
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug repurposing screens identify compounds that inhibit α-synuclein oligomers' membrane disruption and block antibody interactions
Arun Kumar
Somavarapu
,
Giulia
Kleijwegt
,
Madhu
Nagaraj
,
Parvez
Alam
,
Janni
Nielsen
and
Daniel E.
Otzen
 *
*
Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Gustav Wieds Vej 14, 8000 Aarhus C, Denmark. E-mail: dao@inano.au.dk
First published on 21st February 2023
Abstract
Small soluble oligomers of the protein α-synuclein (αSO) have been linked to disruptions in neuronal homeostasis, contributing to the development of Parkinson's Disease (PD). While this makes αSO an obvious drug target, the development of effective therapeutics against αSO is challenged by its low abundance and structural and morphological complexity. Here, we employ two different approaches to neutralize toxic interactions made by αSOs with different cellular components. First, we use available data to identify four neuronal proteins as likely candidates for αSO interactions, namely Cfl1, Uchl1, Sirt2 and SerRS. However, despite promising results when immobilized, all 4 proteins only bind weakly to αSO in solution in microfluidic assays, making them inappropriate for screening. In contrast, the formation of stable contacts formed between αSO and vesicles consisting of anionic lipids not only mimics a likely biological role of αSO but also provided a platform to screen two small molecule libraries for disruptors of these contacts. Of the 7 best leads obtained in this way, 2 significantly impaired αSO contacts with other proteins in a sandwich ELISA assay using αSO-binding monoclonal antibodies and nanobodies. In addition, 5 of these leads suppressed α-synuclein amyloid formation. Thus, a repurposing screening that directly targets a key culprit in PD pathogenesis shows therapeutic potential.
Introduction
The protein α-synuclein (α-syn), widely expressed at presynaptic terminals of neurons, is associated with both sporadic and familial forms of Parkinson's Disease (PD), the second most common neurodegenerative disorder.1 α-Syn is intrinsically disordered in the cytosol and attains a helical conformation when bound to cellular membranes. Aggregation of α-syn to form small soluble cytotoxic oligomers (αSOs) and large fibrillary aggregates, manifested as intracellular inclusions called Lewy Bodies (LBs), are critical in the development of PD2 and are exacerbated by impaired autophagic and lysosomal clearance pathways.3 There is increasing evidence that αSOs are particularly toxic agents in this process.4αSOs exhibit their toxicity through several intracellular mechanisms. They can interact with cell membranes and disrupt their structure, leading to disruption in cellular metabolism and homeostasis, increase in ROS production and ultimately neuronal death.5,6 Recent solution and solid-state NMR studies identify two structural elements essential for membrane disruption, namely a highly exposed lipophilic N-terminal region of α-syn that promotes strong interactions with the lipid membrane and a rigid β-sheet rich core that penetrates the lipid bilayer.7,8 In addition, αSOs impair several cellular pathways. Thus binding of αSOs to the mitochondrial TOM20 receptor inhibits its interaction with the co-receptor TOM22 and impairs mitochondrial protein import, resulting in reduced respiration and increased reactive oxygen species.9 Trapping of neuronal α3-Na+/K+-ATPase by both oligomeric and fibrillar α-syn clusters reduces the pumping efficiency and impairs the Na+ gradient across the plasma membrane.10
Recent electron microscopy and tomography studies of LBs have revealed α-syn-immunoreactive inclusions within the crowded environment of proteins, lipids, lysosomal structures and mitochondria,11 highlighting that these aggregates are likely to have many interaction partners, both proteins and lipids. This is supported by proteomic screen approaches which identify intra-neuronal proteins that can bind preferentially to oligomer species.10,12,13 These studies demonstrate the existence of α-syn conformation-specific (monomer, oligomer or fibril) interactions and further provide opportunities to study pathological processes specific for oligomers. The αSOs studied by us and numerous other groups14,15 are a highly stable species16 which show high cytotoxicity,17,18 form spontaneously during incubation under shaking conditions under physiological conditions at high concentrations of α-synuclein19 and have been shown reproducibly to consist of around 30 monomers.17,20 Consequently, they constitute a very appropriate target for pharmacological investigations.
αSOs are a challenging drug target since they are only formed at low levels (in vitro typically 1–2% of total α-syn used17,21,22) and with a high variety of structural flexibility, ranging from a few well-defined core regions to completely disordered termini and with other segments showing intermediate dynamics.8,17,22 This is not compatible with conventional structure-based drug design approaches. Nevertheless, numerous in vitro and in vivo studies have uncovered a variety of small molecule inhibitors of α-syn aggregation, including antibiotics, polyphenols, curcuminoids, quinones, aminosterol and dopamine analogs.23,24 Besides aggregation inhibitors, natural compounds such as squalamine completely suppress the toxicity of αSOs in human neuroblastoma cells by inhibiting their interactions with lipid membranes.25
Current treatment of PD only targets dopamine-related symptoms, and they do not improve or alter the progression of neuronal toxicity and cell death. We urgently need to identify compounds that protect or restore neuronal health.26,27 Direct targeting of αSOs is a promising avenue for successful PD diagnostics and therapy. To develop a screening strategy which can target and disrupt complexes that involve αSOs, we need to understand the nature of these interactions and their sensitivity to disruption by e.g. small molecule compounds which can curtail αSO toxicity. Besides potential protein binding partners, αSO interactions with membranes are likely to be toxic to cell metabolism and homeostasis, making them important targets for small molecules that could ultimately have a therapeutic effect.
We here used a double-pronged approach. Initially, we focus on interactions of αSOs made with four neuronal proteins (Cfl1, Uchl1, Sirt2 and SerRS) which we identified as potential targets based on previous proteomics studies. Of these four proteins, Cfl1 is an essential regulator of cytoskeletal dynamics in cells. However, its inactivation by α-syn has pathological effects in PD. Neurons incubated with WT and A30P α-syn presented a ∼2-fold increase in inactivate phosphorylated cofilin, respectively, compared to the control.28,29 Uchl1 is abundantly produced in the brain and catalyzes the hydrolysis of ubiquitylated peptides. However, the autosomal dominant missense Ile93Met mutation, which causes a rare familial form of PD, decreases its ubiquitin hydrolase activity. On the other hand, it also exhibits ligase activity towards α-syn, leading to K63-linked polyubiquityl chains which lead to inefficient clearance of α-syn in PD models.30,31 Sirt2 deacetylates histone and non-histone substrates thus regulating a large spectrum of physiological processes. It has been reported to increase α-syn toxicity in PD models by deacetylation at K6 and K10 residues (which are endogenously acetylated in the mouse brain), thus making it more prone to aggregation.32,33 While SerRS catalyzes the attachment of serine to tRNA, its direct role in PD has not been explored. However, it has been reported to recruit Sirt2 to erase prior c-Myc-promoted histone acetylation.34 However, we find that although all 4 protein ligands interact with αSOs when immobilized, they bind weakly to αSOs in solution. This makes them less useful as model systems to identify complex disruptors.
Therefore, our second strategy was to focus on αSOs' ability to form stable complexes with DOPG liposomes. We used the αSO–DOPG system to screen two small-molecule collections of in total 2067 drug compounds from two different collections and identified 7 compounds that reduce membrane disruption by oligomers. Besides inhibiting membrane binding, some of the lead compounds also completely inhibit amyloid fibril formation. Finally, we validate the utility of this approach by demonstrating that two of our top leads disrupt αSO binding to specific antibodies in a novel ELISA sandwich assay. These molecules establish the molecular basis for selective regulation of oligomer toxicity and could be the basis for therapeutic agents to suppress αSO-driven neurodegeneration.
Materials and methods
α-Syn oligomer preparation
WT human α-syn was recombinantly expressed in E. coli Bl21 cells and purified by anion exchange chromatography as described.17 α-Syn fractions were pooled and dialyzed against water, lyophilized and stored at −20 °C. To prepare α-syn oligomers (αSOs), 10 mg of lyophilized α-syn was re-suspended into 1× PBS pH 7.4. The sample was then passed through 0.22 μm filters and incubated at 37 °C for 4 h while shaking continuously at 900 rpm. After incubation, the solution was centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min and the supernatant was loaded on to a Superdex 200 (10/300 GL) column. Oligomer fractions (eluting around 10–12 mL) were collected and stored at −20 °C. Immediately before use, oligomer fractions were concentrated using Centricon filters with a 100 kDa cutoff. Oligomer concentration was determined by UV spectroscopy using an ε280 of 0.412 mg−1 cm−1.
000 rpm for 5 min and the supernatant was loaded on to a Superdex 200 (10/300 GL) column. Oligomer fractions (eluting around 10–12 mL) were collected and stored at −20 °C. Immediately before use, oligomer fractions were concentrated using Centricon filters with a 100 kDa cutoff. Oligomer concentration was determined by UV spectroscopy using an ε280 of 0.412 mg−1 cm−1.
Expression and purification of selected protein partners
Genes for the human proteins Cfl1 (residues M1-L166), Uchl1 (residues M1-A223), Sirt2 (residues M1-Q389), Sirt2_50 (residues S50-Q389) and SerRS (residues M1-A514) were prepared by Genscript (Piscataway, NJ), cloned separately into a pET30a(+) vector with the restriction sites NheI and XhoI and a C-terminal hexa-histidine tag. The Uniprot IDs of all four proteins are provided in Table 1. All genes were codon optimized for maximal expression in E. coli (GenScript). In each case, plasmids were transformed into the E. coli strain BL21 (DE3) cells separately for overexpression. Cells were grown in Luria–Bertani (LB) media and purified by nickel-affinity chromatography as described.35–38 Briefly, bacterial cells were grown in LB media containing 50 μg mL−1 kanamycin at 37 °C with shaking to an OD600 of 0.6–1.0, expression was induced with 1 mM IPTG and the culture was incubated while shaking for 5 h. Cells were then harvested by centrifugation at 4000 rpm for 20 min and re-suspended in buffer A (20 mM Tris–HCl (pH 8.0) and 250 mM NaCl) with 10 mM imidazole, 0.1% v/v Triton X-100 and 1 tablet of EDTA-free protease inhibitor (Roche). The cell pellets from 4 L of culture were lysed by sonication on ice and centrifuged at 12500 rpm for 40 min at 4 °C. The resulting supernatants were loaded onto a Ni-NTA column and the column was washed with 10 column volumes of buffer A with 20 mM imidazole. The bound protein was eluted with buffer A with 250 mM imidazole. The fractions containing the protein were pooled, dialyzed into a buffer containing 10 mM HEPES (pH 7.4), 100 mM NaCl and 1 mM TCEP, and stored at −80 °C.
| Protein name | Gene name | Uniprot ID | # residues | Mass (kDa) | Function |
|---|---|---|---|---|---|
| Cofilin-1 | Cfl1 | P23528 | 166 | 18.5 | Binds to F-actin and regulates actin cytoskeleton dynamics |
| Ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCH-L1) | Uchl1 | P09936 | 223 | 24.8 | Processes ubiquitin precursors and ubiquitinated proteins |
| NAD-dependent protein deacetylase sirtuin-2 | Sirt2 | Q8IXJ6 | 389 (full-length), Sirt2_50 (50–389) lacking the nuclear export signal sequence | 43.2 (38.0 for Sirt2_50) | Deacetylates internal lysines on histones, alpha-tubulin as well as many key transcription factors |
| Serine-tRNA ligase | SerRS | P49591 | 514 | 58.8 | Catalyzes the attachment of serine to tRNASer |
Cell lysate preparation
SH-SY5Y human neuroblastoma cells (∼6 × 106 cells) were washed with ice-cold PBS twice. 2.5 mL of PBS was added to the cell flask and cells were removed using a cell scraper. Cells were transferred to a falcon tube, centrifuged at 4000 rpm for 5 minutes, the supernatant discarded and the cell pellet dissolved by suspension in 300 μL of N-PER™ Neuronal Protein Extraction Reagent (Thermo Fisher Scientific catalog number: 87792) lysis buffer. Subsequently, the suspension was gently mixed by inverting the tube up and down several times for 10 seconds and this process was repeated 3 times for 10 min. Cell debris was then pelleted by centrifuging the lysate for 10 min at 4 °C, 10000 RCF in a tabletop centrifuge. The supernatant (which was used as cell lysate) was stored separately on ice and used within the same day. Cells were kept on ice during the entire process. The lysate concentration was crudely estimated through absorbance at 280 nm (1 Abs unit ≡ 1 mg mL−1).
