DMF molecules intercalated between the POM chains in the crystal structure of 3·DMF and PhCN molecules in 2·PhCN, as shown by elemental analysis, TGA and 1H NMR. On the other hand, the already mentioned shift in the first and most intense peak of the PXRD pattern to the highest 2θ values in the sample containing only water molecules (2·EtOH) could indicate larger chemical pressure induced by the decrease in unit cell volume. This could lead to a stabilization of the LS state, as observed experimentally. In the other samples, a clear trend is not observed as electronic effects due to intermolecular interactions between solvent guest molecules and the coordinated ligands could influence the ligand field strength around the Fe(II) center. Indeed, 1H NMR spectra of freshly filtered crystals of these samples show the presence of the exchanged solvent in all cases except in 2·EtOH (Fig. S11 in the ESI†).
Magnetic properties of filtered crystals of 2·MeCN were measured to study how the collapse of the structure observed by PXRD patterns after filtering affects the spin transition. Magnetic properties change drastically with respect to those of the same sample measured in contact with the mother liquor with an almost constant HS fraction of 70–80% and a loss of the SCO property (see Fig. S17 in the ESI†). Mössbauer spectroscopy measurements in this filtered sample were performed to confirm the oxidation and spin states of Fe(II) in 2 since it was not possible to measure solvated samples due to technical reasons (see Experimental section in the ESI†). In all cases, characteristic spectra of HS or LS Fe(II) were obtained in agreement with magnetic properties and confirming that iron is not oxidized to iron(III) (see Fig. S18 and S19, Table S3 and associated text in the ESI†). A resolvated sample, obtained by soaking the filtered crystals of 2 in MeCN, does not recover the spin transition shown by the solvated sample (see Fig. S17 in the ESI†). This indicates that the initial structure of the solvated sample is not recovered after resolvation if the sample is extracted from the solvent as shown in the PXRD pattern (see above).
Reduction and reoxidation of 2 and 3
In view of the redox properties of POMs, an interesting possibility to explore in these CPs is that of tuning the spin transition by injection of electrons in the POMs since it will be accompanied by insertion of counterions and important structural changes. The possible injection of electrons in the POMs was studied in 2 and 3 by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Nafion 5% and ethanol (1:1) mixtures of 2·MeCN and 3·MeCN were deposited on a glassy carbon disk working electrode and studied at different scan rates in 0.1 M TBABF4/CH3CN solutions. CV experiments show quasi-reversible redox peaks with the mean peak potential of −0.66 V (ΔEp = 0.13 V) for 2 and −0.86 V (ΔEp = 0.18 V) for 3versus Ag/AgCl at a scan rate of 100 mV s−1 (see Fig. 6 and S20 in the ESI†). The first peak agrees with the quasi-reversible one-electron reduction of hexavanadate observed in solution for Lindqvist-type polyoxovanadates.34,35 These results are confirmed by DPV analysis (see Fig. S20 in the ESI†).
 | ||
| Fig. 6 Cyclic voltammetric behavior of 2. | ||
In view of the reversible electrochemical reduction of these samples, they were then reduced chemically by suspending polycrystalline samples of 2·MeCN and 3·MeCN in a solution of cobaltocene in MeCN under an inert atmosphere for 5 hours leading to the reduced samples Red_2 and Red_3. Reoxidized sample of 2 (Reox_2) was obtained by suspending crystals of Red_2 in a MeCN solution of TBABr3 (see Experimental section in the ESI†). The reduction and reoxidation of the hexavanadate unit could be easily detected from the distinctive change in color (red in fully oxidized state and dark green in reduced species, see Fig. 8 and S20d in the ESI†) and by IR spectroscopy (see Fig. S21 in the ESI† and associated text), showing a red shift of the characteristic νV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and νV–O–V bands (from 950 and 790 to 930 and 755 cm−1, respectively) in the reduced sample, in agreement with previous reports.36,37
O and νV–O–V bands (from 950 and 790 to 930 and 755 cm−1, respectively) in the reduced sample, in agreement with previous reports.36,37
EDAX, elemental analysis, X-ray photoelectron spectroscopy (XPS) and 1H NMR of Red_2 and Red_3 were used to study the composition (see Experimental section and Fig. S22–S24 in the ESI† and associated text). They confirmed the expected Fe:V and Zn:V ratios in the pristine polymers, but cobalt appeared with 1.3:1:6 Co:Fe:V and Co:Zn:V ratios in the reduced samples. This suggests the oxidation of cobaltocene to cobaltocenium to compensate the one-electron reduction of the POM and its insertion in the structure leading to the formulas Fe[V6O19(C16H15N6O)2]·[Co(C5H5)2]1.3·8H2O (Red_2) and Zn[V6O19(C16H15N6O)2]·[Co(C5H5)2]1.3·11H2O (Red_3). Elemental analysis and EDAX suggest a small excess of cobaltocenium (0.3 [Co(C5H5)2]+), which could be due to the presence of POMs reduced by more than one electron or to other counteranions (OH−). The majority one-electron reduction of the POM can be inferred from EPR and magnetic data (see below). Finally, control experiments reveal the necessity of cobaltocene for the reduction process (see associated text to Fig. S20 in the ESI†).
