
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent organometallic dyads and triads: establishing spatial relationships†
Yoshinao
Shinozaki‡
,
Stepan
Popov‡
 and
Herbert
Plenio
*
and
Herbert
Plenio
*
Organometallic Chemistry, Technical University of Darmstadt, Alarich-Weiss-Str. 12, 64287 Darmstadt, Germany. E-mail: herbert.plenio@tu-darmstadt.de
First published on 6th December 2022
Abstract
FRET pairs involving up to three different Bodipy dyes are utilized to provide information on the assembly/disassembly of organometallic complexes. Azolium salts tagged with chemically robust and photostable blue or green or red fluorescent Bodipy, respectively, were synthesized and the azolium salts used to prepare metal complexes [(NHC_blue)ML], [(NHC_green)ML] and [(NHC_red)ML] (ML = Pd(allyl)Cl, IrCl(cod), RhCl(cod), AuCl, Au(NTf2), CuBr). The blue and the green Bodipy and the green and the red Bodipy, respectively, were designed to allow the formation of efficient FRET pairs with minimal cross-talk. Organometallic dyads formed from two subunits enable the transfer of excitation energy from the donor dye to the acceptor dye. The blue, green and red emission provide three information channels on the formation of complexes, which is demonstrated for alkyne or sulfur bridged digold species and for ion pairing of a red fluorescent cation and a green fluorescent anion. This approach is extended to probe an assembly of three different subunits. In such a triad, each component is tagged with either a blue, a green or a red Bodipy and the energy transfer blue →green → red proves the formation of the triad. The tagging of molecular components with robust fluorophores can be a general strategy in (organometallic) chemistry to establish connectivities for binuclear catalyst resting states and binuclear catalyst decomposition products in homogeneous catalysis.
Introduction
Linking molecular fragments through chemical bonds to generate more complex species defines synthetic chemistry.1–10 Isolation of newly generated molecules allows their identification using established physical techniques, like NMR spectroscopy. In a non-covalent setting the species generated are characterized by significant dynamics and the rapid interconversion of numerous species may preclude the isolation of a single reaction product whose formation is governed by numerous equilibria involving the individual building blocks.11,12 In such cases it may be difficult to establish connectivities between the different molecular fragments. FRET is a potential tool to study molecular interactions and dynamic changes. Primarily in biochemistry, biology and medicine the use of fluorescent dyes utilizing FRET (fluorescence or Förster resonance energy transfer) is ubiquitous13 and known to provide information about conformational changes involving proteins and DNA14 or intracellular dynamics,15 but also in polymer chemistry16 and supramolecular chemistry.17,18FRET is a non-radiative process in which an excited donor dye transfers energy to an acceptor dye in the ground state through long-range dipole/dipole interactions.19,20 This approach relies crucially on suitable fluorescent dyes (FRET pairs) developed in the last couple of decades, and which are characterized by proper functional groups to enable bioconjugation and compatibility with typical biochemical environments and aqueous solutions.21
Organometallic chemistry mostly takes place in a non-aqueous environment, rendering fluorophores used in biochemistry less suitable. Lewis basic heteroatoms are undesirable in organometallic chemistry, since they potentially coordinate the metal centers. Bodipy based dyes22,23 are proving to be very useful in organometallic chemistry.24–32 Numerous applications of this dye for mechanistic studies of chemical reactions involving single molecules or single particles utilizing fluorescence microscopy have been reported33 – with major contributions by the Blum group.34–40
However, only a few FRET pairs based on Bodipy dyes have been reported in the literature.41 Diederich et al. reported on a FRET pair composed of a green and a red Bodipy FRET pair, which were attached to a resorcin[4]arene cavitand. A Förster radius of approx. 3.7 nm was determined, which is huge compared to the size of typical organometallic complexes.42 Conceptual work from Lim and Blum43 reports on a palladium complex incorporating an allylic ligand tagged with a green fluorophore and a phosphine ligand tagged with an orange fluorescent dye in the coordination sphere. Upon green excitation the close vicinity of the two dyes leads to efficient resonance energy transfer evidenced by an orange fluorescence upon excitation of the green Bodipy. Plenio et al. utilized the Diederich FRET pair in NHC-metal complexes to achieve large virtual Stokes shifts for CO detection.44 The same group showed for closely related fluorophores, that FRET is also observed upon dimerization of NHC-gold complexes.45 However, significant problems for practical applications remain. The synthesis of the previously used red Bodipy is cumbersome and furthermore the chemical stability of the Diederich red Bodipy in an organometallic environment is rather limited and significantly improved red fluorophores are clearly needed.
We wish to report here on the synthesis of organometallic complexes utilizing a stable and readily available red fluorophore with convenient linker groups, and demonstrate organometallic chemistry applications of a new green-red FRET pair with excellent chemical and photochemical stability as well as a related blue-green dyad. The newly established dyad is extended by a blue Bodipy46 to provide a useful triad, in which the blue excitation energy is transferred to the green fluorophore, followed by transfer to the red fluorophore whose emission is observed when spatial vicinity between the three dyes is given.
Results and discussion
Synthesis of red and green NHC-metal complexes47
Red Bodipy-substituted azolium salts were synthesized as outlined in Scheme 1. The literature procedure for the cyclization of the tosylated pyrrole 148 to the spiro-Bodipy was modified. In our hands the original procedure for the Pd/phosphine catalyzed ring–closing reaction generates significant amounts of the undesired β-isomer instead of the desired α-isomer (Scheme 1), whose separation is difficult.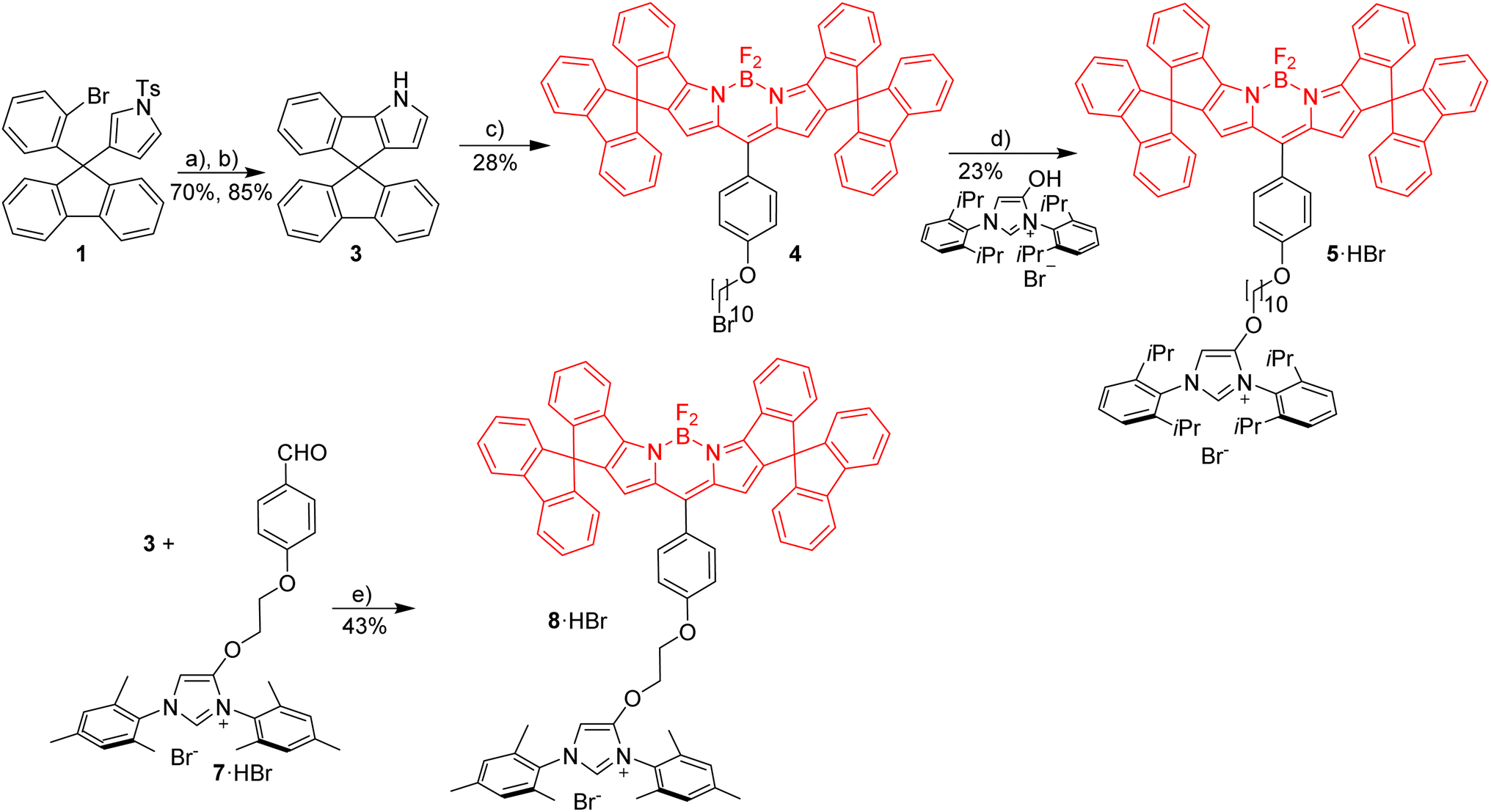 | ||
| Scheme 1 Synthesis of red imidazolium salts with short and long alkyl linker. Reagents and conditions: (a) Pd(OAc)2, DMF, K2CO3, 100 °C, 20 h; (b) Cs2CO3, THF/MeOH, rt, 24 h; (c) alkylation with 1,4-(CHO), (O(CH2)10Br)C6H4, followed by addition of TFA, CH2Cl2, 3 h then DDQ, 1 h then BF3·Et2O, Et3N, 3 h; (d) K2CO3, KI, acetone, reflux, 12 h; (e) TFA, rt, overnight followed by DDQ, 1 h then Et3N, BF3·Et2O, rt, overnight. | ||
Since it is known, that direct arylation reactions do not necessarily require additional phosphine ligands, ligand-free approaches for the cyclization were tested and optimized.49,50 Using Pd(OAc)2 in DMF in the absence of phosphine provides the α-isomer as the predominant cyclization product in 70% yield (β-isomer <1%). The detosylation of spiro-pyrrole was done by simply stirring the protected pyrrole with Cs2CO3 base in THF/MeOH and gave spiro-pyrrole 3 in 85% yield. The reaction of spiro-pyrrole 3 with benzaldehydes provides the respective Bodipy derivatives 4 and 8·HBr following established procedures for related compounds.44 The alkylation of the phenolic hydroxy-imidazolium salt with 4 provides azolium salt 5·HBr tagged with a red spiro-Bodipy.51 This ether forming reaction requires very long alkyl chains (here –(CH2)10Br), since for shorter linkers, like (–(CH2)6Br) significant product formation was not observed. Comparable observations were made before and the lack of reactivity for the shorter linkers is attributed to the steric bulk of the two building blocks (spiro-Bodipy and imidazolium salt).24
It is possible to establish Bodipy-tagged imidazolium salts with shorter linkers using a different order of the synthetic steps (Scheme 1). A –C6H4(CHO) terminated azolium salt 7·HBr was synthesized and this aldehyde condensed with spiro-pyrrole 3 to the respective Bodipy-tagged azolium salt.