SPR analysis
Cfl1, Uchl1 and Sirt2 were immobilized on individual lanes of a CM5 sensor chip using amine coupling chemistry through immobilization solutions (EDC, NHS and EA from an amine coupling kit). The running buffer contained 10 mM HEPES pH 7.4, 100 mM NaCl, 0.5 mM TCEP and 0.01% BSA. αSOs were injected at different dilutions over the active surfaces exposing the three binding proteins on individual lanes, and blank surface control. Assays were performed at 25 °C on a Biacore 2000 instrument (GE Healthcare). Regeneration of the surface to remove the bound analyte was carried out using 0.005% SDS. The sensorgram data were evaluated using the Biacore evaluation software (GE Healthcare). The kinetic data were fitted to a 1:1 binding model to give the equilibrium dissociation constant Kd.
Dot-blot binding assay
0.05–0.4 μg of each neuronal protein ligand was applied as 2 μL spots on three separate nitrocellulose membrane strips and allowed to dry. After drying, the strips were transferred to separate falcon tubes and non-specific sites blocked by soaking in 5 mL of 0.5% BSA for 1 h. The strips were then incubated overnight at 4 °C with 100 μg of either α-syn monomers, oligomers or fibrils in a 3 mL PBS solution. After incubation, the strips were washed three times with TBS-T washing buffer (0.05% Tween 20 in TBS buffer), incubated with 3 mL of 1 μg mL−1 primary antibody (the monoclonal antibody 14-9E7-A1 obtained after immunization of mice with αSOs; this antibody binds strongly to αSOs but also recognizes monomers and fibrils (J. N. and D. E. O., unpublished data)) for 1 h at room temperature, washed three times with TBS-T and incubated with 20000-fold diluted secondary antibody (goat anti-mouse coupled to horseradish peroxidase from Jackson Immunoresearch, Ely, UK) for 1 h at room temperature, washed and incubated with 2–3 mL TMB blotting solution for 5 min, washed with Milli-Q water and dried. The dried blots were imaged with a Geldoc Go imaging system (Biorad, Hercules, CA) and densitometrically analyzed with ImageJ.39
Preparation of DOPG liposomes
To prepare pure liposomes of 100 nm diameter, 5 mg mL−1 of 1,2-dioleoyl-sn-3-phosphatidylglycerol (DOPG) was resuspended in 1× PBS buffer, subjected to 10 freeze–thaw cycles using liquid nitrogen and a 50 °C water bath and then passed through a mini-extruder 21 times using a 100 nm cut-off filter (Avanti Polar Lipids, Alabaster, AL). DOPG liposomes for calcein release experiments were prepared in the presence of 70 mM calcein (self-quenching concentrations). Calcein-filled liposomes were separated from free calcein using a PD-10 desalting column (GE Healthcare).Membrane permeabilization assay
Calcein release from calcein-filled vesicles upon membrane permeabilization was monitored by measuring the fluorescence at time 0 (F0) and after 1 h at 37 °C (F) with excitation at 485 nm and emission at 520 nm for 1 h at 37 °C. Finally, Triton X-100 (0.1% (w/v)) was added to measure fluorescence corresponding to complete calcein release (Fmax). The % of calcein release is then determined as follows:| % of calcein release = (F − F0)/(Fmax − F0) | (1) |
Dose–response calcein release for αSOs was quantified in the presence of each of the lead compounds, and IC50 values were obtained by fitting dose–response curves to a sigmoidal model:
| Y = a + (b − a)/(1 + (x/IC50)) | (2) |
Small molecule libraries
Compound libraries were purchased from the Chemical Biology Consortium Sweden (CBCS) platform, which provides high-quality bioactive chemical compounds. The chemical collection used in the screening contained 2067 unique pharmacologically active and chemically diverse compounds, derived from the Prestwick library (1200 FDA and EMA approved drugs) and the Biomol library (1031 compounds comprising neurotransmitter, nuclear receptor ligand, endocannabinoid and orphan ligand molecules). The total screening set thus contains 2231 compounds, of which 164 were duplicates. The compounds were provided at 10 mM stock concentrations in 100% dimethyl sulfoxide (DMSO) and were stored at −20 °C.Screening libraries using a calcein-based assay
For the primary compound screening, 150 nM αSO (concentration in α-syn monomer units) in PBS buffer was loaded onto a Nunc flat clear bottom 96-well plate (Thermo Fisher Scientific, Roskilde, Denmark), after which 5 μM of compounds diluted in 1× PBS and 0.05% DMSO was added. 5 μM each of EGCG and oleuropein were used as a positive and negative control, respectively. The plates were sealed and incubated in a Clariostar fluorescence plate reader (BMG Labtech, Ortenberg, Germany) for 30 min at 37 °C and 2 s shaking every min. After incubation, F0 was recorded (λexc 485 nm; λem 520 nm) and 100 nm-size calcein-DOPG liposomes were added to each well at a final lipid concentration of 50 μM (monomer lipid units). Calcein release was measured for 1 h at 37 °C with fluorescence recording after 2 s shaking every min (the average of the last 5 measurements was taken as F). Finally, 2 μL of Triton X-100 was added to each well to lyse vesicles, after which the fluorescence signal Fmax was measured.Flow-induced dispersion analysis (FIDA)
Binding affinity studies of αSOs and DOPG liposomes were performed on a FIDA 1 instrument with a UV-LED fluorescence detector (Ex 480 nm/Em > 515 nm) (FidaBio ApS, Søborg, Denmark). Standard non-coated capillaries with an inner diameter of 75 μm, an outer diameter of 375 μm, a total length of 100 cm, and a length to detection window of 84 cm were used for all experiments. The sample compartment and capillary housing are temperature controlled to 25 °C.:3 protein:dye molar ratio for 1 h at room temperature and then desalted on a PD-10 desalting column (GE Healthcare, Brøndby, Denmark), where the free dye and the conjugated Alexa488-αSOs were separated. The NHS Ester group of Alexa-488 reacts with primary amines on proteins. The concentration was calculated according to the absorbance at 280 nm (αSOs) and 488 nm (Alexa-488). The degree of labelling was ∼0.6 labels per monomer protein, corresponding to ∼18 Alexa488 per αSO (for comparison, the recommended level of labelling for IgG molecules, a third of the size of an αSO, is 4–9 per IgG40).
 | (3) |
Ultracentrifugation for measuring compound – vesicle binding
To measure the membrane binding affinity of different compounds, 100 μM of each lead compound was incubated with and without 1 mM of DOPG vesicles for 30 min at room temperature. To separate the vesicle-bound compound (pellet) from the free compound (supernatant), the samples were then centrifuged for 1 h at 40000 rpm using a Beckman Optima MAX-XP ultracentrifuge at 25 °C. Subsequently, the absorption of compounds was measured where they displayed the highest absorption at 224 nm and 260–310 nm using a Nanodrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Roskilde, Denmark). The % of compound bound to vesicles is then determined as follows: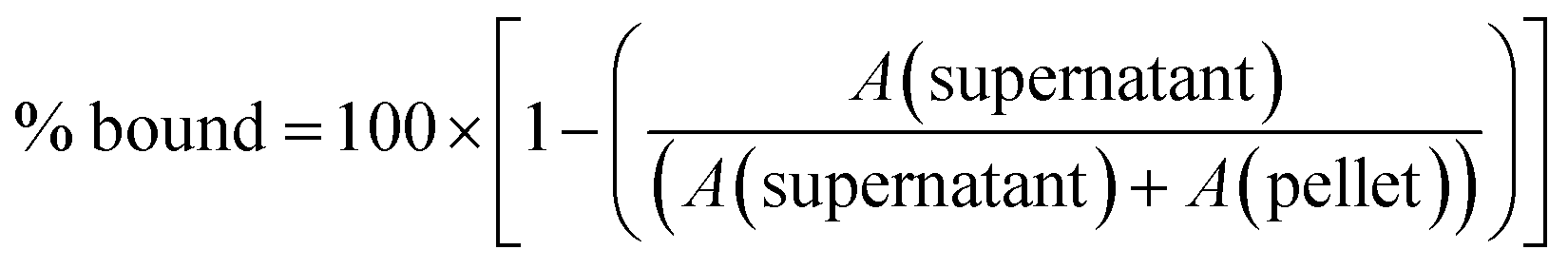 | (4) |
Aggregation kinetics of α-syn in the presence of inhibitors
A stock solution of α-syn was prepared by dissolving freeze-dried α-syn in PBS buffer (pH 7.4), after which it was filtered through a 0.22 μm nylon filter. Stock solutions of lead compounds in DMSO were prepared at 20 mM. Aggregation kinetics assay conditions include 20 μM α-syn, 20 μM compound, 0.1% DMSO, 40 μM ThT, and pH 7.4. 20 μM EGCG at similar assay conditions was used as a positive control. The samples were loaded into a 96-well Nunc plate with 150 μL assay solution in each well. Plates were sealed with clear sealing tape. The change in the ThT fluorescence signal with time was monitored using a Clariostar plate reader under shaking conditions at 300 rpm (10 min shaking in 12 min measurement intervals) and 37 °C. ThT signal was measured with λexc 448 nm and λem 485 nm. All runs were recorded in duplicate and the average run is shown with error bars.Sandwich ELISA assay using specific antibodies and nanobodies
A high bind ELISA plate (Sarstedt, Nümbrecht, Germany) was coated with 60 μL of 5 μg mL−1 anti-αSO nanobody (the nanobody was produced by standard llama immunization with αSOs followed by cloning of B-cells and selection by bacteriophage display41 (J. N. and D. E. O., unpublished results)), incubated overnight at 4 °C, emptied by inversion and gentle tapping on a table and blocked with 75 μL 2% BSA in 1× PBS for 30 min at 37 °C. Then the plate was washed on an Intelispeed Washer IW-8 (BioSan, Riga, Latvia) with 0.05% Tween 20 in 1× PBS three times and incubated with 50 μL 2 μg mL−1 αSO for 1 hour at 37 °C. The plate was again washed, and then 25 μL compound was added (10 mM EGCG (Sigma-Aldrich, ≥95%), 1 mM A1 to A5 (Green Pharma), 1 mM A6–A10 (Merck)) for 30 min at 37 °C (see Table 2 for details on the compounds). Then 25 μL of 1 μg mL−1 primary monoclonal anti-αSO antibody (14-9E7-A1) was added and the plate was incubated for 1 hour at 37 °C, washed three times, and 50 μL secondary Jackson GAM-HRP antibody (1:20000) was added for 1 h at 37 °C. The plate was again washed, and 50 μL TMB One (3,3′,5,5′-tetramethylbenzidine) was added for 30 min at 37 °C while keeping in the dark. The reaction was stopped by adding 100 μL 0.5 M sulfuric acid. The absorption was measured at 450 and 620 nm on a HiPo Microplate Photometer MPP-96 (BioSan, Riga, Latvia).