To determine the structure, the reduction process was tested in single crystals. Single-crystal X-ray diffraction analysis of Red_3, obtained by soaking single crystals of 3·DMF in the cobaltocene MeCN solution without stirring, revealed the preservation of single crystallinity and enabled the solution of the single crystal structure of Red_3. Single-crystal to single-crystal redox transformations in CPs have been observed but are still a relatively rare phenomenon.38 Similar to that of 3·DMF, the structure of Red_3 was solved in the monoclinic space group P21/c with a shortening of ca. 0.2 Å in a and b parameters and an increase of the same order in c parameter with respect to those of 3·DMF. The most remarkable structural change is the presence of cobaltocenium cations running parallel to the polyoxovanadate chains occupying the center of the pores that were previously occupied by DMF molecules in 3·DMF (Fig. 7). The removal of bulky DMF molecules and the presence of cobaltocenium cations shielding the repulsion between the polyoxovanadates of different chains could explain the small decrease of unit cell volume in Red_3 with respect to that of 3·DMF.
 | ||
| Fig. 7 Crystal structure of Red_3 viewed along the [−101] direction (C (black), N (blue), O (red), Zn (gray), Co (dark blue)). Atoms of the Zn[V6O19(C16H15N6O)2]1.3− chains belonging to the same double layer are colored orange or green. Hydrogen atoms have been omitted for clarity. | ||
PXRD patterns of Red_2 and Red_3 measured in contact with the mother liquor are shown in Fig. S7 in the ESI.† They confirm that both compounds are isostructural. They are similar to those of 2·MeCN and 3·MeCN with differences in the relative intensity of the peaks, which could be related to the different pore filling due to the presence of cobaltocenium cations in the reduced samples. An interesting result is that PXRD pattern of dried crystals of Red_2 is quite similar to that of the same sample measured in contact with the mother liquor, in contrast to the initial sample of 2 (see Fig. S25 in the ESI†). This suggests that the inserted cobaltocenium cations stabilize the structure and prevent the collapse of the pores shown by the initial sample.
EDAX, elemental analysis, XPS and 1H NMR of Reox_2 indicate that cobaltocenium is still present but in a less extent, and that Br− enters in the structure after reoxidation. They are consistent with the formula Fe[V6O19(C16H15N6O)2]·[Co(C5H5)2]0.5[Br]0.5·10H2O (see Experimental section, Fig. S22 and S24 and associated text in the ESI†). PXRD pattern of Reox_2 resembles that of Red_2 as expected for the presence of cobaltocenium cations in the pores (Fig. S26 in the ESI†). In this case, PXRD peaks of a minor phase assigned to the [Co(C5H5)2]2[V6O19(C16H15N6O)2]·MeCN salt were detected. This suggests partial decomposition of the polymeric chains upon chemical reduction and reoxidation. EDAX analysis suggests that this impurity contains around 4% of the hexavanadate POM (see Fig. S27 and Table S4 in the ESI† and associated text).