Next it was tested, whether the red azolium salt 8·HBr and the previously synthesized green 9·HBr51 are suitable for the synthesis of a wide range of NHC-metal complexes (Scheme 2). Green fluorescent metal complexes with [(NHC)ML] with ML = AuCl, RhCl(cod), IrCl(cod) and Pd(allyl)Cl and red fluorescent metal complexes [(NHC)ML] with ML = CuBr, AuCl, RhCl(cod), IrCl(cod) and Pd(allyl)Cl were obtained using the weak base approach.52,53 The red meso-aryl Bodipy turns out to be highly stable towards the basic reaction conditions and the same holds true for the green meso-alkyl Bodipy. In stark contrast, the red meso-alkyl Bodipy is highly sensitive towards the basic reagents used in metal complex synthesis and not suitable for the synthesis of NHC-metal complexes.54
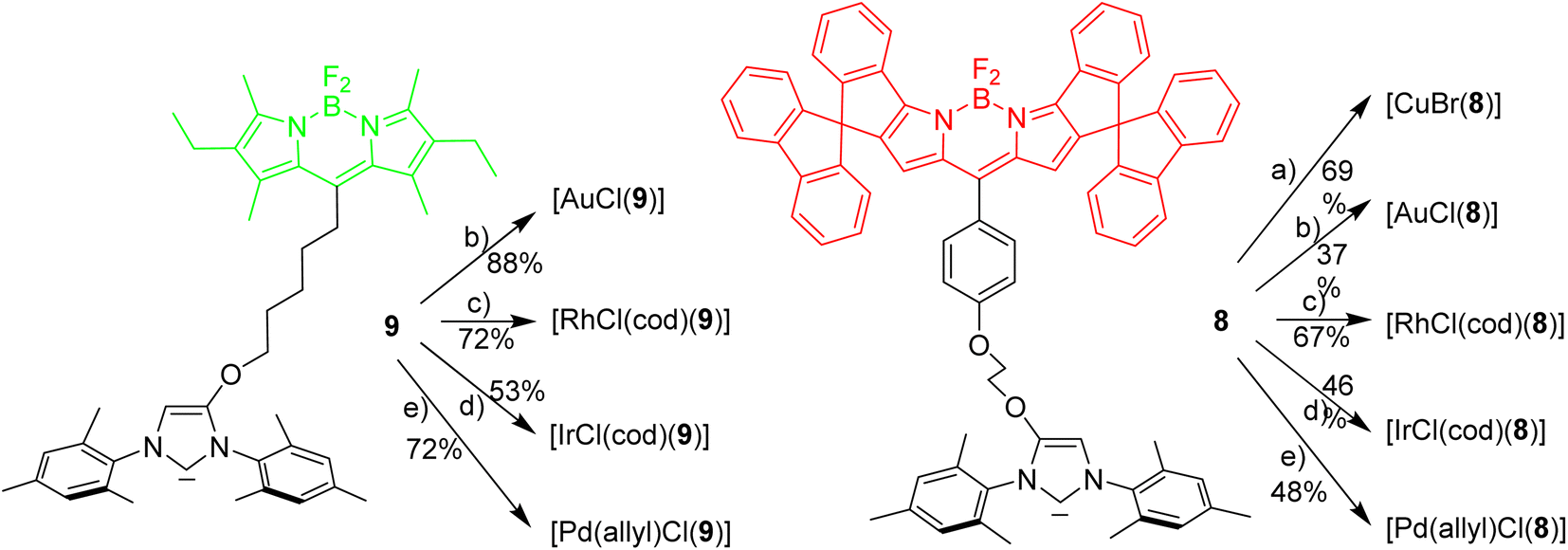 | ||
| Scheme 2 Synthesis of red and green NHC-metal complexes. Reagents and conditions: (a)–(e) metal source [(AuCl(Me2S)] or [RhCl(cod)]2 or [IrCl(cod)]2 or [(Pd(allyl)Cl)]2 or [CuBr(Me2S)] with K2CO3, acetone, reflux, 14 h. | ||
The fluorescence properties of the respective red and green metal complexes [(NHC)ML] with ML = AuCl, Pd(allyl)Cl, RhCl(cod) and IrCl(cod) are identical, since the distance metal-Bodipy is far too large to exert any significant influence on the fluorophore and for the same reason the properties of the metal complexes are not influenced by the nature of the fluorophore. The fluorophores can be considered as remote labels not affecting the metal to any significant degree. The stability of a palladium–pyridine complexes tagged with a related spiro-Bodipy in an organometallic setting was previously demonstrated by Goldsmith et al.55
Evaluation of the red-green FRET
To enable the fine tuning of FRET design, a simple green-red dyad was synthesized, in which the respective fluorophores are connected by a short linker (Scheme 3).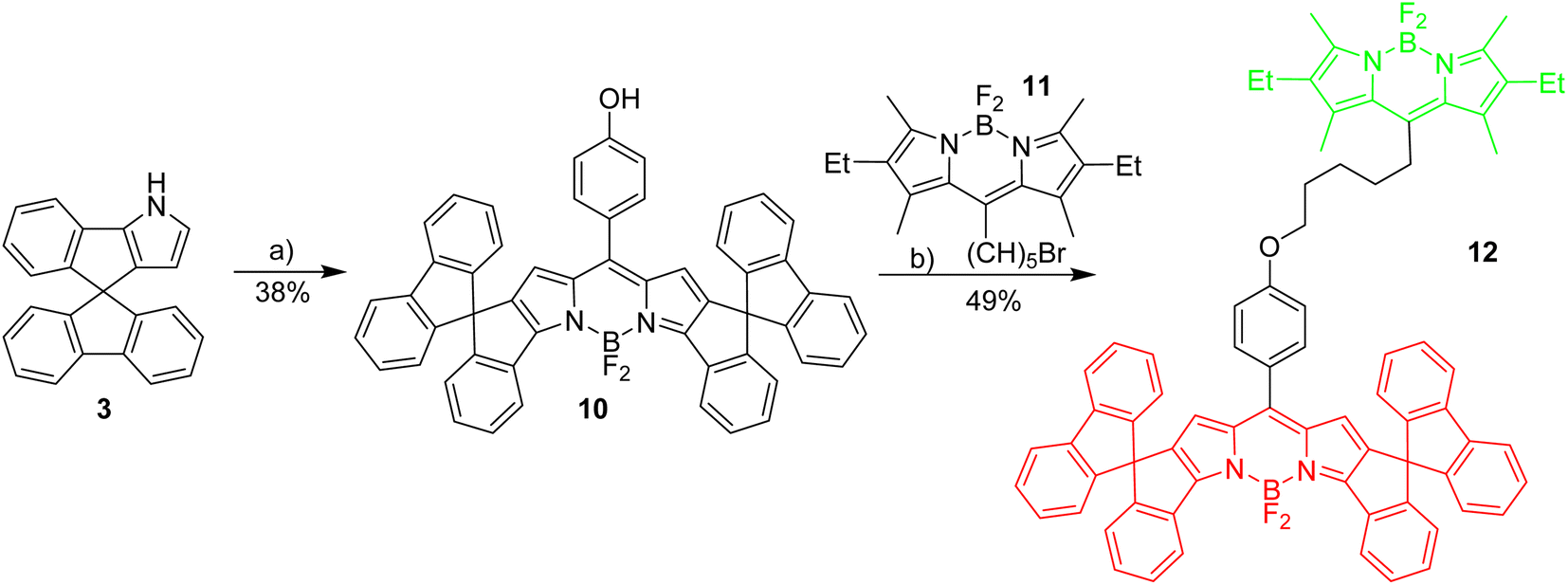 | ||
| Scheme 3 Synthesis of basic red/green dyad. Reagents and conditions: (a) 1,4-hydroxybenzaldehyde, TFA, CH2Cl2, 3 h followed by DDQ, 1 h followed by BF3·Et2O, Et3N, 3 h; (b) K2CO3, KI, acetone, reflux, 12 h. | ||
Initial experiments were done to establish the best parameters for FRET measurements, utilizing the simple dyad 12 and related compounds with the respective green and red fluorophores 11 and 4. First the most useful excitation wavelength for the green-red dyad was determined. Ideally, the excitation of the green donor fluorophore does not lead to excitation of the red fluorophore (cross-talk).56 However, in real systems the excitation of the green fluorophore inevitably leads to some excitation of the red fluorophore, since the absorbance of the red dye in the 500 nm region is small, but different from zero (Fig. 1). The absorption spectrum of dyad 12 is a nearly perfect superposition of the individual spectra of the green and the red Bodipy 11 and 4 suggesting no interaction between the two fluorophores in the respective ground states.
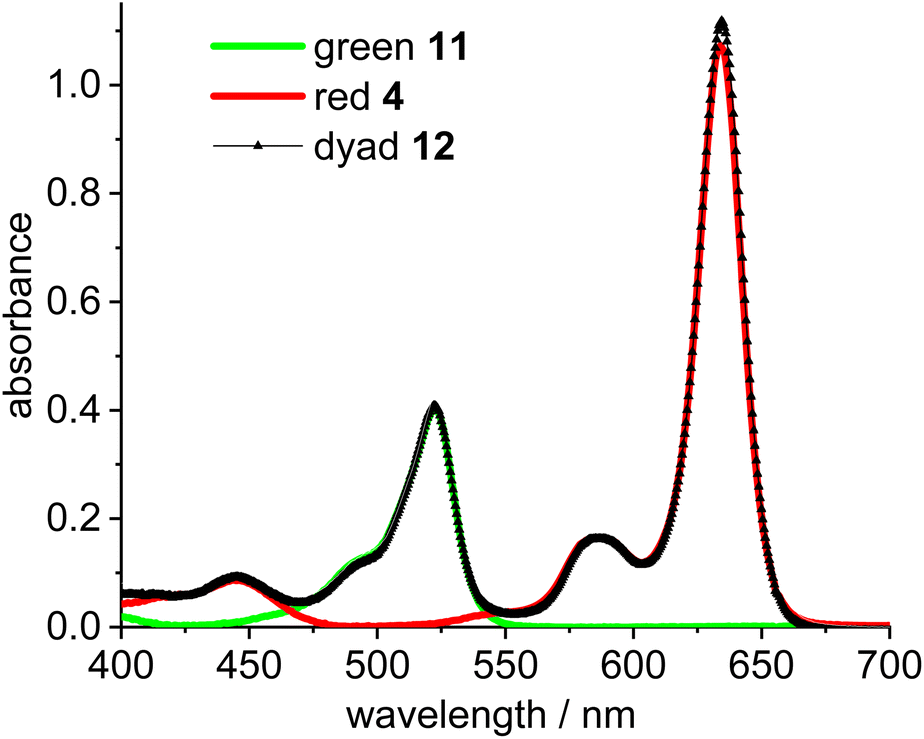 | ||
| Fig. 1 Absorbance spectra of green Bodipy 11 (λmax = 523 nm) and red Bodipy 4 (λmax = 635 nm) and dyad 12 in CH2Cl2 (c = 5.0 × 10−6 mol L−1). | ||
Next, the two individual fluorophores 4 and 11 were probed at different excitation wavelengths and the ratio of the respective emission intensities determined. The highest I535/I648 value is 37 at 505 nm excitation corresponding to a cross-talk intensity of approx. 2.7% (Fig. 2).