| Compounds | Sub-library | Categorya | Compound trivial name | Normalized valuesb | Pharmaceutical activity |
|---|---|---|---|---|---|
| a The lead compounds are categorized as approved or experimental based on the drug bank database (https://go.drugbank.com/). Compounds cisplatin and carboplatin are approved though derived from the Biomol library. b Relative extent of calcein release. 0 indicates complete suppression of calcein release (0% calcein release) while 1 indicates no effect of the compound compared to αSO alone (which leads to 50% calcein release). Thus, the actual extent of calcein release is the normalized value × 50%. | |||||
| A1 | Prestwick | Approved | Chlorhexidine | 0.03 | Antimicrobial and antiseptic |
| A2 | Prestwick | Approved | Methyl benzethonium | 0.03 | Surfactant, antiseptic, and antimicrobial |
| A3 | Prestwick | Approved | Benzethonium | 0.03 | Surfactant, antiseptic, and antimicrobial |
| A4 | Prestwick | Approved | Pinaverium | 0.14 | Spasmolytic agent used for gastrointestinal disorders |
| A5 | Prestwick | Approved | Metacycline | 0.02 | A tetracycline antibiotic, antimicrobial action |
| A6 | Biomol | Experimental | Methoctramine | 0.26 | Antagonize muscarinic receptors, studied to treat bradycardia |
| A7 | Biomol | Experimental | Shikonin | 0.24 | Derived from the roots of the shikonin plant, antibacterial, anti-inflammatory and anti-tumor |
| A8 | Biomol | Approved | Cisplatin | 0.04 | Chemotherapy medication, binds to DNA and inhibits replication |
| A9 | Biomol | Approved | Carboplatin | 0.15 | Chemotherapy medication, binds to DNA and inhibits replication |
| A10 | Biomol | Experimental | Adrenochrome | 0.09 | Produced by the oxidation of adrenaline, the derivative carbazochrome, is an antihemorrhagic |
| A11 | Biomol | Experimental | Tyrphostin 47 | 0.14 | A protein tyrosine kinase inhibitor particularly potent against EGFR kinase activity |
Results
Filtering proteins from proteomics studies to identify suitable binding partners for αSOs
We start by selecting potential binding partners for αSOs based on three proteomics-based studies.10,12,13 In these studies, purified αSOs on agarose beads were used as bait protein to capture potential binding partners in a neuronal lysate, after which the beads were spun down and the binding partners identified by mass spectrometry. The collection of 138 proteins obtained from these independent studies is a useful starting point although no direct information about binding affinity is provided. To select the most appropriate binding partners and to minimize the practical challenges in purifying them, we limited ourselves to proteins < 500 residues which were not membrane-bound or parts of stable multi-subunit complexes. This reduced the data set to 31 proteins. These proteins were then matched against Parkinson's UK annotation datasets to filter disease-relevant genes42 (Fig. 1a). The annotation dataset contains 48 high-priority proteins encoded by familial PD genes or genome-wide association studies, and 330 proteins from an extended list of proteins that interact with the high-priority proteins or play a role in PD-related biological processes. Apart from α-syn itself, none of the 138 proteins from the three proteomic-based studies were matched with the proteins in the high-priority set. On the other hand, we found three matches in the extended Parkinson's annotation list. Of these three proteins, we discarded Non-POU domain-containing octamer-binding protein (NONO) due to a high amount (∼30%) of predicted disordered sequence and its reported tendency to aggregate rapidly after purification,43 leaving cofilin-1 (CFL1)13 and ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1)10 for further studies. We could not identify any ligand from the third study12 that matches Parkinson's annotation list after the first filtering step. However, we chose two proteins from this study, namely the NAD-dependent protein deacetylase sirtuin-2 (SIRT2) and seryl-tRNA synthetase (SerRS) based on their high preferential binding to oligomers compared to monomers12 (gauged as the ratio between the MS peak areas of peptides occurring in immunoprecipitation for monomers and oligomers). The four proteins chosen vary in size (18.5 to 58.8 kDa) (Fig. 1b) and function as summarized in Table 1. Initial expression attempts with full-length 389-residue Sirt2 led to low protein yields, so we made a new construct called Sirt2_50 (residues 50–389) where removal of both the N-terminal disordered and nuclear export signal peptide enabled the production of higher amounts of protein.44 In summary, all 4 binding proteins that are studied here have been implicated in neurodegenerative diseases and in contact with αSOs, but their specific mode of interaction has not been studied, motivating further investigations.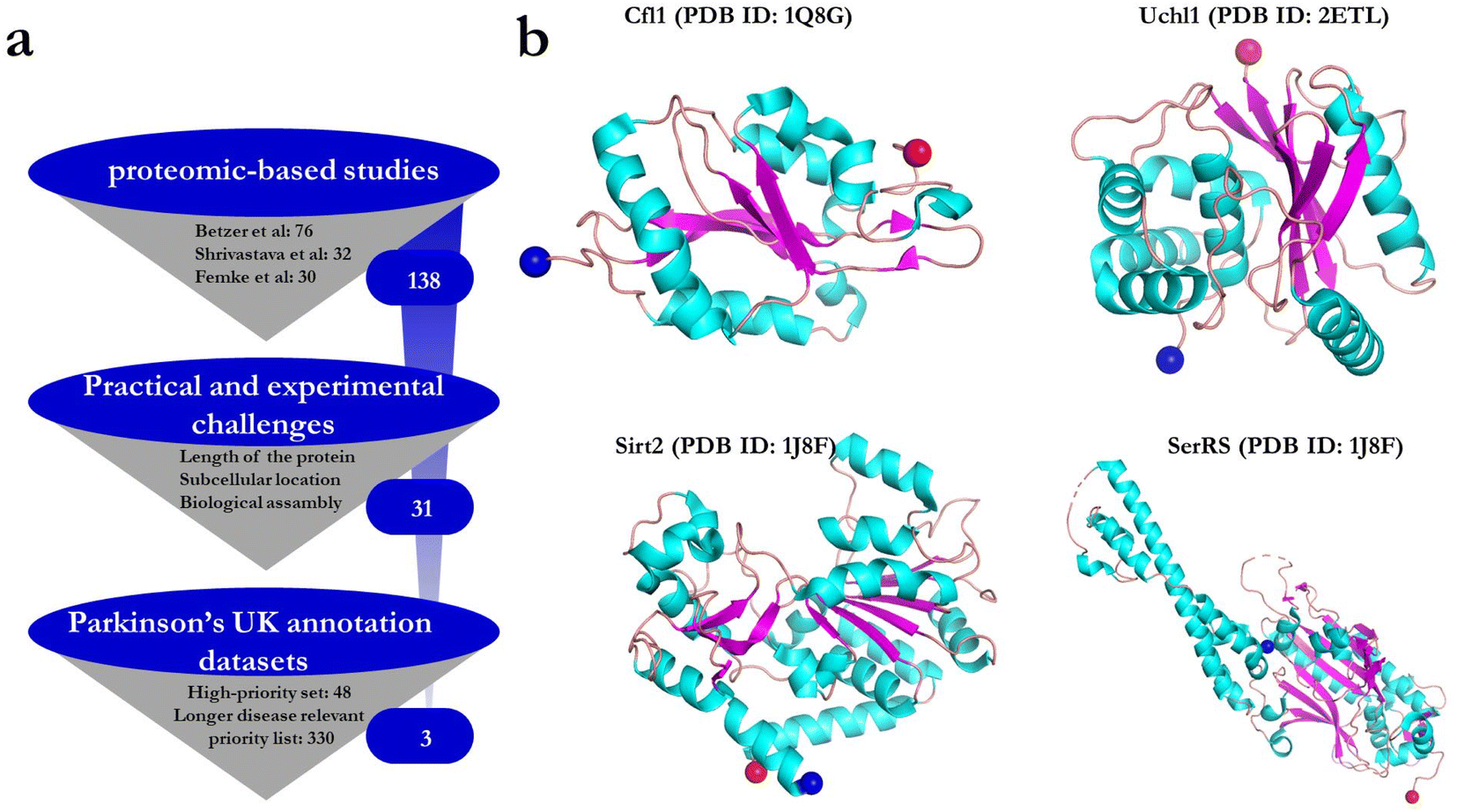 | ||
| Fig. 1 (a) Filtering steps used to select potential protein partners for interaction studies with αSOs. (b) PDB structures of all four protein ligands, colored according to secondary structure. The blue and red spheres represent the N- and C-terminal ends of each structure, respectively. | ||
Immobilized and in-solution assays provide conflicting information about interactions of αSOs with four neuronal proteins
To investigate the binding specificity of the four identified protein ligands for αSOs, we first performed cross-reactivity studies using a dot-blot binding assay. We immobilized increasing amounts (0.05 to 0.4 μg) of ligands on nitrocellulose membranes and then added different species of α-syn (monomer, oligomer and fibril) in the mobile phase. Recombinant human Hsp90, which is known to interact with αSOs,45 was used as a positive control. Fig. 2a shows the concentration-dependent binding of neuronal proteins to different species of α-syn; the darker the dot, the stronger the binding. Using densitometric analysis, we calculated the degree of binding to different species of α-syn, based on the total intensity for all three species combined for each individual ligand (Fig. 2b). Each dot intensity is represented as the percentage of all the measured intensities (monomer + oligomer + fibril) for a particular ligand at that ligand concentration. HSp90, the positive control, showed the expected preferential binding to αSOs. Of the 4 ligands, only Cfl1 and SerRS showed higher binding to αSOs than to α-syn monomers and fibrils. Uchl1 bound most strongly to fibrils while both Sir2 and Sirt2_50 bound to a similar extent to all three species. To confirm this binding profile, we turned to surface plasmon resonance (SPR). We chose Cfl1, Uchl1 and Sirt2 due to their different αSO binding profiles in the dot-blot assay. The 3 proteins were immobilized separately on individual lanes of a CM5 chip, after which we measured the binding kinetics of αSOs at various αSO concentrations (Fig. 2c). All proteins showed a concentration-dependent increase in binding signal (though the signal was weak for Uch1), leading to estimated affinity constants of 0.29 μM, 0.15 μM and 0.6 μM for Cfl1, Sirt2 and Uchl1, respectively.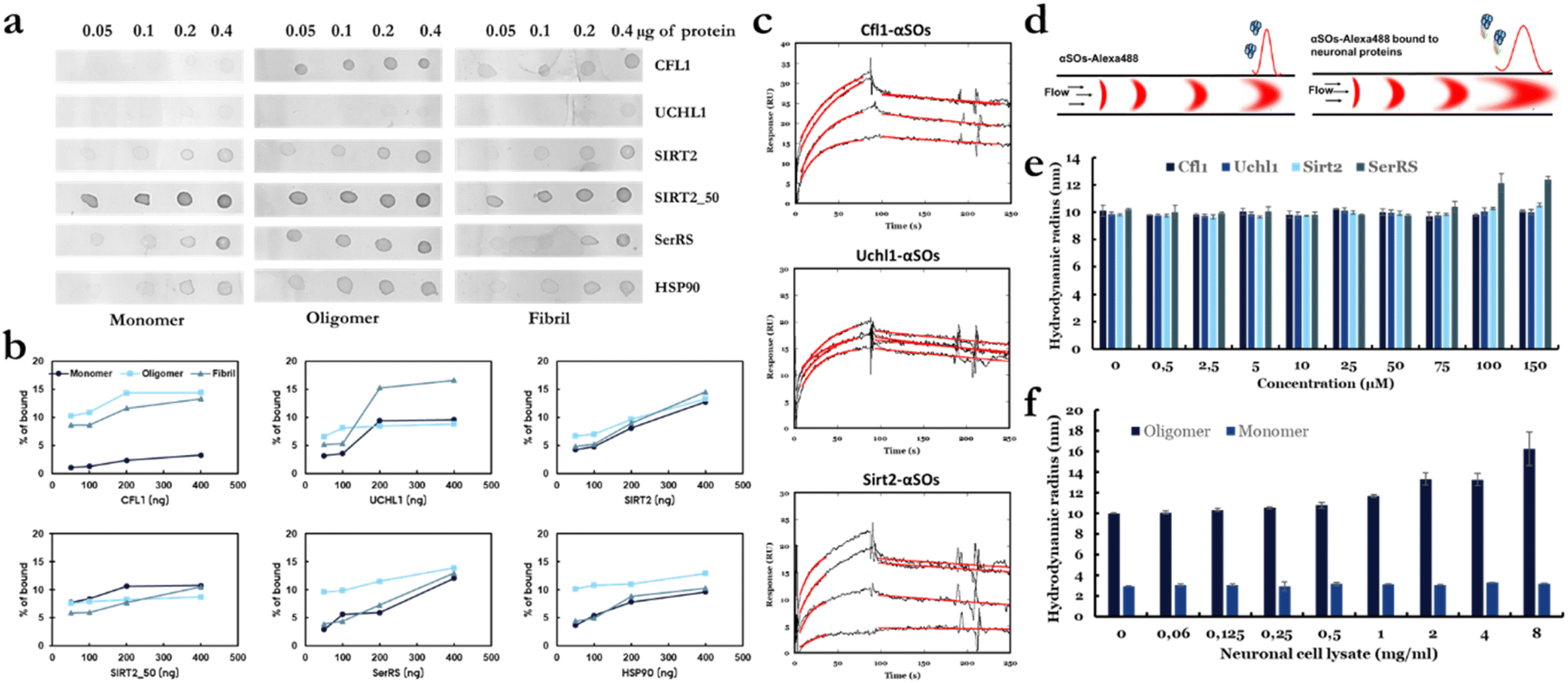 | ||
| Fig. 2 Characterizing the interactions of αSOs with neuronal proteins using various biophysical techniques. (a) Dot blot binding assay using 0.05–0.4 μg of immobilized neuronal proteins exposed to 33 μg mL−1 α-syn in the soluble phase. (b) Binding propensities of each ligand to different species of α-syn were calculated using densitometric analysis (ImageJ software). Values are shown as percentages of the total intensity of all 12 binding dots for each ligand in panel (a). (c) Binding kinetics of αSOs measured by SPR analysis. 1–15 μM of αSOs were passed over sensor surfaces immobilized with Cfl1, Uchl1 and Sirt2 on separate lanes. Sensorgrams show the time curves of the SPR binding signal where the best fits to the binding region using a 1:1 binding model are provided in red. (d) Principle of the use of FIDA to characterize αSOs–ligand interactions. (e) Rh of αSOs-Alexa488 after binding to protein ligands as a function of ligand concentration. (f) Rh of αSOs-Alexa488 and αSMs-Alexa488 after binding to different concentrations of neuronal cell lysate. | ||
Immobilization of proteins potentially complicates the interpretation of binding data. Therefore, to determine the binding affinity of these proteins to αSOs in solution, we turned to Flow-Induced Dispersion Analysis (FIDA), a microfluidic technique in which the hydrodynamic radius Rh of a labelled biomolecule (the indicator) can be obtained from its diffusion behavior (Taylor Dispersion Analysis) in a thin capillary46 (Fig. 2d). Binding to other biomolecules of significant size will increase apparent indicator Rh. The indicator Rh is measured in a titration series with varying concentrations of the unlabeled binding partner (analyte) and the resulting binding curve is used to determine the binding affinity (Kd) and complex size.