Magnetic properties of reduced and reoxidized samples
Temperature dependence of χMT of Red_2 in contact with the mother liquor is shown in Fig. 8. An almost constant χMT value of 3.1 cm3 K mol−1 was observed from 300 to 240 K. At lower temperatures, there is a gradual decrease to reach 1.5 cm3 K mol−1 at 85 K. The gradual SCO of this sample suggests that the intercalation of bulky cobaltocenium molecules between the [FeII(1-bpp)2]2+ centers observed in the structure of Red_2 decreases the cooperativity of the SCO as observed in the samples containing bulky solvent molecules (2·DMF and 2·PhCN). However, the magnetic properties of Reox_2, which contains ca. 50% of the cobaltocenium cations present in Red_2 (see above), could indicate that this is not the only factor, as Reox_2 presents an abrupt and more complete spin transition with thermal hysteresis of 11 K (T1/2↓ = 213 K and T1/2↑ = 224 K at a scan rate of 2 K min−1, see Fig. 8). This behaviour is similar to that observed for the initial 2·MeCN but shifted to higher temperatures by 80 K. This larger stabilization of the LS state in Reox_2 could be a consequence of the increased chemical pressure due to the presence of cobaltocenium cations and Br− species in the channels of the compound since the interchain distances in 2·MeCN and Reox_2 are similar, as shown by similar 2θ values (see Fig. S26 in the ESI†). Furthermore, other factors such as the interactions of the Fe(II) complexes with the Br− counteranions could also contribute. Single crystal structure of the reoxidized sample is needed to better understand this behaviour. First attempts to reach this goal, however, were not successful due to the loss of single crystallinity during the reoxidation process. | ||
| Fig. 8 Thermal variation of χMT of 2·MeCN (red circles), Red_2 (green circles) and Reox_2 (orange circles) measured in contact with the solvent. Full circles: data recorded without irradiation; empty circles: data recorded after irradiation at 10 K. | ||
Finally, light was used as an external stimulus to tune the spin state of these compounds. These samples were protected with a grease to prevent desolvation. After irradiation with a 633 nm laser at 10 K, a drastic increase of the magnetic signal was observed in the three samples. Once saturation was reached, the irradiation was switched off and the samples were heated at a scan rate of 0.3 K min−1 to determine the relaxation process (see Fig. 8). In the 10–30 K temperature range, an increase of χMT was observed consistent with LS to HS photoconversions close to 50% for 2·MeCN, 100% for Red_2 and 40% for Reox_2 of the LS centers at this temperature range. The T(LIESST), defined as the minimum of the derivative of χMT after irradiation with temperature, is 75 K for 2·MeCN, 40 K for Red_2 and 65 K for Reox_2.
Magnetic measurements on a reduced sample of 3 (Red_3) in contact with the mother liquor were performed to evaluate the contribution of unpaired electrons of the reduced POM chain since it contains the diamagnetic Zn(II) ion. χMT shows an almost constant value of 0.3 cm3 K mol−1 from 300 to 2 K. This value is consistent with one electron reduction of the sample and lower than that reported for 2-, 4- and 6-electron reduced functionalized hexavanadates (see Fig. S28 in the ESI†).34
The reduction of the POM is evidenced by EPR measurements of Red_2 and Red_3 (see Fig. 9 and S29 in the ESI† and associated text). The spectra below 80 K exhibit the eight lines expected for the hyperfine interaction between an electron localized on a single I = 7/2 51V nucleus as observed for the electrochemically-generated trans-[VV5VIVO13[(OCH2)3CNO2]2]3− (ref. 34) or with cis-[VV5VIVO13[(OCH2)3CCH2OH]2]3−.39 Above 40 K, a broadening of these characteristic 8-line spectra is observed together with a decrease of the splitting between the two outermost features (see Fig. 9 and S29 in the ESI†). This is indicative of electron delocalization between various vanadium centers at increasing temperatures, which is consistent with the mixed valence character of these compounds.34 Therefore, the use of this hexavanadate ligand for the preparation of a SCO complex leads to the coexistence of two properties, SCO and mixed valence, that normally do not appear together in the same compound. In this case, the temperature at which thermal SCO takes place (above 80 K) coincides with the temperature range of a delocalized unpaired electron within the POM framework. The relatively long distance between the SCO center and the POM (minimum interchain and intrachain Zn–V distances of 5.8 and 11.5 Å, respectively, in the structure of 3·DMF) and the lack of electronic communication could prevent the synergy between these two properties in these compounds.
 | ||
| Fig. 9 EPR spectra of Red_2 in contact with the mother liquor. | ||
Conclusions
In this work, 1D CPs with alternating hexavanadate POM and SCO FeII(1-bpp)2 units have been prepared. The neutral character of this 1D network has proven to be crucial to obtain crystalline compounds in contrast to the previous charged CPs so far obtained with [FeII(1-bpp)2]2+ complexes. The remarkable SCO properties of this family of complexes (thermal spin transitions with hysteresis and LIESST effect), which have been obtained typically in salts with small counteranions and strong intermolecular interactions between the complexes, are preserved in these polymeric neutral compounds. Thus, they are the first POM-based materials to show complete and abrupt spin transitions. In addition to this, the organization of these SCO units in this neutral 1D polymer leads to a flexible and stable structure with two main advantages with respect to previous SCO CPs:(i) It enables reversible exchange of solvents into the channels formed between the POM chains. This allows reversible tuning of the abruptness and temperature of the thermal spin transition.