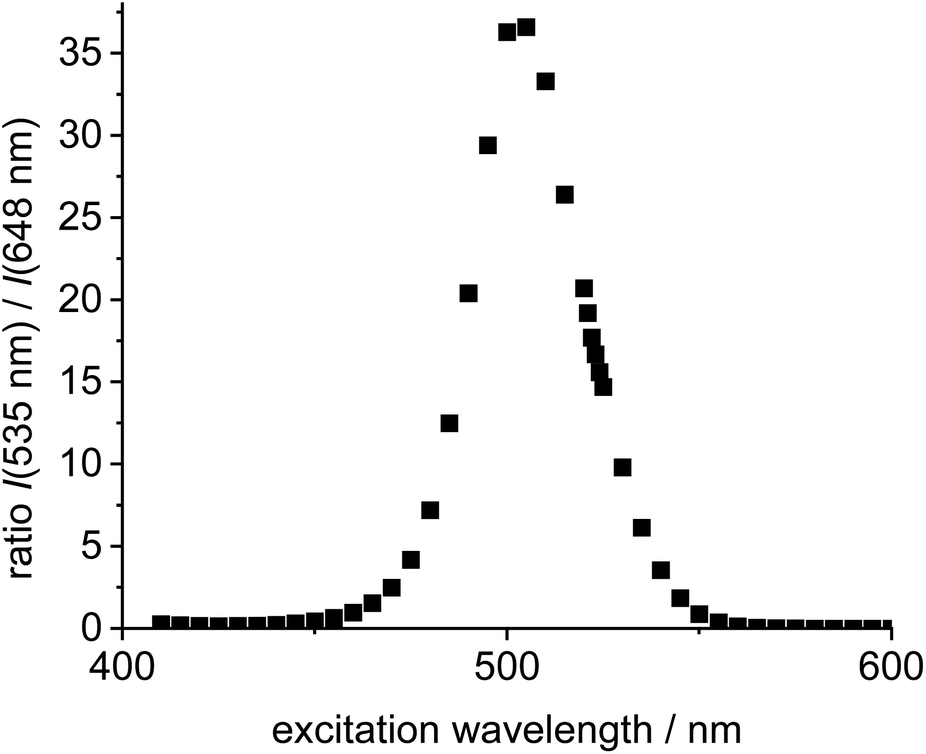 | ||
| Fig. 2 Plot of excitation wavelength vs. ratio of emission intensities of the individual green Bodipy 11 (λem,max = 535 nm) and the individual 4 (λem,max = 648 nm) in DCE (c = 5.0 × 10−6 mol L−1). | ||
In dyad 12 (Scheme 3) the excitation energy is transferred from the green donor fluorophore to the red acceptor fluorophore. Consequently, the excitation of the dyad at 505 nm leads to insignificant emission at 535 nm, and full emission at 648 nm. This is indicative of virtually complete energy transfer from the green to the red fluorophore, which is not surprising with a view to the short distance between the green and the red fluorophore.
The formation of dinuclear species from two individual species containing fluorophores for FRET experiments depends on the concentration of the reactants. Obviously, the position of the equilibrium as well as the rate for such reactions are more favorable at higher concentrations. However, due to the small Stokes shift the emission wavelength is close to the maxima of the absorption of the respective fluorophore and at higher concentration of the fluorophore a significant portion of the emitted light is reabsorbed. Based on experiments at different concentrations of fluorophore 4 and 11 (c = 1–50 × 10−6 mol L−1) the brightest fluorescence is observed between c = 5–10 × 10−6 mol L−1. However, already at c = 5 × 10−6 mol L−1 significant self-absorbance occurs and the molar emission for the green fluorophore is attenuated by ca. 20% (based on the extrapolated emission at c = 1.0 × 10−6 mol L−1), and by ca. 50% for the red fluorophore (extrapolated from c = 1.0 × 10−6 mol L−1). When measuring the FRET at higher concentrations the 648 nm emission intensity at c = 10.0 × 10−6 mol L−1 is lower than at c = 5.0 × 10−6 mol L−1 and at c = 50 × 10−6 mol L−1 the absolute fluorescence intensity is less than 10% of the emission intensity at c = 5.0 × 10−6 mol L−1. However, despite significant signal attenuation, fluorescence can be easily determined due to the excellent brightness of the Bodipy fluorophore. Based on this, FRET measurements over a wide concentration range are possible.
FRET studies on green-red NHC-gold complexes
The reaction of [Au(CCPh)(NHC)] with [Au(NTf2)(NHC)] is known to provide the respective digold–acetylide complex with a σ- and a π-bound NHC–gold complex (Widenhoefer dimer).57 The analogous reaction is reported here using the green and red fluorescent gold complexes 13 and 14 to provide the dinuclear complex 16 (Scheme 4).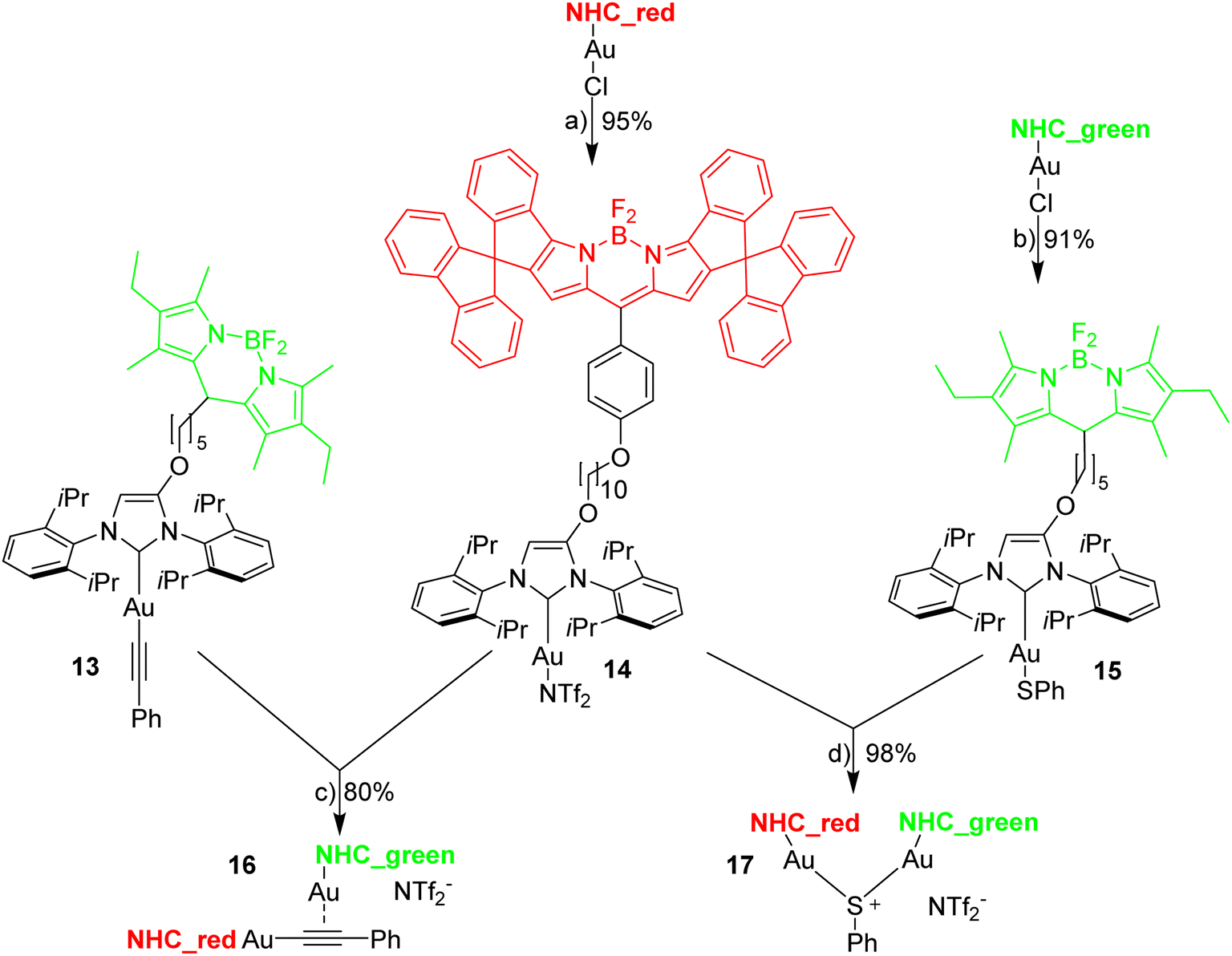 | ||
| Scheme 4 Synthesis of two red-green dinuclear gold complexes. Reagents and conditions: (a) [Ag(NTf2)], CH2Cl2, rt, 30 min, (b) 5 equiv. PhSH, Et3N, EtOH/CH2Cl2; (c) CH2Cl2, rt, 40 min; (d) CH2Cl2, 30 min. | ||
A solution of the red gold complex 14 in 1,2-dichloroethane was added to a solution of the green gold complex 13 in the same solvent in a fluorescence cuvette at t = 7 min. The reaction mixture was irradiated with 505 nm light and the fluorescence in the red channel (648 nm) and in the green channel (535 nm) observed (Fig. 3). The reaction ensues after mixing both reactants and the formation of the dimeric gold complex is evidenced by a pronounced decrease in the green 535 nm emission and a corresponding increase in the red 648 nm emission due to FRET (Fig. 3). The green emission decreases to 14% of the initial value.58 In contrast to dyad 12 the resonance energy transfer is not quantitative, which is hardly surprising, because the distance between the green and the red fluorophore is much larger.59 Considering the flexibility of the red and green subunits of the molecules it is likely, that the FRET is isotropic and orientation factors need not be considered. Because of this flexibility it is also difficult to obtain precise information on the average distance between the green and the red fluorophore. Based on the observed FRET the distance between the red and the green Bodipy in 16 and 17 is estimated to be at least 25% shorter than the Förster radius of the green/red FRET pair.60 Obviously, the interaction between the two fluorophores is sufficiently long range to be suitable for the observations of FRET in molecules of this size. The chemical transformation appears to be complete within ca. 30 min and after that both emission signals in the red and in the green channel remain constant–despite continuous irradiation – thus providing evidence for the photostability of the respective fluorophores. In order to verify this approach, the complex 16 was also synthesized (Scheme 4), isolated and fully characterized by NMR-spectroscopy and high-resolution mass spectrometry.