We used Alexa488-labelled αSOs as an indicator in combination with unlabeled ligands. Pre-incubation mixing mode of samples was used where αSOs and a given ligand were pre-incubated for 15 min prior to the measurements and subsequently analyzed. In practice the solution was injected onto the capillary as a plug surrounded by a buffer containing the corresponding concentrations of ligand sample, allowing the maintenance of a constant concentration of ligands, which is required for proper equilibrium measurements. The Rh of αSOs alone was 10 ± 0.2 nm, which is similar to the Rh of ∼11 nm that we previously reported based on SEC-MALS and SAXS17 and confirms the structural integrity of the αSO. Using FIDAbio's PDB Rh predictor tool, we predicted the Rh of the four ligands, which were 2.3 nm (Cfl1), 2.4 nm (Uchl1), 2.8 nm (Sirt2) and 3.6 nm (SerRS). The Rh of αSO is expected to increase to 12–13 nm upon binding to ligands, and this increase is well within the limits of detection (since increases as low as 0.5 nm should be measurable). However, we saw little change in Rh when titrated with up to 150 μM Cfl1, Uchl1 and Sirt2 (Fig. 2e). Only for SerRS was there a small but significant increase in the size of oligomer (from 10 to ∼12.5 nm) and this only took place at high (100–150 μM) concentrations. This observation suggests that the binding affinity of ligands to αSO in solution is very weak and well below that indicated by dot-blot assays and measured by SPR.
FIDA reveals cross-reactivity of αSOs with functional neuronal proteins in cell lysates
While the protein ligands studied here did not show strong binding to αSOs in solution, we investigated whether there might be other neuronal components that could bind to αSOs in solution under physiological conditions, i.e. in a cell-like environment crowded with neuronal proteins. Different concentrations (0.06–8 mg mL−1) of freshly prepared human neuroblastoma cell lysate were incubated with Alexa488 labelled αSOs and Alexa488 labelled α-syn monomer (αSMs-Alexa488) and the Rh values were determined by FIDA. Remarkably, while there is no change in the size of the monomer, a significant increase in the average size of the oligomer (10 nm to 16.2 nm) was observed (Fig. 2f). While FIDA does not allow us to identify the binding partner(s), our data clearly show that there are in-solution interaction partners for αSOs in the lysate, consistent with the ability to identify interaction partners in proteomic studies.10,12,13Protein ligands either inhibit or stimulate α-syn aggregation
Despite the weak interactions of αSOs with the 4 binding partners in solution and the caveats associated with their identification by immobilization techniques, their ability to interact with immobilized αSOs implies a potential to affect α-syn aggregation. We therefore followed aggregation kinetics of 20 μM α-syn monomer in the presence of different concentrations of the four ligands (Cfl1, Uchl1, Sirt2_50 and SerRS) by ThT fluorescence (Fig. 3). On its own, α-syn alone at 20 μM concentration showed an aggregation half-time of 44 h. As little as 1 μM Cfl1 led to a dramatic reduction in aggregation. The effect reached saturation at sub-stoichiometric concentrations of 10 μM. In contrast, the other three ligands (Uchl1, Sirt2_50 and SerRS) promoted aggregation of α-syn. Thus 5 μM Sirt2_50 and Uchl1 SerRS reduced half-times of aggregation 2 and 3-fold respectively, while as little as 1 μM of SerRS ligand reduced half-time of α-syn fibrillation more than 3-fold. This indicates a strong impact of the binding partners on α-syn fibrillation which is not related to αSO formation, given that αSOs are off-pathway to fibrillation.17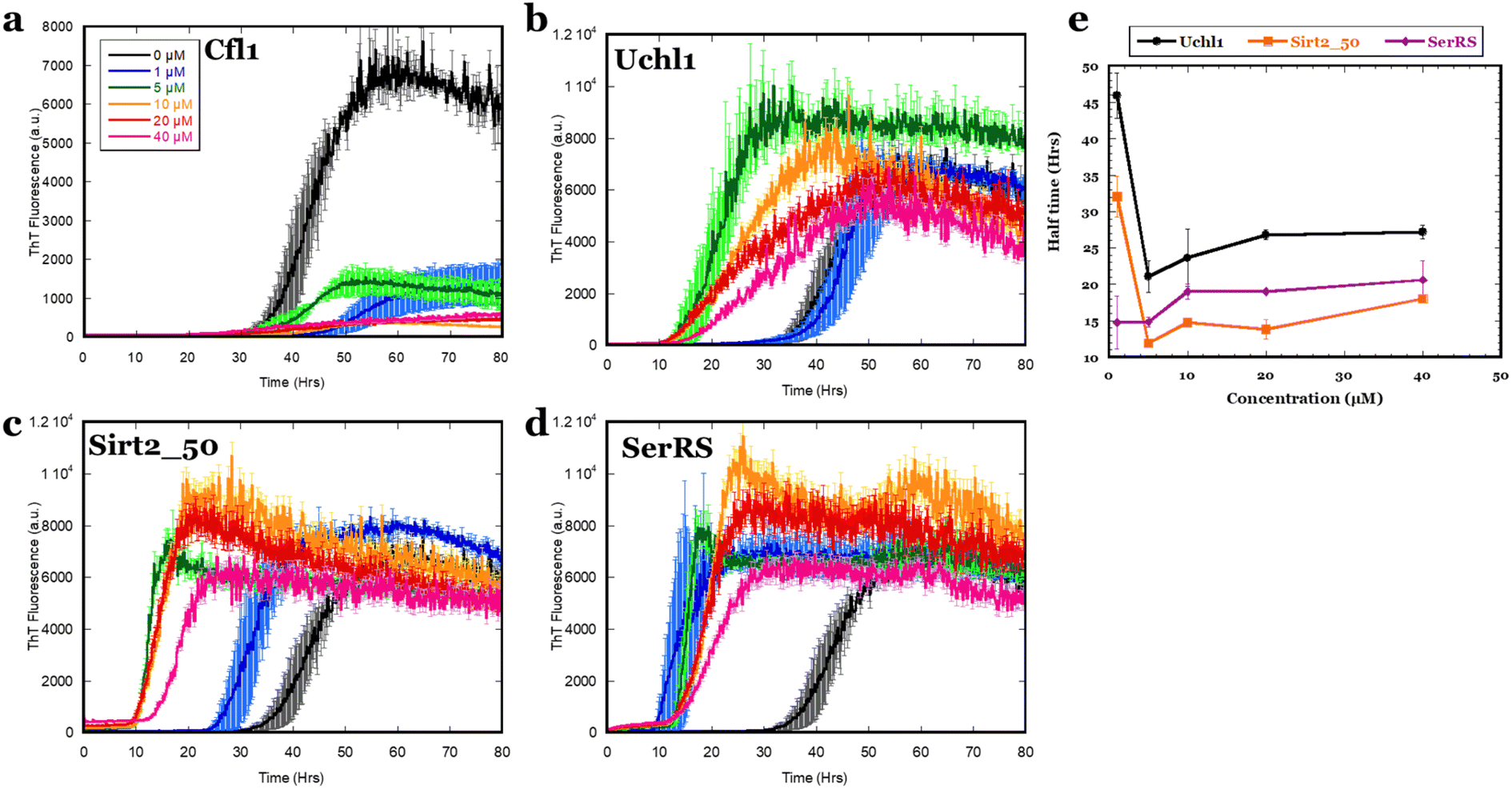 | ||
| Fig. 3 Tht aggregation kinetics of α-syn in the presence of four different protein ligands (a) Cfl1, (b) Uchl1, (c) Sirt2_50 and (d) SerRS. (e) t1/2 of aggregation as a function of ligand concentration for the 3 αSO ligands which show variation in lag times rather than ThT end-point levels. Colour codes in panels (b–d) as indicated in panel (a). | ||
Using FIDA to characterize oligomer–membrane interactions
Given the weak interactions of the proposed binding partners to αSOs, we instead turned to the αSO–membrane system as an approach to identify small molecules that disrupt αSO interactions. To elucidate the cytotoxic interactions of αSOs bound to cell membranes, we prepared membrane-mimicking liposomes made of DOPG lipids and titrated them against αSOs using FIDA. As for the protein ligand studies, αSOs-Alexa488 and DOPG liposomes were co-incubated for 15 min prior to the measurements and subsequently analyzed with corresponding concentrations of liposome sample in the surrounding solvent (Fig. 4a). | ||
| Fig. 4 (a) Principle of the use of FIDA to characterize αSOs–DOPG liposome interactions. (b) Rh of αSOs-Alexa488 after binding to DOPG liposomes as a function of DOPG lipid concentration. (c) The apparent dissociation constant Kd (where lipid concentration is expressed in monomer units) was obtained using data from panel (b) which were transformed and fitted with eqn (3) (including a term for unspecific binding) as described. (d) Extent of the release of calcein from DOPG vesicles incubated with different concentrations of αSO (concentration expressed in α-syn monomer units). Data fitted to a binding isotherm with an amplitude of 100. | ||
R
h of αSOs-Alexa488 in the absence of liposomes is 10.15 ± 0.02 nm (consistent with our previous measurements), while that of the α-syn-Alexa488 monomer is 3.0 ± 0.03 nm, in good agreement with previous single-molecule fluorescence correlation spectroscopy (FCS) measurements.47 DOPG liposomes extruded to an expected Rh of 50 nm were then titrated against αSOs-Alexa488. As the concentration of liposomes is increased, the apparent size of αSOs-Alexa488 increases due to binding. The apparent Rh of liposome bound αSOs-Alexa488 was determined using three-species taylorgram fitting where the size of the one unknown species (here αSOs) is determined by fixing the sizes of two known species in the sample, i.e. free label (0.6 nm) and monomers (3.0 nm). Fitting the FIDA data thus provides the average αSO size, i.e. a weighted average of free and liposome-bound αSOs (Fig. 4b). Furthermore, to differentiate the oligomer species into liposome bound and non-bound, we fixed the size of pre-determined αSOs as 10.2 nm and liposome bound αSO as 60 nm (assuming 1:1 binding stoichiometry) and fitted the FIDA data again. Thanks to the weighting inherent in the average αSO size, this allows us to calculate the fraction-bound oligomers. The resulting plot of the percentage bound species as a function of lipid concentration (Fig. 4c) shows a steep rise at low lipid concentrations followed by a more shallow increase at higher. When fitted with a model that includes specific binding (a hyperbolic relationship) along with a weaker and more unspecific binding (linear relationship), we obtain an apparent lipid binding affinity (KDOPGd) of around 40 μM (Fig. 4c) (based on concentrations in lipid monomer units). It would be more correct to express vesicle concentration in binding sites (or even vesicle concentration), but that is not possible to gauge here. Nevertheless, the large number of lipid molecules involved in contact with αSO makes the actual affinity of oligomers for vesicles likely much higher (see below and Fig. 4d).