(ii) The presence of the redox active POM unit as spacer between the SCO units affords an additional tuning capability to our materials, related to the structural changes associated with the entrance or removal of the products of the bulky reductant (cobaltocenium) and oxidant species (bromide) acting as counterions. To our knowledge, there are only few very recent reports of SCO CPs with redox active ligands containing TTF or [AuI(CN)2]−. Still, in these compounds the changes of the SCO properties after postsynthetic oxidation with I2 or Br2 are much less drastic.14,17,40 The use of electrochemical methods or less bulky reductant or oxidant species could improve the reversibility of the process.
In conclusion, we have prepared a multifunctional material in which SCO switching of the Fe(II) complexes coexists with the redox properties of the POM, which can switch between a diamagnetic state and a paramagnetic state by injection of one electron. These two switching properties can be tuned by a variety of stimuli such as temperature, light, solvent exchange or redox processes, making them attractive materials for their future implementation in optoelectronic devices or sensors.
Author contributions
MPC synthesized and characterized the compounds under the supervision of MCL and EC (structural, spectroscopic, electrochemical and magnetic characterization) with the help of VGL. JCW and BJCV performed and interpretated Mössbauer measurements and wrote the Mössbauer section of the manuscript. GME supervised the structural characterization, solved the structures and interpretated the powder diffraction results. MPC, MCL and EC wrote the manuscript. All authors contributed to the final interpretation of the experimental results and critically revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the EU (ERC Advanced Grant MOL-2D 788222), the Spanish MCIN (Grants PID2020-117264GB-I00 and PID2020-117152RB-I00 and Unidad de Excelencia María de Maeztu CEX2019-000919-M) and the Generalitat Valenciana (PROMETEO/2021/027), FCT (Portugal) through contracts UID/Multi/04349/2019 and PTDC/QUI-QIN/32240/2017 is acknowledged. This study is part of the Advanced Materials program and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana. We all thank J. Romero-Pascual, A. Soriano-Portillo, J. M. Martínez-Agudo and G. Agustí for PXRD, electrochemical and magnetic measurements.Notes and references
- (a) Spin Crossover in Transition Metal Compounds, Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer Verlag, Berlin-Heidelberg-New York, 2004, vol. 233–235 Search PubMed; (b) Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013 Search PubMed.
- (a) K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176 CrossRef CAS; (b) A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS; (c) E. Coronado, Nat. Rev. Mater., 2020, 5, 87 CrossRef; (d) K. Senthil Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502 CrossRef PubMed.
- (a) M. A. Halcrow, Coord. Chem. Rev., 2005, 249, 2880 CrossRef CAS; (b) M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS; (c) J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203 CrossRef; (d) M. A. Halcrow, New J. Chem., 2014, 38, 1868 RSC; (e) L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2 CrossRef CAS.
- (a) S. Decurtins, P. Gütlich, C. P. Köhler, H. Spiering and A. Hauser, Chem. Phys. Lett., 1984, 105, 1 CrossRef CAS; (b) A. Hauser, Top. Curr. Chem., 2004, 234, 155 CrossRef CAS.
- (a) L. J. Kershaw Cook, F. L. Thorp-Greenwood, T. P. Comyn, O. Cespedes, G. Chastanet and M. A. Halcrow, Inorg. Chem., 2015, 54, 6319 CrossRef CAS PubMed; (b) V. García-López, M. Palacios-Corella, S. Cardona-Serra, M. Clemente-León and E. Coronado, Chem. Commun., 2019, 55, 12227 RSC.
- L. J. Kershaw Cook, R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Chem.–Eur. J., 2016, 22, 1789 CrossRef PubMed.
- R. Kulmaczewski, E. Trzop, L. J. Kershaw Cook, E. Collet, G. Chastanet and M. A. Halcrow, Chem. Commun., 2017, 53, 13268 RSC.
- V. García-López, M. Palacios-Corella, A. Abhervé, I. Pellicer-Carreño, C. Desplanches, M. Clemente-León and E. Coronado, Dalton Trans., 2018, 47, 16958 RSC.
- I. Capel Berdiell, R. Kulmaczewski, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. Commun., 2021, 57, 6566 RSC.
- (a) C. Rajadurai, O. Fuhr, R. Kruk, M. Ghafari, H. Hahn and M. Ruben, Chem. Commun., 2007, 2636 RSC; (b) J. Elhaïk, C. M. Pask, C. A. Kilner and M. A. Halcrow, Tetrahedron, 2007, 63, 291 CrossRef.