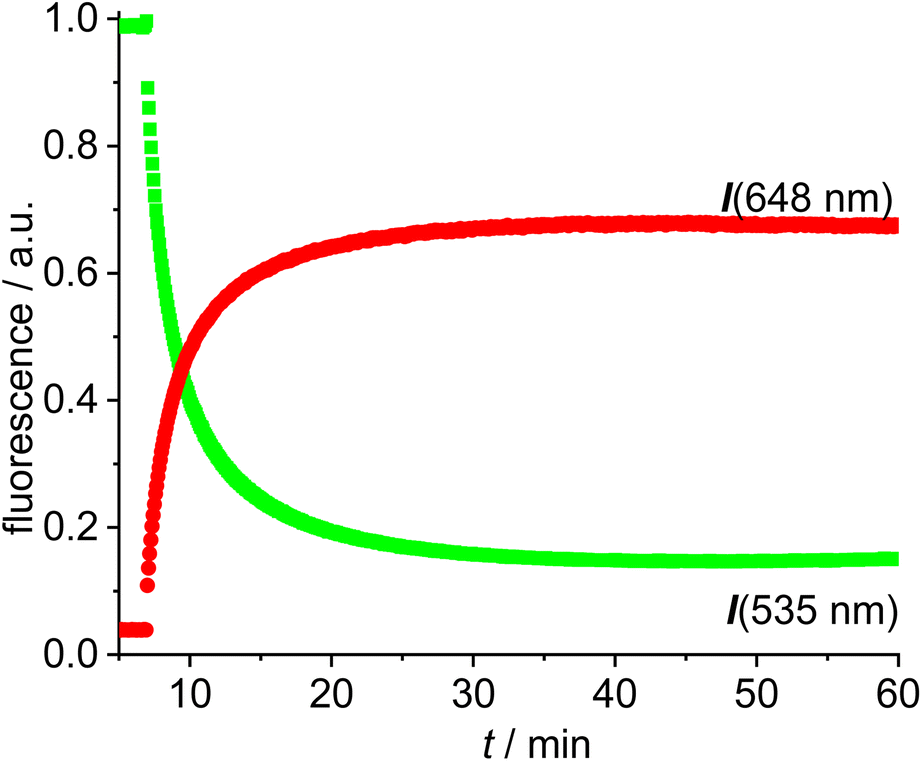 | ||
| Fig. 3 Time-dependent evolution of the fluorescence (green fluorescence renormalized to 1.0) for the reaction of [Au(CCPh)(NHC_green)] 13 with [Au(NTf2)(NHC_red)] 14 in DCE (c = 5.0 × 10−6 mol L−1). | ||
The next experiments involved mixing 1,2-dichloroethane solutions of [Au(SPh)(NHC_green)] 15 with [Au(NTf2)(NHC_red)] 14 in the fluorescence cuvette and monitoring the changes in the red (648 nm) and the green (535 nm) channel (Fig. 4). As evidenced by the rapid change in the fluorescence signal this reaction is significantly faster and within a few minutes an emission plateau in the red and the green channel is reached. The reaction leads to a sulfur-bridged species with an Au–S–Au core.61,62 Again, the outcome of the reaction was verified by synthesizing and characterizing the sulfur-bridged dimer 17. The fast formation of the sulfur-bridged dimer is followed by a much slower secondary reaction as evidenced by correspondingly slow changes (over the course of several hours) in the red and the green emission (Fig. 4).
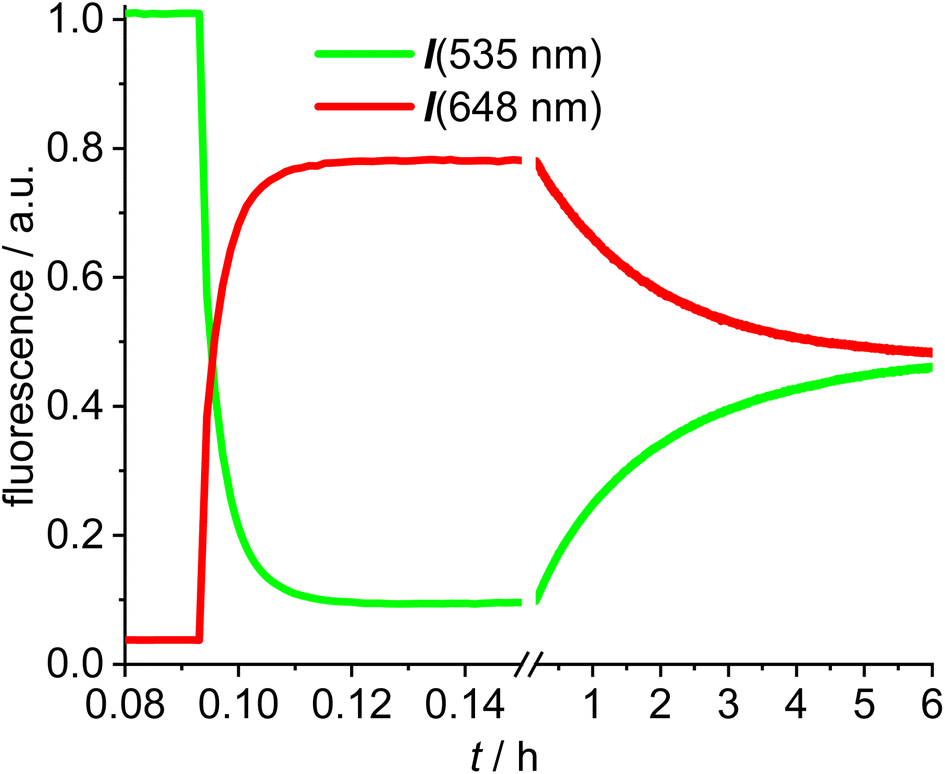 | ||
| Fig. 4 Time-dependent evolution of the fluorescence (green fluorescence renormalized to 1.0) for the reaction of [Au(NTf2)(NHC_red)] 14 with [Au(SPh)(NHC_green)] 15 (break region of the graph from 0.15 h to 0.16 h followed by different scaling in DCE (c = 5.0 × 10−6 mol L−1). | ||
The initial reaction results in a rapid increase in the 648 nm emission and a simultaneous decrease in emission at 535 nm, which is typical for FRET pair formation. The green fluorescence decreases to 9% of the initial value, indicating that the FRET in the sulfur bridged dimer 17 is more efficient than in the alkyne-dimer 16. Based on an estimate of the spacing of the respective red-green pairs, it is likely that the inter-Bodipy distance will be smaller in the sulfur-bridged 17 than in the alkyne-bridged 16, which would be in accord with the observed decrease of the green emission. Following this initial change in the fluorescence, the emission intensities are reverted during the slow secondary reaction until after 10 h the green and red signals remain nearly constant. Considering the excellent photostability of both fluorescent labels (Fig. 3), photodegradation of the red label can hardly be the explanation for the slow change in the emission properties. Instead, the slow response is attributed to scrambling of the respective fluorescent labels and finally the reaction mixture contains three different permutations in a statistical ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, i.e. [(NHC_green)Au(SPh)Au(NHC_green)]+, [(NHC_green)Au(SPh)Au(NHC_red)]+, [(NHC_red)Au(SPh)Au(NHC_red)]+ – each with a NTf2− counterion. Of those three complexes, only the mixed red/green species displays a FRET signal and consequently the intensity of the red fluorescence signal halves during ligand mixing. Upon 505 nm excitation the green/green complex displays green emission, while the red/red complex shows negligible red fluorescence (originating only from cross-talk). The slow decrease in the concentration of the FRET complex initially formed, leads to a decrease in the red signal and an increase in the green signal. Interestingly this ligand scrambling cannot be observed by NMR spectroscopy because the NHC ligands of the red and green complexes are identical (except for the remote fluorescent labels). The observed ligand shuffling does not give rise to observable changes in the 1H-NMR spectrum since the distance between the two fluorophores is well above 1 nm.
:2:1, i.e. [(NHC_green)Au(SPh)Au(NHC_green)]+, [(NHC_green)Au(SPh)Au(NHC_red)]+, [(NHC_red)Au(SPh)Au(NHC_red)]+ – each with a NTf2− counterion. Of those three complexes, only the mixed red/green species displays a FRET signal and consequently the intensity of the red fluorescence signal halves during ligand mixing. Upon 505 nm excitation the green/green complex displays green emission, while the red/red complex shows negligible red fluorescence (originating only from cross-talk). The slow decrease in the concentration of the FRET complex initially formed, leads to a decrease in the red signal and an increase in the green signal. Interestingly this ligand scrambling cannot be observed by NMR spectroscopy because the NHC ligands of the red and green complexes are identical (except for the remote fluorescent labels). The observed ligand shuffling does not give rise to observable changes in the 1H-NMR spectrum since the distance between the two fluorophores is well above 1 nm.