Screening small molecule libraries to inhibit oligomer–membrane interactions
To identify compounds that inhibit αSO permeabilization of membranes, we first optimized the calcein assay conditions (illustrated in Fig. 5a) for screening. When we add increasing amounts of αSO (3.9–1000 nM) to a constant concentration of DOPG liposomes (50 μM in monomer units), there is a gradual increase in calcein signal with ∼50% calcein release around 150 nM αSO (Fig. 4d). The data can be fitted to a binding curve with an amplitude of 100% to give an apparent KαSOd of 143 nM. We therefore decided to use 150 nM αSO and 50 μM DOPG as standard conditions under which to screen two datasets of compounds that cover both FDA-approved (Prestwick library) and clinical-stage biologically active drugs (Biomol library). We used EGCG and oleuropein as positive and negative controls, given that EGCG completely inhibits calcein release,48 whereas oleuropein displays weak or no inhibitory effect.49 | ||
| Fig. 5 (a) Illustration of the membrane permeabilization assay using calcein-filled DOPG liposomes, (b) boxplot and (c) histogram showing the distribution of normalized calcein release values from primary compound screening. See the text for the definition of normalized values. | ||
Calcein release values of all the compounds were normalized according to control (αSOs alone without compounds) (Fig. 5b and c) i.e. ∼50% of calcein release. Here, 0 refers to complete suppression of calcein release (that is, 0% calcein release), 1 refers to no effect of the compound compared to αSO alone (that is, 50% calcein release) and >1 refers to an increase in calcein release caused by the compound (>50% calcein release). Out of 2067 unique compounds studied, 24 compounds inhibited the calcein release by at least 50% (that is, they led to 25% calcein release or less) compared to the absence of compounds. To identify the most potent compounds for further studies, we set the threshold at 15% calcein release (i.e. a reduction by 35% or more compared to the absence of compounds), resulting in 11 (0.5%) leads (Table 2 and Fig. 6). Of these, five were from the Prestwick library and six from Biomol. While most of the remaining compounds did not significantly affect the calcein release, ∼12 compounds (those with normalized values of >1.5) increased the release of calcein by more than 50% on top of the existing 50% by αSOs alone, i.e. at least 75% calcein release.
 | ||
| Fig. 6 The 11 compounds which reduce αSO-driven membrane permeabilization to at most 15% calcein release. Details are provided in Table 2. | ||
Dose–response curves rank the different lead compounds
We constructed dose–response curves for 10 of the 11 best compounds with our calcein assay, (compound A11 was not available for purchase from vendors). Six compounds (A1–A4, A6 and A8) maintained the inhibition of vesicle permeabilization as expected from primary compound screening. Of the remaining 4, compound A10 displayed lower efficacy at concentrations used for primary screening (5 μM), while compounds A5, A7 and A9 failed to show any response even at 20 μM, indicating that they were false positives in terms of membrane interactions. For the six compounds which showed an effect, we obtained IC50 values by fitting data to eqn (2) (Fig. 7a and b). Compounds A1–A4 displayed similar IC50 values (1.65–2.65 μM); the highest inhibitory effect was shown by A6 and A8 with IC50 values of 0.32 μM and 1.05 μM, respectively. Both EGCG and PGG plant polyphenols that were included in the study for comparative analysis exhibited a much more pronounced inhibitory effect than the lead compounds, giving IC50 values of 0.08 μM and 0.01 μM respectively. Overall, the lead compounds inhibit membrane permeabilization of αSOs in the ranking order of A6 > A8 > A1–A4 > A10 (Fig. 7c). | ||
| Fig. 7 Quantifying the potency of lead compounds to inhibit membrane permeabilization. Dose–response calcein release for αSOs in the presence of each of the lead compounds. All the values were normalized against calcein release due to αSOs alone in the absence of compounds. IC50 values were obtained by fitting dose–response curves to eqn (2). Note the logarithmic x-axes in panels (a and b) and the logarithmic y-axis in panel (c). | ||
Using FIDA to elucidate the mechanism of binding inhibition
To investigate the underlying mechanism by which the compounds inhibit membrane permeabilization, we performed FIDA on αSOs–DOPG liposome mixtures in the presence of the lead compounds. For these experiments, 150 nM αSOs-Alexa488 and 5 μM compounds were pre-incubated for 15 min, and 50 μM DOPG liposomes were then added after which the Rh was measured (Fig. 8). Using the pre-determined αSO size (10.2 nm) as one of the species during taylorgram data fitting, we computed the percentages of liposome-bound and unbound species of oligomers based on the measured Rh values. | ||
| Fig. 8 Interaction of αSOs with compounds in the presence and absence of liposomes analyzed by FIDA. (a) The apparent size (Rh) of the largest species of αSOs-Alexa488 in the presence of DOPG liposomes and lead compounds. The values of Rh and the percentage of species can be read on the left and right y-axes respectively. (b) Rh of free αSOs-Alexa488 (i.e. without liposomes) in the presence of the lead compounds. | ||
The apparent size computed for αSOs-Alexa488 bound to liposomes was ∼60 nm. Remarkably, compounds A1, A2, A3, A4 and A6 completely prevented the formation of αSO:liposome complexes and retained the original size of αSOs (∼10 nm), indicating 0% bound oligomers (Fig. 8a). The remaining 5 compounds (A5, A7, A8, A9 and A10) did not completely prevent αSO–DOPG complex formation but showed a reduction in the percentage of bound species by 15–20% compared to control samples in the absence of lead compounds (26%).
While EGCG is more potent than all lead compounds in inhibiting calcein release during membrane permeabilization assay (IC50 = 0.08 μM), we note that the EGCG (unlike the 5 compounds A1–A4 and A6) could not completely displace αSOs from binding to DOPG liposomes, leading to ∼15% bound species (consistent with our previous observations48). The apparent size of liposome-bound αSOs in the presence of EGCG was also reduced to 50 nm, which is slightly smaller than the apparent size of the αSO-vesicle complex (∼60 nm) computed in the absence of lead compounds.
It is possible that the inhibition of membrane permeabilization could also be a side effect due to the complete dissociation of oligomers into monomers, or aggregation of small soluble oligomers into less toxic larger oligomers in the presence of lead compounds. We investigated this possibility in a new FIDA assay, where we incubated αSOs in the presence of all 10 lead compounds along with two controls separately, and measured the sizes of labelled αSOs-Alexa488 (Fig. 8b). None of the compounds changed the size of the oligomer, demonstrating that the αSOs are intact and the inhibitory effect of compounds was not due to the dissociation or aggregation of αSOs. However, in the presence of compounds A2, A3 and EGCG, the percentage of α-syn monomers was increased from 13% to 23%, 18% and 22% respectively (Fig. 8b).
To investigate the compounds' own interactions with membranes, we incubated 100 μM of each compound (A1–A4, A6, A8 and A10) with and without 1 mM of DOPG vesicles for 30 minutes, ultracentrifuged the samples (1 h at 40000 rpm), and subsequently determined the percentage of the compound that is bound to vesicles using eqn (4). The five compounds A1, A2, A3, A4 and A6 that inhibited membrane permeabilization in the calcein assay and also prevented the formation of αSO:liposome complexes in FIDA analysis showed significant binding affinity to the membrane with ∼60–95% of the compound pelleted along with vesicles (Fig. 9). On the other hand, compounds A8, A10 and EGCG that did not prevent the αSO–DOPG complex formation in FIDA assay but still inhibited membrane permeabilization in calcein assay showed no significant affinity to a membrane, giving only ∼0–2% of bound compound in the pellet.
 | ||
| Fig. 9 Percentage of compounds bound to DOPG vesicles. Data are based on the change in absorption of 100 μM compounds in the presence and absence of 1 mM DOPG vesicles measured after incubating for 30 min and ultracentrifugation for 1 h at 40000 rpm (eqn (4)). Purple and blue refer to use of absorption at 224 nm and 260–310 nm respectively. | ||
The lead compounds show varying abilities to inhibit fibril formation
We finally evaluated whether the compounds identified from primary screening affect α-syn aggregation in ThT assays using 20 μM each of α-syn and compound. Among 10 lead compounds, compounds A5, A7–A10 completely inhibited fibril formation similar to EGCG control at 20 μM. The 5 compounds that completely inhibited αSO binding to membranes (A1, A2, A3, A4 and A6) also altered the course of α-syn aggregation. Compounds A1, A2 and A3 decreased the half-time of fibrillation ∼2-fold compared to α-syn alone, while A4 and A6 modestly increased the lag phase (Fig. 10). | ||
| Fig. 10 Efficiency of lead compounds in inhibiting fibrillation of 20 μM α-syn at an equimolar ratio of compounds at 300 rpm shaking and 37 °C, monitored using ThT fluorescence. | ||
Sandwich ELISA identifies compounds that block αSO interactions with other proteins
To further demonstrate the utility of these lead compounds, sandwich ELISA of αSOs was performed. In this assay, nanobody 1 (NB1) (which is specific for αSO with a Kd of ca. 3 nM according to FIDA measurements and does not bind the monomer, data not shown) was first immobilized on the plate which was then blocked, after which 1 μg mL−1 αSOs was added with or without 2 mM of each of the 10 lead compounds (A1–A10). The concentration of αSO remaining bound after washing (which reflects the ability of the compound to inhibit αSO binding to either NB1, the antibody or both) was measured by adding an antibody raised against αSOs (14-9E7-A1, which like NB1 has an affinity of around 3 nM for the oligomer). Fig. 11a summarizes the results. The positive control (absence of compounds) shows significant binding of αSOs to immobilized NB1, but several compounds (particularly A1 and A2, also A3 and A6 to a smaller extent) show a decrease in binding which indicate inhibition of oligomer contacts to either nanobody or antibody (or both). Compounds A4, A5, A7, and A10 lead to a slightly higher absorption than the positive control, due to the background absorption of the compounds themselves upon binding. To investigate the best leads in more detail, we carried out a dose–response curve with A1 and A2 using NB1 and the monoclonal antibody 14-9E7-A1 (Fig. 11b). In both cases, there is a decline in absorption with increasing compound concentration as would be expected from a competition experiment, where the compound competes with NB1 or antibody for binding to αSOs. NB1 leads to a less scattered signal as well as a larger signal change, due both to a higher binding of αSOs in the absence of compound and (for A1) a lower level of binding at high compound concentrations. Fitting the NB1 data using an inverted binding isotherm leads to an apparent Kd of 0.13 ± 0.04 and 0.12 ± 0.09 mM for A1 and A2, respectively. Note that although these affinities are significantly lower than those between αSOs, NB1 and the monoclonal antibody, simple mass-action effects will eventually favor displacement of antibody and/or NB1 from the αSOs at sufficiently high concentrations of A1 or A2.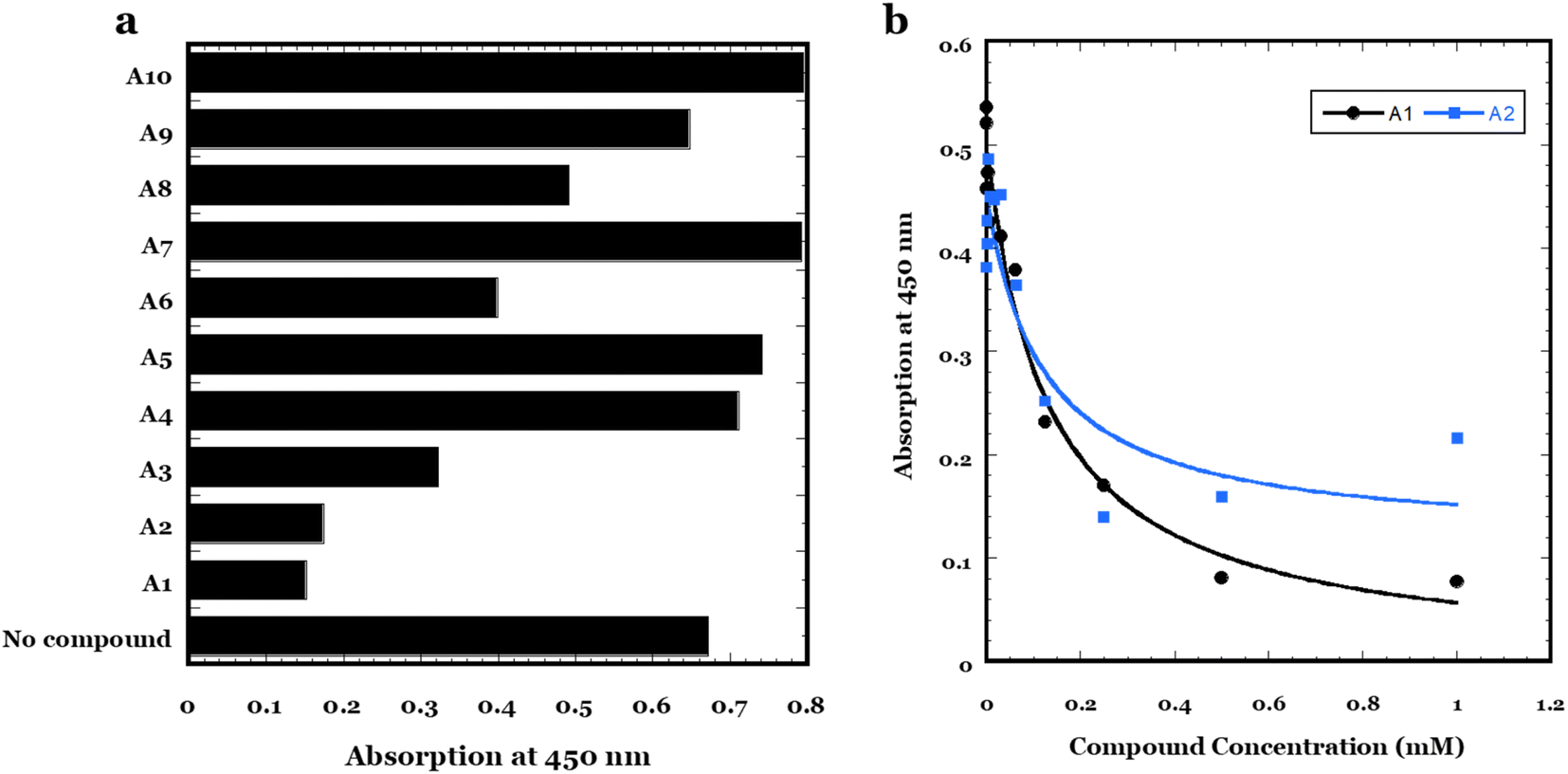 | ||