- M. Attwood and S. S. Turner, Coord. Chem. Rev., 2017, 353, 247 CrossRef CAS.
- I. Galadzhun, N. Shahid, I. Capel Berdiell and M. A. Halcrow, CrystEngComm, 2019, 21, 6330 RSC.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC.
- H. Y. Wang, J. Y. Ge, C. Hua, C. Q. Jiao, Y. Wu, C. F. Leong, D. M. D'Alessandro, T. Liu and J. L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465 CrossRef CAS PubMed.
- Y. R. Qiu, L. Cui, P.-Y. Cai, F. Yu, M. Kurmoo, C. F. Leong, D. M. D'Alessandro and J. L. Zuo, Chem. Sci., 2020, 11, 6229 RSC.
- S. Schönfeld, K. Dankhoff, D. Baabe, M. K. Zaretzke, M. Bröring, K. Schötz, A. Köhler, G. Hörner and B. Weber, Inorg. Chem., 2020, 59, 8320 CrossRef PubMed.
- L. Zappe, S. Schönfeld, G. Hörner, K. A. Zenere, C. F. Leong, C. J. Kepert, D. M. D'Alessandro, B. Weber and S. M. Neville, Chem. Commun., 2020, 56, 10469 RSC.
- S. Schönfeld, G. Hörner, F. W. Heinemann, A. Hofmann, R. Marschall and B. Weber, Z. Anorg. Allg. Chem., 2021, 647, 295 CrossRef.
- Y. R. Qiu, L. Cui, J. Y. Ge, M. Kurmoo, G. Ma and J. Su, Front. Chem., 2021, 9, 692939 CrossRef CAS PubMed.
- (a) M. T. Pope, Comprehensive Coordination Chemistry II, Elsevier Ltd, Oxford, UK, 2004, vol. 4, p. 635 Search PubMed; (b) C. L. Hill, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed (special issue: polyoxometalates)..
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed.
- M. P. Santoni, G. S. Hanan and B. Hasenknopf, Coord. Chem. Rev., 2014, 281, 64 CrossRef CAS.
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395 CrossRef CAS.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef.
- J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- K. P. Sullivan, W. A. Neiwert, H. Zeng, A. K. Mehta, Q. Yin, D. A. Hillesheim, S. Vivek, P. Yin, D. L. Collins-Wildman, E. R. Weeks, T. Liu and C. L. Hill, Chem. Commun., 2017, 53, 11480 RSC.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382 RSC.
- J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. Mall Haidaraly, G. N. Newton and G. Izzet, Chem. Soc. Rev., 2022, 51, 293 RSC.
- S. Kuramochi, T. Shiga, J. M. Cameron, G. N. Newton and H. Oshio, Inorganics, 2017, 5, 48 CrossRef.
- Y. L. Hong, X. X. Pan, W. W. Cheng, M. X. Yang, Y. M. Han, R. K. Kang, H. Mei and Y. Xu, J. Mol. Struct., 2020, 1219, 128566 CrossRef CAS.
- A. Abhervé, M. Palacios-Corella, J. M. Clemente-Juan, R. Marx, P. Neugebauer, J. van Slageren, M. Clemente-León and E. Coronado, J. Mater. Chem. C, 2015, 3, 7936 RSC.
- N. Bridonneau, L. Rigamonti, G. Poneti, D. Pinkowicz, A. Forni and A. Cornia, Dalton Trans., 2017, 46, 4075 RSC.
- Q. Chen, D. P. Gosborn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667 CrossRef CAS.
- C. Allain, S. Favette, L. M. Chamoreau, J. Vaissermann, L. Ruhlmann and B. Hasenknopf, Eur. J. Inorg. Chem., 2008, 2008, 3433 CrossRef.
- J. W. Han, K. I. Hardcastle and C. L. Hill, Eur. J. Inorg. Chem., 2006, 2006, 2598 CrossRef.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2005, 127, 13978 CrossRef CAS PubMed.
- J. A. DeGayner, I. R. Jeon, L. Sun, M. Dinca and T. D. Harris, J. Am. Chem. Soc., 2017, 139, 4175 CrossRef CAS PubMed.
- A. Müller, J. Meyer, H. Bögge, A. Stammler and A. Botar, Z. Anorg. Allg. Chem., 1995, 621, 1818 CrossRef.
- S. G. Wu, L. F. Wang, Z. Y. Ruan, S. N. Du, S. Gómez-Coca, Z. P. Ni, E. Ruiz, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2022, 144, 14888 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2213981–2213984. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05800f |
| This journal is © The Royal Society of Chemistry 2023 |