Monitoring ion pairing/ion separation using green-red FRET
Dissociation of close ion pairs in solution leads to spatial separation of the respective anions and cations. It should be possible to monitor this process via FRET if appropriate fluorescent labels are used. We have recently studied ion pairing/dissociation processes based on changes in fluorescence of the anion,63 but this approach only works in a specific setting because it relies on the quenching effect of the transition metal (iridium) cation towards the anionic Bodipysulfonate (bdpSO3−).63 Consequently, close ion pairs are characterized by weak fluorescence and separated ion pairs by strong fluorescence. Here we will present a general approach independent of specific properties of metals, in which both the anion and the cation are labeled with red and green fluorescent dyes, respectively. This should lead to a pronounced FRET signal for close ion pairs. A salt consisting of a Bodipy-labeled NHC gold cation and a Bodipysulfonate anion was synthesized in the reaction of [AuCl(NHC_red)] with the silver salt of Bodipysulfonate (Scheme 5).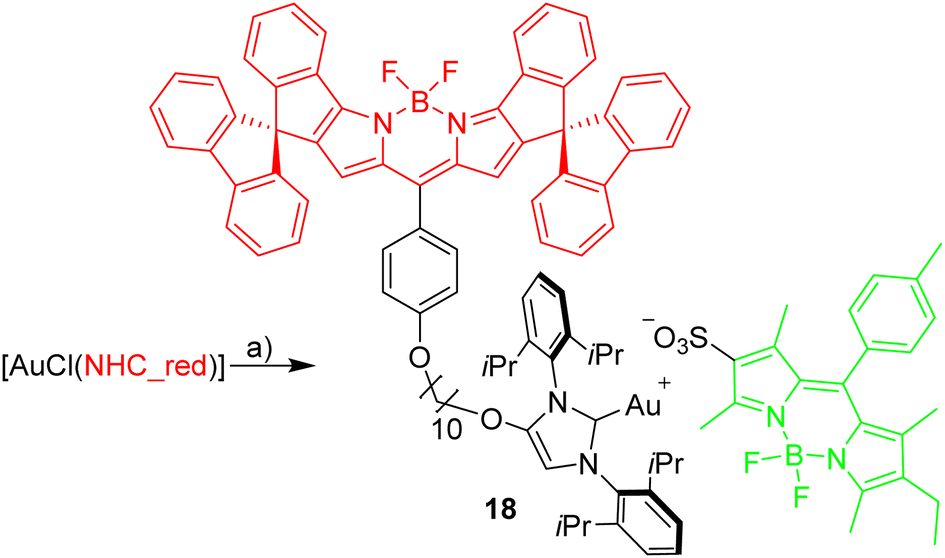 | ||
| Scheme 5 Synthesis of [Au(bpdSO3)(NHC_red)]. Reagents and conditions: (a) [Ag(bdpSO3)], CH2Cl2. | ||
A solution of complex 18 in toluene displays a strong FRET signal, indicating the presence of close ion pairs in the weakly solvating aromatic solvent (excitation spectra red and green circles, Fig. 5). The close ion pairs can be separated by addition of a polar solvent, such as DMAc (excitation spectra red and green triangles, Fig. 5). The increase in the dielectric constant of the solvent mixture and the weak interaction of DMAc with gold lead to solvent-separated ion pairs,64,65 which are characterized by a weak 648 nm signal (red triangles, Fig. 5) and a strong 535 nm signal upon excitation at 500 nm. Following the addition of DMAc to the toluene solution (1.4%-vol. DMAc) of 18 the average distance between the fluorophores corresponds to the Förster radius of the green/red pair. Upon formation of the close ion pair the intensity of the 648 nm signal increases 21-fold while the green signal decreases by a factor of 6.6 (Fig. 6). The close ion pair can be disconnected by addition of a ligand to gold, such as phenyl acetylene. At c = 1.0 × 10−6 mol L−1 of the salt in toluene approx. 104 equivalents of phenyl acetylene are needed to fully separate the ion pair. The method presented here is fairly general and allows the investigation of ion pairing in numerous different settings. Understanding ion pairing is highly relevant for catalytic olefin polymerization using cationic transition metal complexes66,67 or for cationic gold catalyzed hydration and alkoxylation of alkynes68 and the methodology presented here should be highly useful.
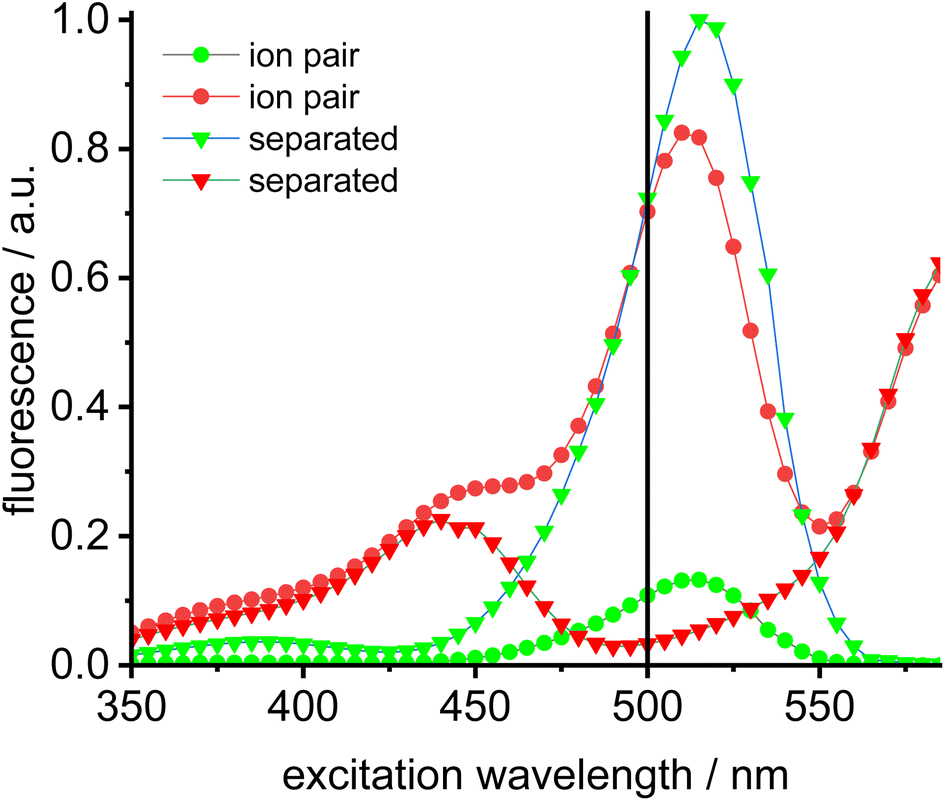 | ||
| Fig. 5 Excitation spectra of Bodipy ion pair dyad 18 in toluene (c = 2.0 × 10−6 mol L−1) (green dots = 535 nm emission, red dots = 648 nm emission, green fluorescence renormalized to 1.0) and of the separated dyad in toluene solution (c = 2.0 × 10−6 mol L−1) (green triangles = 535 nm emission, red triangles = 648 nm emission) after addition of concentrated NBu4Br solution (vertical black line denotes preferred excitation wavelength 500 nm). | ||
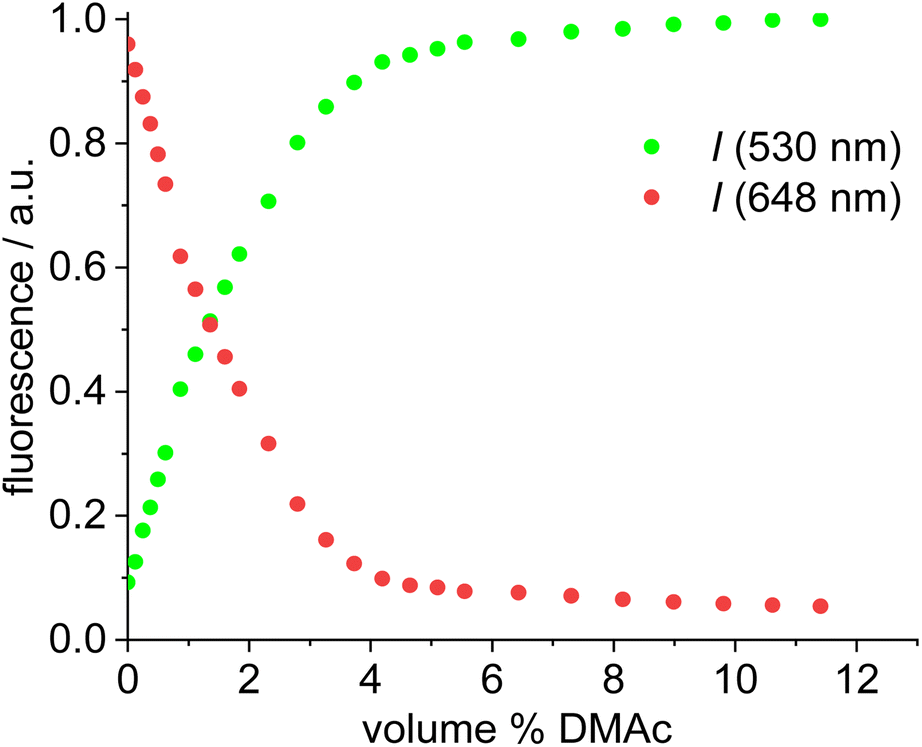 | ||
| Fig. 6 Plot of the fluorescence of red/green (acceptor/donor) (fluorescence renormalized to 1.0) for complex 19vs. volume-% of DMAc in toluene solution (c = 1.0 × 10−6 mol L−1) upon 500 nm excitation. | ||
Evaluation of the blue-green dyad
Prior the construction of the triad, we tested, whether it is possible to synthesize NHC-gold complexes incorporating a blue Bodipy. This may not be straightforward, since the NH-unit attached directly to the Bodipy-core is slightly acidic and may react with the base needed for the formation of the carbene from the imidazolium salt. Nonetheless, a blue imidazolium salt 19 was synthesized as described in Scheme 1. The synthesis of the NHC–gold complex was not straightforward and different approaches were tested (weak base approach, via free carbene, NHC–Ag as NHC transfer reagent) and only the latter approach turned out to be successful. The NHC–Ag complex was in situ generated from the imidazolium salt using Ag2O and used directly used for the reaction with [AuCl(Me2S)]. The conversion appears to be near quantitative, but due to the small scale on which the synthesis was done, the isolated yield of pure material is only 55% (Scheme 6).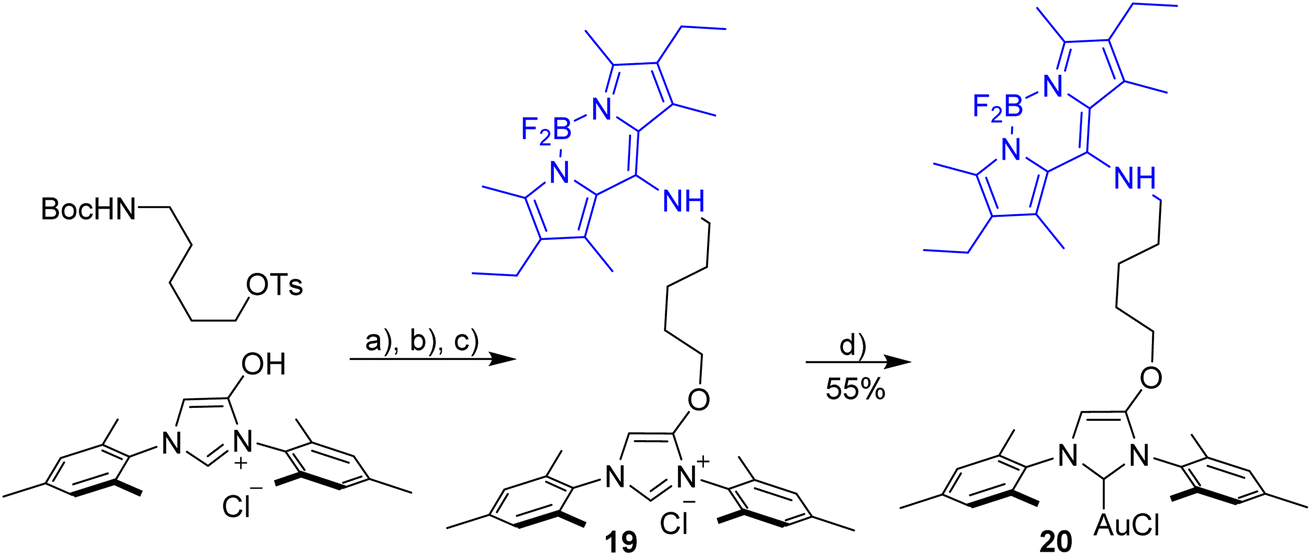 | ||
| Scheme 6 Synthesis [AuCl(NHC_blue)] 20. Reagents and conditions: (a) K2CO3, KI, acetone, reflux, 12 h; (b) HCl in dioxane, CH2Cl2, overnight; (c) meso-Cl-Bodipy, Et3N, CH3CN/CH2Cl2, 12 h; (d) Ag2O, DCE, T = 60 °C, 60 min then [AuCl(SMe2)], 12 h. | ||
To form a triad involving three different components each molecule requires an individual fluorescent tag and the FRET conditions need to be fulfilled to enable the efficient blue → green → red energy transfer. The green/red dyad has already been established and the next step is to extend the established dyad by a third dye to a triad. To retain the emission of the triad in the visible region of the spectrum, the third dye should be on the short-wave side of the established dyad.69 Meso-amino substituted Bodipy constitute a synthetically variable and easily available group of fluorophores,70–73 which display absorption/emission properties in the 400–450 nm region and which appear to be suitable for a Bodipy triad. In order to select the most appropriate dye the absorption and emission spectra of four different amino-Bodipy B1, B2, B3 and B4 were measured (Scheme 7). The initial aim of the selection process was to find a blue Bodipy with a suitable excitation spectrum with minimal cross-talk concerning the green dye G1, which serves as a simple model compound for the green complexes synthesized.