| Fig. 11 Sandwich ELISA to measure the displacement of αSO from binding to nanobody NB1 and 14-9E7-A1 antibody. (a) Screening of all 10 lead compounds using 2 mM compounds and 1 μg mL−1 αSOs. (b) Dose–response curves of compounds A1 and A2 at 0–1 mM concentrations. Data are fitted to an inverted binding isotherm to give an apparent Kd of 0.13 ± 0.04 and 0.12 ± 0.09 mM for A1 and A2, respectively. | ||
Discussion
Existing treatments of PD mainly target dopamine-related symptoms by either increasing the amount of dopamine using dopamine agonists or inhibiting endogenous enzymes that break down dopamine. Therefore, drugs that can reduce neuronal toxicity and increase neuronal viability are vital in treating PD. Since αSOs are more pathogenic on a per-mass basis than α-syn monomers and fibrils, identification of small molecules that can target oligomer-derived cytotoxicity might eventually reduce the pathogenesis of PD. Here, we established a simple assay to screen small molecules that prevent interactions of αSOs with cell membranes as a proxy for the toxic mechanisms of αSOs in the cell.Protein ligands bind transiently to αSOs but significantly promote monomer aggregation
We first identified a small collection of potential αSO binding ligands based on previous proteomics studies. We then investigated the strength of αSOs interactions with these proteins using different biophysical techniques to evaluate their potential use in a small molecule screening assay. It is evident from both dot blot and SPR studies (where α-syn species were applied at concentrations of 7 μM, i.e. well below the 150 μM used in solution studies) that these protein ligands exhibit strong binding interaction when one of the binding partners is immobilized on a surface. In contrast, the affinity of ligands to αSOs in solution is so weak as not to be detected when measured by FIDA. We note that all the ligands that are identified using co-immunoprecipitation have αSOs immobilized on agarose beads using α-syn-specific antibodies. This suggests in line with other reports that immobilization can lead to artifactual results, both false negatives50 and false positives51 caused e.g. by surface-assisted avidity effects52 or hydrophobic contacts to the surface.53 In addition, structural differences and changes in conformational flexibility of the protein ligands when they are in solution and bound to a surface may also determine the binding of αSOs.In summary, our FIDA data suggest that the interactions between the oligomer and the four ligands (Cfl1, Uchl1, Sirt2 and SerRS) are very weak when both binding partners are in solution. In addition, it is evident from dot-blot densitometric analysis that the α-syn monomer shows similar or weaker binding to all four ligands when compared to αSOs. It is noteworthy that three of the four protein ligands dramatically increased α-syn aggregation during shaking-induced aggregation assays. It is well known that weakly transient bound protein complexes play a crucial role in cell metabolism as well as in regulatory and signaling pathways with affinities in the μM-mM range.54,55 The implication is that the protein ligands do not form stable complexes with αSOs but change the course of aggregation through transient interactions with other α-syn species, most likely the monomer.
Investigating the pathological interactions of αSOs and cell membranes
The physiological role of α-syn–membrane interactions is strongly associated with the ability of α-syn to cluster synaptic vesicles and chaperones SNARE complex formation to maintain neurotransmitter release.56 These interactions are primarily driven by the lipophilic initial 25 residues in the N-terminal region, where the first 14 residues penetrate into lipid head groups as an anchor, leaving the remaining residues folded as an α-helix on the membrane surface.7,57 Such αSOs–membrane interactions can turn pathological in the case of the αSO, where N-terminal binding is accompanied by the insertion of a rigid β-sheet rich oligomer core into the lipid membrane, thereby disrupting its integrity. This further triggers a channel-like pore formation and disrupts cellular calcium ion homeostasis, leading to cell death.5,8FIDA and the calcein assay allowed us to quantify binding affinity between αSOs-Alexa488 and DOPG vesicles, resulting in an apparent KDOPGd of ∼40 μM and Koligomerd of ∼143 nM respectively. The apparent discrepancy reflects both the different types of assays (direct in FIDA and indirect in the calcein assay) and the fact that KDOPGd is based on monomeric lipid units; it would probably be more appropriate to express lipid concentration in terms of available binding sites but that is not possible to gauge. However, the ratio between the two Kd values (ca. 280) would at face value suggest a ratio of 280 lipid molecules per oligomer which is not an unreasonable figure, given that 280 DOPG molecules have a molecular weight of 217 kDa which is in the same ball-park region as the αSO (450 kDa).
Different conformations of α-formed during the course of α-syn aggregation have a different binding affinity towards cell membranes. For example, the later-stage α-syn oligomers (“type B”) identified by Fusco et al. showed the greatest membrane affinity, followed by α-syn monomers, early-stage (“type A”) oligomers and finally fibrils.8 Type A and B oligomers have similar sizes and morphologies but different abilities to disrupt lipid bilayers, clearly linked to their different structural features. We exploited this disruptive αSO:membrane interaction to establish a screening assay and screened two datasets of small molecules.
General considerations about lead compounds
Both compound sets used in our primary screening are quite diverse, and the promising lead compounds are found in different structural classes. Nevertheless, some generalizations are possible. Compounds A1–A3 and A6 share the central feature of having two aromatic rings connected by an alkyl chain. These compounds show a broad range of activity in their original application, including antimicrobial and surfactant properties (chlorhexidine, methyl benzethonium, benzethonium, metacycline and shikonin), antispasmodics that block calcium channels (pinaverium), antagonists of acetylcholine receptors (methoctramine), chemotherapy medication which binds DNA and stops replication (cisplatin and carboplatin) and a compound produced by the oxidation of adrenaline which affects mood and thought processes (adrenochrome). Though none of these compounds have been studied directly in relation to αSOs before, some of them have been reported with reference to other aspects of neurodegeneration. For example, methoctramine competitively antagonizes acetylcholine receptors (M2 muscarinic receptors), thus improving memory in cognitively impaired aged rats.58 Chlorhexidine is an inhibitor of the Keap1 regulator, thus boosting the antioxidant potential of dopaminergic neurons.59 Cisplatin, a PtII containing an antitumor drug, has been shown to inhibit α-syn aggregation by coordinating platinum to side chains of methionine and histidine residues.60 In addition, metacycline is an antibiotic, shikonin is a quinone and adrenochrome has also been shown to reduce α-syn aggregation.61,62Lead compounds and their various courses of action in preventing membrane disruption of αSOs
Out of 11 lead compounds identified in the primary screen, we confirmed 7 compounds (ranked by impact as A6 > A8 > A1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) A2A3A4 > A10) in a more detailed dose–response study, while A5, A9 and A10 failed to show any effect and A11 could not be obtained in sufficient amounts for analysis. A1, A2, A3, A4 and A6 all inhibited αSOs binding to membranes to an equal extent (whereas the remaining 5 compounds had no effect compared to the oligomer alone) in an orthogonal FIDA assay based on oligomer size. The different performances of the 11 compounds reflect differences in the mode of action screened in the various assays. The calcein release assay screens for the compounds' ability to block oligomers' deleterious effects on membrane integrity, whereas the FIDA assay simply gauges the extent of membrane interactions and the sandwich ELISA screens for their ability to block oligomer interactions with other compounds. The 5 effective compounds effective in the FIDA assay all showed significant binding affinity towards membranes on their own (ranked A1A2 > A4A6 > A3). This suggests that these compounds compete with oligomers for membrane binding, enabling them to either displace them from, or (equivalently) prevent them from binding to, the surface of membranes. The 5 compounds also block αSO contacts with anti/nanobodies in our ELISA sandwich assay. This implies that they inhibit interactions either at the oligomer–antibody or oligomer–nanobody interface (or both). Together with their membrane-binding and leakage-blocking activity, this highlights their versatility, i.e. multiple mechanisms of αSO containment. The membrane-blocking phenomenon has previously been observed for several aminosterol compounds such as squalamine and trodusquemine, which in this way are thought to suppress the toxicity of Aβ and αSOs.25,63,64 Claramine, a blood–brain barrier permeable small molecule from the aminosterol class, has also been shown to prevent the toxicity of pore-forming agents including melittin from honeybee venom and α-hemolysin from Staphylococcus aureus.65 Compounds A8 and A10 reduced the amount of calcein release but did not affect binding to DOPG liposomes, suggesting that the compounds could perhaps reduce oligomer membrane permeabilization without preventing binding (e.g. by leading to more superficial membrane attachment), as observed for the control compound EGCG.48 However, with both compounds the oligomers retain the same overall size, indicating that they do not cause either dissociation or aggregation of oligomers (Fig. 12).