 | ||
| Scheme 7 List of blue Bodipy dyes tested with green Bodipy G1. | ||
The NMR spectra of blue series compounds B1, B2, B3 and B4 display interesting features. In B1 the six protons attached to Bodipy display unique (slightly broadened) signals in the 1H-NMR and nine distinct 13C signals for the Bodipy unit plus three signals for the propyl chain. Based on this observation, the C–N(propyl) bond appears to have significant double bond character with inhibited rotation, decreasing the symmetry of the molecule and leading to two different Bodipy sides. In B2 this partial double bond character appears to be reduced (probably due to the slightly electron-richer Bodipy unit due to the two electron-releasing methyl groups) since fewer signals in the 13C-NMR are observed, some of which are severely broadened. In B3 and B4 the alkyl groups on both sides of the N-propyl chain prevent the planarization of the propyl nitrogen atom and a symmetric NMR spectrum is observed.
The absorption maxima of the blue Bodipy B1, B2, B3 and B4 are 413 nm, 422 nm, 426 nm and 431 nm, respectively (Fig. 7). The four blue fluorophores possess the same backbone but the additional alkyl substituents attached to the Bodipy core shift the absorbance towards the longer wavelengths, while the extinction of the dyes remains nearly constant. The absorption spectrum of the green dye displays a 523 nm absorbance and another (weaker) one at 375 nm, while the 415–435 nm region between those two maxima is characterized by weak absorbance. To characterize the spectral behavior, the ratio of the absorbances for the four blue dyes relative to the green dye are shown in Fig. 7. The most favorable ratio of 29 is observed for B2, meaning that the absorbance of B2 at 422 nm is 29 times stronger than that of G1 at the same wavelength. From a chemical point-of-view B2 is also preferred over B1, since the two methyl groups replace two fairly acidic protons next to the NBF2N -unit, which renders B1 (and derivatives) base-sensitive.
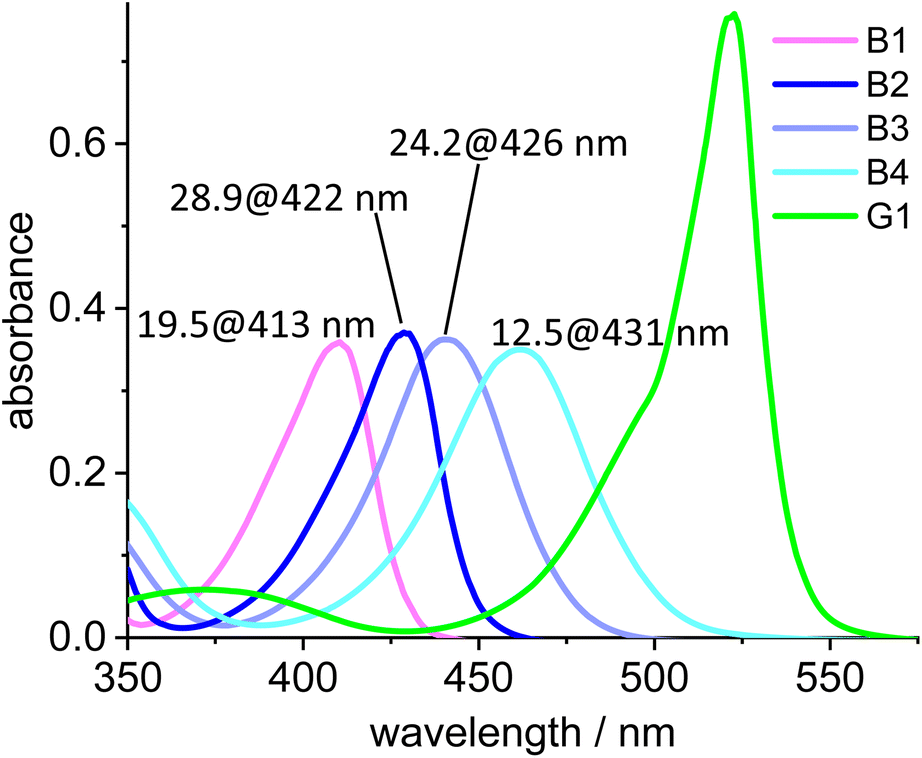 | ||
| Fig. 7 Absorption spectra of B1, B2, B3, B4 and the green dye G1. The numbers on top of the peaks denote the ratio of the absorption of B1, B2, B3 and B4, respectively, relative to the absorbance of G1 at the given wavelengths (c = 2.0 × 10−6 mol L−1). | ||
To further aid the selection of a favorable dye for the blue-green FRET, the cross-talk intensities of the dyes B1, B2, B3 and B4 are plotted relative the fluorescence intensity of the green at the respective excitation wavelength (Fig. 8). B2 turns out to be the most favorable FRET-partner for the green dye, since at 422 nm excitation the emission from the blue dye is 9.5 stronger than the emission from the green dye at the same excitation wavelength.
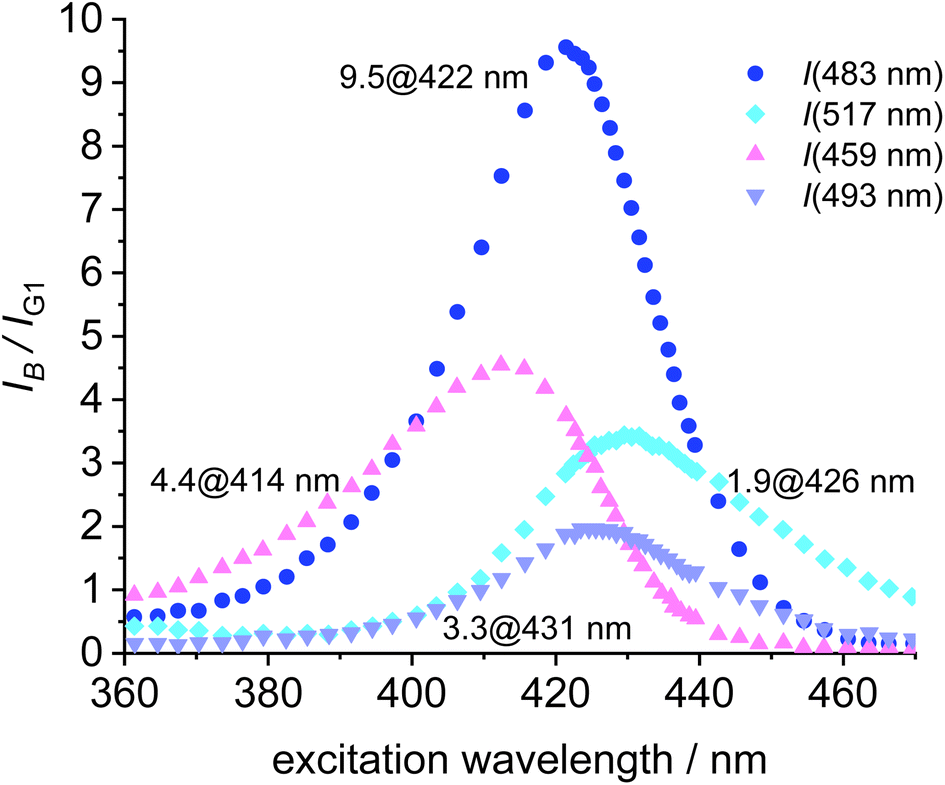 | ||
| Fig. 8 Plot of the ratio of fluorescence intensities (IB/IG1) vs. the excitation wavelength for B1, B2, B3, B4 and G1. 9.5 @ 422 nm denotes the best ratio I(B2)/I(G1) at 422 nm excitation for the 483 nm fluorescence (c = 2.0 × 10−6 mol L−1). | ||
Following the selection of the blue Bodipy B2 the gold–acetylide complex 22 representing the respective blue-green dyads was synthesized (Scheme 8). The identity of the gold-acetylide subunit was established via NMR-spectroscopy and by high resolution mass spectrometry (FRET experiments, see below). The resulting material is a 1:1-mixture of 22 and dbu (1,8-diazabicyclo[5.4.0]undec-7-ene used as the base for alkyne deprotonation and gold-acetylide formation). This adduct could not be separated since the material decomposes upon attempted purification. In the 1H-NMR spectrum of 22·dbu the NH-proton of the blue Bodipy unit cannot be observed and based on this it is assumed that the dbu closely interacts with the NH-unit and the respective proton is shared by the nitrogen atom attached to Bodipy and the nitrogen atom of dbu enhancing adduct formation. The dbu molecule is also observed in the mass spectrum of 22·dbu.
 | ||
| Scheme 8 Synthesis of the blue-green dyad and the blue-green-red triad. Reagents and conditions: (a) [AuCl(NHC_green)], dbu, CH2Cl2; (b) + [Au(NTf2)(NHC_red)] 14 in 1,2-dichlorethane; (c) [AuCl(NHC_red)], dbu, CH2Cl2. | ||
Excitation of the triad
The three fluorophores in the potential triad 23 enable multiple fluorescence information channels reporting on the status of the molecule during the triad forming reaction. The respective signals either the blue, the green or the red fluorescence from 423 nm excitation of the blue fluorophore or alternatively from 500 nm excitation of the green fluorophore.A solution of 22·dbu in 1,2-dichloroethane in a fluorescence cuvette was converted into the triad 23 by adding a solution of [Au(NTf2)(NHC_red)] 14 and the fluorescence changes are monitored. Based on the design of the triad, irradiation at 423 nm should give rise to the efficient energy transfer from the blue Bodipy to the green Bodipy and then to the red Bodipy in a cassette type fashion.41 The individual emission spectra of the blue, the green and the red Bodipy (Fig. S9†), show that 423 nm excitation also leads to significant direct excitation of the red Bodipy and consequently to red cross-talk emission, while the excitation of the green Bodipy at 423 nm gives rise to a weak cross-talk fluorescence signal. This blue-red cross-talk occurs because the red Bodipy displays a significant absorbance in the 400–450 nm region. The only visible spectral region in which the red Bodipy displays only weak absorbance is between 480 – 530 nm but irradiation in this spectral region leads to some direct excitation of the green fluorophore, A solution of 22·dbu (c = 5.0 × 10−6 mol L−1) was treated with one equivalent of [AuCl(NHC_red)] and irradiated at 423 nm (Fig. 9). This leads to an instantaneous increase of the red emission,74 but it does not produce a significant change in the green emission. Obviously, the 423 nm light leads to the excitation of the blue Bodipy and the excitation energy is then transferred to the green dye via FRET. However, since the green emission experiences only small changes after addition of the first equivalent of 14, it can be concluded that FRET to the red Bodipy does not occur to a significant extent. Consequently, the addition of one equiv. of 14 does not lead to the formation of the triad. The observed red fluorescence originates from the direct excitation of the red Bodipy at 423 nm (cross-talk). Additional evidence for this interpretation comes from the fact that this red fluorescence intensity is virtually the same as the one produced by a c = 5.0 × 10−6 mol L−1 solution of 14 in 1,2-dichloroethane.75 Consequently, in the cross-talk and self-absorbance corrected data set, the red emission is almost unchanged (Fig. S28†). To better understand the absence of triad formation after addition of one equiv. of 14, the reaction of [Au(NTf2)(IPr)] with a single equivalent of dbu was probed by 1H-NMR spectroscopy. Upon addition of one equivalent of dbu to a solution of [Au(NTf2)(IPr)] in CDCl3 several 1H and 13C-NMR signals of the dbu experience significant shifts. Based on this, it is concluded that dbu forms a complex with the cationic gold, which explains why the triad is not formed after addition one equivalent of complex 14.
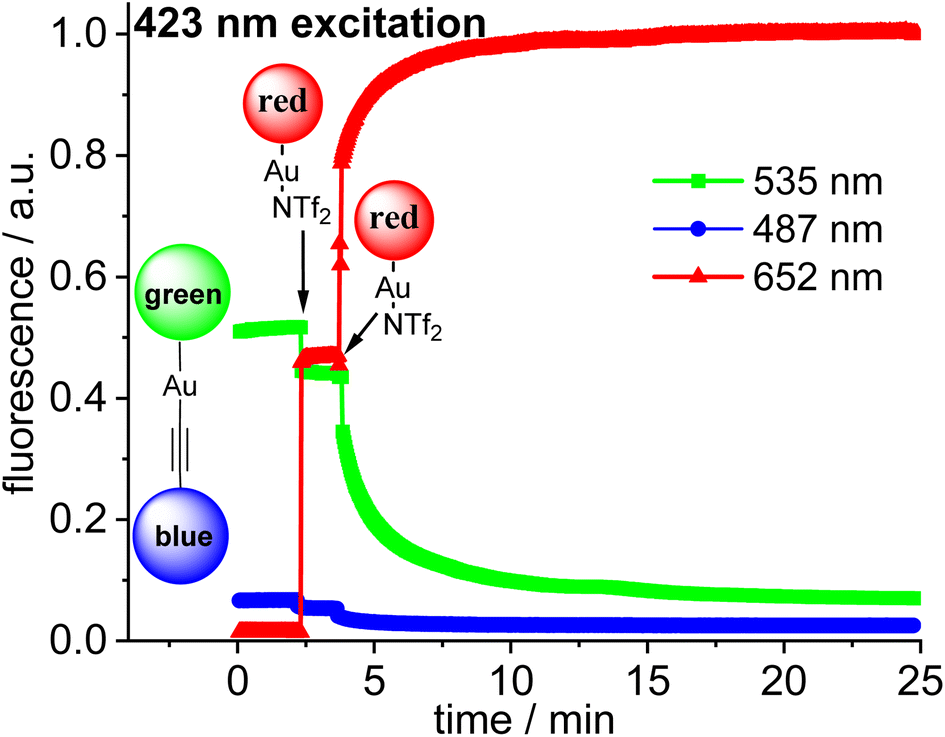 | ||
| Fig. 9 Plot of the time-dependent fluorescence intensities for the blue Bodipy (λem,max = 487 nm), green Bodipy (λem,max = 535 nm) and red Bodipy (λem,max = 652 nm) at λexc = 423 nm within the blue-green-red acetylide triad with addition of n equiv. (n = 1, 2) of 14 [Au(NTf2)(NHC_red)] leading finally to the formation of 23. Initial concentration of complexes in 1,2-dichloroethane c = 5.0 × 10−6 mol L−1 (uncorrected data, red fluorescence renormalized to 1.0). | ||
Addition of a second equivalent of 14 (resulting in c = 10.0 × 10−6 mol L−1 of the red fluorophore) leads to a pronounced, but slow change in the fluorescence during the following minutes both upon 423 nm excitation (Fig. 9) and upon 500 nm excitation (Fig. 10). The slow change in the red fluorescence indicates a chemical reaction with a corresponding reaction rate. This is in contrast to the immediate change in fluorescence after addition of the first equivalent of 14. The addition of the second equivalent of 14 now also leads to a very pronounced drop in the green fluorescence. Obviously, the triad is formed and the 423 nm excitation energy is predominantly (supporting p. 56) transferred from the blue to the green and finally to the red Bodipy. The formation of the triad can also be followed, by switching to 500 nm excitation of the green fluorophore. As shown before, 500 nm light does not lead to a significant cross-talk with the red fluorophore. Therefore, the observation of red fluorescence upon 500 nm excitation is primarily the result of resonance energy transfer from the green to the red Bodipy (Fig. 10) providing additional evidence for triad formation.
 | ||
| Fig. 10 Plot of the time-dependent fluorescence intensities for the blue Bodipy (λem,max = 487 nm), green Bodipy (em,max = 535 nm) and red Bodipy (λem,max = 652 nm) at λexc = 500 nm within the blue-green-red acetylide triad with addition of n equiv. (n = 1,2) of 14 [Au(NTf2)(NHC_red)] leading finally to the formation of 23. Initial concentration of complexes is (c = 5.0 × 10−6 mol L−1) in 1,2-dichloroethane (green fluorescence renormalized to 1.0). | ||
Conclusions
FRET pairs of fluorescent dyes are useful for establishing connectivities in small molecule chemistry. Three types of Bodipy-based fluorescent dyes for this purpose have been established for applications in organometallic complexes. The dyes reported here are conveniently synthesized, are chemically and photochemically stable, characterized by the absence of Lewis basic heteroatoms and are equipped with convenient functional groups for conjugation. Two different FRET pairs involving NHC-metal complexes based on blue-green and green-red dyads, respectively, are demonstrated in binuclear Widenhoefer type gold complexes, in sulfur bridged digold complexes and in an organometallic ion pair. A molecule composed of three different subunits, each tagged with different dyes (blue, green, red) was synthesized. The excitation of the blue dye initiates energy transfer blue → green → red proving the assembly of the three different tagged subunits in a single molecule as well as a minor blue-red pathway.Based on highly sensitive fluorescence spectroscopy FRET pairs are shown to be a powerful tool to establish connectivities. This will also be useful for the detection of dinuclear intermediates or dinuclear decomposition products in homogeneous catalysis, which likely occur in several very important transition metal catalyzed reactions: For the copper-catalyzed click reactions Fokin et al.76 demonstrated the intermediate formation of dinuclear copper complexes. In olefin metathesis Grubbs et al.77 were able to identify a dinuclear ruthenium complex as a catalyst decomposition product and bimolecular decomposition appears to be a major catalyst decomposition pathway in olefin metathesis.78,79 In Pd-catalyzed cross coupling reactions di- and polynuclear precatalysts and intermediates appear to be highly significant.80 The small amount of catalyst used in such reactions and/or the small amount of binuclear species formed, renders their direct observation difficult – especially in the absence of a unique spectroscopic signature of such bimetallic species. The methodology presented here, should enable the direct observation of such intermediates via FRET and should facilitate the determination of the respective kinetics.
This methodology complements the extensive applications of fluorescent dyes in cell biology, biochemistry and medicine and opens the doors for a better understanding of dynamic interactions in solution organometallic chemistry – in a concentration range less easily available to many other spectroscopic techniques.
Data availability
All experimental procedures, characterization data, experimental spectra (UV/vis, fluorescence, mass spectrometry, NMR).Author contributions
Y. S.: conceptualization, data curation, formal analysis, and investigation (experimental studies). S. P.: conceptualization, data curation, formal analysis, and investigation (experimental studies). H. P.: conceptualization, project administration, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) via grant Pl 178/18-2.Notes and references
- K. C. Nicolaou and J. S. Chen, Classics in Total Synthesis: Targets, Strategies, Methods, Wiley-VCH, Weinheim 1996 Search PubMed.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T. Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk, B. B. Sanchez, S. Yang, M. A. Perry, G. M. Gallego, J. J. Mousseau, M. R. Collins, R. J. Cherney, P. S. Lebed, J. S. Chen, T. Qin and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS PubMed.
- P. Bellotti, M. Koy, C. Gutheil, S. Heuvel and F. Glorius, Chem. Sci., 2021, 12, 1810–1817 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- T. T. Nguyen, M. J. Koh, X. Shen, F. Romiti, R. R. Schrock and A. H. Hoveyda, Science, 2016, 352, 569–575 CrossRef CAS PubMed.
- J. W. Yang, C. Chandler, M. Stadler, D. Kampen and B. List, Nature, 2008, 452, 453–455 CrossRef CAS PubMed.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed., 1995, 34, 1143–1170 CrossRef CAS.
- A. A. Nagarkar, S. E. Root, M. J. Fink, A. S. Ten, B. J. Cafferty, D. S. Richardson, M. Mrksich and G. M. Whitesides, ACS Cent. Sci., 2021, 7, 1728–1735 CrossRef CAS PubMed.
- Y. Wu, M. Frasconi, W.-G. Liu, R. M. Young, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 11835–11846 CrossRef CAS PubMed.