A2A3A4 > A10) in a more detailed dose–response study, while A5, A9 and A10 failed to show any effect and A11 could not be obtained in sufficient amounts for analysis. A1, A2, A3, A4 and A6 all inhibited αSOs binding to membranes to an equal extent (whereas the remaining 5 compounds had no effect compared to the oligomer alone) in an orthogonal FIDA assay based on oligomer size. The different performances of the 11 compounds reflect differences in the mode of action screened in the various assays. The calcein release assay screens for the compounds' ability to block oligomers' deleterious effects on membrane integrity, whereas the FIDA assay simply gauges the extent of membrane interactions and the sandwich ELISA screens for their ability to block oligomer interactions with other compounds. The 5 effective compounds effective in the FIDA assay all showed significant binding affinity towards membranes on their own (ranked A1A2 > A4A6 > A3). This suggests that these compounds compete with oligomers for membrane binding, enabling them to either displace them from, or (equivalently) prevent them from binding to, the surface of membranes. The 5 compounds also block αSO contacts with anti/nanobodies in our ELISA sandwich assay. This implies that they inhibit interactions either at the oligomer–antibody or oligomer–nanobody interface (or both). Together with their membrane-binding and leakage-blocking activity, this highlights their versatility, i.e. multiple mechanisms of αSO containment. The membrane-blocking phenomenon has previously been observed for several aminosterol compounds such as squalamine and trodusquemine, which in this way are thought to suppress the toxicity of Aβ and αSOs.25,63,64 Claramine, a blood–brain barrier permeable small molecule from the aminosterol class, has also been shown to prevent the toxicity of pore-forming agents including melittin from honeybee venom and α-hemolysin from Staphylococcus aureus.65 Compounds A8 and A10 reduced the amount of calcein release but did not affect binding to DOPG liposomes, suggesting that the compounds could perhaps reduce oligomer membrane permeabilization without preventing binding (e.g. by leading to more superficial membrane attachment), as observed for the control compound EGCG.48 However, with both compounds the oligomers retain the same overall size, indicating that they do not cause either dissociation or aggregation of oligomers (Fig. 12).
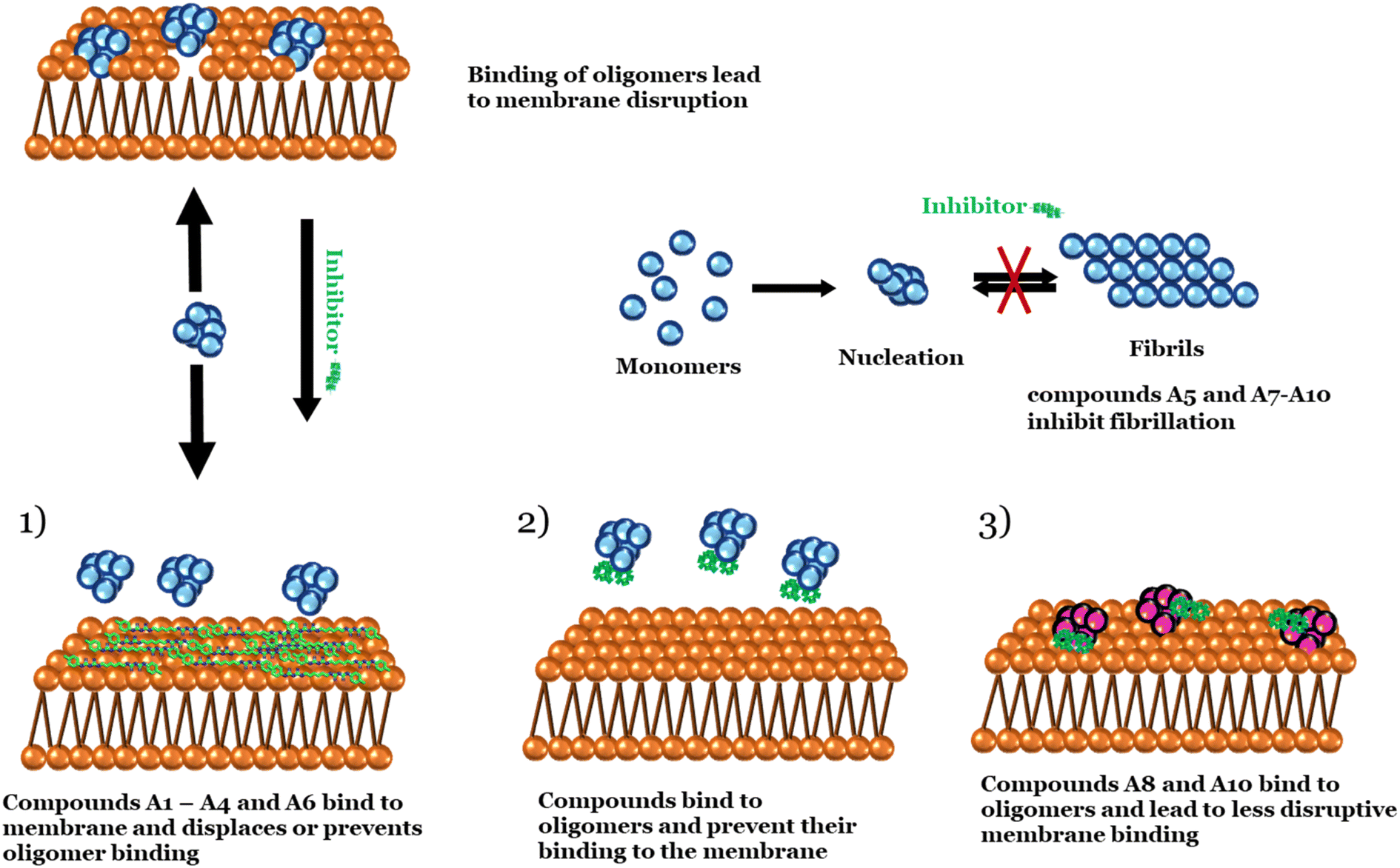 | ||
| Fig. 12 Schematic representation of lead compounds inhibiting membrane permeabilization and fibrillation. | ||
Finally, it is noteworthy that the roles are completely reversed in our fibrillation assays: here compounds showing poor performance against αSO (A5 and compounds A7–A10) are very effective at inhibiting fibrillation while the remaining 5 compounds show only a modest increase in the lag phase (A4 and A6) or even an acceleration of fibrillation (A1–A3). This complementarity of action suggests that different mechanisms are required to target the αSO and the α-syn fibril, again implying that the two targets have substantially different structural properties and thus different binding interfaces. Such molecular insight may be useful in guiding future therapeutic strategies. It will be very interesting to obtain further insight into the pharmacological properties of these compounds in the brain using appropriate animal models.
Data availability
All data are provided in figures and tables. Raw data can be provided upon request.Abbreviations
| E. coli | Escherichia coli |
| α-syn | α-Synuclein monomer |
| αSO | α-Synuclein oligomer |
| FIDA | Flow-induced dispersion analysis |
| SPR | Surface plasmon resonance |
| ThT | Thioflavin T |
Author contributions
A. K. S.: conceptualization, methodology, formal analysis, investigation, writing-original, writing-review & editing, project administration, funding acquisition; G. K.: methodology, investigation; M. N.: methodology, investigation; P. A.: investigation; J. N.: methodology, investigation; D. E. O.: conceptualization, methodology, formal analysis, writing-original, writing-review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. S. is supported by the Lundbeck Foundation (grant R287-2018-1836). D. E. O. is supported by the Independent Danish Research Council|Natural Sciences (grant 8021-00208B, funding M. N.), Novo Nordisk Foundation (grant NNF17OC0028806, funding P. A.) and the Lundbeck Foundation (grant R276-2018-671).References
- R. L. Nussbaum and C. E. Ellis, Alzheimer's Disease and Parkinson's Disease, N. Engl. J. Med., 2003, 348, 1356–1364, DOI:10.1056/NEJM2003ra020003.
- M. Ingelsson, Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders, Front. Neurosci., 2016, 10, 408, DOI:10.3389/fnins.2016.00408.
- C. R. Fields, N. Bengoa-Vergniory and R. Wade-Martins, Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease, Front. Mol. Neurosci., 2019, 12, 299, DOI:10.3389/fnmol.2019.00299.
- B. Winner, et al., In vivo demonstration that alpha-synuclein oligomers are toxic, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4194–4199, DOI:10.1073/pnas.1100976108.
- P. R. Angelova, M. H. Ludtmann, M. H. Horrocks, A. Negoda, N. Cremades, D. Klenerman, C. M. Dobson, N. W. Wood, E. V. Pavlov, S. Gandhi and A. Y. Abramov, Ca2+ is a key factor in α-synuclein-induced neurotoxicity, J. Cell Sci., 2016, 129, 1792–1801, DOI:10.1242/jcs.180737.
- E. Deas, et al., Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson's Disease, Antioxidants Redox Signal., 2016, 24, 376–391, DOI:10.1089/ars.2015.6343.
- E. Cholak, K. Bugge, A. Khondker, K. Gauger, E. Pedraz-Cuesta, M. E. Pedersen, S. Bucciarelli, B. Vestergaard, S. F. Pedersen, M. C. Rheinstädter, A. E. Langkilde and B. B. Kragelund, Avidity within the N-terminal anchor drives α-synuclein membrane interaction and insertion, Faseb. J., 2020, 34, 7462–7482, DOI:10.1096/fj.202000107R.
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades, L. Ying, C. M. Dobson and A. De Simone, Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers, Science, 2017, 358, 1440–1443, DOI:10.1126/science.aan6160.
- R. D. Maio, P. J. Barrett, E. K. Hoffman, C. W. Barrett, A. Zharikov, A. Borah, X. Hu, J. McCoy, C. T. Chu, E. A. Burton, T. G. Hastings and J. T. Greenamyre, α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease, Sci. Transl. Med., 2016, 8, 342ra378, DOI:10.1126/scitranslmed.aaf3634.
- A. N. Shrivastava, V. Redeker, N. Fritz, L. Pieri, L. G. Almeida, M. Spolidoro, T. Liebmann, L. Bousset, M. Renner, C. Léna, A. Aperia, R. Melki and A. Triller, α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient, EMBO J., 2015, 34, 2408–2423, DOI:10.15252/embj.201591397.
- S. H. Shahmoradian, et al., Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes, Nat. Neurosci., 2019, 22, 1099–1109, DOI:10.1038/s41593-019-0423-2.
- C. Betzer, A. J. Movius, M. Shi, W. P. Gai, J. Zhang and P. H. Jensen, Identification of synaptosomal proteins binding to monomeric and oligomeric α-synuclein, PLoS One, 2015, 10, e0116473, DOI:10.1371/journal.pone.0116473.
- F. van Diggelen, S. A. Frank, A. K. Somavarapu, C. Scavenius, M. M. Apetri, J. Nielsen, A. W. J. W. Tepper, J. J. Enghild and D. E. Otzen, The interactome of stabilized α-synuclein oligomers and neuronal proteins, FEBS J., 2020, 287, 2037–2054, DOI:10.1111/febs.15124.
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades, L. Ying, C. M. Dobson and A. De Simone, Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers, Science, 2017, 358, 1440–1443, DOI:10.1126/science.aan6160.
- H. A. Lashuel, B. M. Petre, J. Wall, M. Simon, R. J. Nowak, T. Walz and P. T. Lansbury, alpha-synuclein, especially the Parkinson's Disease-associated mutants, forms pore-like annular and tubular protofibrils, J. Mol. Biol., 2002, 322, 1089–1102 CrossRef CAS PubMed.
- W. Paslawski, M. Andreasen, S. B. Nielsen, N. Lorenzen, K. Thomsen, J. D. Kaspersen, J. S. Pedersen and D. E. Otzen, High stability and cooperative unfolding of cytotoxic α-synuclein oligomers, Biochemistry, 2014, 53, 6252–6263 CrossRef CAS PubMed.
- N. Lorenzen, et al., The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation, J. Am. Chem. Soc., 2014, 136, 3859–3868 CrossRef CAS PubMed.
- N. Lorenzen, S. B. Nielsen, Y. Yoshimura, C. B. Andersen, C. Betzer, B. S. Vad, J. D. Kaspersen, G. Christiansen, J. S. Pedersen, P. H. Jensen, F. A. Mulder and D. E. Otzen, How epigallogatechin gallate can inhibit α-synuclein oligomer toxicity in vitro, J. Biol. Chem., 2014, 289, 21299–21310 CrossRef PubMed.
- W. Paslawski, N. Lorenzen and D. E. Otzen, Formation and characterization of α-synuclein oligomers, Methods Mol. Biol., 2016, 1345, 133–150 CrossRef CAS PubMed.