- X. Li, A. H. G. David, L. Zhang, B. Song, Y. Jiao, D. Sluysmans, Y. Qiu, Y. Wu, X. Zhao, Y. Feng, L. Mosca and J. F. Stoddart, J. Am. Chem. Soc., 2022, 144, 3572–3579 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- B. T. Bajar, E. S. Wang, S. Zhang, M. Z. Lin and J. Chu, Sensors, 2016, 16, 1488 CrossRef PubMed.
- M. Sustarsic and A. N. Kapanidis, Curr. Opin. Struct. Biol., 2015, 34, 52–59 CrossRef CAS PubMed.
- P. Rajdev and S. Ghosh, J. Phys. Chem. B, 2019, 123, 327–342 CrossRef CAS PubMed.
- R. K. Castellano, S. L. Craig, C. Nuckolls and J. Rebek, J. Am. Chem. Soc., 2000, 122, 7876–7882 CrossRef CAS.
- M. J. Mayoral, D. Serrano-Molina, J. Camacho-García, E. Magdalena-Estirado, M. Blanco-Lomas, E. Fadaei and D. González-Rodríguez, Chem. Sci., 2018, 9, 7809–7821 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer-Verlag, New York, 2010 Search PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhi, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- R. P. Haughland, The Molecular Probes Handbook − A Guide to Fluorescent Probes and Labeling Technologies, 2005, https://www.probes.com Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- P. Kos and H. Plenio, Chem.–Eur. J., 2015, 21, 1088–1095 CrossRef CAS PubMed.
- P. Kos and H. Plenio, Angew. Chem., Int. Ed., 2015, 54, 13293–13296 CrossRef CAS PubMed.
- R. Vasiuta and H. Plenio, Chem.–Eur. J., 2016, 22, 6353–6360 CrossRef CAS PubMed.
- O. Halter, J. Spielmann, Y. Kanai and H. Plenio, Organometallics, 2019, 38, 2138–2149 CrossRef CAS.
- M. Navarro, S. Wang, H. Müller-Bunz, G. Redmond, P. Farràs and M. Albrecht, Organometallics, 2017, 36, 1469–1478 CrossRef CAS.
- D. Wang, R. Malmberg, I. Pernik, S. K. K. Prasad, M. Roemer, K. Venkatesan, T. W. Schmidt, S. T. Keaveney and B. A. Messerle, Chem. Sci., 2020, 11, 6256–6267 RSC.
- P. Irmler and R. F. Winter, Organometallics, 2018, 37, 235–253 CrossRef CAS.
- J.-H. Sohn, K. H. Kim, H.-Y. Lee, Z. S. No and H. Ihee, J. Am. Chem. Soc., 2008, 130, 16506–16507 CrossRef CAS PubMed.
- J. Lee, K. H. Kim, O. S. Lee, T.-L. Choi, H.-S. Lee, H. Ihee and J.-H. Sohn, J. Org. Chem., 2016, 81, 7591–7596 CrossRef CAS PubMed.
- A. Kiel, J. Kovacs, A. Mokhir, R. Krämer and D.-P. Herten, Angew. Chem., Int. Ed., 2007, 46, 3363–3366 CrossRef CAS PubMed.
- T. Cordes and S. A. Blum, Nat. Chem., 2013, 5, 993–999 CrossRef CAS PubMed.
- C. Feng, D. W. Cunningham, Q. T. Easter and S. A. Blum, J. Am. Chem. Soc., 2016, 138, 11156–11159 CrossRef CAS PubMed.
- Q. T. Easter, V. Trauschke and S. A. Blum, ACS Catal., 2015, 5, 2290–2295 CrossRef CAS.
- A. Garcia, S. J. Saluga, D. J. Dibble, P. A. Lopez, N. Saito and S. A. Blum, Angew. Chem., Int. Ed., 2021, 60, 1550–1555 CrossRef CAS PubMed.
- Q. T. Easter and S. A. Blum, Acc. Chem. Res., 2019, 52, 2244–2255 CrossRef CAS PubMed.
- S. J. Saluga, D. J. Dibble and S. A. Blum, J. Am. Chem. Soc., 2022, 144, 10591–10598 CrossRef CAS PubMed.
- Q. T. Easter and S. A. Blum, Angew. Chem., Int. Ed., 2018, 57, 1572–1575 CrossRef CAS PubMed.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- I. Pochorovski, B. Breiten, W. B. Schweizer and F. Diederich, Chem.–Eur. J., 2010, 16, 12590–12602 CrossRef CAS PubMed.
- S.-G. Lim and S. A. Blum, Organometallics, 2009, 28, 4643–4645 CrossRef CAS.
- O. Halter, I. Fernández and H. Plenio, Chem.–Eur. J., 2017, 23, 711–719 CrossRef CAS PubMed.
- O. Halter and H. Plenio, Chem. Commun., 2017, 53, 12461–12464 RSC.
- Z. Wu, H. Fujita, N. C. M. Magdaong, J. R. Diers, D. Hood, S. Allu, D. M. Niedzwiedzki, C. Kirmaier, D. F. Bocian, D. Holten and J. S. Lindsey, New J. Chem., 2019, 43, 7233–7242 RSC.
- The descriptions “green”, “red” and “blue” refer to the color of the fluorescence light of the respective Bodipy dye.
- Y. Hayashi, N. Obata, M. Tamaru, S. Yamaguchi, Y. Matsuo, A. Saeki, S. Seki, Y. Kureishi, S. Saito, S. Yamaguchi and H. Shinokubo, Org. Lett., 2012, 14, 866–869 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 3308–3311 CrossRef CAS PubMed.
- Y. Kanai, D. Müller-Borges and H. Plenio, Adv. Synth. Catal., 2022, 364, 679–688 CrossRef CAS.
- O. Halter, R. Vasiuta, I. Fernández and H. Plenio, Chem.–Eur. J., 2016, 22, 18066–18072 CrossRef CAS PubMed.
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC.
- R. Savka and H. Plenio, Dalton Trans., 2015, 44, 891–893 RSC.
- Y. Shinozaki and H. Plenio, unpublished results.
- J. D. Ng, S. P. Upadhyay, A. N. Marquard, K. M. Lupo, D. A. Hinton, N. A. Padilla, D. M. Bates and R. H. Goldsmith, J. Am. Chem. Soc., 2016, 138, 3876–3883 CrossRef CAS PubMed.
- N. K. Lee, A. N. Kapanidis, Y. Wang, X. Michalet, J. Mukhopadhyay, R. H. Ebright and S. Weiss, Biophys. J., 2005, 88, 2939–2953 CrossRef CAS PubMed.
- T. J. Brown and R. A. Widenhoefer, Organometallics, 2011, 30, 6003–6009 CrossRef CAS.
- The residual green fluorescence is not the result of incomplete conversion of the starting materials into the products, since re-dissolved samples from the preparative synthesis also display residual green fluorescence of comparable intensity. Very fast scrambling of the green/red labels is also unlikely since it should lead to much higher green fluorescence.
- Intermolecular gold shuffling with mixing of fluorescent labels does not take place during the observation time.
- Estimate is based on the extrapolation of the near linear central segment of the plot of distance vs. energy transfer efficiency.
- T. J. Robilotto, J. Bacsa, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2012, 51, 12077–12080 CrossRef CAS PubMed.
- R. Visbal, R. P. Herrera and M. C. Gimeno, Chem.–Eur. J., 2019, 25, 15837–15845 CrossRef CAS PubMed.
- S. Popov and H. Plenio, Eur. J. Inorg. Chem., 2021, 22, 3708–3718 CrossRef.
- A. Macchioni, Chem. Rev., 2005, 105, 2039–2074 CrossRef CAS PubMed.
- N. Salvi, L. Belpassi, D. Zuccaccia, F. Tarantelli and A. Macchioni, J. Organomet. Chem., 2010, 695, 2679–2686 CrossRef CAS.
- F. Zaccaria, L. Sian, C. Zuccaccia and A. Macchioni, in Adv. Organomet. Chem, ed. P. J. Pérez, Academic Press, 2020, vol. 73, pp. 1–78 Search PubMed.
- L. Sian, A. Macchioni and C. Zuccaccia, ACS Catal., 2019, 10, 1591–1606 CrossRef.
- D. Zuccaccia, A. Del Zotto and W. Baratta, Coord. Chem. Rev., 2019, 396, 103–116 CrossRef CAS.
- G. Duran-Sampedro, A. R. Agarrabeitia, I. Garcia-Moreno, L. Gartzia-Rivero, S. de la Moya, J. Banuelos, I. Lopez-Arbeloa and M. J. Ortiz, Chem. Commun., 2015, 51, 11382–11385 RSC.
- J. Bañuelos, V. Martín, C. F. A. Gómez-Durán, I. J. A. Córdoba, E. Peña-Cabrera, I. García-Moreno, Á. Costela, M. E. Pérez-Ojeda, T. Arbeloa and I. L. Arbeloa, Chem.–Eur. J., 2011, 17, 7261–7270 CrossRef PubMed.
- A. M. Costero, M. L. Betancourt-Mendiola, P. Gaviña, L. E. Ochando, S. Gil, K. Chulvi and E. Peña-Cabrera, Eur. J. Org. Chem., 2017, 2017, 6283–6290 CrossRef CAS.
- T. V. Goud, A. Tutar and J.-F. Biellmann, Tetrahedron, 2006, 62, 5084–5091 CrossRef CAS.
- C. A. Osorio-Martínez, A. Urías-Benavides, C. F. A. Gómez-Durán, J. Bañuelos, I. Esnal, I. López Arbeloa and E. Peña-Cabrera, J. Org. Chem., 2012, 77, 5434–5438 CrossRef PubMed.
- The sudden change in the fluorescence indicates that the change in fluorescence is less likely caused by a chemical reaction (which should take some time), but is the result of the sudden increase in the concentration of the red Bodipy after addition of the solution of the red dye.
- This experiment was also used to correct the observed red fluorescence in the triad experiment for cross-talk intensity.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- S. H. Hong, A. G. Wenzel, T. T. Salguero, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 7961–7968 CrossRef CAS PubMed.
- V. Thiel, K.-J. Wannowius, C. Wolff, C. M. Thiele and H. Plenio, Chem.–Eur. J., 2013, 19, 16403–16414 CrossRef CAS PubMed.
- D. L. Nascimento, M. Foscato, G. Occhipinti, V. R. Jensen and D. E. Fogg, J. Am. Chem. Soc., 2021, 143, 11072–11079 CrossRef CAS PubMed.
- N. Jeddi, N. W. J. Scott and I. J. S. Fairlamb, ACS Catal., 2022, 12, 11615–11638 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization (NMR spectra, UV/Vis spectra, fluorescence spectra, mass spectrometry). See DOI: https://doi.org/10.1039/d2sc04869h |
| ‡ Both authors (Y. S. and S. P.) contribute equally. |
| This journal is © The Royal Society of Chemistry 2023 |