- N. Zijlstra, C. Blum, I. M. Segers-Nolten, M. M. Claessens and V. Subramaniam, Molecular composition of sub-stoichiometrically labeled alpha-synuclein oligomers determined by single-molecule photobleaching, Angew. Chem., Int. Ed. Engl., 2012, 51, 8821–8824, DOI:10.1002/anie.201200813.
- L. Giehm, D. I. Svergun, D. E. Otzen and B. Vestergaard, Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3246–3251, DOI:10.1073/pnas.1013225108.
- W. Paslawski, S. Mysling, K. Thomsen, T. J. Jørgensen and D. E. Otzen, Co-existence of two different α-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry, Angew. Chem., Int. Ed. Engl., 2014, 53, 7560–7563, DOI:10.1002/anie.201400491.
- D. S. Pena and S. Ventura, One ring is sufficient to inhibit alpha-synuclein aggregation, Neural Regener. Res., 2022, 17, 508–511, DOI:10.4103/1673-5374.320973.
- M. Kurnik, et al., Novel α-synuclein aggregation inhibitors, identified by HTS, mainly target the monomeric state, Cell Chem. Biol., 2018, 25, 1389–1402 CrossRef CAS PubMed.
- M. Perni, et al., A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1009–E1017, DOI:10.1073/pnas.1610586114.
- C. J. Van der Schyf, Rational drug discovery design approaches for treating Parkinson's disease, Expet Opin. Drug Discov., 2015, 10, 713–741, DOI:10.1517/17460441.2015.1041495.
- P. H. Jensen, M. G. Schlossmacher and L. Stefanis, Who Ever Said It Would Be Easy? Reflecting on Two Clinical Trials Targeting α-Synuclein, Mov. Disord., 2023 DOI:10.1002/mds.29318.
- J. N. Namme, A. K. Bepari and H. Takebayashi, Cofilin Signaling in the CNS Physiology and Neurodegeneration, Int. J. Mol. Sci., 2021, 22, 10727, DOI:10.3390/ijms221910727.
- M. I. Oliveira da Silva and M. A. Liz, Linking Alpha-Synuclein to the Actin Cytoskeleton: Consequences to Neuronal Function, Front. Cell Dev. Biol., 2020, 8, 787, DOI:10.3389/fcell.2020.00787.
- A. E. Cartier, K. Ubhi, B. Spencer, R. A. Vazquez-Roque, K. A. Kosberg, L. Fourgeaud, P. Kanayson, C. Patrick, E. Rockenstein, G. N. Patrick and E. Masliah, Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy, PLoS One, 2012, 7, e34713, DOI:10.1371/journal.pone.0034713.
- Y. Liu, L. Fallon, H. A. Lashuel, Z. Liu and P. T. Lansbury Jr, The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility, Cell, 2002, 111, 209–218, DOI:10.1016/s0092-8674(02)01012-7.
- R. M. de Oliveira, et al., The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease, PLoS Biol., 2017, 15, e2000374, DOI:10.1371/journal.pbio.2000374.
- Y. Liu, Y. Zhang, K. Zhu, S. Chi, C. Wang and A. Xie, Emerging Role of Sirtuin 2 in Parkinson's Disease, Front. Aging Neurosci., 2019, 11, 372, DOI:10.3389/fnagi.2019.00372.
- Y. Shi, X. Xu, Q. Zhang, G. Fu, Z. Mo, G. S. Wang, S. Kishi and X. L. Yang, tRNA synthetase counteracts c-Myc to develop functional vasculature, Elife, 2014, 3, e02349, DOI:10.7554/eLife.02349.
- J. X. Lu, Y. F. Xiang, J. X. Zhang, H. Q. Ju, Z. P. Chen, Q. L. Wang, W. Chen, X. L. Peng, B. Han and Y. F. Wang, Cloning, soluble expression, rapid purification and characterization of human Cofilin1, Protein Expr. Purif., 2012, 82, 186–191, DOI:10.1016/j.pep.2012.01.002.
- S. L. Rydning, P. H. Backe, M. M. L. Sousa, Z. Iqbal, A. M. Øye, Y. Sheng, M. Yang, X. Lin, G. Slupphaug, T. H. Nordenmark, M. D. Vigeland, M. Bjørås, C. M. Tallaksen and K. K. Selmer, Novel UCHL1 mutations reveal new insights into ubiquitin processing, Hum. Mol. Genet., 2017, 26, 1031–1040, DOI:10.1093/hmg/ddw391.
- Y. B. Teng, H. Jing, P. Aramsangtienchai, B. He, S. Khan, J. Hu, H. Lin and Q. Hao, Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies, Sci. Rep., 2015, 5, 8529, DOI:10.1038/srep08529.
- C. Wang, Y. Guo, Q. Tian, Q. Jia, Y. Gao, Q. Zhang, C. Zhou and W. Xie, SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis, Nucleic Acids Res., 2015, 43, 10534–10545, DOI:10.1093/nar/gkv996.
- C. A. Schneider, W. S. Rasband and K. W. N. I. H. Eliceiri, Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9, 671–675, DOI:10.1038/nmeth.2089.
- Invitrogen (ThermoFisher Scientific) User Guide: Protein Labeling Kits (For Alexa Fluor™, Pacific Blue™, Fluorescein-EX, and Oregon Green™ 488), Rev A.0, 2021.
- C. E. Gavira-O'Neill, J. X. Dong and J. S. Trimmer, Development, Screening, and Validation of Camelid-Derived Nanobodies for Neuroscience Research, Curr. Protoc. Neurosci., 2020, 94, e107, DOI:10.1002/cpns.107.
- R. E. Foulger, P. Denny, J. Hardy, M. J. Martin, T. Sawford and R. C. Lovering, Using the Gene Ontology to Annotate Key Players in Parkinson's Disease, Neuroinformatics, 2016, 14, 297–304, DOI:10.1007/s12021-015-9293-2.
- G. J. Knott, S. Panjikar, A. Thorn, A. H. Fox, M. R. Conte, M. Lee and C. S. Bond, A crystallographic study of human NONO (p54(nrb)): overcoming pathological problems with purification, data collection and noncrystallographic symmetry, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 761–769, DOI:10.1107/s2059798316005830.
- C. Sun, D. Song, P. A. Marcotte, P. L. Richardson and P. J. Hajduk, High-level bacterial expression and purification of human SirT2 protein for NMR studies, Protein Expr. Purif., 2006, 48, 56–60, DOI:10.1016/j.pep.2005.12.006.
- S. Daturpalli, C. A. Waudby, S. Meehan and S. E. Jackson, Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers, J. Mol. Biol., 2013, 425, 4614–4628, DOI:10.1016/j.jmb.2013.08.006.
- D. E. Otzen, A. K. Buell and H. Jensen, Microfluidics and the quantification of biomolecular interactions, Curr. Opin. Struct. Biol., 2021, 70, 8–15, DOI:10.1016/j.sbi.2021.02.006.
- X. Li, C. Dong, M. Hoffmann, C. R. Garen, L. M. Cortez, N. O. Petersen and M. T. Woodside, Early stages of aggregation of engineered α-synuclein monomers and oligomers in solution, Sci. Rep., 2019, 9, 1734, DOI:10.1038/s41598-018-37584-6.
- N. Lorenzen, S. B. Nielsen, Y. Yoshimura, B. S. Vad, C. B. Andersen, C. Betzer, J. D. Kaspersen, G. Christiansen, J. S. Pedersen, P. H. Jensen, F. A. Mulder and D. E. Otzen, How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro, J. Biol. Chem., 2014, 289, 21299–21310, DOI:10.1074/jbc.M114.554667.
- H. Mohammad-Beigi, F. Aliakbari, C. Sahin, C. Lomax, A. Tawfike, N. P. Schafer, A. Amiri-Nowdijeh, H. Eskandari, I. M. Møller, M. Hosseini-Mazinani, G. Christiansen, J. L. Ward, D. Morshedi and D. E. Otzen, Oleuropein derivatives from olive fruit extracts reduce α-synuclein fibrillation and oligomer toxicity, J. Biol. Chem., 2019, 294, 4215–4232, DOI:10.1074/jbc.RA118.005723.
- A. W. Butch, Dilution protocols for detection of hook effects/prozone phenomenon, Clin. Chem., 2000, 46, 1719–1721 CrossRef CAS.
- S. M. H. Qadri and K. K. Smith, Nonspecificity of the Anda A60-tb ELISA test for serodiagnosis of mycobacterial disease, Can. J. Microbiol., 1992, 38, 804–806, DOI:10.1139/m92-131.
- G. R. Pullen, M. G. Fitzgerald and C. S. Hosking, Antibody avidity determination by ELISA using thiocyanate elution, J. Immunol. Methods, 1986, 86, 83–87, DOI:10.1016/0022-1759(86)90268-1.
- J. Homola, Present and future of surface plasmon resonance biosensors, Anal. Bioanal. Chem., 2003, 377, 528–539, DOI:10.1007/s00216-003-2101-0.
- P. Chien and L. M. Gierasch, Challenges and dreams: physics of weak interactions essential to life, Mol. Biol. Cell, 2014, 25, 3474–3477, DOI:10.1091/mbc.E14-06-1035.
- J. R. Perkins, I. Diboun, B. H. Dessailly, J. G. Lees and C. Orengo, Transient protein-protein interactions: structural, functional, and network properties, Structure, 2010, 18, 1233–1243, DOI:10.1016/j.str.2010.08.007.
- J. Diao, J. Burré, S. Vivona, D. J. Cipriano, M. Sharma, M. Kyoung, T. C. Südhof and A. T. Brunger, Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2, Elife, 2013, 2, e00592, DOI:10.7554/eLife.00592.
- G. Fusco, A. De Simone, P. Arosio, M. Vendruscolo, G. Veglia and C. M. Dobson, Structural Ensembles of Membrane-bound α-Synuclein Reveal the Molecular Determinants of Synaptic Vesicle Affinity, Sci. Rep., 2016, 6, 27125, DOI:10.1038/srep27125.
- A. Lazaris, S. Cassel, J. Stemmelin, J. C. Cassel and C. Kelche, Intrastriatal infusions of methoctramine improve memory in cognitively impaired aged rats, Neurobiol. Aging, 2003, 24, 379–383, DOI:10.1016/s0197-4580(02)00067-2.
- S. Unni, P. Deshmukh, G. Krishnappa, M. M. S. Bharath and B. Padmanabhan, Chlorhexidine as a Keap1-Nrf2 inhibitor: a new target for an old drug for Parkinson's disease therapy, J. Biomol. Struct. Dyn., 2022, 1–15, DOI:10.1080/07391102.2022.2086175.
- B. B. Pan, Y. Yang, H. Z. Liu, Y. H. Li and X. C. Su, Coordination of Platinum to α-Synuclein Inhibits Filamentous Aggregation in Solution, Chembiochem, 2019, 20, 1953–1958, DOI:10.1002/cbic.201900224.
- J. Li, M. Zhu, A. B. Manning-Bog, D. A. Di Monte and A. L. Fink, Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson's and Alzheimer's disease, Faseb. J., 2004, 18, 962–964, DOI:10.1096/fj.03-0770fje.
- M.-m. Xu, P. Ryan, S. Rudrawar, R. J. Quinn, H.-y. Zhang and G. D. Mellick, Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein, Acta Pharmacol. Sin., 2020, 41, 483–498, DOI:10.1038/s41401-019-0304-y.
- S. Errico, et al., Making biological membrane resistant to the toxicity of misfolded protein oligomers: a lesson from trodusquemine, Nanoscale, 2020, 12, 22596–22614, 10.1039/D0NR05285J.
- R. Limbocker, et al., Squalamine and Its Derivatives Modulate the Aggregation of Amyloid-β and α-Synuclein and Suppress the Toxicity of Their Oligomers, Front. Neurosci., 2021, 15, 680026, DOI:10.3389/fnins.2021.680026.
- R. P. Kreiser, et al., A Brain-Permeable Aminosterol Regulates Cell Membranes to Mitigate the Toxicity of Diverse Pore-Forming Agents, ACS Chem. Neurosci., 2022, 13, 1219–1231, DOI:10.1021/acschemneuro.1c00840.
| This journal is © The Royal Society of Chemistry 2023 |